

Inhibiting the BK channel SLO-1 in the aging nervous system promotes both life span and health span in C. elegans.
Abstract
As animals and humans age, the motor system undergoes a progressive functional decline, leading to frailty. Age-dependent functional deteriorations at neuromuscular junctions (NMJs) contribute to this motor aging. However, it is unclear whether one can intervene in this process to slow motor aging. The Caenorhabditis elegans BK channel SLO-1 dampens synaptic transmission at NMJs by repressing synaptic release from motor neurons. Here, we show that genetic ablation of SLO-1 not only reduces the rate of age-dependent motor activity decline to slow motor aging but also surprisingly extends life span. SLO-1 acts in motor neurons to mediate both functions. Genetic knockdown or pharmacological inhibition of SLO-1 in aged, but not young, worms can slow motor aging and prolong longevity. Our results demonstrate that genetic and pharmacological interventions in the aging motor nervous system can promote both health span and life span.
INTRODUCTION
Aging is characterized by a progressive decline in physiological functions of multiple tissues and organs, leading to an increased probability of death (1). Age-dependent motor function decline occurs in diverse organisms ranging from worms to humans, representing one of the most prominent features of normal aging (1). For example, the nematode Caenorhabditis elegans begin to lose their motor activity in early adult life (2–7). Humans develop deficits in motor functions beginning in mid-life (8, 9). This aging process ultimately leads to frailty, resulting in falling that causes injury and mortality (8, 9). However, it remains a challenge to develop strategies to delay or reduce the rate of motor aging.
C. elegans has been widely used as a genetic model organism for the study of aging (1). Previous efforts have characterized age-dependent motor activity decline in C. elegans. For example, it has been shown that motor neurons at neuromuscular junctions (NMJs) in aging worms undergo functional deterioration beginning in early life (2). Subsequently in mid-life, muscle cells begin to develop functional deficits, culminating in sarcopenia in late life (2, 10). Morphological abnormalities are also detected at NMJs in aged worms (11–13). Acute pharmacological stimulation of synaptic transmission at NMJs in old worms can transiently potentiate motor activity (2). Nevertheless, no pharmacological interventions have been developed to persistently decrease the rate of age-dependent motor activity decline to slow motor aging. It is also unclear whether these interventions, if successful, benefit longevity.
In this study, we sought to identify a molecular target whose function can be manipulated pharmacologically and/or genetically to slow motor aging and possibly promote longevity. SLO-1 is the C. elegans ortholog of the BK channel, a large-conductance, calcium-activated potassium channel that dampens neuronal excitability (14). SLO-1 is known to repress synaptic transmission at NMJs by suppressing motor neuron excitability and thereby blunting synaptic release from these neurons (14). We found that slo-1 mutant worms display a reduced rate of motor activity decline during aging and exhibit a higher motor activity in mid-late life compared to wild-type (WT) animals. slo-1 mutant worms are also long lived. We further showed that SLO-1 acts in motor neurons to regulate both motor aging and longevity. Genetic knockdown or pharmacological inhibition of SLO-1 can both slow motor aging and extend life span in aged, but not young, worms. These results reveal an important role for SLO-1 in aging, demonstrating that genetic and pharmacological interventions in aging motor neurons at NMJs can promote both health span and life span.
RESULTS
slo-1 mutant worms exhibit a slower rate of age-dependent motor activity decline and are long lived
Synaptic release from motor neurons at NMJs is known to undergo a progressive functional decline beginning in early life, which contributes to age-dependent motor activity decline in C. elegans (2). To identify a molecular target whose function can be manipulated to slow this motor aging, we considered the genes that function to dampen synaptic release from motor neurons at NMJs. We reasoned that blunting the function of these “negative regulators” of synaptic transmission might boost synaptic release from aging motor neurons, thereby promoting synaptic transmission at NMJs and increasing motor activity in aged worms. SLO-1 thus came to our attention, as it has been reported to dampen synaptic release from motor neurons at NMJs (14).
We recorded locomotion behavior of WT and slo-1 null mutant worms throughout life span (Fig. 1A and fig. S1). As reported previously (2, 3), WT worms exhibited a progressive decline in locomotion speed, beginning in early life (Fig. 1A). Although the locomotion speed of slo-1 mutant worms was lower than that of WT worms in early life (e.g., day 3 in Fig. 1B), these mutant worms displayed a slower rate of motor activity decline with age (Fig. 1, A and B). Consequently, slo-1 mutant worms maintained a higher motor activity than WT worms in mid-late life, revealing a beneficial role of slo-1 mutation in motor aging (Fig. 1, A and B).
Fig. 1. slo-1 mutant worms exhibit a slower rate of motor activity decline during aging and are long lived.
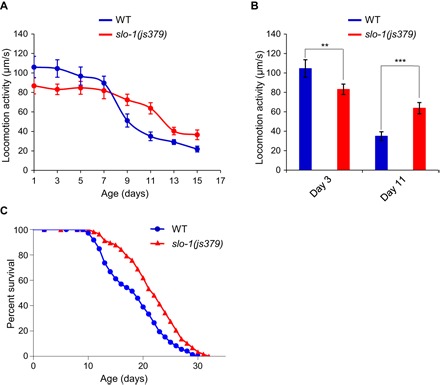
(A and B) slo-1(js379) mutant worms exhibit a slower rate of motor activity decline during aging. (A) slo-1(js379) and WT (N2) worms were analyzed for locomotion behavior every other day, and their locomotion speed was quantified. (B) Data summary for young (day 3) and aged (day 11) worms. Error bars represent SEM. n ≥ 15. **P < 0.005 and ***P < 0.0005 (t test). (C) slo-1(js379) mutant worms are long lived. See table S1 for details.
