

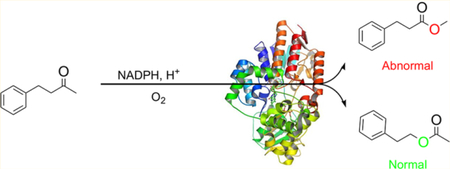
Abstract
Controlling the regioselectivity of Baeyer− Villiger (BV) reactions remains an ongoing issue in organic chemistry, be it by synthetic catalysts or enzymes of the type Baeyer−Villiger monooxygenases (BVMOs). Herein, we address the challenging problem of switching normal to abnormal BVMO regioselectivity by directed evolution using three linear ketones as substrates, which are not structurally biased toward abnormal reactivity. Upon applying iterative saturation mutagenesis at sites lining the binding pocket of the thermostable BVMO from Thermocrispum municipale DSM 44069 (TmCHMO) and using 4-phenyl-2-butanone as substrate, the regioselectivity was reversed from 99:1 (wild-type enzyme in favor of the normal product undergoing 2-phenylethyl migration) to 2:98 in favor of methyl migration when applying the best mutant. This also stands in stark contrast to the respective reaction using the synthetic reagent m-CPBA, which provides solely the normal product. Reversal of regioselectivity was also achieved in the BV reaction of two other linear ketones. Kinetic parameters and melting temperatures revealed that most of the evolved mutants retained catalytic activity, as well as thermostability. In order to shed light on the origin of switched regioselectivity in reactions of 4-phenyl-2-butanone and phenylacetone, extensive QM/MM and MD simulations were performed. It was found that the mutations introduced by directed evolution induce crucial changes in the conformation of the respective Criegee intermediates and transition states in the binding pocket of the enzyme. In mutants that destabilize the normally preferred migration transition state, a reversal of regioselectivity is observed. This conformational control of regioselectivity overrides electronic control, which normally causes preferential migration of the group that is best able to stabilize positive charge. The results can be expected to aid future protein engineering of BVMOs.
Graphical Abstract
INTRODUCTION
The Baeyer−Villiger (BV) reaction, first reported in 1899,1 constitutes a powerful method for converting ketones into the respective esters or lactones by insertion of an O atom into a C−C bond adjacent to the carbonyl function.2 Stoichiometric amounts of alkylhydroperoxides ROOH or peracids RCO3H commonly serve as oxidants, the transformation being catalyzed by acids, bases, transition metal complexes or organocatalysts. When enantioselectivity is involved, as in the desymmetrization of symmetrically substituted cyclic ketones or oxidative kinetic resolution of racemic ketones, chiral transition metal catalysts3 or organocatalysts4 have been used successfully. Mechanistically, oxidants such as ROOH add to the carbonyl function with formation of a short-lived tetrahedral species called the Criegee-intermediate, which then undergoes fragmentation and rearrangement to the product. The control of regioselectivity in the reaction of unsymmetrical ketones leading to two different constitutionally isomeric products continues to be a central synthetic and mechanistic issue (Scheme 1).2
Scheme 1.
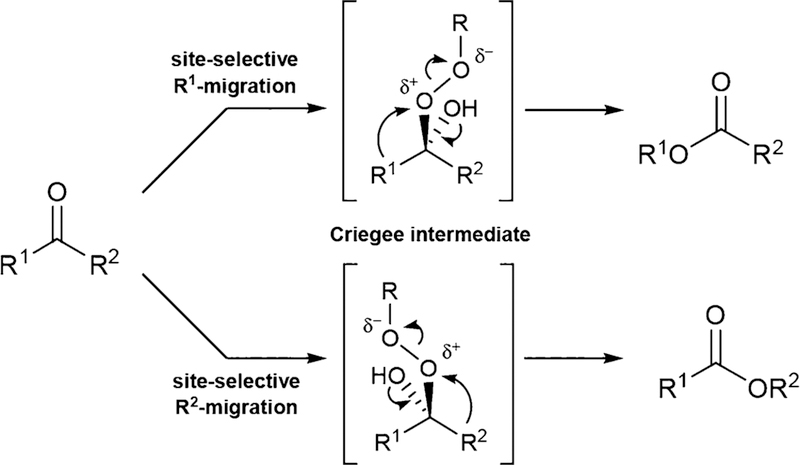
Baeyer−Villiger Reaction of Unsymmetrical Acyclic Ketones with Potential Formation of Two Different Esters
Stereoelectronic effects play a dominant role in many organic transformations,5 BV reactions being a prominent example.2−4 As shown by a number of computational and mechanistic studies, these effects play a crucial role in determining the regioselectivity of BV reactions of unsymmetrical ketones.6 The most generally accepted mechanistic model calls for a conformation in the Criegee-intermediate in which the migrating group is antiperiplanar to the fragmenting peroxy O−O bond. This so-called primary stereoelectronic requirement enables the preferred reaction due to an optimal interaction (overlap) of the C−R σ-bond with the O−O σ*-orbital. Since the O−O is broken heterolytically as shown in Scheme 1, the best migrating groups in unsymmetrical ketones are those that stabilize the partial positive charge best. Therefore, as documented experimentally many times, the following migratory tendency has been established: tert-Bu > Phe ∼ iso-Pr > Et > Me.2 Thus, in reactions of unsymmetrical ketones, regioselectivity and electronic effects are intimately connected. A secondary stereoelectronic effect has also been postulated in some cases, which requires the migrating group to be antiperiplanar to one of the lone electron pairs of the hydroxyl group in the Criegee-intermediate.6 Exceptions to the generally observed migratory aptitude with formation of so-called abnormal products occur whenever structural peculiarities are involved, as for example in the case of strained bior tricylic ketones,2 or certain cyclic α-keto-amides.7 Ketones bearing CF3-groups at the α-position also react anomalously, which was explained by DFT computations.8 In the most recent experimental and computational study, β-ketoesters were subjected to BV reactions using H2O2/BF3, resulting in the formation of stable Criegee intermediates.6i
Biocatalytic BV oxidations catalyzed by Baeyer−Villiger monooxygenases (BVMOs) constitute an alternative to synthetic catalysts.9 Nature likewise adheres to the stereo-electronic effects which govern the migratory aptitude in synthetic BV reactions, but the protein environment in the binding pockets of these enzymes may lead to the formation of abnormal products or product mixtures, depending upon which BVMO is used. Examples of abnormal product formation involve BVMO-catalyzed reactions of terpenones such as cis-dihydrocarvone, carvomenthone and menthone.9 Interestingly, one study found that (+)-trans-dihydrocarvone also leads to the abnormal product, in contrast to the enantiomeric (−)-trans-dihydrocarvone.10 Other examples refer to reactions of certain β-hydroxy- and β-aminoketones.11 Abnormal reactivity was also identified in the biosynthesis of the antibiotic pentalenolactone in which a tricyclic ketone precursor undergoes a BVMO-catalyzed reaction.12 In all of these studies, the reasons for the formation of abnormal products have not been fully elucidated.
