

Selective activation of local mast cells promotes healing of bacterial skin infections and protects against reinfection.
Abstract
Mast cells (MCs) are strategically distributed at barrier sites and prestore various immunocyte-recruiting cytokines, making them ideal targets for selective activation to treat peripheral infections. Here, we report that topical treatment with mastoparan, a peptide MC activator (MCA), enhances clearance of Staphylococcus aureus from infected mouse skins and accelerates healing of dermonecrotic lesions. Mastoparan functions by activating connective tissue MCs (CTMCs) via the MRGPRX2 (Mas-related G protein-coupled receptor member X2) receptor. Peripheral CTMC activation, in turn, enhances recruitment of bacteria-clearing neutrophils and wound-healing CD301b+ dendritic cells. Consistent with MCs playing a master coordinating role, MC activation also augmented migration of various antigen-presenting dendritic cells to draining lymph nodes, leading to stronger protection against a second infection challenge. MCAs therefore orchestrate both the innate and adaptive immune arms, which could potentially be applied to combat peripheral infections by a broad range of pathogens.
INTRODUCTION
Although mast cells (MCs) are best known for their role in promoting immunoglobulin E (IgE)–mediated allergic reactions, there is a growing consensus that these cells play a critical physiologic role in immune surveillance and are pivotal in modulating early immune responses to microorganisms such as bacteria, viruses, and parasites (1). Several properties of MCs make them especially effective in immune surveillance. First, their strategic location at the host-environment interface, such as skin and mucosa, makes them one of the first immune cells to encounter pathogens that invade these integumental barriers. Second, upon activation, MCs degranulate, releasing into the extracellular space a plethora of prestored mediators such as tumor necrosis factor (TNF), histamine, serotonin, heparin, and proteases, which is followed by de novo synthesis and secretion of a wide range of proinflammatory mediators such as eicosanoids (prostaglandins and leukotrienes), cytokines, and chemokines (various interleukins and TNF). Because these MCs are also in close proximity to the vasculature, the impact of these mediators is especially profound in inducing vascular leakage and recruitment of various pathogen-fighting immune cells and serum components, such as complement, to the site of infection. Third, MC surfaces are equipped with a wide range of pattern recognition receptors (PRRs) that can directly recognize pathogen-associated molecular patterns (PAMPs) found on pathogens or their products, as well as receptors for host-generated antimicrobial peptides, complement components, and danger signals such as ATP (adenosine 5′-triphosphate) (1–3). Studies in multiple MC-deficient mouse models have revealed that in the absence of MCs, the ability of the host to clear various bacterial and viral pathogens is severely compromised (3–5). MCs have also been shown to promote initiation of adaptive immune responses to infection by orchestrating antigen-presenting dendritic cell (DC) trafficking (3). It is perhaps in recognition of this critical role played by MCs that several host-adapted pathogens such as Salmonella and Yersinia express effector proteins that inactivate MC degranulation by specifically inhibiting critical signaling events preceding degranulation (6).
In view of their potential to drive antimicrobial reactions, we hypothesized that purposeful activation of MCs at sites of infection could augment the host’s natural immune responses and accelerate resolution of infection. With the rapid emergence of multiresistant bacteria and the lack of new antibiotics in the pipeline (7), there is growing interest in identifying inducers of innate immunity that can safely be used as an alternative approach to combat infections (8). Nonspecific immunostimulants such as bacterial extracts or ligands to specific PRRs such as monophosphoryl lipid A have been shown to exhibit varying beneficial effects when applied to treat infections (9, 10). However, since they directly activate multiple effector cell types simultaneously, the nature and severity of the inflammatory responses they evoke can be excessive and unpredictable. Therefore, selectively targeting a single immunoregularory cell type such as MCs for activation at a local site may be more prudent and less harmful, as these cells have their own controlled program to release different immunomodulators with different time courses and coordinate both innate and adaptive immunity (1). A number of naturally occurring MC activators (MCAs) identified in the literature are cationic peptides including substance P, cathelicidin, β-defensin, and mastoparan (1). The receptor for these MC secretogogues is the Mas-related G protein–coupled receptor (GPCR) member X2 (MRGPRX2) (11), whose ortholog in mice is Mrgprb2, which is found exclusively in connective tissue MCs (CTMCs) (12). The nature of MRGPRX2-mediated activation of MCs is distinct from that by the IgE receptor FcεRI. Unlike the sustained inflammatory reactions mediated by the IgE receptor during allergy, the inflammatory responses evoked by MCs following activation of MRGPRX2 are transient and markedly more tempered (13).
Skin and soft tissue infections, which range from simple folliculitis to severe necrotic wounds, are now a major therapeutic challenge due to the ever-increasing antimicrobial resistance (14). Since MCs are abundant in skin and have the capacity to recruit neutrophils that contribute to bacterial clearance (15), we reasoned that boosting their activation at the site of infection will enhance bacterial clearance with minimal inflammatory side effects. To validate this notion, in this study we tested mastoparan, a 14-mer peptide MCA (16) in a mouse dermonecrotic Staphylococcus aureus infection model and identified multifaceted therapeutic benefits of MCAs in treating skin infections as well as the key immunomodulatory pathways behind them.
RESULTS
CTMCs control skin infections via neutrophil recruitment
We chose S. aureus as our model pathogen since this bacterial species is responsible for the majority of skin infections (17, 18). Although MCs have been reported to mediate the clearance of various pathogenic bacteria (1), their role in S. aureus–mediated skin infections is not known. To determine the innate role of MCs during skin S. aureus infections, we used an MC-deficient mouse model (Mcpt5-Cre+ iDTR+ mice) in which CTMCs, the MC type that populates skin, could selectively be depleted (Fig. 1A and fig. S1) (19). We induced a localized dermonecrotic infection by intradermal injection of 108 bacteria into the dorsocaudal area and observed that MC-depleted mice had significantly less neutrophil influx at 4 hours and more bacterial burden by 24 hours after infection compared with MC-sufficient littermate controls (Fig. 1, B and C). The lesion sizes, which correlated with bacterial numbers (fig. S2), were also twofold larger in MC-depleted mice, as measured on day 1 after infection (Fig. 1D). As expected, the larger initial lesions in the absence of MCs resulted in a much slower resolution of infection (Fig. 1D). However, this increase of lesion size in MC-depleted mice was no longer observed when we also depleted neutrophils prior to the infection (Fig. 1E and fig. S3), indicating that the protective role of CTMCs was primarily via neutrophils. Cumulatively, these data demonstrate that CTMCs play a major role in neutrophil recruitment at the very early stage of skin infection, which is crucial in limiting the infection.
Fig. 1. CTMCs control skin infection via neutrophil recruitment.

(A) Schematic plan for CTMC depletion, dermonecrotic infection, and lesion measurement. (B) Neutrophil recruitment at 4 hours after intradermal infection of MC-sufficient (Cre−) or MC-deficient (Cre+) mice with 108 S. aureus, assessed by myeloperoxidase (MPO) assay (n = 4 to 5). (C) Quantification of bacteria in the infected skin tissues at 24 hours after infection (n = 5). Colony forming units (CFUs) were determined by plating on LB agar. (D) Representative images of skin lesions taken on indicated days after infection. Black dotted lines delineate area of infection on day 1. The graph at the bottom represents size of skin lesions in different mouse groups measured on indicated days and area calculated using ImageJ (n = 5). (E) Initial lesion sizes in neutrophil-depleted MC-sufficient (Cre−) or neutrophil-depleted MC-deficient (Cre+) mice, measured 24 hours after infection with S. aureus (n = 7). Note that initial lesions in neutrophil-depleted mice were larger compared with those in neutrophil-sufficient mice shown in (D). Data are representative of two independent experiments. Data were analyzed via unpaired two-tailed Student’s t test. Error bars represent SEM. *P < 0.05, **P < 0.01, n.s., not significant. DT, diphtheria toxin.
CTMC activation via MRGPRX2 receptor recruits neutrophils
Following the identification of CTMC as a critical recruiter of neutrophils during skin infections, we next sought to establish whether receptor-specific activation of these cells by an MCA would also result in local neutrophil recruitment, even in the absence of an infection. However, before undertaking this study, it was important to establish the receptor-binding specificity and distinct MC signaling activity of the MCA used. Mastoparan is an MCA, originally isolated from wasp venom (16) and reported to activate both human and murine MCs via the MRGPRX2 receptor (12). We examined the in vitro capacity of mastoparan to mediate degranulation of human and murine MC lines (ROSA and MC/9, respectively) by measuring extracellular release of β-hexosaminidase, a component of MC granules. Mastoparan evoked degranulation in a dose-dependent manner in both human and murine MC lines (Fig. 2A). As a negative control, we exposed mastoparan to another well-known MC line, rat basophilic leukemia cell line (RBL-2H3), which, however, does not express MRGPRX2 receptors (20). As expected, the degranulation responses evoked by mastoparan in RBL-2H3 cells were minimal even at high dosages (Fig. 2A). To further establish the receptor specificity of mastoparan, we transfected the RBL-2H3 cells with human MRGPRX2 receptor and examined their degranulation response to the MCA. We observed significant degranulation responses even at very low concentrations (0.17 μM) but not in the mock-transfected cells (Fig. 2B).