The observation that slo-1 mutant worms exhibited a slower rate of motor aging prompted us to test their life span. Strikingly, slo-1 mutant worms were long lived (Fig. 1C). We also examined a gain-of-function slo-1 mutant (ky389) and found that this mutant had a very short life span (fig. S2), indicating that the mutant is quite unhealthy. We thus decided to focus on slo-1 null mutant worms for further characterizations. Nonetheless, this gain-of-function phenotype appears to be opposite to that observed with slo-1 null mutant worms. These results demonstrate that loss of SLO-1 not only slows motor aging but also promotes longevity.
SLO-1 acts in motor neurons to regulate both longevity and motor aging
SLO-1 is broadly expressed in neurons and muscles (14). To ascertain in which tissues SLO-1 functions to regulate life span and motor aging, we attempted to rescue the phenotypes of slo-1 mutant worms using cell type–specific promoters. Transgenic expression of slo-1 WT gene in neurons under a pan-neuron promoter rescued the long-lived life-span phenotype of slo-1 mutant worms, while a muscle-specific slo-1 transgene did not (Fig. 2A). Thus, SLO-1 appears to act in neurons to regulate life span.
Fig. 2. SLO-1 acts in motor neurons to regulate both longevity and motor aging.
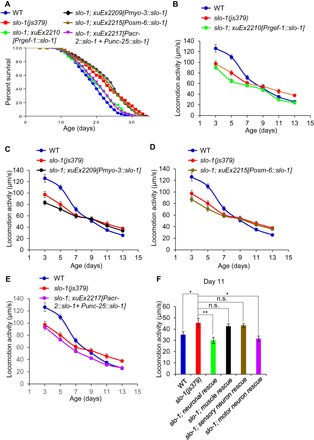
(A) SLO-1 acts in motor neurons to regulate life span. Transgenic expression of slo-1 complementary DNA (cDNA) in neurons using a pan-neuronal promoter (rgef-1) and in motor neurons using a combination of acr-2 and unc-25 promoters rescued the longevity phenotype of slo-1 mutant worms, whereas such expression in muscles (myo-3 promoter) or sensory neurons (osm-6 promoter) did not. See table S1 for details. (B to F) SLO-1 acts in motor neurons to regulate motor aging. Transgenic expression of slo-1 cDNA in all neurons (B) and motor neurons (E), but not in the muscle (C) or sensory neurons (D), rescued the motor aging phenotype of slo-1 mutant worms in mid-late life. (B) to (E) share the same WT and slo-1 traces, which are listed for ease of comparison. (F) Bar graph summarizing the data in (B) to (E) of day 11 animals. Error bars represent SEM. n ≥ 15. *P < 0.05 and **P < 0.005 [analysis of variance (ANOVA) with Dunnett’s test]. n.s., not significant.
Ventral cord motor neurons at NMJs drive locomotion behavior in C. elegans (15). We found that a slo-1 transgene expressed in these motor neurons rescued the longevity phenotype of slo-1 mutant worms (Fig. 2A). As a control, the longevity phenotype was not rescued by a slo-1 transgene expressed in sensory neurons (Fig. 2A). These data demonstrate that SLO-1 can act in motor neurons to regulate longevity.
We then attempted to rescue the locomotion phenotypes associated with slo-1 mutant worms. Similarly, slo-1 mutant worms, which carried a slo-1 rescuing transgene expressed in neurons but not in muscles, no longer exhibited a slower rate of motor activity decay in mid-late life and consequently no longer maintained a higher motor activity in mid-late life, indicating that their slow motor aging phenotype was rescued (Fig. 2, B, C, and F). The same slo-1 neuronal transgene failed to rescue the reduced motor activity phenotype of slo-1 mutant worms in their early life (Fig. 2B). Namely, these transgenic worms still displayed lower motor activity in early life compared to WT worms (Fig. 2B). This suggests that unlike the slow motor aging phenotype, the reduced motor activity phenotype in young slo-1 mutant worms may be caused by a different mechanism. Alternatively, although our rescuing experiments focused on slo-1a, the primary slo-1 isoform, there are a dozen of other alternatively spliced slo-1 transcripts; thus, it is possible that the latter phenotype might be mediated by a different slo-1 isoform(s). Nevertheless, these data indicate that SLO-1 acts in neurons to regulate motor aging in mid-late life. Additional rescuing experiments further showed that a slo-1 transgene expressed in motor neurons, but not in sensory neurons, was sufficient to rescue the slow motor aging phenotype of slo-1 mutant worms in mid-late life (Fig. 2, D to F). Thus, similar to longevity, SLO-1 also acts in motor neurons to regulate motor aging in mid-late life.
Genetic knockdown of slo-1 in aged, but not young, worms slows motor aging and extends life span
As described above, one interesting phenomenon is that, while slo-1 mutant worms exhibited a slower rate of motor aging and maintained a higher motor activity in mid-late life (Fig. 1, A and B), the locomotion speed of these mutant worms was actually lower than that of WT worms in their early life (Fig. 1A). This suggests that genetic ablation of slo-1 may have a detrimental effect on motor activity in early life, although loss of slo-1 in mid-late life may be beneficial, pointing to a model that the role of SLO-1 in motor aging may be stage dependent. This model is consistent with our rescuing data showing that the slo-1 transgenes, which rescued the slower motor aging phenotype in mid-late life, were unable to rescue the lower motor activity phenotype observed in slo-1 mutant worms in early life (Fig. 2, B, E, and F). To further test this model, we knocked down slo-1 by RNA interference (RNAi) in WT worms at different ages (Fig. 3, A to D). As SLO-1 acts in neurons to regulate life span and motor aging, we performed RNAi experiments using a neuron-specific RNAi strain (16). RNAi of slo-1 in young worms beginning at day 1 or day 3 failed to increase their motor activity in mid-late life (Fig. 3, A and B). By contrast, RNAi of slo-1 at a later stage starting from day 5 or day 7 decreased the rate of motor activity decline (Fig. 3C), and consequently, these worms maintained a higher motor activity in mid-late life (Fig. 3D). These data suggest that loss of slo-1 in mid-late, but not early, life benefits the motor system.
Fig. 3. Genetic knockdown of slo-1 in aged, but not young, worms slows motor aging and extends life span.