Protein engineering based on rational design or directed evolution provides a means to control, inter alia, stereo- and/or regioselectivity. The first example of BVMO directed evolution involved the stereoselective desymmetrization of 4-hydroxy-cyclohexanone, catalyzed by the cyclohexanone monooxyge-nase from Acinetobacter sp. NCIMB 9871 (AcCHMO).13 Later the result was rationalized by QM/MM computations14 using the insights gained by a previous theoretical study concerning the mechanism of the WT as catalyst in the desymmetrization of 4-methylcyclohexanone.15 Other protein engineering studies of BVMOs have been reviewed.16 Whereas the primary focus was on stereoselectivity, efforts aimed at improving or reversing regioselectivity (sometimes called site-selectivity)17 have increased recently. A seminal example concerns the switch from abnormal to normal product in the BV reaction of (+)-trans-dihydrocarvone catalyzed by an evolved CHMO mutant from Arthrobacter sp.18 Earlier it had been shown that the reaction of enantiomeric (−)-trans-dihydrocarvone catalyzed by WT also delivers the normal product.10 It should be mentioned that in the same reaction the use of a synthetic Sn-containing zeolite provides the normal lactone irrespective of the absolute configuration of trans-dihydrocarvone,19 which means that in this particular case the synthetic problem is solved.
Mutations introduced by rational design can also influence regioselectivity in BVMO-catalyzed reactions12,20 although these examples involve strained bi- and tricyclic ketones with special electronic and steric properties. As reported by the Fraaije group, a unique case of a nonbiased ketone substrate concerns the reaction of 2-butanone which was found to result in a mixture of normal product (ethyl acetate) and abnormal product (methyl propanoate) in a ratio of ∼3:1 at low conversion using several different CHMOs.21 With the aim of increasing regioselectivity in favor of the abnormal product, a semirational mutagenesis strategy was applied using Ac-CHMO.22 A moderate shift to the abnormal product was induced, from 26% (WT) to 40% (mutant I491A).
In the present study we addressed the problem of switching normal to abnormal BVMO regioselectivity using three linear ketones, 1, 4, and 7 (Scheme 2). This is particularly Scheme 2. Baeyer−Villiger Oxidation of Model Ketones 1, 4, and 7 Catalyzed by TmCHMO Used in This Study challenging because, unlike the above-mentioned ke-tones,9−11,18 these linear substrates are not structurally biased. The plan was to introduce effective point mutations by directed evolution, flanked by a theoretical study in the quest to unveil the structural and mechanistic reasons for such a switch. Upon testing several WT BVMOs or synthetic reagents in the BV reaction of these ketones, complete or extremely high preference for the normal products 2, 5 and 8, respectively, was observed. We anticipated that laboratory evolution of BVMO mutants was necessary that override the traditional stereoelectronic effect. Experimentally, we applied an unconventional variation of our previously developed semirational directed evolution technique based on combinatorial active-site saturation test (CAST) and iterative saturation mutagenesis (ISM) at sites lining the binding pocket,23 flanked by protein QM/MM computations.24
Scheme 2.
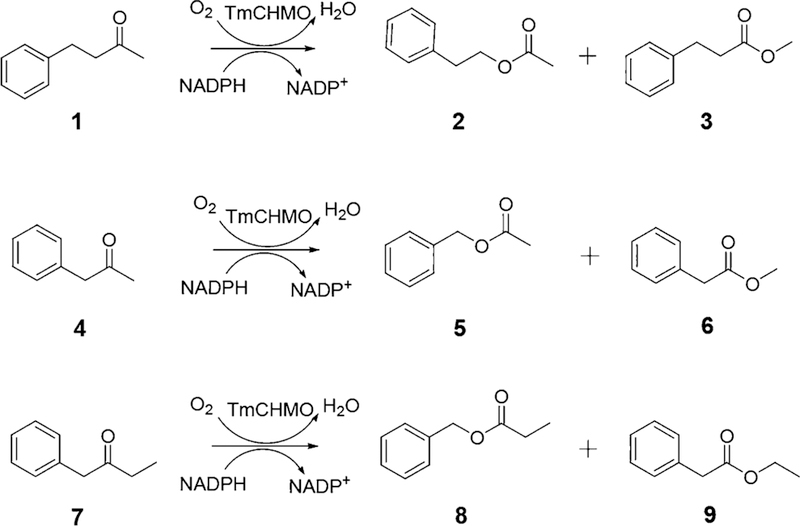
Baeyer−Villiger Oxidation of Model Ketones 1, 4, and 7 Catalyzed by TmCHMO Used in This Study
RESULTS AND DISCUSSION
Directed Evolution Experiments and Characterization of Mutants.
As the model BVMO in this study, we chose the recently discovered thermostable cyclohexanone mono-oxygenase from Thermocrispum municipale DSM 44069 (TmCHMO), because (1) a crystal structure was available, and (2) it had been shown that it displays similar kinetic properties when compared with AcCHMO, i.e., it has sufficient activity at room temperature.25 In an initial directed evolution study of this enzyme, we had already applied CAST/ISM, in that case with the aim of inverting enantioselectivity in the desymmetrization of 4-methylcyclohexanone.26 By first performing NNK-based saturation mutagenesis individually at 11 selected residues lining the binding pocket (L145, L146, F248, F279, R329, F434, T435, N436, L437, W492 and F507) as already reported in our previous study,26 5 hotspots were identified on the basis of the stereoselectivity criterion. Mutant L437A proved to be the best one. This mutant was then employed as the template for NDT-based saturation mutagenesis at the remaining four hotspots in the form of two randomization sites, A (F434/T435) and B (L146/F507) and an ISM scheme leading to reversal of enantioselectivity.26 Anticipating that in the present case reversal of regioselectivity would be more difficult, we developed a different strategy. We first docked substrate 1 into the crystal structure of TmCHMO (Figure 1) and found that all 11 previously selected amino acid positions remain aligned to the binding pocket, i.e., the binding process does not cause major conformational changes of these residues. Therefore, we screened by automated GC the already available original 11 mini-NNK libraries26 in the BV trans-formations of all three ketones 1, 4, and 7 (Scheme 2), but subsequently tested a technique not reported previously. The chemical reactions using m-CPBA as oxidant led to exclusive or nearly exclusive formation of the normal products 2 (>99:1), 5 (>99:1), and 8 (95:5) (Figure S4, S10 and S16).
Figure 1.
