

Abstract
Multiplex isobaric tags have become valuable tools for high-throughput quantitative analysis of complex biological samples in discovery-based proteomics studies. Hybrid labeling strategies that pair stable isotope mass difference labeling with multiplex isobaric tag-based quantification further facilitate these studies by greatly increasing multiplexing capability. In this work, we present a cost-effective chemical labeling approach that couples duplex stable isotope dimethyl labeling with our custom 12-plex N,N-dimethyl leucine (DiLeu) isobaric tags in a combined precursor isotopic labeling and isobaric tagging (cPILOT) strategy that is compatible with a wide variety of biological samples and permits 24-plex quantification in a single LC-MS/MS experiment. We demonstrate the utility of the DiLeu cPILOT approach by labeling yeast digests and performing proof-of-principle quantification experiments on the Orbitrap Fusion Lumos.
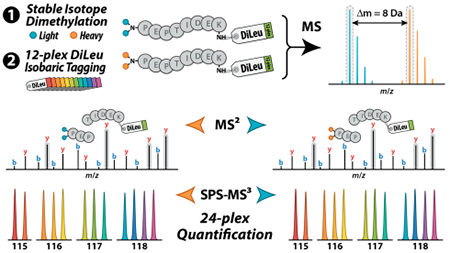
Stable-isotope labeling permits multiplexed comparative analysis of proteins and peptides by liquid chromatography–mass spectrometry (LC-MS). Heavy isotopes are differentially incorporated either metabolically or chemically onto two or more samples that are pooled for parallel relative quantitative analysis in a single LC-MS experiment. In mass difference approaches, which include stable-isotope labeling by amino acids in cell culture (SILAC)1–3 and dimethyl labeling,4 heavy- and light-labeled peptide ions are detected in MS precursor scans, and the areas of their extracted ion chromatogram signal intensities are compared to determine relative peptide abundance. In isobaric labeling techniques, utilizing amine-reactive chemical tags such as tandem mass tags (TMT)5,6 and isobaric tags for relative and absolute quantification (iTRAQ),7,8 differentially labeled peptides incur the same mass shift and are measured as single precursors in the MS1 scan. Upon tandem MS (MS/MS) analysis, discrete reporter ions are generated in the low m/z region of MS2 spectra, the intensities of which reflect the labeled peptides’ abundances in each sample for quantitative comparison. Traditional mass difference approaches are generally limited to duplex and triplex comparisons due to increased mass spectral complexity, while reporter ion-based methods yield much greater multiplexing capacity and analytical throughput. However, reporter ion-based methods suffer from distortion of quantitative ratios in complex samples due to coisolation and cofragmentation of interfering precursors alongside target precursors during MS/MS acquisition.9 MS3 acquisition has proven to be an effective way of obtaining more accurate reporter ion spectra,10 and modern Orbitrap platforms with MSn acquisition modes are equipped with synchronous precursor selection (SPS) of multiple MS2 fragment ions to ensure adequate reporter ion signal intensities.11,12
Hybrid labeling strategies combine isotopic mass difference labeling with isobaric tag labeling to offer a straightforward means of substantially increasing multiplexing. These “hyper-plexing” techniques employ a mass difference at the MS1 level to further parallelize two or three sets of isobaric tag-labeled samples within a single pooled sample, effectively doubling or tripling the number of samples quantified via reporter ions at the MS2 or MS3 level in a single LC-MS experiment. This was initially demonstrated by pairing triplex SILAC with 6-plex TMT to achieve 18-plex quantification.13 By also employing custom medium and heavy TMT variants, 54-plex quantification was shown with a targeted proteomics experiment.14 Another technique called combined precursor isotopic labeling and isobaric tagging (cPILOT) combines duplex stable isotope dimethyl labeling of peptide N-termini at low pH with 6-plex TMT labeling of lysine residues at high pH to permit 12-plex quantification.15–19 An advantage of the cPILOT strategy is that the chemical labeling steps are compatible with samples from any origin, enabling studies of human fluids and tissues or other organisms for which metabolic labeling is impractical. The isotopic reagents used for dimethylation are inexpensive compared to SILAC amino acids, and this rapid peptide derivatization step with cleanup can be completed in less than an hour, in contrast to the several days necessary for metabolic incorporation with heavy amino acids. On the other hand, it has been observed that SILAC outperforms dimethyl labeling in numbers of protein and peptide identifications by ~20% due to losses of hydrophobic peptides following the dimethylation step.20 With any hyperplexing approach, the inherent necessity of performing MS/MS acquisition on two or three precursors for each peptide to achieve full multiplexing reduces proteomic depth; modern instrumentation with high sampling rates is preferred in order to alleviate this trade-off.