Fig. 2. CTMC activation via MRGPRX2 receptor recruits neutrophils.
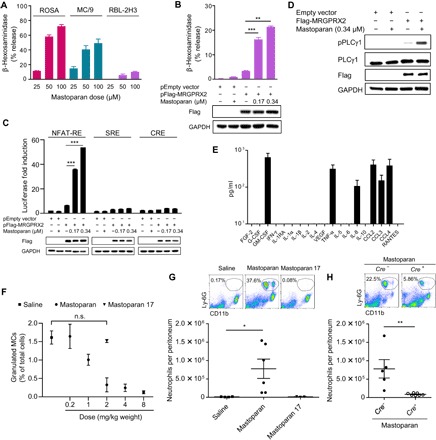
(A) Degranulation of a human MC line (ROSA), a murine CTMC line (MC/9), and a rat MC-like cell line (RBL-2H3) in vitro expressed as β-hexosaminidase release by increasing concentrations of mastoparan. (B) Degranulation of RBL-2H3 cells transfected with expression construct encoding MRGPRX2 or empty vector followed by mastoparan stimulation. Immunoblots show the introduction of MRGPRX2 in the cells and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. (C) Luciferase reporter activities triggered by mastoparan in mock- or MRGPRX2-transfected HEK293 cells coexpressing NFAT-RE, SRE, or CRE (representing the Gαq, Gi/o, or Gαs pathway, respectively). Immunoblots show the MRGPRX2 expression and loading control GAPDH. (D) Immunoblot analysis of lysates prepared from RBL-2H3 cells (mock or MRGPRX2 transfected) stimulated with mastoparan for 5 min and probed for phospho-PLCγ1, PLCγ1, Flag, and GAPDH. Data are presented as the mean of triplicate values in one experiment, representative of at least two independent experiments. (E) Cytokines (prestored or de novo synthesized) secreted by a human MC line (LAD2) upon stimulation with mastoparan (25 μM) for 20 hours, measured by a Luminex multiplex fluorescence immunoassay. Data represent mean of triplicate values in a single experiment. (F) Quantification of granulated MCs in the peritoneal lavage of mice 30 min after intraperitoneal injection with various doses of mastoparan (0.2 to 8 mg/kg bodyweight; n = 3) or controls. (G) Numbers of neutrophils per peritoneum 2 hours after intraperitoneal injection with saline or 2 mg/kg bodyweight of mastoparan. Data represent two independent experiments (n = 3 to 6). Flow cytometry plots represent percentages of CD11b+Ly6G+ neutrophils in the peritoneums of each group of mice. (H) Numbers of neutrophils per peritoneum 2 hours after intraperitoneal injection 2 mg/kg bodyweight of mastoparan. Flow cytometry plots represent percentages of neutrophils in peritoneums of MC-sufficient or MC-deficient mice. Data represent two independent experiments (n = 5 to 7). Data were analyzed via unpaired two-tailed Student’s t test or one-way analysis of variance (ANOVA). Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant. IFN, interferon.
Since little is known with regard to the downstream G protein–signaling pathways triggered by mastoparan-mediated activation of MRGPRX2 on MCs, we investigated this question. Depending on the activating ligand, GPCRs such as MRGPRX2 can interact with multiple Gα subunits to initiate distinct signaling pathways (21, 22). To identify which specific Gα pathways are activated by mastoparan, we used a reporter gene assay involving human embryonic kidney (HEK) 293 cells, which do not endogenously express MRGPRX2 but have a full complement of G protein–directed signaling components. We transfected these cells with empty plasmids or plasmids encoding human MRGPRX2 along with plasmids encoding a luciferase-conjugated response element (RE) known to be activated by a particular Gα subunit: 3′,5′-cyclic adenosine monophosphate RE (CRE) for Gαs, nuclear factor of activated T cell RE (NFAT-RE) for Gαq, or serum RE (SRE) for Gi/o. Upon incubation with mastoparan, we observed specific activation of NFAT-RE but not SRE-RE or CRE (Fig. 2C), indicating that mastoparan specifically activates Gαq, the subunit known to activate phospholipase C (PLC). We detected MRGPRX2-specific phosphorylation of PLCγ1 (Fig. 2D), which in turn results in inositol triphosphate (IP3) production, the critical second messenger that mobilizes Ca2+ required for MC degranulation. Collectively, these data indicate that mastoparan activates a single pathway downstream to MRGPRX2, which specifically triggers MC degranulation. To investigate whether mastoparan could activate other receptors, especially PRRs such as Toll-like receptors (TLRs) or NOD-like receptors (NLRs) commonly found in both immunocytes (including MCs) and nonimmune cells, we conducted a commercial TLR/NLR screening assay (see Materials and Methods). We observed that mastoparan did not activate any of the tested TLRs or NLRs (fig. S4), indicating that mastoparan does not target common innate immune receptors.
To investigate whether mastoparan-triggered activation of MCs was associated with the release of immune-mediators known to influence neutrophil recruitment, we examined various cytokines in the human MC line LAD2 by a multiplex assay. The cytokines that were released from human MCs specifically upon mastoparan treatment include TNF, GM-CSF (granulocyte-macrophage colony-stimulating factor), IL-8 (interleukin-8), CCL2, and CCL3 [chemokine (C-C motif) ligand 3] (Fig. 2E). While TNF has been shown to mediate MC-dependent neutrophil recruitment following infection (15), the other mediators are known to play critical roles in recruitment of neutrophils in different inflammatory states (23–25). So far, our data indicate that mastoparan can activate human MCs to secrete neutrophil-recruiting cytokines, and its action is highly restricted to a single MC-specific receptor and downstream pathway, indicating the potential of MCAs as selective immunomodulators to combat infection.
Upon determining the capacity of mastoparan to activate MCs in vitro and trigger release of neutrophil-recruiting cytokines, we investigated mastoparan’s capacity to activate MCs and mediate neutrophil recruitment in vivo. Since the mouse peritoneum is rich in CTMCs and relatively easy to assess for MC degranulation and leukocyte recruitment, we injected increasing doses of mastoparan into the peritoneal cavities of mice to determine the minimum concentration of mastoparan required for maximum MC degranulation. We have previously shown using MC-specific markers (FcεRIα and c-Kit) that activating skin MCs with Mrgprb2-binding activator results in fewer granulated MCs, while the total number of MCs remains the same (26). Therefore, with the use of toluidine blue, which metachromatically binds to heparin in MC granules, we could distinguish granulated MCs from other cells in peritoneal lavage using brightfield microscopy and were able to readily ascertain their degree of degranulation (fig. S5) (6). Upon administration of concentrations ranging from 0.2 to 8 mg/kg body weight, we observed dose-dependent degranulation until the 2 mg/kg dose when maximum degranulation was attained (Fig. 2F). When we examined the effect of mastoparan 17, an inactive analog of mastoparan (27), we did not detect any degranulation at a dose equimolar to mastoparan (2 mg/kg).
Next, we investigated whether mastoparan-mediated MC activation in the peritoneum was accompanied by a neutrophil influx. We treated mice intraperitoneally with mastoparan (2 mg/kg) and 2 hours thereafter collected the peritoneal lavage. Assessment of the lavages by flow cytometry revealed substantial recruitment of neutrophils into the peritoneal cavity following administration of mastoparan but not its inactive counterpart (Fig. 2G). To confirm that the neutrophil recruitment by mastoparan was dependent on the presence of MCs and not by its effect on some other cell type, we assessed neutrophil recruitment in CTMC-depleted mice. In contrast to Cre− iDTR+ control mice, neutrophil influx upon mastoparan injection was minimal in MC-depleted Mcpt5-Cre+ iDTR+ mice (Fig. 2H). Cumulatively, these data show that mastoparan specifically activates MCs via the MRGPRX2 receptor and that this results in MC degranulation, which, in vivo, can result in significant neutrophil recruitment even in the absence of infection.
MCA-mediated neutrophil recruitment accelerates bacterial clearance in the skin
We found that it was necessary to dissolve mastoparan in a skin-permeating cocktail (vehicle) composed of dimethyl sulfoxide (DMSO) and olive oil to achieve penetrance of the stratum corneum with adequate amounts of mastoparan. Examination of high-magnification images of mouse skin cross sections upon topical application of mastoparan–fluorescein isothiocyanate (FITC) revealed that the peptide would readily penetrate the epidermis and reach the dermis (Fig. 3A), where skin MCs reside. The numbers of granulated MCs were significantly reduced in the skin treated with 10 μg of mastoparan in 10 μl of vehicle, compared with the skin treated with vehicle alone (Fig. 3B), suggesting that topical application of mastoparan results in activation of skin MCs. The recruitment of neutrophils into the dermis upon topical application of mastoparan was detectable but not substantial (Fig. 3C); presumably in the absence of additional cues from an infection or injury, most of the recruited neutrophils immediately left the skin by reverse migration (28). Mice were monitored for local adverse events such as irritation, inflammation, or mutilation of the treatment site after repeated daily treatments of the skin by mastoparan. Signs for systemic adverse events were also monitored by observing for any changes in body temperature and behavior (e.g., distress, discomfort, and activity level). We did not observe any local or systemic adverse effects in the mice, indicating that the topical application of low-dose MCA does not cause any significant local or systemic adverse effects.