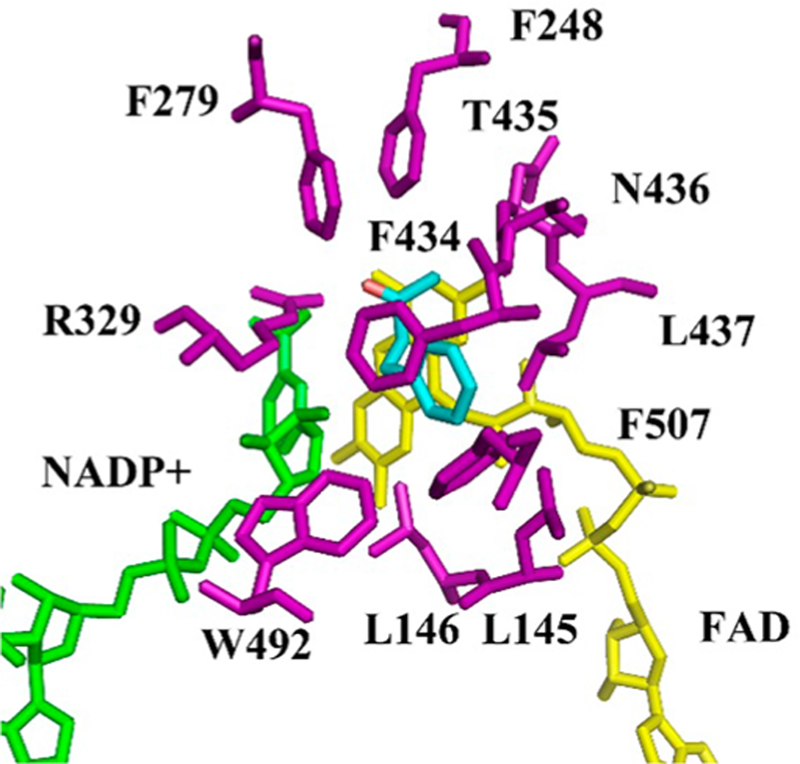
TmCHMO structure model showing docked 4-phenyl-butan-2-one (1) (in cyan) based on the crystal structure of wild-type TmCHMO (PDB code 5M10),25 and the 11 CAST amino acids lining 1 displayed in purple. Green: NADP+; Yellow: FAD.
In the case of ketone 1, several variants were identified by this initial exploration that showed moderately decreased selectivity for the normal product 2, namely L145A, L145G, L145V, L437T and L437A, thus pointing the way toward possible reversal of regioselectivity. The best hit proved to be mutant L145G, which was chosen as a template to visit position L437, leading to two variants with reversed regioselectivity, LGY2-B12 (L145G/L437T, 2:3 = 17:83) and LGY2-D8 (L145G/L437V, 2:3 = 23:77). Thereafter, LGY2-B12 (L145G/L437T) was used as a template to test ISM by considering the remaining 9 residues (L146, F248, F279, R329, F434, T435, N436, W492 and F507), which were grouped into five randomization sites: A (F279, F507), B (L146, N436), C (F248, W492), D (F434, T435) and E (R329). In contrast to our earlier stereoselectivity study,26 this meant that residues were considered that were not hotspots in the initial 11 exploratory NNK-based mutagenesis experiments. In order to minimize the screening effort, NDT codon degeneracy encoding only 12 amino acids (Phe, Leu, Ile, Val, Tyr, His, Asn, Asp, Cys, Arg, Ser and Gly) was chosen for constructing libraries at the 2-residue sites (A, B, C and D). This requires in each case the screening of 4 microtiter plates for 95% coverage assuming the absence of bias, and NNK for the single amino acid library E one plate only. Several mutants were discovered showing significantly enhanced regioselectivity favoring the abnormal product 3 in library D, namely LGY3-D-A9 (L145G/F434I/T435I/L437T, 2:3 = 3:97), LGY3-D-E1 (L145G/F434G/T435F/L437T, 2:3 = 2:98), LGY3-D-B7 (L145G/F434N/T435G/L437T, 2:3 = 8:92) and LGY3-D-E9 (L145G/F434G/T435Y/L437T, 2:3 = 7:93). The libraries created at the other four sites did not harbor any positive variants (Table S1). The best results are summarized in Figure 2a.
Figure 2.
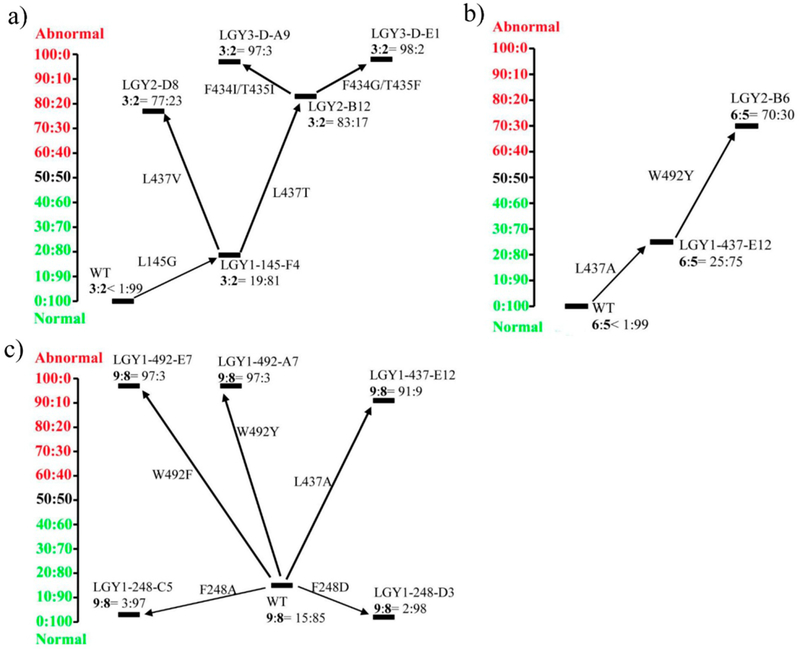
Iterative saturation mutagenesis (ISM) exploration of TmCHMO as catalyst in the reactions with focus on reversal of regioselectivity in favor of the abnormal product using substrate 1 (a), 4 (b), or 7 (c).
An analogous procedure was applied to ketone 4, the best results being summarized in Figure 2b (see also Table S2 for full details). As can be seen, regioselectivity favoring the abnormal product 6 was achieved starting from an initial complete preference by wild-type TmCHMO for the normal product 5 formed by benzyl migration, but the degree of reversal remained at the incomplete stage of 5:6 = 30:70. Nevertheless, in view of the BV reaction using m-CPBA, which occurs with exclusive benzyl migration, this is a synthetically remarkable result.
In the case of ketone 7, WT TmCHMO proved to be selective for the normal product 8 produced by preferential 2-phenylethyl migration, but a small amount of the abnormal product 9 was also formed (8:9 = 85:15). Upon screening the original 11 mini-NNK libraries (see Figure 2c and Table S3 for details), it was possible to improve the initial outcome to nearly exclusive preference for the normal ester (8:9 = 98:2), and also to completely reversed regioselectivity in favor of the abnormal product (8:9 = 3:97). Surprisingly, it required less steps to evolve mutants that allow either benzyl migration or ethyl migration on an optional basis, in contrast to the outcome of the reaction of ketone 4 in which benzyl competes with the smaller methyl group.