These highly multiplexed isobaric tag-based approaches facilitate high-throughput analysis of many biological conditions and replicates to greatly decrease instrument run-time, but the high cost of commercial isobaric tag reagents presents a prohibitive financial barrier. For example, a 10-plex TMT set suitable for 30 samples costs a few thousand dollars. The need for a cost-effective alternative led us to develop our own custom N,N-dimethyl leucine (DiLeu) isobaric tags that can be synthesized in-house at high yield with readily available isotopic starting materials.21 Originally developed as a 4-plex set, the current generation of DiLeu reagents permits 12-plex quantification via high-resolution MS/MS acquisition.22 The 12-plex DiLeu reporter ions generated at 115–118 m/z differ in mass by a minimum of ~6 mDa and are baseline resolved at resolving powers of 30K (at 400 m/z) or 50K (at 200 m/z) on Orbitrap platforms. The DiLeu tags are structurally similar to commercial tags and feature comparable performance at a cost of just a couple of dollars per sample. Other notable merits include the small mass of the tag, the greater signal intensity of generated reporter ions compared to iTRAQ and TMT, and enhanced collision-induced fragmentation of labeled peptides at reduced collision energies.23
In this work, we pair our custom DiLeu isobaric tags with stable isotope dimethyl labeling in a cost-effective cPILOT approach to achieve 24-plex quantification in a single LC-MS/MS experiment. Yeast tryptic digests are subjected to light and heavy dimethyl labeling of peptide N-termini at acidic pH followed by labeling of lysine residues with 12-plex DiLeu isobaric tags at basic pH. Proof-of-principle DiLeu cPILOT experiments are performed on the Orbitrap Fusion Lumos using an SPS-MS3 acquisition method.
MATERIALS AND METHODS
Chemicals.
Heavy isotopic reagents used for the synthesis of labels were purchased from Isotec (Miamisburg, OH). Yeast protein extract and MS-grade enzymes were purchased from Promega (Madison, WI). ACS grade and Optima LC/MS grade solvents were purchased from Fisher Scientific (Pittsburgh, PA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Yeast Protein Extract Digestion.
S. cerevisiae protein extracts were digested by trypsin/Lys-C mix according to the manufacturer’s protocol and desalted via C18 SPE. Digested peptides were divided into two equal aliquots and dried in vacuo.
Stable Isotope Dimethyl Labeling of Peptide N-Termini.
Duplex dimethyl labeling of peptide N-termini was performed in 1.25% acetic acid pH < 3 by addition of CH2O (to 60 mM) and NaBH3CN (to 24 mM) or 13C2H2O and NaB2H3CN for light and heavy dimethylation, respectively. Following incubation at ambient temperature for 10 min, labeling was quenched by addition of 1% NH3 and incubation for 5 min. Samples were acidified with 10% TFA to pH < 3 and desalted via C18 SPE. Light and heavy samples were each divided into 12 equal aliquots and dried in vacuo.
DiLeu Labeling of Lysine Residues.
12-plex DiLeu isobaric tags were synthesized in-house as reported previously.22 Following N-terminal-specific dimethyl labeling, 12-plex DiLeu labeling of lysine residues was performed in 60:40 ACN/0.5 M triethylammonium bicarbonate buffer pH 8.5 by addition of activated label in dry DMF at a label to protein digest ratio of 15:1 (w/w). Following incubation with vortexing at ambient temperature for 30 min, the labeling reaction was quenched by addition of hydroxylamine to 0.25% v/v. DiLeu-labeled peptide samples were combined in known ratios of 1:1:1:1:1:1:1:1:1:1:1:1 (1) and 16:8:4:2:1:10:10:1:2:4:8:16 (16) for each of the light and heavy dimethylated samples. The light (L) and heavy (H) dimethylated samples were then combined in equal amounts to create four samples: (1) L1:H1; (2) L16:H16; (3) L16:H1; (4) L16:H1. Pooled samples were fractionated into five fractions by SCX SPE and desalted via C18 SPE.