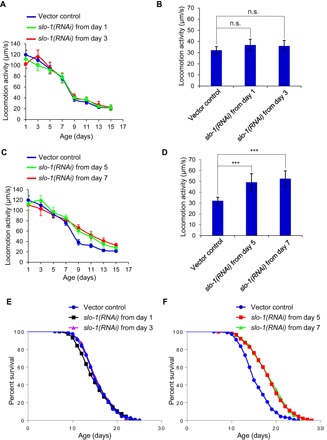
(A to D) RNAi of slo-1 in young worms has no notable effect on motor aging (A and B), whereas this treatment in aged worms reduces the rate of motor activity decline in mid-late life (C and D). WT worms were fed bacteria expressing RNAi against slo-1 gene beginning at days 1 and 3 (A) or days 5 and 7 (C). (A) and (B) as well as (C) and (D) share the same vector control. (B) and (D) summarize the data in (A) and (C) for aged (day 11) animals, respectively. Error bars represent SEM. n ≥ 15. ***P < 0.0005 (ANOVA with Dunnett’s test). (E and F) slo-1 in young worms has no notable effect on their life span (E), whereas this treatment in aged worms extends life span (F). (E) and (F) share the same WT trace. RNAi was performed on TU3401, a neuron-specific RNAi strain (16). See table S1 for details.
We then assayed life span by repeating the same RNAi knockdown experiment on WT worms. Similarly, RNAi of slo-1 in young worms beginning at day 1 or day 3 did not elicit an effect on life span (Fig. 3E), while treating worms with RNAi starting from a later age (e.g., days 5 and 7) extended life span (Fig. 3F). Thus, genetic knockdown of slo-1 in aged, but not young, worms not only slows motor aging but also promotes longevity.
SLO-1 regulation of motor aging and longevity requires DAF-16
As aging pathways tend to converge on a handful of transcription factors (1), we went on to search for a transcription factor, if present, that mediates SLO-1’s function in longevity and motor aging. DAF-16, a FOXO transcription factor, is a master regulator of longevity (17, 18). Loss of the daf-16 gene suppressed the long-lived phenotype of slo-1 mutant worms (Fig. 4A), indicating that DAF-16 is required for SLO-1 to regulate life span. We checked sod-3, a well-characterized DAF-16 target gene, and found that it was up-regulated in slo-1 mutant worms (fig. S3, A to C), consistent with a role for DAF-16 in acting downstream to mediate the effect of SLO-1. DAF-16 is best known to be regulated by insulin/insulin-like growth factor 1 (IGF-1) signaling (IIS). We thus asked whether there is a potential genetic interaction between SLO-1 and IIS. Loss of slo-1 can further extend the life span of daf-2 and age-1 mutant worms (fig. S4, A and B). daf-2 and age-1 encode the worm ortholog of insulin/IGF-1–like receptor and phosphatidylinositol 3-kinase, respectively, the two key players in IIS. This suggests that SLO-1 and IIS act independently to regulate DAF-16. IIS regulates DAF-16 by inhibiting its translocation to the nucleus (19, 20), while some other longevity pathways regulate DAF-16 function within the nucleus without affecting its nuclear translocation (21–23). While daf-2 deficiency promoted nuclear translocation of DAF-16 (fig. S3, F and G), no such nuclear translocation was detected in slo-1 mutant worms (fig. S3, D to G), consistent with the notion that SLO-1 and IIS act independently to regulate DAF-16. These results reveal an essential role for DAF-16 in mediating the function of SLO-1 in longevity.
Fig. 4. SLO-1 regulation of life span and motor aging requires the FOXO transcription factor DAF-16.
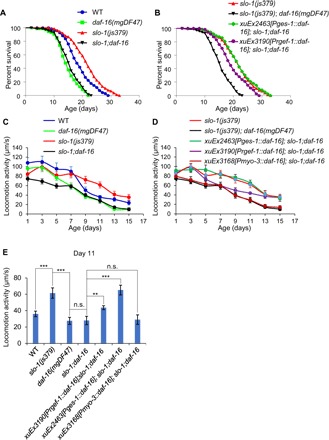
(A) SLO-1 regulation of life span requires DAF-16. Loss of daf-16 blocked the long-lived phenotype of slo-1 mutant worms. (B) Transgenic expression of daf-16 cDNA in the intestine using the ges-1 promoter can fully rescue the daf-16 phenotype, whereas a neuron-specific transgene (rgef-1 promoter) only has a partial effect. No rescuing effect was observed with a muscle transgene (see fig. S5). (A) and (B) share the same slo-1 and slo-1;daf-16 curves. See table S1 for details. (C to E) SLO-1 regulation of motor aging requires DAF-16. Loss of daf-16 blocked the higher motor activity phenotype observed in aged slo-1 mutant worms (C), and this phenotype can be rescued by an intestinal daf-16 transgene (ges-1 promoter), only partially by a neuronal transgene (rgef-1 promoter), but not by a muscle transgene (myo-3 promoter) (D). (C) and (D) share the same slo-1 and slo-1;daf-16 curves. (E) Bar graph summarizing the data in (C) and (D) for aged (day 11) worms. Error bars represent SEM. n ≥ 15. **P < 0.005 and ***P < 0.0005 (ANOVA with Dunnett’s test).
DAF-16 has been shown to act in both the intestine and neurons to regulate life span (24). To determine in which tissues DAF-16 functions to mediate the role of SLO-1 in life-span control, we set out to rescue the daf-16 mutant phenotype by expressing WT daf-16 cDNA as a transgene in the intestine or neurons using tissue-specific promoters (Fig. 4, A and B). An intestine-specific daf-16 transgene fully rescued the daf-16 mutant phenotype (Fig. 4B), while a neuron-specific daf-16 transgene only had a partial effect (Fig. 4B). We also examined the muscle, but found that transgenic expression of daf-16 cDNA in the muscle showed no rescuing effect (fig. S5). Thus, while DAF-16 can act in both the intestine and neurons, it primarily acts in the intestine to mediate the function of SLO-1 in life-span regulation.