Due to the possibility of ester hydrolysis catalyzed by endogenous lipases or esterases in E. coli cells, the BV oxidations of the three substrates 1, 4, and 7 were repeated using purified enzyme of the best TmCHMO mutants. The results turned out to be the same (Table S4 and Figure S1− S19), or even slightly better, e.g., LGY3-D-E1 (2:3 = 2:98), LGY2-B6 (5:6 = 26:74), and LGY1–248-D3 (8:9 = 99:1). A previous report also noticed a possible effect of substrate concentration on the regioselectiviy,27 thus we also tested the WT and the mutant LGY3-D-E1 in the conversion of 1 at 50 mM and 100 mM concentrations. In our system, all reactions retained a high regioselectivity as shown Table S6, WT leading to 2:3 > 99:1 and LGY3-D-E1 leading to 2:3 < 3:97.
To establish whether the mutants are catalytically competent, we next determined the kinetic parameters for the wild-type enzyme and the most promising mutants with the corresponding substrates as shown in Table 1 and Figure S20. Using purified enzymes, we spectrophotometrically measured the rate of oxidation of the NADPH cofactor at varying substrate concentrations. The uncoupling rate, namely the unproductive decay of the peroxyflavin without substrate oxidation, was measured in control reactions without substrate. As revealed in Table 1, the uncoupling rate (kunc) was found to be very low for the WT and most mutant enzymes (0.02−0.10 s−1) except for mutant LGY3-D-A9 (0.20 s−1), prohibiting accurate determination of its kcat and Km values for 1. For the WT enzyme, 4 was found to be the best of the three substrates, with a kcat of 0.73 s−1 and a Km of 0.86 mM. The evolved mutant LGY2-B6 showed a reduced kcat of 0.16 s−1, and with a Km of 1.5 mM also a somewhat lower affinity for the substrate than WT TmCHMO. On the other hand, mutant LGY3-D-E1 illustrated a drastically increased affinity to substrate 1 as compared to WT (kcat = 0.31 s−1, Km = 12 mM versus kcat = 0.16 s−1/Km = 0.02 mM). For substrate 7, the mutant LGY1-492-A7 displayed similar kinetic parameters relative to WT. LGY1–248-D3 had a very low Km (0.02 mM), but also obviously decreased kcat and probably suffers from substrate inhibition. Clearly the best mutant is LGY1–437-E12, showing a slightly lower Km than WT but the same kcat.
Table 1.
Kinetic Parameters of WT TmCHMO and Best Mutants Evolved for the Abnormal BV Product of Three Substrates
| enzyme | substrate | mutations | kunc (s−1) | Km (mM) | kcat (s−1) | Tm (°C) |
|---|---|---|---|---|---|---|
| WT | 1 | − | 0.016± 0.0030 | 12 ± 7.0 | 0.31± 0.11 | 52.67± 0.76 |
| LGY3-D-A9 | 1 | L145G/F434I/T435I/L437T | 0.20 ± 0.019 | n.d. | n.d. | 50.83 ± 0.76 |
| LGY3-D-E1 | 1 | L145G/F434G/T435F/L437T | 0.10 ± 0.0096 | 0.024 ± 0.0041 | 0.16±0.0028 | 51.33 ± 0.76 |
| WT | 4 | − | 0.067± 0.034 | 0.86 ± 0.060 | 0.73± 0.014 | |
| LGY2-B6 | 4 | L437A/W492Y | 0.067± 0.0053 | 1.5± 0.46 | 0.16± 0.014 | 55.33 ± 0.29 |
| WT | 7 | − | 0.015 ± 0.00036 | 4.4± 1.0 | 0.37± 0.033 | |
| LGY1–492-A7 | 7 | W492Y | 0.029± 0.015 | 5.5± 1.7 | 0.18± 0.026 | 53.5 ± 0.5 |
| LGY1–248-D3 | 7 | F248D | 0.026± 0.0034 | 0.020 ± 0.016 | 0.065 ± 0.0048 | 47.83± 2.02 |
| LGY1–437-E12 | 7 | L437A | 0.014± 0.0020 | 2.8± 0.36 | 0.36 ± 0.018 | 55.33± 0.29 |
To probe thermostability, we measured the melting temperatures (Tm) of WT and all the evolved mutants, using a fluorescence-based thermal shift assay (ThermoFAD).28 It was found that the thermostability of most mutants is maintained as compared to WT TmCHMO, except for LGY1–248-D3 showing a 5 °C drop in Tm (Table 1).
We also tested the best mutant LGY3-D-E1 for semipreparative scaled transformation of substrate 1 (50 mg) in 25 mL of reaction volume. Full conversion was achieved within 24 h, while maintaining the originally evolved high regioselectivity as analyzed by GC. After isolation using column chromatography, 46 mg of pure product 3 (83% isolated yield) were obtained (Figure S21−22).
In order to check whether the present ISM strategy is accompanied by nonadditive cooperative effects rather than traditional additive effects,29 we deconvoluted the quadruple mutant LGY3-D-E1 (L145G/F434G/T435F/L437T) and tested the respective four single mutants L145G, F434G, T435F, and L437T separately as catalysts in the BV reaction of ketone 1 (Table S5). This procedure revealed dramatically pronounced cooperative mutational effects, since all four single mutants were found to show very high or even complete normal reactivity with formation of ester 2 (L145G: 2:3 = 81:19; F434G: 2:3 = 99:1; T435F: 2:3 = 99:1; L437T: 2:3 = 97:3), yet together they orchestrate opposite regioselectivity with complete preference for ester 3.
Unraveling the Origin of Regioselectivity by Computational Modeling.
To gain more insights into the regioselectivity switch through evolution, we used DFT computations to model the intrinsic preferences of the reaction and used QM/MM and MD simulations to model the enzymatic reactions.
Using DFT calculations (see SI for computational details), we first studied the BV reaction involving substrates 1, 4, and 7 using m-CPBA as oxidant. It has been previously reported that the rate-limiting and also the regioselectivity-determining step corresponds to the migration step after the formation of the protonated Criegee intermediate,6 thus we focused on analyzing that particular reaction step. Our optimized transition states (TSs) demonstrated that the intrinsically preferred normal migration TS is favored over the abnormal one in substrate 1 (ΔΔG‡ = −3.3 kcal·mol−1), substrate 4 (ΔΔG‡ = −5.2 kcal·mol−1), and 7 (ΔΔG‡ = −1.9 kcal·mol−1), due to the better stabilization of the partial positive charge generated on the migrating carbon atom at the TS. The smallest differences in activation barriers are found for substrate 7 (predicted 8:9 ratio 96:4), which is the experimentally less selective case when m-CPBA is used as oxidant (95:5). On the other hand, the larger energy barrier difference favoring the normal migration is found for ketone 4, the substrate for which protein directed evolution could not achieve a complete inversion of regioselectivity toward abnormal product formation (Figure 2b), presumably because of this intrinsic strong preference toward normal migration of the benzylic group. These results are in excellent agreement with the experimental ratios observed when m-CPBA is used as oxidant. The optimized TS structures also highlight the required antiperiplanar conformation of the migrating group and the peroxy O−O bond (see Figure S23), needed to reach the SN2-like TS.