Fig. 3. MCA-mediated neutrophil recruitment accelerates bacterial clearance in the skin.
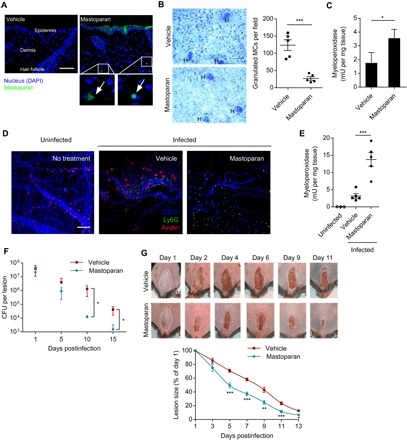
(A) Representative images of mouse dorsal skin cross sections collected at 2 hours after topical application of 10 μg of mastoparan-FITC or vehicle (n = 2 to 3). Arrows point to the fluorescent peptide detected within dermis. Scale bar, 50 μm. (B) Representative whole-mount images showing granulated MCs in vehicle- and mastoparan-treated ear tissue 2 hours after treatment. “H” indicates hair follicle. Scale bar, 50 μm. Graph shows quantification of granulated MCs in each treatment group (n = 5). (C) Quantification of neutrophils in dorsal skins of each treatment group using MPO assay 6 hours after treatment (n = 3). (D) Representative whole mount of the dorsal skin following no infection or treatment (left), intradermal infection (108 S. aureus) and topical treatment with vehicle (middle), or mastoparan (right). The infected mice were treated twice daily and sacrificed 4 hours after treatment on day 5; skin tissues adjacent to scabs were stained for avidin (red), Ly6G (green), and CD31 (blue). Notice that in the mastoparan-treated tissue, MCs are not visible because they are degranulated, and numerous neutrophils appear in the image. Scale bar, 100 μm. (E) Quantification of neutrophils (MPO) in the skins of each group of mice 4 hours after treatment on day 7 (n = 3 to 5). (F) Quantification of bacteria in the infected skin tissues at different time points after infection (n = 4 to 5). (G) Representative images of skin lesions taken on indicated days after infection and treatment. White dotted lines delineate area of infection on day 1. The graph at the bottom represents size of skin lesions in different treatment groups measured on indicated days and area calculated using ImageJ (n = 10). Data are representative of at least two independent experiments. Data were analyzed via unpaired two-tailed Student’s t test or ANOVA. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we proceeded to investigate the impact of mastoparan treatment in our dermonecrotic model of S. aureus skin infection. Beginning 12 hours after infection when a palpable swelling at the site of infection had formed, mice were treated on and around the infected site with mastoparan or vehicle every 12 hours for 2 weeks. To visualize MC degranulation and neutrophil recruitment into the dermis adjacent to the lesion, we sacrificed a subset of mice on day 5 after infection. Since avidin binds to heparin, we used avidin conjugated with a fluorochrome to specifically visualize MC granules by confocal microscopy of whole skin mounts. We observed that many MCs remained granulated during the course of infection in the vehicle-treated mice, and there was a limited recruitment of neutrophils (Fig. 3D, middle). On the other hand, in mastoparan-treated mice, almost no granulated MCs were detected, whereas numerous neutrophils were readily detectable (Fig. 3D, right). To quantitate neutrophil influx, another subset of mice was sacrificed on day 7, approximately halfway to the resolution of infection. Using MPO assay, we observed a significant increase of neutrophil recruitment in infected mice treated with mastoparan compared with vehicle (Fig. 3E). These data indicated that exogenous activation of local MCs by mastoparan recruited additional neutrophils throughout the course of infection. As a result, the bacterial burden in mastoparan-treated mice was trending lower than that in vehicle-treated mice by day 5 and became significantly lower by day 10 and continued to remain so until end of the study period (Fig. 3F). In parallel with the reduced bacterial number, the lesion size of the mastoparan-treated animals was significantly smaller than that of the other group beginning from day 5 (Fig. 3G). We included an additional control group of mice, which were infected and then treated with 10 μl of a cocktail of three over-the-counter antibiotics commonly used for topical treatment of skin infections (neomycin/polymyxin B/bacitracin). The rate of healing induced by mastoparan was very similar to that mediated by the antibiotics (fig. S6). Notably, mastoparan-mediated control in lesion size and bacterial burden was completely abolished in neutrophil-depleted mice (fig. S7), indicating that mastoparan’s therapeutic effects were neutrophil dependent. To summarize, topical treatment with mastoparan significantly accelerated bacterial clearance and reduction of lesion size by enhancing neutrophil recruitment, indicating its strong therapeutic effect on resolution of local infection.
Therapeutic effect of mastoparan is specific to MC activation
Next, we investigated the specificity of MCA-mediated therapeutic effects by multiple approaches. First, using the previously described CTMC depletion model, we asked whether these cells are essential for the therapeutic effect observed during skin infection. When we treated MC-depleted mice with mastoparan, it failed to improve healing (Fig. 4A) compared with vehicle treatment, indicating that the therapeutic effects of mastoparan require CTMCs.
Fig. 4. Therapeutic effect of mastoparan is specific to MC activation.
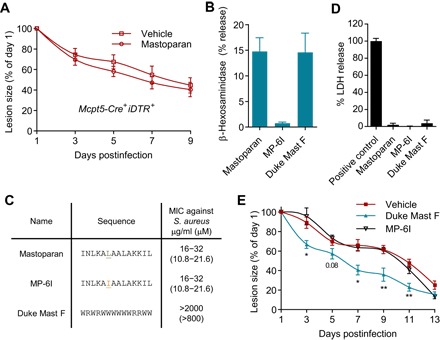
(A) Quantification of lesion sizes at indicated days after infection of MC-depleted mice and vehicle or mastoparan treatment (n = 6 to 7). Note that healing rates in MC-depleted mice were slower than that in MC-sufficient mice due to larger initial lesions. Data represent two independent experiments. (B) Degranulation of MC/9 cells expressed as β-hexosaminidase release by 25 μM mastoparan, MP-6I, and Duke Mast F. (C) Amino acid sequence and MIC (minimum inhibitory concentration) for mastoparan or each of its analogs. (D) Cytotoxicity of mastoparan or its analogs measured by LDH assay 4 hours after incubation of L929 cells at a concentration of 50 μM. (E) Quantification of lesion sizes in each treatment group (n = 5). Data were analyzed via unpaired two-tailed Student’s t test or one-way ANOVA. Error bars represent SEM. *P < 0.05, **P < 0.01.
A common feature of many peptide and nonpeptide MCAs is the direct antimicrobial capacity, attributed to their cationic amphiphilic nature, which allows the MCA to bind and disrupt negatively charged bacterial membranes (29, 30). Since mastoparan is a cationic peptide, it can also lyse bacterial membrane, and we attempted to ascertain whether part of the therapeutic effects of mastoparan was related to its direct antibacterial activity. We made various alterations to the amino acids of mastoparan to generate a mastoparan derivative that retained its antimicrobial activities but lost its MC-degranulating capacity. By systematic substitutions of single amino acids on the mastoparan peptide, we identified such a derivative “MP-6I” (see Materials and Methods) in which leucine at position 6 was substituted with isoleucine. This single change was sufficient to cause complete loss in MC-degranulating activity while retaining comparable antibacterial activity against S. aureus (Fig. 4, B and C, and table S1). However, identifying a mastoparan derivative that retained its MC-degranulating property while losing its antibacterial activity was much more challenging. We therefore developed a tryptophan-rich 14-mer peptide named “Duke Mast F” (see Materials and Methods), whose amino acid sequence is unrelated to mastoparan but has a comparable MC-degranulating capacity without any antibacterial activity (Fig. 4, B and C). Having selected mastoparan derivatives that exhibited either MC-degranulating activity or antibacterial activity without any significant toxicity (Fig. 4D), we investigated their ability to resolve S. aureus skin infections at quantities equimolar to native mastoparan. To our surprise, we found that MP-6I, despite its inherent antibacterial activity in vitro, did not significantly reduce lesion size compared with vehicle treatment following S. aureus infections (Fig. 4E). Conversely, when we examined the therapeutic ability of Duke Mast F, we found that it was significantly better than the vehicle in reduction of lesion size (Fig. 4E). Note that the lesion sizes in mice treated by mastoparan (Fig. 3G) were also significantly smaller than that by MP-6I (P ≤ 0.001 for days 5, 7, and 9) but comparable to that by Duke Mast F. Cumulatively, these initial structure-activity relationship studies on mastoparan suggest that its therapeutic effects are dependent on its MC-activating capacity rather than on direct antibacterial properties and attributable to other MC-activating peptides.