We then assessed the role of DAF-16 in motor aging. Mutations in daf-16 suppressed the higher motor activity phenotype observed in aged slo-1 mutant worms in mid-late life (Fig. 4, C and E). Namely, the motor activity of slo-1;daf-16 double mutant in mid-late life was indistinguishable from that of daf-16 single mutant (Fig. 4, C and E). This observation indicates that DAF-16 is required for SLO-1 to regulate motor aging in mid-late life. Notably, the motor activity of the double mutant was lower than that of slo-1 and daf-16 single mutants in early life (Fig. 4C), indicating an additive effect. Thus, SLO-1 and DAF-16 may function in separate pathways in early life, consistent with our model that SLO-1 acts in mid-late life, but not in early life, to regulate motor aging. We further rescued the daf-16 phenotype using tissue-specific transgenes and found that transgenic expression of daf-16 WT gene in the intestine of slo-1;daf-16 double mutant can fully restore the higher motor activity phenotype observed in slo-1 single mutant worms (Fig. 4, D and E), while a neuron-specific transgene only had a partial effect and a muscle-specific transgene displayed no effect (Fig. 4, D and E). Thus, while DAF-16 can function in both the intestine and neurons, it primarily acts in the intestine to mediate the function of SLO-1 in motor aging, a phenomenon similar to that observed in life-span experiments. This set of data identifies DAF-16 as a key transcription factor that mediates SLO-1’s function in both motor aging and longevity.
Pharmacological blockade of SLO-1 in aged, but not young, worms slows motor aging and extends life span
Encouraged by our data from genetic studies, we wondered if pharmacological blockade of SLO-1 would also be able to slow motor aging and extend life span. We tested paxilline, a potent BK channel blocker (25). Paxilline (10 nM) treatment of young worms beginning at days 1 and 3 did not have a notable effect on life span (Fig. 5A). Treating worms with paxilline (10 nM) starting at a later age (days 5 and 7) prolonged life span (Fig. 5B). It is possible that when paxilline was applied in early life (e.g., day 1), worms were exposed to paxilline for a longer period and thus might accumulate more paxilline in their body, which might potentially exert a negative effect on life span. This possibility is unlikely, as lower concentrations of paxilline (e.g., 1 and 5 nM) also lacked a life span–extending effect when applied to day 1 worms (fig. S6A), whereas treating worms at a later stage (day 5) with much higher concentrations of paxilline (e.g., 100 and 1000 nM) can still extend life span (fig. S6B). The life span–extending effect of paxilline depended on SLO-1, as no such effect was detected in slo-1 mutant worms (Fig. 5C), consistent with the view that paxilline is a BK channel blocker. These results indicate that pharmacological blockade of SLO-1 in aged, but not young, worms promotes longevity.
Fig. 5. Pharmacological blockade of SLO-1 in aged, but not young, worms extends life span and slows motor aging.
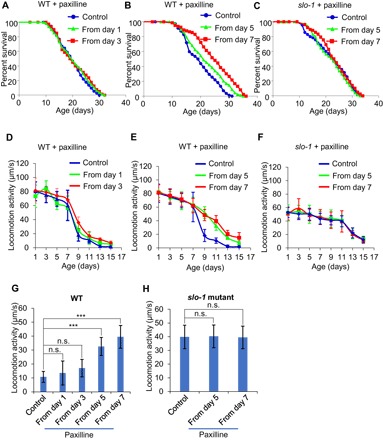
(A and B) Paxilline treatment of aged, but not young, worms extends life span. (A) Paxilline (10 nM) treatment from early age (days 1 and 3) did not extend life span (P < 0.348, log-rank test). (B) Paxilline (10 nM) treatment from aged worms (days 5 and 7) extended life span. See table S1 for statistics. (C) Paxilline extension of life span depends on SLO-1. No life-span extension by paxilline was detected in slo-1 mutant worms. See table S1 for details. (D to F) Paxilline treatment of aged, but not young, worms improves motor activity in mid-late life, which depends on SLO-1. (D) Paxilline treatment (10 nM) from early life (days 1 and 3) had no notable effect on age-dependent motor activity decline. (E) Paxilline (10 nM) treatment in aged worms (starting from days 5 and 7) reduced the rate of age-dependent motor activity decline and improved motor activity in mid-late life. (F) The effect of paxilline on motor aging depended on SLO-1, as no effect was detected in slo-1 mutant worms. (D) and (E) share the same WT curve. Error bars represent SEM. (G and H) Bar graphs in (G) and (H) summarizing the data in (D) and (E), and (F) of aged (day 11) WT and slo-1 mutant worms, respectively. Error bars represent SEM. n ≥ 15. ***P < 0.0005 (ANOVA with Dunnett’s test).
We then tested the effect of paxilline on motor aging. While paxilline (10 nM) treatment of young worms beginning at days 1 and 3 did not affect age-dependent motor activity decline (Fig. 5, D and G), treating worms at a later age starting from days 5 and 7 reduced the rate of motor activity decline, and these worms maintained a higher motor activity in mid-late life (Fig. 5, E and G). As was the case with life-span experiments, lower concentrations of paxilline (e.g., 1 nM), when applied to day 1 worms, did not reduce the rate of motor activity decline, whereas treating older worms (day 5) with much higher concentrations of paxilline (e.g., 1000 nM) was still able to slow motor aging (fig. S6C), consistent with the notion that paxilline is more effective on aged worms. The effect of paxilline on motor aging relied on SLO-1, as this effect was absent in slo-1 mutant worms (Fig. 5, F and H). This supports the view that paxilline is a BK channel blocker (25). Thus, similar to the case with genetic knockdown, pharmacological inhibition of SLO-1 in aged, but not young, worms can also promote longevity and slow motor aging.