Previously, Thiel and co-workers studied the BV reaction mechanism catalyzed by a flavin-dependent CHMO using QM/MM calculations.15 They showed that the enzymatic reaction proceeds through the formation of a high energy flavin-Criegee intermediate; the migration step is rate- and regioselectivity-determining. They also showed the importance of an active site arginine in stabilizing the negatively charged Criegee intermediate, although no proton transfer occurs from the protonated arginine (R329 in the current TmCHMO enzyme) to the Criegee intermediate; this is different from the peracid catalyzed mechanism where the protonation of the Criegee intermediate occurs.15 More recently, a similar computational strategy was applied by Huang, Moody and co-workers to study the BV reaction mechanism in the WT Thermobifida fusca phenylacetone monooxygenase (PAMO) toward its natural substrate, phenylacetone, and 2-octanone.30 They showed, in line with Thiel studies, the importance of stabilizing the enzymatic Criegee intermediate and the migration transition state for enzyme activity.
On the basis of this, we hypothesized that the role played by the enzyme in controlling the selectivity of the BV reaction may be due to the imposition of conformational restrictions to the Criegee intermediate, favoring one particular bound conformation that could lead to one migration TS while preventing the other migration from occurring. In principle, one could expect two relevant conformations to be possible, where the corresponding migrating groups should be placed antiperiplanar to the peroxyl O−O bond to reach the corresponding TSs with little further distortion. This hypothesis implies that newly introduced mutations during evolution may alter the conformation adopted by the Criegee intermediate.
Thus, we analyzed the conformations adopted by the critical Criegee intermediate when formed in the enzyme active site in its deprotonated form by using MD simulations. We ran 500 ns MD simulations on the Criegee intermediate bound into the different enzyme variants and monitored the dihedral angles formed by the O(peroxy1)−O(peroxy2)−C(carbonyl)−C- (migrating) atoms during the trajectories. A dihedral angle value close to ±180° indicates that the migration is possible because the considered migrating group is antiperiplanar to the O−O peroxy bond in that bound conformation. We chose the bulkiest ketone 1 for our computational modeling because experimentally it exhibits the largest degree of selectivity (Figure 2a).
When the Criegee intermediate is formed, a new quaternary carbon center is generated. We considered the possibility of forming both R and S stereoisomers when the peroxyflavin addition to ketone 1 occurs. Based on the docking results (see Figure 1) and on MD simulations carried out (see Figure S24), we concluded that only the R-Criegee stereoisomer may be formed in order to keep a less strained conformation of the phenylethyl group and the strong H-bonding interaction with R329 needed for stabilizing the negative charge generated on the former carbonyl oxygen atom. MD simulations for the 1-R-Criegee intermediate bound into the WT BVMO showed that it mainly explores one conformation in which the dihedral angle corresponding to the phenylethyl group migration (dihed1-normal) is ca. 150−180° during the entire simulation time, as shown in Figure 3a. The equivalent dihedral angle for the methyl migration (dihed1-abnormal) is ca. −60 to −100°. On the basis of this, only the phenylethyl migration would be possible since the conformation that the Criegee intermediate adopts in the enzyme active site only allows the phenylethyl group to be antiperiplanar to the peroxyl O−O bond, which is required for the migration to occur. This group is also most effective in stabilizing the positive charge at the peroxy O atom in the transition state, in line with the traditional mechanistic view of normal BV reactions. These results are fully consistent with the exclusive formation of the normal BV product 2 by the WT enzyme (Figure 2a).
Figure 3.
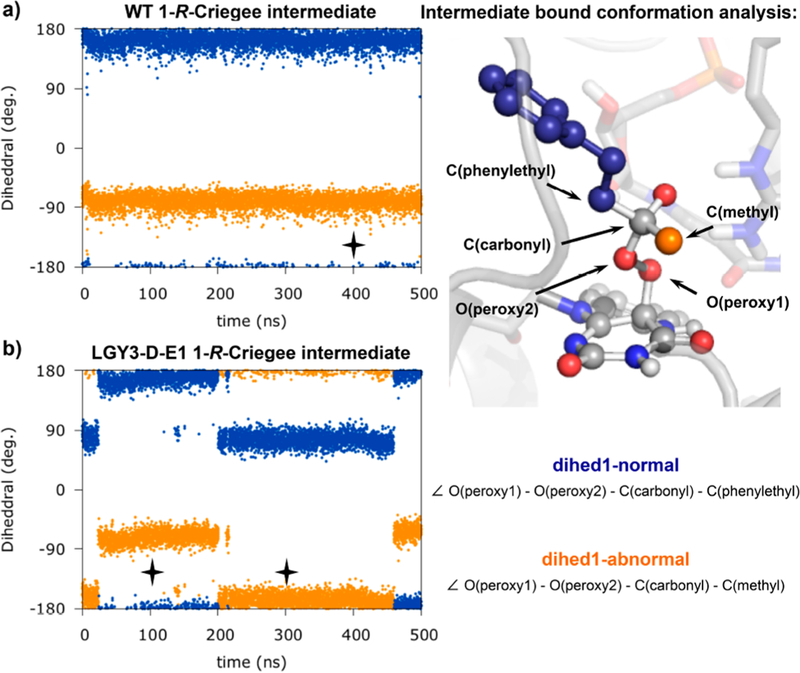
Analysis of 1-R-Criegee intermediate conformations though 500 ns MD simulations when bound in the (a) WT enzyme; and (b) LGY3-D-E1 variant. Black symbols denote selected snapshots used for further analysis in Figure 4.
However, when the LGY3-D-E1 variant is considered, the measured dihedral angles for the phenylethyl migration (dihed1-normal) and methyl migration (dihed1-abnormal) indicate that two interconverting conformations of the Criegee intermediate can be explored (Figure 3b). From these results, one may expect to observe the formation of both possible BV products, although experimentally this variant is highly selective toward abnormal product formation (Figure 2a). In addition, MD simulations on the Criegee intermediate bound into individual single mutants (L145G, F434G, T435F, and L437T, see SI Figures S25 and S26) were carried out. These simulations showed that some of the mutations introduced, when considered individually, also allow the intermediate to explore conformations that may lead to the abnormal migration, although these single mutants exhibit high selectivities toward normal product 2 formation. This indicates that the abnormal product will not be formed just by allowing this conformation to be explored, presumably because the normal migration is intrinsically preferred energetically, as described earlier. Why then, does LGY3-D-E1 give the abnormal product?