Treatment with MCA promotes regenerative healing
The lesions in dermonecrotic infections mimic wounds that arise upon loss of the scab, and reduction of lesion size involves proliferation and reepithelialization similar to that occurring in wound healing. Since persistence of bacteria at wound sites can significantly delay natural wound healing and increase scarring (31), we speculated that mastoparan-mediated acceleration in bacterial clearance could result in not only faster but also more regenerative healing of skin lesions, that is, less scarring. To investigate this possibility, we allowed the lesions in various groups of mice to close completely (which typically takes 2 to 3 weeks after infection) and then waited an additional 2 weeks to allow all of the proliferative processes in the skin to reach steady-state levels. Then, the scars along with the surrounding skins were harvested, and the levels of scarring at each of the infection sites were visualized by trichrome staining of the cross sections. The scar area was significantly smaller in the mastoparan-treated mice compared with controls (Fig. 5, A and B). The reduced scarring could be primarily due to accelerated bacterial clearance by mastoparan; nonetheless, we investigated whether MCs had any additional role in healing of these infectious wounds other than promoting bacterial clearance. We have recently identified the critical roles of dermal CD301b+ cells in promoting reepithelialization of sterile wounds (32). In our current study, we observed that the CD301b+ dermal DC (DDC) population significantly decreased in the infected skin area by 24 hours after infection (Fig. 5C and fig. S8A). We also did not detect any increase in CD301b+ cells within the dead cell population in the infected skin (fig. S8B), indicating that the disappearance of these cells was due to their migration and not cell death. However, the population was restored at the later stage of infection (Fig. 5C) when bacterial burden was minimal. This phenomenon could be unique to CD301b+ cells, as we did not observe a similar reduction in the case of the other major DDC population (CD103+ cells) by 24 hours (fig. S8C). Since we previously identified that MCs could recruit CD11b+ DCs early during E. coli skin infection (33) and CD301b+ DCs were shown to be a major subset of CD11b+ DDCs (34), we determined whether MCs had a role in the repopulation of these cells during the healing stage of infection. Depletion of CTMC results in higher bacterial burden (Fig. 1C), which may also contribute to the reduction of CD301b+ cells in the dermis. Therefore, we adjusted the bacterial inoculum to obtain similar lesions in MC-sufficient and MC-depleted mice. We observed that the number of CD301b+ DCs on day 12 was significantly lower in the absence of MCs (Fig. 5C), indicating that MCs contributed to restoring the skin CD301b+ DC population to homeostatic levels. Note that absence of MCs did not affect the CD301b+ cell population in the uninfected distal skin area of infected mice (fig. S8D). These data suggest that MCs contribute to the healing of infectious wounds not only by clearing bacteria via neutrophil recruitment but also by restoring the CD301b+ DC population, which promotes reepithelialization.
Fig. 5. Treatment with MCA promotes regenerative healing.
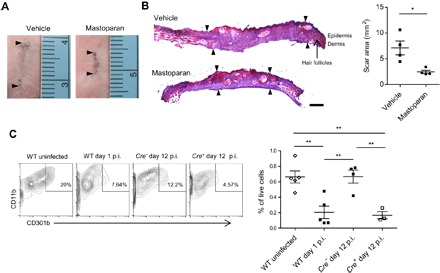
(A) Representative images of skin scars from different mouse groups taken on day 28 after infection. Arrowheads indicate the length of scars. (B) Representative cryosections from scarred regions. The sections taken were perpendicular to the long axis of the scar. Arrowheads define the area of scar regions devoid of hair follicles. Scale bar, 500 μm. The graph on the right represents area of the scar in each group measured using ImageJ (n = 4). (C) Representative flow cytometry plots depicting the CD11b+CD301b+ DDC population in skins of mice of indicated groups. Cells within the CD45+CD64loCD11c+ gate are shown. Graph shows skin CD301b+ DDCs as percentage of total live cells (n = 3 to 5). Data are representative of at least two independent experiments. Data were analyzed via unpaired two-tailed Student’s t test or ANOVA. Error bars represent SEM. *P < 0.05, **P < 0.01. p.i., postinfection; WT, wild type.
MCA boosts adaptive immunity and controls reinfection
Concurrent with recruiting innate immune cells to clear pathogens at the site of infection, MC mediators also modulate the trafficking of key immune cells involved in antigen presentation, the critical immune activity responsible for initiating adaptive immunity (1). Recent studies have revealed that MCs are responsible for recruiting antigen-presenting DCs to the site of infection and promoting their migration from the inflamed site to the draining lymph nodes (DLN), the epicenter of the adaptive immune response (3). In this study, we observed that mastoparan triggered release of several cytokines from the human LAD2 MCs such as TNF, CCL2, CCL3, and CCL4 (Fig. 2E), which had previously been implicated to be involved in either MC-dependent or MC-independent mobilization of DCs (33, 35, 36). To determine whether mastoparan can mobilize DCs to DLNs, we injected mastoparan or its inactive counterpart (mastoparan 17) intradermally into rear footpads of mice since this site is drained by a single DLN, the popliteal node (PN). We observed that by 24 hours, mastoparan elicited lymph node hypertrophy (Fig. 6A) in a CTMC-dependent manner. We detected significant levels of various migratory skin DC subsets recruited specifically by mastoparan into PNs, which include CD103+CD207± DDCs and CD207+CD103− Langerhans cells (LCs) (Fig. 6B and fig. S9) (37). Therefore, we speculated that mastoparan treatment could provide a powerful adjuvant effect during infection and augment bacterial antigen-specific antibody production. To test the hypothesis, dermonecrotic infections were topically treated with vehicle, mastoparan, or mastoparan 17 for 2 weeks, as described before, and sera from each group of mice were collected on day 21 and assayed for S. aureus–specific total IgG. IgG titers were significantly higher in the mastoparan-treated group compared with mice treated with vehicle or mastoparan 17 (Fig. 6C). Note that the antimicrobial effect of mastoparan, which accelerated bacterial clearance (Fig. 3F) and therefore reduced the source of immunogens, did not cancel out its adjuvant effect. To determine whether increased antibody response is associated with protection from reinfection, we infected the dorsorostral area of mice with S. aureus on day 28 (1 week after complete resolution of initial infections in all mice) and measured the lesion size after 24 hours. We observed that lesions upon reinfection in mice previously infected and treated with mastoparan were significantly smaller than that in mice previously infected and treated with vehicle or mastoparan 17 or that in previously uninfected (naive) mice (Fig. 6D). These data demonstrate that along with expediting resolution of infection, the MCA is capable of boosting adaptive immunity, which provides protection against reinfection.
Fig. 6. MCA boosts adaptive immunity and controls reinfection.
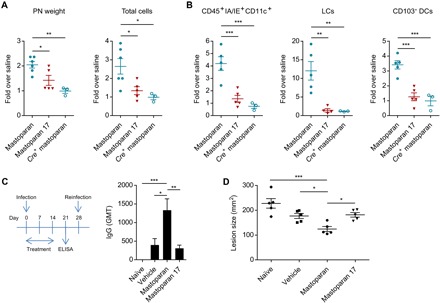
(A) Weight of PNs and total number of cells in the PN 24 hours after footpad injection of WT mice with saline, 10 μg of mastoparan, or mastoparan 17, or MC-depleted mice with mastoparan (Cre+ mastoparan group). (B) Numbers of CD11c+ IA/IE+ antigen-presenting cells, LCs, and CD103+ DCs in PNs 24 hours after various treatments described above. Data are representative of two independent experiments. (C) Schematic showing experimental plan and graph depicting IgG geometric mean titer (GMT) against whole S. aureus cells in sera collected on day 21 after infection from mice infected with S. aureus and treated with vehicle, mastoparan, or mastoparan 17 for 2 weeks or in sera of uninfected (naïve) mice (n = 5). (D) Lesion size 24 hours after reinfection with 108 S. aureus of mouse groups described above (n = 5). Note that the lesion sizes were larger since the bacteria were not coated with microbeads (see Materials and Methods). Data were analyzed via unpaired two-tailed Student’s t test or ANOVA. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001. ELISA, enzyme-linked immunosorbent assay.
DISCUSSION
Over 20 years ago, we demonstrated the critical role of MCs in modulating bacterial clearance in the lungs and peritoneal cavities of mice through recruitment of neutrophils (15). Since then, data have emerged from various laboratories, implicating MCs as critical modulators of defense against a wide variety of bacterial and viral infections at different body sites (3). Since MCs can be activated exogenously by natural or synthetic MCAs, we reasoned that administering small amounts of MCAs locally, at sites of infection, could boost bacterial clearance through enhanced neutrophil recruitment without evoking any systemic side effects. Unlike other sentinel immunocytes, MCs prestore granules loaded with chemoattractants and other proinflammatory mediators and are therefore ideal cellular targets for rapid triggering of the immune system. Furthermore, MCs recruit immunocytes of both innate and adaptive pathways; therefore, coordination between them could be achievable. Topical application of MCAs was found to be highly effective in reducing S. aureus skin infections and in resolving the resulting lesions. That this effect was via neutrophil recruitment was deduced from the observations that bacterial clearance was closely associated with neutrophil influx into the infection site (Fig. 3, D to F), and the effect was abolished if neutrophils were depleted (fig. S7).