Genetic ablation of SLO-1 slows the functional aging of motor neurons at NMJs
To obtain further evidence, we recorded synaptic transmission at NMJs in the ventral nerve cord by patch clamp. NMJs in this area are formed between body wall muscles and ventral cord motor neurons, which drive locomotion behavior (15). We quantified both the frequency and amplitude of endogenous postsynaptic currents (PSCs). The frequency of PSCs measures the frequency of neurotransmitter release from presynaptic motor neurons and thus reports the functional status of these neurons (2, 26). A decrease in this parameter would indicate a functional decline in these motor neurons (2, 26). On the other hand, the amplitude of PSCs is determined by both postsynaptic muscle cells and presynaptic motor neurons (2, 26). We found that the frequency of PSCs at NMJs was markedly reduced in aged worms (day 9) compared to young worms (day 3) (Fig. 6, A and B), whereas the amplitude of PSCs in these two groups of worms was similar to each other (Fig. 6, A and C). This result demonstrates that motor neurons already underwent a functional decline in day 9 worms, while muscle cell receptors were functionally normal at this age, consistent with our previous data (2). We did not record later stages of worms, as muscle cells display functional deficits in older worms (2), and it also becomes more technically challenging to record them then. Neither the frequency nor the amplitude of PSCs in slo-1 mutant worms showed a significant decrease in aged (day 9) worms compared to young worms (day 3) (Fig. 6, D to F). These data indicate that genetic ablation of slo-1 greatly reduced the rate of functional aging in motor neurons at NMJs, revealing a potential mechanism by which SLO-1 modulates aging.
Fig. 6. Pharmacological inhibition of SLO-1 in aged worms promotes synaptic release from motor neurons at NMJs.
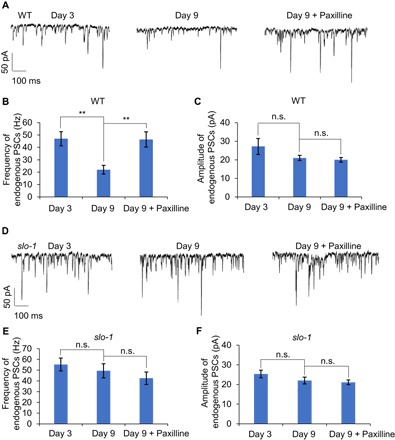
(A) Synaptic release from motor neurons at NMJs is greatly reduced in aged worms and paxilline inhibition of SLO-1 can promote such release in aged worms. Sample traces of endogenous PSCs recorded from NMJs at the ventral nerve cord in young (day 3) and aged (day 9) worms with or without paxilline treatment (10 nM). Worms were treated with paxilline beginning at day 7, and their NMJs were recorded at day 9. Membrane voltage was clamped at −60 mV during recording. (B and C) Bar graphs summarizing the data in (A). (B) Frequency of endogenous PSCs. (C) Amplitude of endogenous PSCs. Error bars represent SEM. n ≥ 7. **P < 0.005 (ANOVA with Dunnett’s test). (D) Motor neurons show robust synaptic release at NMJs in aged slo-1 mutant worms, and paxilline treatment is unable to promote synaptic release from motor neurons in these mutant worms. Sample traces of endogenous PSCs recorded from NMJs at the ventral nerve cord in young (day 3) and aged (day 9) slo-1 worms with or without paxilline treatment (10 nM). Membrane voltage was clamped at −60 mV during recording. (E and F) Bar graphs summarizing the data in (D). (E) Frequency of endogenous PSCs. (F) Amplitude of endogenous PSCs. Error bars represent SEM. n ≥ 7.
Pharmacological blockade of SLO-1 promotes synaptic release from motor neurons at NMJs in aged worms
Last, we examined the effect of paxilline treatment on synaptic transmission at NMJs in aged worms, given that pharmacological inhibition of SLO-1 by paxilline slows motor aging and extends life span. We found that the frequency of PSCs in aged worms treated with paxilline was much higher than that in untreated worms (Fig. 6, A and B). In contrast, the amplitude of PSCs was unaltered by paxilline in these worms (Fig. 6, A and C). Thus, paxilline treatment greatly enhanced synaptic release from motor neurons while having no notable effect on postsynaptic muscle cell receptors. As a control, paxilline treatment had no effect on the frequency or amplitude of PSCs at NMJs in slo-1 mutant worms (Fig. 6, D to F). Thus, the effect of paxilline depends on SLO-1, consistent with the view that paxilline targets SLO-1 (25). These results demonstrate that pharmacological blockade of SLO-1 can promote synaptic release from motor neurons at NMJs in aged worms, suggesting a potential mechanism underlying paxilline-induced beneficial effects on aging.
DISCUSSION
Age-dependent motor activity decline is a prominent feature of normal aging (1). Motor deficits represent one of the main risk factors for falling in elderly humans, which leads to injury and mortality (8, 9). It would be beneficial to delay or slow motor aging to improve the quality of life and, ideally, to extend life span. This, however, has remained a challenge. In the present study, using C. elegans as a model, we showed that genetic and pharmacological targeting of SLO-1 in aging motor neurons at NMJs can slow motor aging and promote longevity.
We found that SLO-1 acts in motor neurons to mediate its function in aging. SLO-1 dampens synaptic release from motor neurons at NMJs (14). As genetic knockdown and pharmacological blockade of SLO-1 in aged worms slow motor aging and extend life span, these results suggest that the reduced synaptic release from aged motor neurons at NMJs may contribute to the observed motor deficits in mid-late life. This set of data identifies synaptic transmission at NMJs in aging worms as a potential site for intervention to promote health span and life span. Our results also raise the possibility that other neuronal proteins with a function similar to SLO-1 in synaptic transmission at NMJs may also be targeted genetically and pharmacologically to slow motor aging and extend life span.