To analyze this, we carried out QM/MM calculations (see SI for computational details) to estimate the relative energy barrier heights for these two possible migrations in the LGY3-D-E1 variant. We selected two representative snapshots from the MD trajectory where the two different bound conformations are visited, respectively (see Figure 3b, and Figure 4e,f). QM/MM calculated energy barriers are 17.5 kcal·mol−1 for the methyl migration TS (1-TS-abnormal, snapshot at 300 ns) and 20.8 kcal·mol−1 for the phenylethyl migration TS (1-TS-normal, snapshot at 100 ns). Thus, although a conformation of the Criegee intermediate leading to the normal BV product 2 (i.e., phenylethyl migration) can be visited, the higher in energy 1-TS-normal prevents this pathway and favors instead the formation of the abnormal product 3, through a much lower energy 1-TS-abnormal. The consequence is preferred methyl migration, although in the traditional view of BV reactions this group is least effective in stabilizing the incipient positive charge in the transition state (Scheme 1).
Figure 4.
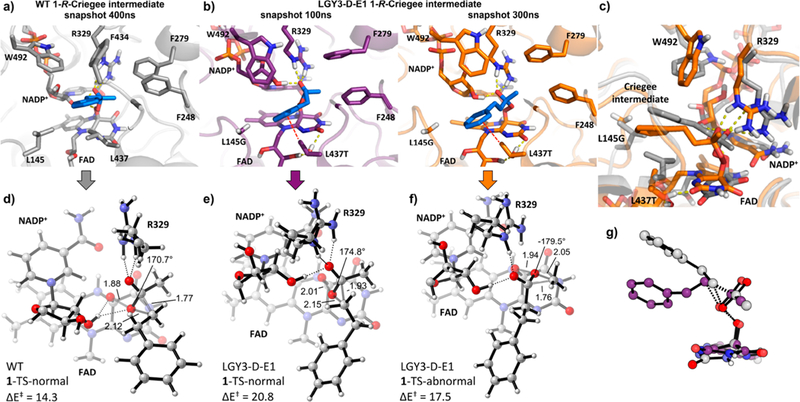
Active site arrangement in selected snapshots obtained from 500 ns MD trajectories of the 1-R-Criegee intermediate bound into the (a) WT enzyme (snapshot at 400 ns in gray); and (b) LGY3-D-E1 variant (snapshots at 100 ns in purple, and 300 ns in orange). The active sites are shown from the same perspective. Substrate 1 in the 1-R-Criegee intermediate is shown in blue. (c) Superimposition of the WT (snapshot at 400 ns in gray) and LGY3-D-E1 (snapshot at 300 ns in orange) 1-R-Criegee intermediate bound active sites. QM/MM optimized transition state geometries (only atoms included in the QM-region are shown) for selected snapshots obtained from (d) WT 1-TS-normal phenylethyl migration (snapshot at 400 ns); (e) LGY3-D-E1 1-TS-normal phenylethyl migration (snapshot at 100 ns); and (f) LGY3-D-E1 1-TS-abnormal methyl migration (snapshot at 300 ns). Energies are given in kcal·mol−1, distances in Å, and O(peroxy1)−O(peroxy2)−C(carbonyl)−C(migrating) dihedral angles in degrees. (g) Superimposition of the 1-R-Criegee intermediate QM/MM optimized 1-TS-normal structures (only Criegee intermediate atoms are shown) in the WT (snapshot at 400 ns in gray) and LGY3-D-E1 (snapshot at 100 ns in purple).
In order to understand the origins of the high energy barrier for the normal product formation in LGY3-D-E1 variant, we analyzed the equivalent phenylethyl migration TS but in the WT enzyme using QM/MM (Figure 4d). In this case, the QM/MM computed energy barrier is only 14.3 kcal·mol−1, being more than 5 kcal·mol−1 lower. When comparing the optimized TS geometries (Criegee intermediate atoms included in the QM region, Figure 4g), it is found that the phenylethyl migrating group in LGY3-D-E1 is more distorted than in the WT TS due to the new conformation that the Criegee intermediate is forced to adopt in LGY3-D-E1. The LGY3-D-E1 1-TS-normal is a late TS as compared to the WT 1-TS-normal and the LGY3-D-E1 1-TS-abnormal (Figure 4d− f), where the main differences arise from the breaking C−C bond distances (1.93 vs 1.77 and 1.76 Å, respectively).
A closer look into the enzyme active site revealed the profound changes cooperatively induced by the set of mutations introduced by directed evolution. As shown in Figure 4a−c, L145G and L437T mutations create more space to accommodate the phenyl ring in this region of the active site pocket. At the same time, the side chain of the newly introduced L437T is H-bonding with the FAD cofactor, displacing it from its original position. The methyl group of L437T side chain can also effectively create new CH-π interactions with the phenyl ring of the Criegee intermediate further stabilizing this conformation. Finally, F434G and T435F mutations allow W492 to move closer to the Criegee intermediate phenyl ring, pushing it down, and forcing it to keep that particular orientation. All these effects increase the strain on the phenylethyl group when it is migrating, resulting in a worse orbital overlap and a destabilization of this normal migration TS. On the other hand, the new conformation that the Criegee intermediate explores, allows the methyl migration to form the abnormal BV product 3. Consequently, evolu-tionary pressure enables the LGY3-D-E1 enzyme to stabilize a bound conformation and a TS that could efficiently lead to the abnormal product formation. And more importantly, the new introduced set of mutations also significantly destabilizes the intrinsically preferred migration pathway by increasing the barrier of the normal migration TS due to cooperative effects of the different mutated residues. These prevent the normal migration TS from occurring, leading to the regioselectivity observed with LGY3-D-E1.
Simulation studies of the reactions of phenylacetone substrate 4 catalyzed by the WT and mutant LGY2-B6 are reported in the SI.
CONCLUSION AND PERSPECTIVES
In summary, we have addressed the challenging issue of reversing the regioselectivity of a Baeyer−Villiger monoox-ygenase as the biocatalyst in reactions of three structurally unbiased linear ketones. The switch involves the preferred formation of the abnormal product resulting from selective group migrations in the respective Criegee intermediates, which are normally disfavored due to stereoelectronic effects. This was accomplished by applying an unconventional strategy characterized by exploratory saturation mutagenesis at 11 sites lining the binding pocket (CAST-type residues), identifying 5 hotspots, and performing iterative saturation mutagenesis (ISM) at the remaining residues which were not hotspots in the thermally stable TmCHMO. Deconvolution of one of the best quadruple mutants with formation of the respective four single mutants uncovered strong cooperative mutational effects, not simply additivity as might be expected.
The experimental results are of synthetic significance because the standard reagent for Baeyer−Villiger oxidations, m-CPBA, was shown to provide the normal products in all three cases. The evolved mutants suffered essentially no trade-off in thermostability and activity as shown by Tm measurements and kinetics, respectively. In terms of mutagenic “hot spots” in TmCHMO, our study reveals, inter alia, that position L437 plays an especially critical role in controlling the regioselectivity in reactions of two of the three model ketones, but other positions are also of distinct importance. Recently a review listing hotspots observed in protein engineering of other BVMOs appeared.31 Adding the present data to this list provides potentially useful information in further directed evolution of BVMOs. In future protein engineering efforts, simply combining some of the known mutations may prove to be a simple and fast way to obtain selective BVMOs with little or no high-throughput screening, provided the respective “rational” choices are guided by structural and theoretical information.