Since MCs can recruit heterogeneous DC populations and release a panoply of growth factors, some of which can promote wound healing processes (32, 33, 38), there was also a possibility that these factors might contribute to the beneficial effects of MCAs. This study identified a novel role of CTMCs in restoring the wound-healing CD301b+ DDC population, which disappeared from the skin upon infection. The CD301b+ DDC is also a key transporter of antigens to skin-draining lymph nodes (34). Therefore, the low abundance of CD301b+ DDC we observed after infection was presumably due to their continuous migration to lymph nodes upon exposure to bacterial antigens, which continued until the bacterial burden became low. The MC mediators involved in DC recruitment may be prestored and de novo synthesized, since it has previously been shown that PAMPs that do not induce MC degranulation can still trigger MCs to secrete DC-recruiting cytokines (2).This wound-healing role of MCs does not appear to occur in the absence of infection as dermal CD301b+ cell frequency does not alter during sterile wounding (32). Several recent studies have also shown MCs as dispensable in sterile wound healing (39, 40).
Conventional MC knockout models involve mutations in or upstream of c-Kit, which have been shown to display confounding abnormalities such as neutrophilia or neutropenia (41, 42). In this study, we verified MCs’ role in neutrophil recruitment and control of skin infections by using an inducible CTMC depletion system (Mcpt5-Cre+ iDTR+ mice) (Fig. 1), which does not affect levels of other immune cells (19). Since the MRGPRX2 receptor is expressed primarily on CTMCs and not on other immune cells, the actions of MCAs can be assumed to target only this subset of MCs. The protective actions of the MCA were critically dependent on CTMCs since the MCA had limited efficacy in recruiting neutrophils or resolving infections in CTMC-depleted mice (Figs. 2H and 4A). Therefore, the inflammation that MCAs evoke is likely to be more controlled, as opposed to TLR ligands or other PAMPs, which can activate PRRs on various immune and nonimmune cells. In addition, a recent study has revealed that the inflammatory response evoked by the MRGPRX2 receptor, although rapid, had a short duration compared with the response evoked by FcεRI, the IgE receptor on MCs (13). Another important property of the MRGPRX2 receptor on MCs is that unlike FcεRI, it is resistant to desensitization (11) upon activation by peptide MCAs, and therefore, it is likely to remain responsive, allowing for successive MCA treatments.
Like many natural cationic peptides, mastoparan had previously been reported in several in vitro studies to have direct antibacterial activity. The bactericidal action of this peptide was attributed in part to its net positive charge, which facilitated penetration and disruption of the negatively charged bacterial surface membranes (29, 43). When we examined a mutant derivative of mastoparan, MP-6I, which had lost its ability to activate MCs but still retained its antibacterial activity, for its capacity to resolve S. aureus infections, we found very little activity. Furthermore, when we examined the ability of mastoparan to resolve dermonecrotic infections in MC-depleted mice where the immune stimulating activity of mastoparan should not occur, we found no significant therapeutic benefit (Fig. 4). Together, the protective actions of mastoparan in vivo are primarily attributable to its innate immunostimulatory activity rather than to any antibacterial actions. This result is consistent with findings that many host defense peptides with antimicrobial properties based on in vitro studies have limited activity in vivo. The positive charge of these compounds causes many soluble host proteins to adsorb to these peptides and interfere with their ability to intercalate with bacterial membranes (44), presumably without impeding their ability to activate MCs. Therefore, it is possible that the cationic antimicrobial peptides secreted from neutrophils (e.g., cathelicidin and β-defensin) may also contribute to bacterial clearance via MRGP receptors, providing a positive feedback to MC-mediated neutrophil recruitment.
While our studies here have primarily dwelled on the use of MCs in boosting innate immune responses, MCAs have previously been found to be highly efficacious in boosting adaptive immune responses. These MC-targeting molecules could function as highly effective adjuvants when coadministered with subunit vaccine antigens at mucosal surfaces or the skin (45). MCAs at sites of vaccine administration caused extensive MC degranulation (45), with the exteriorized granules draining into lymph nodes, where they released their cargo of mediators (46). The result was a sharp up-regulation of chemoattractant CCL21 expression in the nodes (33), which enhanced coordinated trafficking of antigen-presenting DCs from the vaccination site into the lymph node (45) and T cells from the circulation (26). This synchronous recruitment of critical immune cells resulted in a highly enhanced antibody response to the vaccine antigen (45). Our current study demonstrates that MCA treatment of skin can also enhance DC recruitment and protective antibody response during infection (Fig. 6, B and C), indicating that MCs can be targeted to induce both innate and adaptive immunity to pathogens simultaneously, which would be the most advantageous means to develop host-directed therapy.
The potential for anaphylaxis is often a concern whenever the medical use of MCAs is considered. However, our studies involving topical MCAs failed to reveal any harmful side effects either at the site of application or systemically, even after multiple applications. Arguably, harmful systemic reactions were avoided because both the dose and the volume of the administered MCA were low; thus, very little of the MCAs could become systemic. However, it is also likely that even if these MCAs become absorbed systemically, this absorption is unlikely to result in anaphylaxis. This notion is based on the fact that two peptide antibiotics, polymyxin B and colistin, which activate MCs (47), are U.S. Food and Drug Administration approved for human use since 1964 and do not induce anaphylaxis in patients even when applied systemically. This inability to provoke systemic anaphylaxis is perhaps because, as described above, the MRGPRX2-dependent MC responses induced by peptide MCAs are distinctively less intense and more transient than IgE-mediated MC activation (13). In addition, although the MCAs were found to elevate IgG and IgA responses, very little IgE was found to be evoked (45).
In conclusion, our findings highlight MCs as ideal cellular targets for boosting innate and adaptive immune responses during infection. By using a peptide that targets a receptor found almost exclusively on CTMCs, we are able to specifically target this subset of cells for activation without evoking any off-target effects. Since the MCAs are directed at host cells, there is less likelihood that bacterial pathogens will develop resistance to this mode of treatment. Furthermore, since the MCAs are boosting the general innate immune response, it will be effective regardless of the identity of the pathogen. Since the mode of action of MCA is distinct from that of conventional antibiotics, they can readily be applied as adjuncts to current standard-of-care antibiotics, possibly resulting in a synergistic outcome.
MATERIALS AND METHODS
Study design
The primary objective of this study was to test whether application of exogenous MCAs improves resolution of bacterial infections and mechanism of their function. All mice were infected and then randomly divided into treatment or control groups. Blinding was not deemed necessary for the animal infection models, and all animals were used. Power analyses were performed to determine minimum numbers of animals per group needed to attain significance of P < 0.05 with a 90 or 95% probability. At least 10 mice per group were used for lesion size measurement upon mastoparan treatment and 5 to 7 mice for the other experiments. End points to assess different parameters of resolution of infection were designed on the basis of various established assays. All histomorphometric analyses were performed by a blinded observer. Three cell cultures per group were used for in vitro experiments.
Mice
Eight- to 10-week-old C57BL/6 mice (male or female) were purchased from the National Cancer Institute Animal Production Area or the Jackson laboratory. Mcpt5-Cre+ iDTR+ mice were generated by crossing Mcpt5-Cre mice (a gift from Axel Roers, University of Technology, Dresden) and iDTR mice. To achieve and maintain depletion of CTMCs, Mcpt5-Cre+ iDTR+ mice or littermates were treated every 72 hours with 20 ng of diphtheria toxin per gram bodyweight divided into intraperitoneal and subcutaneous injections. All animal studies were carried out at Duke University and approved by the Duke University Institutional Animal Care and Use Committee.
Cell lines and transfections
The mouse MC line MC/9 [American Type Culture Collection (ATCC) CRL-8306] was maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) containing 10% fetal bovine serum (FBS; HyClone) at 37°C with 5% CO2. RBL-2H3 (ATCC CRL-2256) cells were maintained in MEM with Earle’s salts, l-glutamine, 1 mM sodium pyruvate, and 15% heat-inactivated FBS. The ROSA and LAD2 cell lines were maintained in IMDM (Iscove’s modified Dulbecco’s medium) and StemPro-34 SFM (serum-free media) media, respectively. For transfections, human MRGPRX2 was amplified from LAD2 cells, and cloned in pCMV-Flag–tagged plasmid (Takara), termed Flag-MRGPRX2. RBL-2H3 cells were transfected with empty plasmid or plasmid encoding Flag-MRGPRX2 using P2 primary solution and program DN-100 in 4D-Nucleofector (Lonza). Cells were assayed 36 hours after transfection. HEK293 cells were cotransfected with Renilla luciferase plasmid (Promega), Flag-MRGPRX2, or empty plasmid and one of the following plasmids: NFAT [pGL4 (NFAT-RE/minP/luc2P)], CRE [pGL4 (CRE/minP/luc2P)], or SRE [pGL4 (luc2/Hygro) SRE] (Promega), using JetPrime reagent (Polyplus). All transfections were verified by Western blot.