One interesting observation is that blunting the function of SLO-1 in aged, but not young, worms slows motor aging and extends life span. In addition, slo-1 mutant worms, although maintaining a higher motor activity in mid-late life, have a lower motor activity than WT worms in early life. These observations suggest that SLO-1 may play an important role in motor functions in early life. In this case, blunting its activity in early life would not be beneficial for motor functions or life span. The transgenes, which rescue the motor aging phenotype observed in mid-late life of slo-1 mutant worms, are unable to rescue the lower motor activity phenotype in their early life, indicating that the functions of SLO-1 in the motor system in early and mid-late life are likely to be mediated by distinct mechanisms. These findings reveal a complex role of SLO-1 and perhaps NMJs in motor aging and longevity, underscoring that timing is an important factor for consideration when designing strategies to modulate aging. Future work is needed to delineate the detailed mechanisms underlying the differential roles of SLO-1 in aging in early versus mid-late life. In summary, our results identify a molecular target (i.e., SLO-1), a potential site (i.e., motor neurons at NMJs), and timing (i.e., aged, but not young, worms) for genetic and pharmacological interventions to influence motor aging and life span.
We found that SLO-1 regulation of motor aging and life span requires the FOXO transcription factor DAF-16, a master regulator of aging. While SLO-1 acts in motor neurons, DAF-16 functions in both the intestine and neurons and primarily in the intestine, revealing a cell nonautonomous mechanism. Currently, it is not clear exactly how SLO-1 in motor neurons signals DAF-16 in the intestine cell nonautonomously. Presumably, it may do so through intercellular signaling molecules. These brain-gut communications have recently been reported to underlie a number of longevity pathways (27–30). We found that loss of slo-1 cannot extend the life span of those neurotransmission mutants that fail to secrete signaling molecules (e.g., unc-31 and unc-13 mutants; fig. S7, A and B). Future studies may help to elucidate the detailed mechanisms.
BK channels are evolutionarily conserved (31). Upon activation, mammalian BK channels also dampen neuronal excitability and synaptic transmission (31). Mammalian BK channels may become overly activated under pathophysiological conditions (32, 33). For example, BK channel overactivation leads to depression of synaptic transmission in the early stage of Alzheimer’s disease (AD) in a mouse model and has thus been suggested to contribute to the progression of AD (34). Notably, such a symptom can be mitigated by BK channel blockers such as paxilline (34). Reactive oxygen species (ROS) can potentiate BK channel activity in mice (32, 33). As ROS accumulate during aging, redox modulation of BK channels has been suggested as a potential mechanism underlying age-related loss of brain functions in mice (32, 33). Similar to slo-1 mutant worms, BK channel knockout mice are viable and display mild phenotypes in motor functions at young ages (35, 36); nevertheless, older knockout mice were not examined for motor functions, nor was their life span. The fact that BK channels play a modulatory rather than essential role in neuronal excitability and synaptic transmission offers an advantage for targeting these channels for potential therapeutic interventions. Our results would encourage researchers to examine the role of BK channels in aging in mammals.
MATERIALS AND METHODS
Strains and molecular genetics
WT: N2. TQ6025: slo-1(js379) 6x outcrossed. TQ9085: slo-1(ky389). TQ6361: xuEx2210[Prgef-1::slo-1::SL2::CFP]; slo-1(js379). TQ6360: xuEx2209[Pmyo-3::slo-1::SL2::CFP]; slo-1(js379). TQ6366: xuEx2215[Posm-6::slo-1::SL2::CFP]; slo-1(js379). TQ6368: xuEx2217[Pacr-2::slo-1::sl2::CFP+Punc-25::slo-1::SL2::CFP]; slo-1(js379). TU3401: uIs69 [pCFJ90 (myo-2p::mCherry) + unc-119p::sid-1]; sid-1(pk3321). TQ1654: daf-16(mgDF47). TQ8062: slo-1(js379); daf-16(mgDF47). TQ8063: xuEx2463[Pges-1::daf-16::sl2::mCherry]; slo-1(js379); daf-16(mgDF47). TQ8673: xuEx3190[Prgef-1::daf-16::sl2::mCherry]; slo-1(js379); daf-16(mgDF47). TQ8888: xuEx3168[Pmyo-3::daf-16::sl2::mCherry]; slo-1(js379); daf-16(mgDF47). TQ2620C: slo-1(js379);muIS84[Psod-3::gfp]. TJ356: zIs356[daf-16::gfp + rol-6]. TQ2617C: slo-1(js379); zIs356[daf-16::gfp + rol-6]. TQ6697: age-1(hx546). TQ9084: age-1(hx546); slo-1(js379). TQ2548C: daf-2(e1368). TQ2615C: daf-2(e1368); slo-1(js379). TQ2178: unc-13(e51). TQ2622C: unc-13(e51); slo-1(js379). TQ1280: unc-31(e169). TQ2616C: unc-31(e169); slo-1(js379).
For the experiments involving transgenes, the plasmid DNA was injected at a concentration of 50 ng/μl. Three independent transgenic lines were tested to confirm the results. slo-1a cDNA was cloned by reverse transcription polymerase chain reaction from total RNA isolated from WT (N2) worms. The expression of the transgene was verified by cyan fluorescent protein or mCherry expression, which is driven by SL2 from the same transcript.
Behavioral assay
Worms were cultured on NGM (nematode growth medium) plates. Locomotion assays were performed every other day throughout life span at 20°C under a relative humidity of ~35% using a WormLab system (MBF Bioscience), as described previously (37). A thin layer of freshly grown OP50 bacteria was spread on assay plates 10 min before recording. Worms were tracked every other day throughout life span. Images were recorded under a Nikon 60-mm micro lens (Nikon Inc.) and an AVT Stingray F-504B digital camera (Allied Vision Technologies) for 10 min. For consistency, the last 3.5 min of video clips were used for analyzing locomotion speed using software from WormLab (MBF Bioscience). Specifically, to quantify the mean locomotion speed, the travel distance of the centroid of the worm (in micrometers) was divided by the entire duration of the recorded video clip (in seconds). To test the effect of paxilline (catalog no. 35417, Sigma-Aldrich) on locomotion, paxilline was spread on NGM plates 1 day before the experiment. The working concentration of paxilline was 10 nM unless specified otherwise.