A model for explaining the molecular phenomenon behind the reversal of regioselectivity was constructed by MD simulations and QM/MM computations. The mutations introduced by directed evolution result in changes in the conformation of the respective Criegee intermediates in the binding pocket of the enzyme, which allow the abnormal TS to occur at the same time that largely destabilizes the normal migration TS, thereby setting the stage for selective stereo-electronically controlled fragmentation in favor of abnormal product formation. This novel conformational control leads to the violation of the traditional rule that those migrating groups are preferred that stabilize the incipient positive charge at the peroxy O atom in the Criegee intermediate most effectively.
In conclusion, our work has shown how mutations can cause distortions of a favored transition state such that a previously disfavored transition state becomes predominant. The steric effects that we observed overcame otherwise favorable stereoelectronic effects. Such a novel mechanism of control adds another way that enzymes can control selectivity, and complements the electrostatic factors that Major has shown can control competing pathways in terpene cyclases.32 We hope that the mechanistic lessons learned and the insights gained from our theoretical study will provide a basis for making future protein engineering of BVMOs easier and faster.
Supplementary Material
ACKNOWLEDGMENTS
M.T.R. acknowledges generous support from the Max-Planck-Society and the LOEWE research cluster SynChemBio. K.N.H. acknowledges funding from NSF (CHE-1361104) and financial support from the National Institutes of Health, National Institute for General Medical Sciences and GM-124480. G.L. thanks the Chinese Academy of Agriculture Science for the fund of Elite Youth Program and the Max-Planck-Society for a postdoc stipend. M.G.-B. thanks the Ramón Areces Foundation for a postdoctoral fellowship. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575). The research for the work of M.J.L.J.F. received funding from the European Union (EU) project ROBOX (grant agreement 635734) under EU’s Horizon 2020 Programme Research and Innovation actions H2020-LEIT BIO-2014–1.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Baeyer A; Villiger V Ber. Dtsch. Chem. Ges 1899, 32, 3625–3633. [Google Scholar]
- (2).Reviews of Baeyer−Villiger reactions: (a) Krow GR Org. React 1993, 43, 251–256. [Google Scholar]; (b) Renz M; Meunier B Eur. J. Org. Chem 1999, 4, 737–750. [Google Scholar]; (c) Ten Brink G-J; Arends IWCE; Sheldon RA Chem. Rev 2004, 104, 4105–4124. [DOI] [PubMed] [Google Scholar]; (d) Mihovilovic MD; Rudroff F; Grötzl B Curr. Org. Chem 2004, 8, 1057–1069. [Google Scholar]; (e) Jiménez-Sanchidriań C; Ruiz JR Tetrahedron 2008, 64, 2011–2026. [Google Scholar]; (f) Uyanik M; Ishihara K ACS Catal 2013, 3, 513–520. [Google Scholar]
- (3).Asymmetric BV reactions using synthetic transition metal catalysts: (a) Bolm C; Schlingloff G; Weickhardt K Angew. Chem., Int. Ed. Engl 1994, 33, 1848–1849. [Google Scholar]; (b) Ito K; Ishii A; Kuroda T; Katsuki T Synlett 2003, 643–646. [Google Scholar]; (c) Strukul G Angew. Chem., Int. Ed 1998, 37, 1198–1209. [DOI] [PubMed] [Google Scholar]; (d) Frison J-C; Palazzi C; Bolm C Tetrahedron 2006, 62, 6700–6706. [Google Scholar]; (e) Malkov AV; Friscourt F; Bell M; Swarbrick ME; Kocovsky P J. Org. Chem 2008, 73, 3996–4003. [DOI] [PubMed] [Google Scholar]; (f) Zhou L; Liu X; Ji J; Zhang Y; Hu X; Lin L; Feng XJ Am. Chem. Soc 2012, 134, 17023–17026. [DOI] [PubMed] [Google Scholar]; (g) Zhou L; Liu X; Ji J; Zhang Y; Wu W; Liu Y; Lin L; Feng X Org. Lett 2014, 16, 3938–3941. [DOI] [PubMed] [Google Scholar]; (h) Poudel PP; Arimitsu K; Yamamoto K Chem. Commun 2016, 52, 4163–4166. [DOI] [PubMed] [Google Scholar]; (i) Russo A; De Fusco C; Lattanzi A ChemCatChem 2012, 4, 901–916. [Google Scholar]
- (4).Asymmetric BV reactions using chiral organocatalysts: (a) Murahashi S; Ono S; Imada Y Angew. Chem., Int. Ed 2002, 41, 2366–2370. [DOI] [PubMed] [Google Scholar]; (b) Imada Y; Iida H; Murahashi SI; Naota T Angew. Chem., Int. Ed 2005, 44, 1704–1706. [DOI] [PubMed] [Google Scholar]; (c) Wang B; Shen YM; Shi YJ Org. Chem 2006, 71, 9519–9521. [DOI] [PubMed] [Google Scholar]; (d) Peris G; Miller SJ Org. Lett 2008, 10, 3049–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xu S; Wang Z; Zhang X; Ding K Angew. Chem., Int. Ed 2008, 47, 2840–2843. [DOI] [PubMed] [Google Scholar]; (f) Abascal NC; Miller SJ Org. Lett 2016, 18, 4646–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Yadav VK Steric and Stereoelectronic Effects in Organic Chemistry; Springer, 2016. [Google Scholar]; (b) Alabugin IV Stereoelectronic Effects: A Bridge between Structure and Reactivity; John Wiley & Sons, 2016. [Google Scholar]; (c) Deslongchamps P Stereoelectronic Effects in Organic Chemistry; Pergamon Press: New York, 1983. [Google Scholar]