NanoLC-MS2.
Samples were analyzed by nanoLC-MS/MS using a Dionex UltiMate 3000 UPLC system coupled to a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer. Labeled peptide samples were dried in vacuo and dissolved in 3% ACN, 0.1% formic acid in water. Peptides were loaded onto a 75 μm inner diameter microcapillary column fabricated with an integrated emitter tip and packed with 15 cm of BEH C18 particles (1.7 μm, 130 Å, Waters). Mobile phase A was composed of water and 0.1% formic acid. Mobile phase B was composed of ACN and 0.1% formic acid. Separation was performed using a gradient elution of 5% to 35% mobile phase B over 120 min at a flow rate of 300 nL/min. FT-MS survey scans of peptide precursors from 350 to 1500 m/z were performed in the Orbitrap at RP 120K (at 200 m/z) with an AGC target of 2 × 105 and maximum injection time of 50 ms. Using a data-dependent acquisition (DDA) cycle time of 3 s, the most intense precursors were selected by quadrupole isolation for turbo scan CID IT-MS2 analysis in the linear ion trap with an isolation width of 0.5 Da, an NCE of 35, an AGC target of 1 × 104, and a maximum injection time of 100 ms. Light and heavy precursor partners separated by 8.044 Da (±25 ppm tolerance) within 20–100% intensity of the opposite partner were targeted for selection. DDA was not restricted only to precursors with partners. Selected precursors were subject to dynamic exclusion for 20 s with a mass tolerance of ±10 ppm. Following each MS2 scan, the top four fragment ions within 400–1200 m/z were coisolated in the ion trap using synchronous precursor selection for HCD FT-MS3 acquisition in the Orbitrap at RP 60K with an isolation width of 2.0 Da, an NCE of 55, an AGC target of 5 × 104, a maximum injection time of 120 ms, and detection mass range of 100–400 m/z. The precursor was excluded from MS3 selection with a tolerance of −18 to +5 m/z. Current Fusion tune control software, made available after these experiments were performed, permits acquisition at RP 50K for resolving 6 mDa mass differences between reporter ions.
Data Analysis.
Mass spectra were processed using Proteome Discoverer (PD; version 2.1, Thermo Scientific) to identify and quantify proteins and peptides. Raw files were searched against the UniProt Saccharomyces cerevisiae complete database using Sequest HT. Searches were performed with a precursor mass tolerance of 25 ppm and a fragment mass tolerance of 0.6 Da. Static modifications consisted of carbamidomethylation of cysteine residues (+57.02146 Da), either light (+28.03130 Da) or heavy (+36.07567 and +35.06940 Da) dimethylation on peptide N-termini, and DiLeu (+145.12801 Da) on lysine (K) residues. Dynamic modifications consisted of oxidation of methionine residues (+15.99492 Da), deamidation of asparagine and glutamine residues (+0.98402 Da), and acetylation (+42.01057 Da) of protein N-termini. Peptide spectral matches (PSMs) were validated on the basis of q-values to 1% FDR using percolator. Quantification of reporter ions in MS3 spectra was performed in PD using an integration tolerance of 20 ppm for the most confident centroid. Only the PSMs that contained all 12 reporter ions were considered, and protein quantitative ratios were determined using a minimum of one quantified peptide. Reporter ion ratio values for protein groups were exported to Excel workbook format, and isotopic interference correction factors, determined using the measured isotopic abundances for each channel and with respect to the mixing ratios, were applied as described previously.22 Relative abundances were calculated by dividing each channel’s intensity by the sum of intensities.