For the experiments involving RNAi, empty vector L4440 and RNAi plasmids were transformed into OP50(xu363), an RNAi-compatible OP50 bacterial strain (38). As described previously (38), RNAi bacteria were cultured for 24 hours at 37°C in LB with carbenicillin (100 μg/ml) by picking freshly streaked single colonies and then diluted into fresh LB [with carbenicillin (100 μg/ml)] and cultured for a few hours to reach the OD600 (optical density at 600 nm) value of 0.5 to 0.6. Then, isopropyl-β-D-thiogalactopyranoside (IPTG) (200 μM) was added to the culture to grow for four more hours, followed by seeding on NGM plates containing carbenicillin (25 μg/ml) and 1 mM IPTG 2 days before experiment.
Life-span assay
Life-span experiments were performed at 20°C, as previously described (27, 39). Worms were age synchronized by picking L4 hermaphrodites, and the next day was scored as day 1. Each worm was scored for viability every day, and the worms that crawled off the plate, exploded, or bagged were censored at the time of the event. The life-span data were processed using the Prism 7 and SPSS Statistics software. P values were calculated with the log-rank (Kaplan-Meier) method and Cox proportional hazard regression, as indicated. RNAi experiments were done using OP50(xu363), an RNAi-compatible OP50 bacterial strain, as described above and previously (38). The detailed life-span data were described in table S1.
Electrophysiology
Whole-cell patch-clamp recordings were carried out as described previously on an upright microscope (BX51WI) with an EPC 10 amplifier (2, 40). Worms were immobilized and dissected on a Sylgard-coated coverglass to expose muscles. Bath solution contained 145 mM NaCl, 2.5 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 20 mM glucose, and 10 mM Hepes (320 mOsm; pH adjusted to 7.3). Pipette solution contained 115 mM KCl, 15 mM KOH, 1 mM MgCl2, 10 mM Hepes, 0.25 mM CaCl2, 20 mM sucrose, 5 mM EGTA, 5 mM Na2ATP, and 0.5 mM NaGTP (315 mOsm; pH adjusted to 7.2). Series resistance and membrane capacitance were both compensated during recording. Voltage was clamped at −60 mV.
Supplementary Material
Acknowledgments
We thank L. Ronan, Z. Li, W. Zhang, Q. Zhang, and J. Snedeker for technical assistance. Some strains were obtained from the CGC. Funding: This work was supported by the NSFC (31420103909, 81720108031, and 81872945 to Jianfeng Liu), the Program of Introducing Talents of Discipline to the Universities from the Ministry of Education (B08029 to Jianfeng Liu), the Ministry of Science and Technology of China (2018YFA0507003 to Jianfeng Liu), NIA (A.-L.H. and X.Z.S.X.), and NIGMS (X.Z.S.X.). Author contributions: G.L. and J.G. performed most of the experiments and analyzed the data. Jie Liu performed electrophysiological experiments and analyzed the data. Jinzhi Liu and H.L. assisted J.G. in performing some experiments and analyzed the data. A.-L.H. contributed reagents and helped in writing the paper. G.L., J.G., Jianfeng Liu, and X.Z.S.X. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau5041/DC1
Fig. S1. slo-1 mutant worms exhibit slower locomotor activity decline throughout life span.
Fig. S2. slo-1 gain-of-function mutation greatly shortens life span.
Fig. S3. DAF-16 target gene expression and DAF-16 nuclear translocation in slo-1 mutant worms.
Fig. S4. slo-1 and IIS act in parallel to regulate life span.
Fig. S5. Expression of daf-16 cDNA in the muscle fails to rescue the daf-16 mutant phenotype.
Fig. S6. The effects of different concentrations of paxilline on life span and motor aging.
Fig. S7. Loss of slo-1 does not extend the life span of mutants defective in neurotransmission.
Table S1. Summary of life-span data.
REFERENCES AND NOTES
- 1.Kenyon C. J., The genetics of ageing. Nature 464, 504–512 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Liu J., Lei H., Feng Z., Liu J., Hsu A.-L., Xu X. Z. S., Functional aging in the nervous system contributes to age-dependent motor activity decline in C. elegans. Cell Metab. 18, 392–402 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu A.-L., Feng Z., Hsieh M.-Y., Xu X. Z. S., Identification by machine vision of the rate of motor activity decline as a lifespan predictor in C. elegans. Neurobiol. Aging 30, 1498–1503 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dillin A., Hsu A.-L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A. G., Kamath R. S., Ahringer J., Kenyon C., Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Johnson T. E., Conley W. L., Keller M. L., Long-lived lines of Caenorhabditis elegans can be used to establish predictive biomarkers of aging. Exp. Gerontol. 23, 281–295 (1988). [DOI] [PubMed] [Google Scholar]
- 6.Hosono R., Age dependent changes in the behavior of Caenorhabditis elegans on attraction to Escherichia coli. Exp. Gerontol. 13, 31–36 (1978). [DOI] [PubMed] [Google Scholar]
- 7.Hahm J.-H., Kim S., DiLoreto R., Shi C., Lee S.-J. V., Murphy C. T., Nam H. G., C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nat. Commun. 6, 8919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faulkner J. A., Larkin L. M., Claflin D. R., Brooks S. V., Age-related changes in the structure and function of skeletal muscles. Clin. Exp. Pharmacol. Physiol. 34, 1091–1096 (2007). [DOI] [PubMed] [Google Scholar]
- 9.B. R. MacIntosh, P. F. Gardiner, A. J. McComas, Skeletal Muscle (Human Kinetics, ed. 2, 2006), pp. 423. [Google Scholar]
- 10.Herndon L. A., Schmeissner P. J., Dudaronek J. M., Brown P. A., Listner K. M., Sakano Y., Paupard M. C., Hall D. H., Driscoll M., Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419, 808–814 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Tank E. M. H., Rodgers K. E., Kenyon C., Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. 31, 9279–9288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan C.-L., Peng C.-Y., Chen C.-H., McIntire S., Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 9274–9279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toth M. L., Melentijevic I., Shah L., Bhatia A., Lu K., Talwar A., Naji H., Ibanez-Ventoso C., Ghose P., Jevince A., Xue J., Herndon L. A., Bhanot G., Rongo C., Hall D. H., Driscoll M., Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 32, 8778–8790 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z.-W., Saifee O., Nonet M. L., Salkoff L., SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron 32, 867–881 (2001). [DOI] [PubMed] [Google Scholar]