- (6).QM and/or mechanistic studies of synthetic BV reactions: (a) Grein F; Chen A; Edwards D; Crudden CJ Org. Chem 2006, 71, 861–872. [DOI] [PubMed] [Google Scholar]; (b) Alvarez-Idaboy J; Reyes L; Cruz J Org. Lett 2006, 8, 1763–1765. [DOI] [PubMed] [Google Scholar]; (c) Alvarez-Idaboy J; Reyes L; Mora-Diez N Org. Biomol. Chem 2007, 5, 3682–3689. [DOI] [PubMed] [Google Scholar]; (d) Alvarez-Idaboy J; Reyes LJ Org. Chem 2007, 72, 6580–6583. [DOI] [PubMed] [Google Scholar]; (e) Mora-Diez N; Keller S; lvarez-Idaboy. Org. Biomol. Chem 2009, 7, 3682–3690. [DOI] [PubMed] [Google Scholar]; (f) Sever RR; Root TW J. Phys. Chem. B 2003, 107, 10848–10862. [Google Scholar]; (g) Carlqvist P; Eklund R; Brinck TJ Org. Chem 2001, 66, 1193–1199. [DOI] [PubMed] [Google Scholar]; (h) Geibel I; Dierks A; Müller T; Christoffers J Chem. - Eur. J 2017, 23, 7245–7254. [DOI] [PubMed] [Google Scholar]; (i) Vil V; Dos Passos GG; Bityukov O; Lyssenko K; Nikishin G; Alabugin I; Terent’ev A Angew. Chem., Int. Ed 2018, 57, 3372–3376. [DOI] [PubMed] [Google Scholar]
- (7).Badiola E; Olaizola I; Vázquez A; Vera S; Mielgo A; Palomo C Chem. - Eur. J 2017, 23, 8185–8195. [DOI] [PubMed] [Google Scholar]
- (8).Itoh Y; Yamanaka M; Mikami K Org. Lett 2003, 5, 4803–4806. [DOI] [PubMed] [Google Scholar]
- (9).Reviews of Baeyer−Villiger monooxygenases as catalysts in organic chemistry: (a) Kayser MM Tetrahedron 2009, 65, 947–974. [Google Scholar]; (b) Stewart JD Curr. Org. Chem 1998, 2, 195–216. [Google Scholar]; (c) Mihovilovic MD; Drauz K; Grcģer H; May O Enzyme Catalysis in Organic Synthesis, 3rd ed.; Wiley-VCH: Weinheim, 2012; Vol. I− III, pp 1439–1485. [Google Scholar]; (d) Alphand V; Carrea G; Wohlgemuth R; Furstoss R; Woodley JM Trends Biotechnol 2003, 21, 318–323. [DOI] [PubMed] [Google Scholar]; (e) Mihovilovic MD Curr. Org. Chem 2006, 10, 1265–1287. [Google Scholar]; (f) Wohlgemuth R Eng. Life Sci 2006, 6, 577–583. [Google Scholar]; (g) Torres PazmiÇo DE; Dudek HM; Fraaije MW Curr. Opin. Chem. Biol 2010, 14, 138–144. [DOI] [PubMed] [Google Scholar]; (h) Leisch H; Morley K; Lau P Chem. Rev 2011, 111, 4165–4222. [DOI] [PubMed] [Google Scholar]; (i) Hollmann F; Arends IWCE; Buehler K; Schallmey A; Bühler B Green Chem 2011, 13, 226–265. [Google Scholar]
- (10).Černuchová P; Mihovilovic MD Org. Biomol. Chem 2007, 5, 1715–1719. [DOI] [PubMed] [Google Scholar]
- (11).Rehdorf J; Mihovilovic MD; Bornscheuer UT Angew. Chem., Int. Ed 2010, 49, 4506–4508. [DOI] [PubMed] [Google Scholar]
- (12).Chen K; Wu S; Zhu L; Zhang C; Xiang W; Deng Z; Ikeda H; Cane DE; Zhu D Biochemistry 2016, 55, 6696–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Reetz MT; Brunner B; Schneider T; Schulz F; Clouthier CM; Kayser MM Angew. Chem., Int. Ed 2004, 43, 4075–4078. [DOI] [PubMed] [Google Scholar]
- (14).Polyak I; Reetz MT; Thiel WJ Phys. Chem. B 2013, 117, 4993–5001. [DOI] [PubMed] [Google Scholar]
- (15).Polyak I; Reetz MT; Thiel WJ Am. Chem. Soc 2012, 134, 2732–2741. [DOI] [PubMed] [Google Scholar]
- (16).Zhang ZG; Parra LP; Reetz MT Chem. - Eur. J 2012, 18, 10160–10172. [DOI] [PubMed] [Google Scholar]
- (17).Wang J.-b.; Li G; Reetz MT Chem. Commun 2017, 53, 3916–3928. [DOI] [PubMed] [Google Scholar]
- (18).Balke K; Schmidt S; Genz M; Bornscheuer UT ACS Chem. Biol 2016, 11, 38–43. [DOI] [PubMed] [Google Scholar]
- (19).Corma A; Nemeth LT; Renz M; Valencia S Nature 2001, 412, 423–425. [DOI] [PubMed] [Google Scholar]
- (20).Bocola M; Schulz F; Leca F; Vogel A; Fraaije MW; Reetz MT Adv. Synth. Catal 2005, 347, 979–986. [Google Scholar]
- (21).van Beek HL; Winter RT; Eastham GR; Fraaije MW Chem. Commun 2014, 50, 13034–13036. [DOI] [PubMed] [Google Scholar]
- (22).van Beek HL; Romero E; Fraaije MW ACS Chem. Biol 2017, 12, 291–299. [DOI] [PubMed] [Google Scholar]
- (23).(a) Reetz MT Angew. Chem., Int. Ed 2011, 50, 138–174. [DOI] [PubMed] [Google Scholar]; (b) Reetz MT Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology; Wiley-VCH: Weinheim, 2016. [Google Scholar]
- (24).(a) Kiss G; Çelebi-Ölcü̧m N; Moretti R; Baker D; Houk K Angew. Chem., Int. Ed 2013, 52, 5700–5725. [DOI] [PubMed] [Google Scholar]; (b) Romero-Rivera A; Garcia-Borras̀ M; Osuna S Chem. Commun 2017, 53, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Romero E; Castellanos JRG; Mattevi A; Fraaije MW Angew. Chem., Int. Ed 2016, 55, 15852–15855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sheng D; Ballou DP; Massey V Biochemistry 2001, 40, 11156–11167. [DOI] [PubMed] [Google Scholar]
- (26).Li G; Fürst MJ; Mansouri HR; Ressmann AK; Ilie A; Rudroff F; Mihovilovic MD; Fraaije MW; Reetz MT Org. Biomol. Chem 2017, 15, 9824–9829. [DOI] [PubMed] [Google Scholar]
- (27).Zambianchi F; Pasta P; Ottolina G; Carrea G; Colonna S; Gaggero N; Ward J Tetrahedron: Asymmetry 2000, 11, 3653–3657. [Google Scholar]
- (28).Forneris F; Orru R; Bonivento D; Chiarelli LR; Mattevi A FEBS J 2009, 276, 2833–2840. [DOI] [PubMed] [Google Scholar]
- (29).(a) Reetz MT Angew. Chem., Int. Ed 2013, 52, 2658–2666. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z-G; Lonsdale R; Sanchis J; Reetz MT J. Am. Chem. Soc 2014, 136, 17262–17272. [DOI] [PubMed] [Google Scholar]
- (30).Carvalho AT; Dourado DF; Skvortsov T; de Abreu M; Ferguson LJ; Quinn DJ; Moody TS; Huang M Phys. Chem. Chem. Phys 2017, 19, 26851–26861. [DOI] [PubMed] [Google Scholar]
- (31).Balke K; Beier A; Bornscheuer UT Biotechnol. Adv 2018, 36, 247–263. [DOI] [PubMed] [Google Scholar]
- (32).Major DT ACS Catal 2017, 7, 5461–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.