Bacterial culture and infection
A clinical strain of S. aureus (strain ID 10201) was used for all experiments in this study. Bacteria were grown in Luria-Bertani broth (BD Biosciences) overnight at 37°C with shaking and then diluted 1:10,000 and allowed to reach log phase. The mouse dermonecrotic model was modified from skin infection models described previously (48). Briefly, 8- to 10-week-old mice were anesthetized by ketamine and xylazine, and hair was removed from the dorsal area using a clipper and hair removal cream (Veet, Reckitt Benckiser) 2 days prior to infection. To induce dermonecrotic infection, mice were injected intradermally using a 25-gauge needle with 108 log-phase bacteria in 100 μl of phosphate-buffered saline (PBS) complexed with dextran microbeads (Cytodex, Sigma-Aldrich) as carriers. The microbeads were used to ensure localized and uniform lesions for all dermonecrotic infections except for reinfection studies where lesion sizes were expected to be indicators of protective immunity development. The lesions were measured every other day beginning on day 1 by digital planimetry, in which margins of a lesion were traced using acetate transparency films (Staples) and surface area was calculated using ImageJ upon digital rendering of the traces.
Peptides and antibiotics
Mastoparan [molecular weight (MW), 1479.9], originally isolated from the venom of social wasp Vespula lewisii (16), and mastoparan 17 (INLKAKAALAKKLL-NH2; MW, 1494.9) (27) were obtained commercially at >90% purity from CPC Scientific (Sunnyvale, CA). Mastoparan 6I (MW, 1480) and Duke Mast F (MW, 2504.87) were custom made by CPC Scientific for us. The number of molecules used for all other peptides was adjusted to equimolar quantities to that of mastoparan. For the triple antibiotic cocktail, 100 U of polymyxin B, 8 U of bacitracin, and 100 μg of neomycin (all from Sigma-Aldrich) were dissolved into 10 μl of vehicle based on the concentrations of active ingredients in Neosporin, an over-the-counter antibiotic ointment.
Topical treatments
All topical treatments were applied with a pipette, using the pipette tip to facilitate spreading. Ten percent olive oil and DMSO were used to enable the active components to stay in place and facilitate transdermal migration, respectively. In the case of uninfected skins, the area of application was fixed to 2 cm2 and marked with a surgical skin marker (Covidien) for observation or tissue collection.
Skin tissue collection
For CFU determination, the whole infected area of the skin was harvested. For MPO assays and flow cytometry, skin tissues were harvested using an 8-mm biopsy punch (Miltex). For some assays with infected tissues, the diameter of the biopsy punch was aligned to the border of infection so that the harvested tissue includes equal portions of infected area and adjacent uninfected skin.
Enzymatic assays
For β-hexosaminidase assays, 2 × 104 cells seeded in a 96-well plate were incubated in Tyrode’s buffer [5.5 mM glucose, 0.5% bovine serum albumin (BSA), 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.3 mM CaCl2 (pH 7.4)] in presence of various concentrations of mastoparan or its analogs for 30 min at 37°C. Then, β-hexosaminidase activity in the supernatants was measured as described previously (5). Briefly, supernatants or cell lysates (lysed by 0.1% Triton X-100) were incubated with the substrate p-nitrophenyl-N-acetyl-β-d-glucosaminide in 0.1 M sodium citrate (pH 4.5) for 1 hour at 37°C. Then, 0.1 M carbonate buffer (pH 10.0) was added, and the product 4-p-nitrophenol was quantitated by measuring change in absorbance at 405 nm. The percentage degranulation was calculated from total β-hexosaminidase quantitated from cell lysates.
To determine skin MPO levels, skin tissues were collected in 500 μl of potassium phosphate buffer [50 mM (pH 6.5)] with 0.5% hexadecyltrimethylammonium bromide (HTAB) and then homogenized using zirconia silica beads and an automatic homogenizer and lysed by multiple freeze-thaw cycles. To determine peritoneal MPO levels, mouse peritoneums were lavaged using 5 ml of PBS with 10 μM EDTA. Cells from 1 to 2 ml of lavage were then centrifuged and resuspended in 200 μl of HTAB buffer and lysed by multiple freeze-thaw cycles. Then, 20 μl of the lysate supernatants was transferred in triplicate to a 96-well plate, and 200 μl of 50 mM potassium phosphate buffer (pH 6.0) with o-dianisidine dihydrochloride (0.167 mg/ml) and 0.0005% H2O2 was added. The MPO activity was measured three times at 1-min intervals at 450 nm. MPO content was calculated as units per gram tissue or units per peritoneum using a standard curve, which was established using a recombinant MPO standard.
CFU determination
Infected skin tissues were harvested and homogenized in 0.1% Triton X-100 using zirconia silica beads for three cycles of 1.5 min each using an automatic homogenizer. Homogenates were serially diluted and plated on LB agar plates, and the colonies were counted after overnight incubation at 37°C. The limit of detection was 25 CFU.
Immunofluorescence microscopy
Uninfected or infected skin tissues were fixed with 4% paraformaldehyde (PFA) and blocked and permeabilized with 0.3% Triton X-100 in 1% BSA-PBS with 5% goat serum. Then, the tissue samples were stained with anti-Ly6G (clone 1A8) and anti-CD31 antibodies at 4°C overnight, followed by incubation with AF647- and AF488-conjugated secondary antibodies (Jackson ImmunoResearch) and avidin-TRITC (tetramethylrhodamine) for 2 hours at room temperature. Stained whole skins were then mounted on a glass slide using ProLong Gold antifade mounting agent. Confocal images were taken using a Nikon ECLIPSE TE200 microscope.
Toluidine blue staining
To visualize granulated peritoneal MCs, peritoneal lavages were performed using 5 ml of PBS with 10 μM EDTA. Cells from 200 μl of lavage were cytospun on a glass slide, fixed with Carnoy’s fixative, stained with 5% toluidine blue, and viewed under a brightfield microscope. Fully or partially granulated cells were scored as granulated MCs (6, 26). At least 10 random fields were counted for granulated MCs, averaged, and the percentage of total cells was determined for each peritoneum. To visualize granulated MCs in skin tissues, skins from the dorsum or ear pinnae were removed and fixed with Carnoy’s fixative, stained with 5% toluidine blue, washed, and the whole skin was mounted on a glass slide for visualization and counting under microscope.
Mason’s trichrome staining
Skin tissues with scars were harvested 28 days after infection and flash frozen in OCT compound. Fifteen-micrometer sections were prepared from the frozen tissues and stained using a trichrome staining kit (Abcam) as per the manufacturer’s protocol. Scarred area was defined primarily by the absence of hair follicles along with increased collagen density (49).
Flow cytometry
To quantitate peritoneal neutrophils by flow cytometry, cells from 1 ml of lavage were washed with 1% BSA in PBS, incubated with normal serums and Fc receptor block (eBioscience), and stained with anti-Ly6G-PE (clone 1A8) (BD Biosciences) and anti–CD11b-APC (eBioscience) antibodies. To phenotype DCs, skin tissues or popliteal lymph nodes were minced and incubated for 30 min in collagenase A (100 U/ml; Sigma-Aldrich) in Hanks’ balanced salt solution containing FBS and DNase I. Single-cell suspensions were prepared by straining the digested tissues through a 70-μm cell straining filter (BD Biosciences). Cells were then washed with 1% BSA in PBS supplemented with FBS and 10 mM EDTA, blocked, and stained with fluorochrome-tagged antibodies to CD45, CD64, IA/IE, CD11b, CD11c, CD207, CD301b, and CD103 (BioLegend). Dead cells were detected using a Zombie Violet fixable viability kit (BioLegend). Cells stained with a single antibody and isotype controls were used as needed. To detect neutrophils in circulation, peripheral blood was collected in heparin tubes, stained with antibodies, and red blood cells were lysed using FACS lysing solution (BD Biosciences). Upon fixation with PFA, stained cells were analyzed by a BD FACSCalibur or FACSCanto II flow cytometer and FlowJo software version 10.1. Total cells in the peritoneal lavages or lymph nodes were counted using a hemocytometer.
Western blot
Cells were lysed with Western lysis buffer [150 mM NaCl, 0.1% NP-40, 0.5% sodium deoxycholate, 50 mM tris (pH 8.0)] supplemented with complete protease inhibitor cocktail (Roche), and total protein concentrations were determined by Bradford’s assay. Cell lysates were immediately processed for analysis. Lysate (75 μg) was separated on 4 to 15% mini-PROTEAN TGX precast gel and transferred to a nitrocellulose membrane in a semidry electrophoretic transfer cell (Bio-Rad). The membrane was blocked with 5% skimmed milk in TBST [50 mM tris (pH 8), 150 mM NaCl, and 0.05% Tween 20] and incubated with primary antibody in the same blocking solution for 15 hours at 4°C, followed by four washes in TBST. The membrane was then incubated with secondary antibody conjugated to horseradish peroxidase (HRP) for 1 hour at room temperature followed by washing in TBST. The protein bands were detected with an ECL Prime luminol reagent (Pierce, Thermo Fisher Scientific) in a gel documentation system (Chemi-Doc, Bio-Rad). Antibody dilutions were 1:1000 for rabbit–anti–phospho-PLCγ1, anti-PLCγ1, Flag (Cell Signaling Technology), and mouse–anti-rabbit–HRP (Roche), and 1:10,000 for anti-GAPDH-HRP (Proteintech).