- 15.White J. G., Southgate E., Thomson J. N., Brenner S., The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340 (1986). [DOI] [PubMed] [Google Scholar]
- 16.Calixto A., Chelur D., Topalidou I., Chen X., Chalfie M., Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7, 554–559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogg S., Paradis S., Gottlieb S., Patterson G. I., Lee L., Tissenbaum H. A., Ruvkun G., The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Lin K., Dorman J. B., Rodan A., Kenyon C., daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319–1322 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Henderson S. T., Johnson T. E., daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 11, 1975–1980 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Lin K., Hsin H., Libina N., Kenyon C., Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139–145 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Wolff S., Ma H., Burch D., Maciel G. A., Hunter T., Dillin A., SMK-1, an essential regulator of DAF-16-mediated longevity. Cell 124, 1039–1053 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Li J., Ebata A., Dong Y., Rizki G., Iwata T., Lee S. S., Caenorhabditis elegans HCF-1 functions in longevity maintenance as a DAF-16 regulator. PLOS Biol. 6, e233 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao R., Zhang B., Dong Y., Gong J., Xu T., Liu J., Xu X. Z. S., A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell 152, 806–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Libina N., Berman J. R., Kenyon C., Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Strøbæk D., Christophersen P., Holm N.R., Moldt P., Ahring P.K., Johansen T.E., Olesen S.-P., Modulation of the Ca2+-dependent K+ channel, hslo, by the substituted diphenylurea NS 1608, paxilline and internal Ca2+. Neuropharmacology 35, 903–914 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Richmond J. E., Davis W. S., Jorgensen E. M., UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci. 2, 959–964 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang B., Gong J., Zhang W., Xiao R., Liu J., Xu X. Z. S., Brain–gut communications via distinct neuroendocrine signals bidirectionally regulate longevity in C. elegans. Genes Dev. 32, 258–270 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burkewitz K., Morantte I., Weir H. J. M., Yeo R., Zhang Y., Huynh F. K., Ilkayeva O. R., Hirschey M. D., Grant A. R., Mair W. B., Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell 160, 842–855 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leiser S. F., Miller H., Rossner R., Fletcher M., Leonard A., Primitivo M., Rintala N., Ramos F. J., Miller D. L., Kaeberlein M., Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science 350, 1375–1378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durieux J., Wolff S., Dillin A., The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee U. S., Cui J., BK channel activation: Structural and functional insights. Trends Neurosci. 33, 415–423 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peers C., Boyle J. P., Oxidative modulation of K+ channels in the central nervous system in neurodegenerative diseases and aging. Antioxid. Redox Signal. 22, 505–521 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Sesti F., Liu S., Cai S.-Q., Oxidation of potassium channels by ROS: A general mechanism of aging and neurodegeneration? Trends Cell Biol. 20, 45–51 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Ye H., Jalini S., Mylvaganam S., Carlen P., Activation of large-conductance Ca2+-activated K+ channels depresses basal synaptic transmission in the hippocampal CA1 area in APP (swe/ind) TgCRND8 mice. Neurobiol. Aging 31, 591–604 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Typlt M., Mirkowski M., Azzopardi E., Ruettiger L., Ruth P., Schmid S., Mice with deficient BK channel function show impaired prepulse inhibition and spatial learning, but normal working and spatial reference memory. PLOS ONE 8, e81270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sausbier M., Hu H., Arntz C., Feil S., Kamm S., Adelsberger H., Sausbier U., Sailer C. A., Feil R., Hofmann F., Korth M., Shipston M. J., Knaus H.-G., Wolfer D. P., Pedroarena C. M., Storm J. F., Ruth P., Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. U.S.A. 101, 9474–9478 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong J., Yuan Y., Ward A., Kang L., Zhang B., Wu Z., Peng J., Feng Z., Liu J., Xu X. Z. S., The C. elegans taste receptor homolog LITE-1 is a photoreceptor. Cell 167, 1252–1263.e10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao R., Chun L., Ronan E. A., Friedman D. I., Liu J., Xu X. Z. S., RNAi interrogation of dietary modulation of development, metabolism, behavior, and aging in C. elegans. Cell Rep. 11, 1123–1133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B., Xiao R., Ronan E. A., He Y., Hsu A.-L., Liu J., Xu X. Z. S., Environmental temperature differentially modulates C. elegans longevity through a thermosensitive TRP channel. Cell Rep. 11, 1414–1424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L.-B., Lei H., Arey R. N., Li P., Liu J., Murphy C. T., Xu X. Z. S., Shen K., The neuronal kinesin UNC-104/KIF1A is a key regulator of synaptic aging and insulin signaling-regulated memory. Curr. Biol. 26, 605–615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau5041/DC1
Fig. S1. slo-1 mutant worms exhibit slower locomotor activity decline throughout life span.
Fig. S2. slo-1 gain-of-function mutation greatly shortens life span.
Fig. S3. DAF-16 target gene expression and DAF-16 nuclear translocation in slo-1 mutant worms.
Fig. S4. slo-1 and IIS act in parallel to regulate life span.
Fig. S5. Expression of daf-16 cDNA in the muscle fails to rescue the daf-16 mutant phenotype.
Fig. S6. The effects of different concentrations of paxilline on life span and motor aging.
Fig. S7. Loss of slo-1 does not extend the life span of mutants defective in neurotransmission.
Table S1. Summary of life-span data.