MIC assay
The MIC was assayed by the broth microdilution method according to the procedures outlined by the Clinical and Laboratory Standards Institute (CLSI M07, 2012). Bacteria were grown in Mueller Hinton broth (Becton Dickinson) to log phase. The initial inoculum was then adjusted to obtain 5 × 105 CFU/ml and incubated in a microtiter plate, with a twofold serial dilution of the antimicrobial agent ranging from 2 mg/ml to 4 μg/ml in a 100-μl volume. Each concentration was tested in triplicate, and the microtiter plate was incubated at 37°C overnight. The MIC was taken as the lowest concentration of the antibacterial agent that completely inhibits visible bacterial growth.
Luciferase reporter assay
After 36 hours posttransfection, HEK293 cells were stimulated with mastoparan (0.17 and 0.34 μM) or left unstimulated for 4 to 6 hours. The cell lysates were measured for firefly luciferase activities specific to GPCR REs and constitutive Renilla luciferase activity using Dual-Glo luciferase assay kits (Promega) according to the manufacturer’s instructions. The total fold induction was calculated as fold of induction = firefly RLUinduced/Renilla firefly RLU (relative luminescence unit).
Cytotoxicity assay
Cytotoxicity of mastoparan and related peptides was determined using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega) according to the manufacturer’s protocol. Briefly, 2 × 104 L929 fibroblast cells (ATCC CCL-1) were incubated with a test peptide for 4 hours, and release of lactose dehydrogenase (LDH) in culture supernatant was measured. The LDH released spontaneously by cells incubated with media alone was subtracted from all data.
TLR/NLR ligand screening assay
Stimulation of TLR and NLR was tested using a PRR screening service provided by Invivogen (San Diego, CA). In this assay, PRR activation was determined by assessing nuclear factor κB (NF-κB) activation in HEK293 reporter cells expressing a given TLR or NLR. NF-κB activity was assessed by measurement of NF-κB–dependent secreted embryonic alkaline phosphatase at 650 nm. A mastoparan peptide was tested at a concentration of 50 μg/ml on seven different mouse TLRs (TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9) and mouse NOD1 and NOD2. The following ligands were used as positive controls for corresponding receptors: TLR2, HKLM (heat-killed Listeria monocytogenes) at 108 cells/ml; TLR3, poly(I:C) at 1 μg/ml; TLR4, E. coli K12 LPS (lipopolysaccharide) at 100 ng/ml; TLR5, Salmonella Typhimurium flagellin at 100 ng/ml; TLR7, CL097 at 1 μg/ml; TLR8, CL075 at 10 μg/ml + 10 μM poly(dT); TLR9, CpG ODN 1826 at 100 ng/ml; NOD1, C12-iE-DAP at 1 μg/ml; NOD2, L18-MDP at 100 ng/ml.
Depletion of neutrophils
Mice were treated intraperitoneally with 500 μg of anti-Ly6G (1A8) or control antibodies (2A3) (BioXCell) in 100-μl volume 24 hours after infection with S. aureus. Peripheral blood was collected 48 hours after injection of the antibodies to confirm depletion of neutrophils. Neutrophils were also quantitated in infected or uninfected skin by an MPO assay as described above.
Peptide design for structure-function analysis
Mastoparan 17 is a known analog of mastoparan, which has neither MC-degranulating (27) nor antimicrobial activity (MIC against S. aureus >2000 μg/ml). Since mastoparan 17 has substitutions at positions 6 and 13 (INLKAKAALAKKLL-NH2), we first generated analogs with one substitution (either position 6 or position 13) to determine which position is more critical. While MP-13L peptide (leucine in place of isoleucine at position 13) retained both antimicrobial and MC-degranulating activities, MP-6K (lysine in place of leucine at position 13) completely lost them (table S1). Finding position 6 to be critical, we then synthesized a series of mastoparan analogs with various single amino acid substitutions at this position with an aim to obtain a peptide with no degranulating but comparable antimicrobial activity. When the degranulating capacities of these peptide analogs were compared with mastoparan, MP-6W (tryptophan at position 6) was slightly better, degranulation by MP-6I (isoleucine at position 6) was significantly reduced but still detectable (Fig. 4B), and most of the others had no detectable activity (table S1). On the other hand, when the analogs were tested for MIC against S. aureus, only MP-6I retained antimicrobial activity similar to mastoparan (Fig. 4C). Other substitutions tested either reduced or completely lost antimicrobial activity (table S1). Therefore, MP-6I was chosen as the mastoparan analog with no degranulating but similar antimicrobial activity. Single amino acid substitutions did not yield to the alternate mastoparan analog, which has no antimicrobial activity but similar MC-degranulating activity. Therefore, we took another strategy to generate a series of completely unrelated MCA peptides, which were rich in tryptophans and arginines and devoid of any antimicrobial activity. It has previously been demonstrated that while arginines facilitate interaction with membranes, lysines disrupt them (50, 51). In Vespula mastoparan, there is no arginine; however, there are single or multiple arginines present in human cationic peptide MCAs such as kallidin, LL-37, or substance P. Therefore, we developed multiple mastoparan-inspired peptides, where lysines were replaced with arginines to nullify the membrane-disrupting activity. The asparagine at position 2 was also replaced with arginine to increase cationic property of the peptide. Other amino acids were replaced with tryptophans to provide ample motifs close to tetrahydroisoquinoline [which has recently been speculated to be the motif critical for MRGPRX2 activation (12)] and to increase hydrophobicity. These modifications resulted in various novel peptides including Duke Mast F (mastoparan, INLKALAALAKKIL→INLRALAALARRIL→IRLRALAALARRIL→WRWRWWWWWWRRWW). Once tested for MC-degranulating and antimicrobial properties, Duke Mast F was selected as the peptide with similar degranulating but no antimicrobial activity (Fig. 4, B and C).
Statistical analyses
Statistical significance was determined by unpaired two-tailed Student’s t tests where only two groups existed, or by one-way ANOVA with Dunnett’s or Tukey’s posttest. Differences between groups were considered significant at P < 0.05. Analyses were performed using GraphPad Prism 5.0 and Microsoft Excel 2010.
Supplementary Material
Acknowledgments
We thank A. Roers for the Mcpt5-Cre+ iDTR+ mice and M. Cook for support with flow cytometry. Funding: This work was funded by NIH grants U01-AI082107, R01-AI096305, and R56-DK095198 and a block grant from Duke-NUS Graduate Medical School, Singapore. This work was funded by the National Medical Research Council, Ministry of Health, Singapore (grant numbers NMRC/CIRG/1357/2013 and NMRC/CIRG/1467/2017). Author contributions: M.A., Y.R.M., and S.N.A. conceived the project. M.A. designed and performed majority of the experiments and data analyses. Y.R.M. conducted the initial study design and experiments. H.W.C. assisted with generating Mcpt5-Cre+ iDTR+ mice, cytokine profiling, and confocal microscopy. C.A.S. assisted in various in vitro and in vivo studies. P.B. and Z.D.B. performed several in vitro experiments. M.A. and S.N.A. wrote the manuscript, and H.W.C., H.F.S., and S.L.C. provided insightful suggestions. All authors contributed to manuscript review. Competing interests: S.N.A. and H.F.S. are inventors on a patent related to this work (USPTO #9,301,999; filed, 28 September 2010; issued, 5 April 2016). S.N.A. is the cofounder and chief scientific officer for Mastezellen Bio Inc. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaav0216/DC1
Table S1. Antibacterial and MC-activating capacities of mastoparan analog peptides.
Fig. S1. Depletion of CTMCs.
Fig. S2. Correlation between CFU and lesion size.
Fig. S3. Depletion of neutrophils.
Fig. S4. Receptor specificity of mastoparan.
Fig. S5. Detection of granulated MCs by toluidine blue staining.
Fig. S6. Comparison of mastoparan with triple antibiotic.
Fig. S7. Mastoparan treatment in neutrophil-depleted mice.
Fig. S8. Flow cytometry plots from skin samples.
Fig. S9. Flow cytometry plots from lymph node samples.
REFERENCES AND NOTES
- 1. Abraham S. N., St. John A. L., Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10, 440–452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall J. S., Mast-cell responses to pathogens. Nat. Rev. Immunol. 4, 787–799 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Shelburne C. P., Abraham S. N., The mast cell in innate and adaptive immunity. Adv. Exp. Med. Biol. 716, 162–185 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Gendrin C., Vornhagen J., Ngo L., Whidbey C., Boldenow E., Santana-Ufret V., Clauson M., Burnside K., Galloway D. P., Waldorf K. M. A., Piliponsky A. M., Rajagopal L., Mast cell degranulation by a hemolytic lipid toxin decreases GBS colonization and infection. Sci. Adv. 1, e1400225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. St. John A. L., Rathore A. P. S., Yap H., Ng M.-L., Metcalfe D. D., Vasudevan S. G., Abraham S. N., Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc. Natl. Acad. Sci. U.S.A. 108, 9190–9195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi H. W., Brooking R., Neupane S., Lee C.-J., Miao E. A., Staats H. F., Abraham S. N., Salmonella typhimurium impedes innate immunity with a mast-cell-suppressing protein tyrosine phosphatase, SptP. Immunity 39, 1108–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silver L. L., Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hancock R. E. W., Nijnik A., Philpott D. J., Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 10, 243–254 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Del-Rio-Navarro B. E., Espinosa Rosales F., Flenady V., Sienra-Monge J. J., Immunostimulants for preventing respiratory tract infection in children. Cochrane Database Syst Rev. CD004974 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero C. D., Varma T. K., Hobbs J. B., Reyes A., Driver B., Sherwood E. R., The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection. Infect. Immun. 79, 3576–3587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramanian H., Gupta K., Guo Q., Price R., Ali H., Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: Resistance to receptor phosphorylation, desensitization, and internalization. J. Biol. Chem. 286, 44739–44749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNeil B. D., Pundir P., Meeker S., Han L., Undem B. J., Kulka M., Dong X., Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519, 237–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaudenzio N., Sibilano R., Marichal T., Starkl P., Reber L. L., Cenac N., McNeil B. D., Dong X., Hernandez J. D., Sagi-Eisenberg R., Hammel I., Roers A., Valitutti S., Tsai M., Espinosa E., Galli S. J., Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Invest. 126, 3981–3998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daum R. S., Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N. Engl. J. Med. 357, 380–390 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Malaviya R., Ikeda T., Ross E., Abraham S. N., Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature 381, 77–80 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Hirai Y., Yasuhara T., Yoshida H., Nakajima T., Fujino M., Kitada C., A new mast cell degranulating peptide “mastoparan” in the venom of Vespula lewisii. Chem. Pharm. Bull. 27, 1942–1944 (1979). [DOI] [PubMed] [Google Scholar]
- 17.McCaig L. F., McDonald L. C., Mandal S., Jernigan D. B., Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg. Infect. Dis. 12, 1715–1723 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran G. J., Krishnadasan A., Gorwitz R. J., Fosheim G. E., McDougal L. K., Carey R. B., Talan D. A.; EMERGEncy ID Net Study Group , Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355, 666–674 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Dudeck A., Dudeck J., Scholten J., Petzold A., Surianarayanan S., Köhler A., Peschke K., Vöhringer D., Waskow C., Krieg T., Müller W., Waisman A., Hartmann K., Gunzer M., Roers A., Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Subramanian H., Kashem S. W., Collington S. J., Qu H., Lambris J. D., Ali H., PMX-53 as a dual CD88 antagonist and an agonist for Mas-related gene 2 (MrgX2) in human mast cells. Mol. Pharmacol. 79, 1005–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobilka B. K., G protein coupled receptor structure and activation. Biochim. Biophys. Acta 1768, 794–807 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian H., Gupta K., Lee D., Bayir A. K., Ahn H., Ali H., β-Defensins activate human mast cells via Mas-related gene X2. J. Immunol. 191, 345–352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez-Cambronero J., Horn J., Paul C. C., Baumann M. A., Granulocyte-macrophage colony-stimulating factor is a chemoattractant cytokine for human neutrophils: Involvement of the ribosomal p70 S6 kinase signaling pathway. J. Immunol. 171, 6846–6855 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Maus U. A., Waelsch K., Kuziel W. A., Delbeck T., Mack M., Blackwell T. S., Christman J. W., Schlondorff D., Seeger W., Lohmeyer J., Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: Role of the CCL2-CCR2 axis. J. Immunol. 170, 3273–3278 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Sadik C. D., Kim N. D., Luster A. D., Neutrophils cascading their way to inflammation. Trends Immunol. 32, 452–460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLachlan J. B., Hart J. P., Pizzo S. V., Shelburne C. P., Staats H. F., Gunn M. D., Abraham S. N., Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat. Immunol. 4, 1199–1205 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Higashijima T., Burnier J., Ross E. M., Regulation of Gi and Go by mastoparan, related amphiphilic peptides, and hydrophobic amines. Mechanism and structural determinants of activity. J. Biol. Chem. 265, 14176–14186 (1990). [PubMed] [Google Scholar]
- 28.de Oliveira S., Rosowski E. E., Huttenlocher A., Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 16, 378–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katsu T., Kuroko M., Morikawa T., Sanchika K., Yamanaka H., Shinoda S., Fujita Y., Interaction of wasp venom mastoparan with biomembranes. Biochim. Biophys. Acta 1027, 185–190 (1990). [DOI] [PubMed] [Google Scholar]
- 30.Zasloff M., Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Singer A. J., McClain S. A., Persistent wound infection delays epidermal maturation and increases scarring in thermal burns. Wound Repair Regen. 10, 372–377 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Yang B., Suwanpradid J., Sanchez-Lagunes R., Choi H. W., Hoang P., Wang D., Abraham S. N., MacLeod A. S., IL-27 facilitates skin wound healing through induction of epidermal proliferation and host defense. J. Invest. Dermatol. 137, 1166–1175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shelburne C. P., Nakano H., St John A. L., Chan C., McLachlan J. B., Gunn M. D., Staats H. F., Abraham S. N., Mast cells augment adaptive immunity by orchestrating dendritic cell trafficking through infected tissues. Cell Host Microbe 6, 331–342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumamoto Y., Linehan M., Weinstein J. S., Laidlaw B. J., Craft J. E., Iwasaki A., CD301b+ dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 39, 733–743 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osterholzer J. J., Curtis J. L., Polak T., Ames T., Chen G. H., McDonald R., Huffnagle G. B., Toews G. B., CCR2 mediates conventional dendritic cells recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J. Immunol. 181, 610–620 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sokol C. L., Luster A. D., The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 7, a016303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henri S., Guilliams M., Poulin L. F., Tamoutounour S., Ardouin L., Dalod M., Malissen B., Disentangling the complexity of the skin dendritic cell network. Immunol. Cell Biol. 88, 366–375 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Artuc M., Steckelings U. M., Henz B. M., Mast cell-fibroblast interactions: Human mast cells as source and inducers of fibroblast and epithelial growth factors. J. Invest. Dermatol. 118, 391–395 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Antsiferova M., Martin C., Huber M., Feyerabend T. B., Forster A., Hartmann K., Rodewald H. R., Hohl D., Werner S., Mast cells are dispensable for normal and activin-promoted wound healing and skin carcinogenesis. J. Immunol. 191, 6147–6155 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Nauta A. C., Grova M., Montoro D. T., Zimmermann A., Tsai M., Gurtner G. C., Galli S. J., Longaker M. T., Evidence that mast cells are not required for healing of splinted cutaneous excisional wounds in mice. PLOS ONE 8, e59167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chervenick P. A., Boggs D. R., Decreased neutrophils and megakaryocytes in anemic mice of genotype W/W. J. Cell. Physiol. 73, 25–30 (1969). [DOI] [PubMed] [Google Scholar]
- 42.Nigrovic P. A., Gray D. H., Jones T., Hallgren J., Kuo F. C., Chaletzky B., Gurish M., Mathis D., Benoist C., Lee D. M., Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 173, 1693–1701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li M. L., Liao R. W., Qiu J. W., Wang Z. J., Wu T. M., Antimicrobial activity of synthetic all-D mastoparan M. Int. J. Antimicrob. Agents 13, 203–208 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Svenson J., Brandsdal B. O., Stensen W., Svendsen J. S., Albumin binding of short cationic antimicrobial micropeptides and its influence on the in vitro bactericidal effect. J. Med. Chem. 50, 3334–3339 (2007). [DOI] [PubMed] [Google Scholar]
- 45.McLachlan J. B., Shelburne C. P., Hart J. P., Pizzo S. V., Goyal R., Brooking-Dixon R., Staats H. F., Abraham S. N., Mast cell activators: A new class of highly effective vaccine adjuvants. Nat. Med. 14, 536–541 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Kunder C. A., St John A. L., Li G., Leong K. W., Berwin B., Staats H. F., Abraham S. N., Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J. Exp. Med. 206, 2455–2467 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshino N., Endo M., Kanno H., Matsukawa N., Tsutsumi R., Takeshita R., Sato S., Polymyxins as novel and safe mucosal adjuvants to induce humoral immune responses in mice. PLOS ONE 8, e61643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bunce C., Wheeler L., Reed G., Musser J., Barg N., Murine model of cutaneous infection with gram-positive cocci. Infect. Immun. 60, 2636–2640 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peranteau W. H., Zhang L., Muvarak N., Badillo A. T., Radu A., Zoltick P. W., Liechty K. W., IL-10 overexpression decreases inflammatory mediators and promotes regenerative healing in an adult model of scar formation. J. Invest. Dermatol. 128, 1852–1860 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Mitchell D. J., Kim D. T., Steinman L., Fathman C. G., Rothbard J. B., Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56, 318–325 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Fischer D., Li Y., Ahlemeyer B., Krieglstein J., Kissel T., In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 24, 1121–1131 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaav0216/DC1
Table S1. Antibacterial and MC-activating capacities of mastoparan analog peptides.
Fig. S1. Depletion of CTMCs.
Fig. S2. Correlation between CFU and lesion size.
Fig. S3. Depletion of neutrophils.
Fig. S4. Receptor specificity of mastoparan.
Fig. S5. Detection of granulated MCs by toluidine blue staining.
Fig. S6. Comparison of mastoparan with triple antibiotic.
Fig. S7. Mastoparan treatment in neutrophil-depleted mice.
Fig. S8. Flow cytometry plots from skin samples.
Fig. S9. Flow cytometry plots from lymph node samples.