RESULTS AND DISCUSSION
The cPILOT workflow offers a simple and accessible chemical labeling approach to double the throughput of isobaric tag-based quantitative proteomics experiments. To further expand the multiplexing capacity of our DiLeu isobaric tags, we apply this strategy in tandem with 12-plex DiLeu to achieve 24-plex quantification. The DiLeu cPILOT experimental workflow is shown in Figure 1. Protein tryptic digest samples first undergo stable isotope dimethyl labeling at pH < 3 to selectively derivatize N-terminal amines with either light [L; –(CH3)2] or heavy [H; –(13C2H3)2] dimethyl groups and impart an 8 Da mass difference between the labeled peptides. Light and heavy dimethylated peptides are then labeled at their lysine side chain amines with 12-plex DiLeu isobaric tags at pH ~8. Samples are pooled for LC-MS/MS analysis on the Orbitrap Fusion Lumos. Targeted MS2 acquisition is employed to sequentially isolate and fragment both precursor partners of each peptide in FT-MS spectra, based on their mass difference of 8 Da, in an effort to collect complete quantitative data, and SPS-MS3 acquisition mitigates distortion of reporter ion abundances caused by precursor coisolation. Peptides are identified by their CID fragment ions in IT-MS2 spectra, and quantification is performed via reporter ions in FT-MS3 spectra acquired at RP 60K generated by HCD of y ions containing the DiLeu tag.
Figure 1.
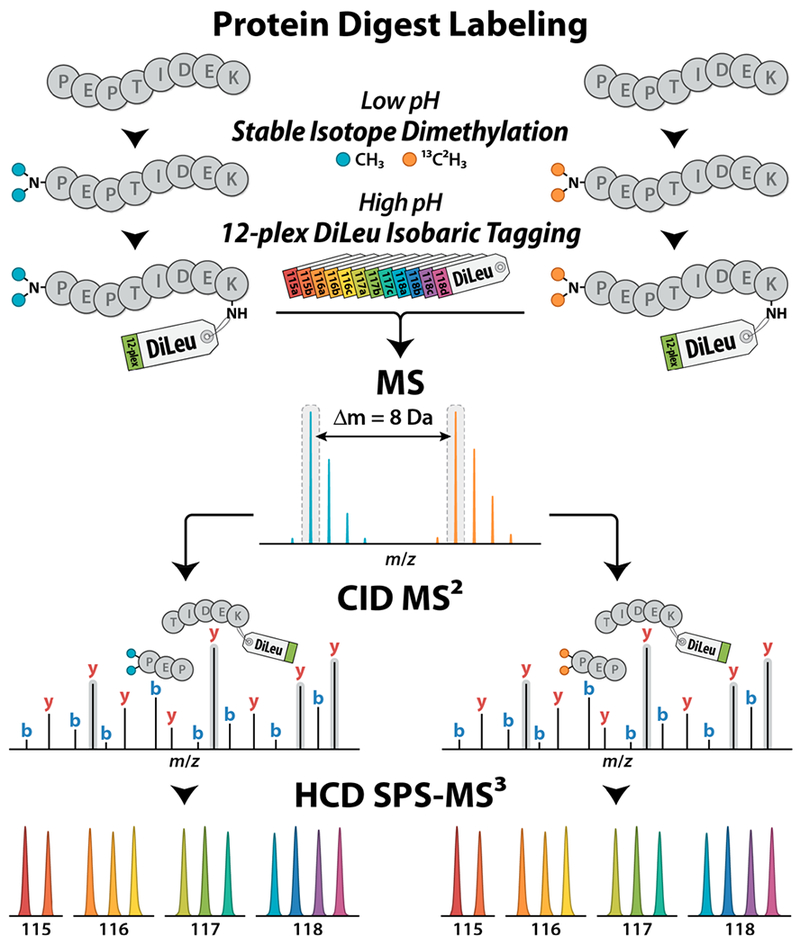
DiLeu cPILOT experimental workflow. Protein digest samples undergo stable isotope dimethyl labeling of peptide N-termini with either light [–(CH3)2] or heavy [–(13C2H3)2] dimethyl groups at low pH followed by labeling of lysine residues with 12-plex DiLeu isobaric tags at high pH. Pooled samples are analyzed by LC-MS/MS on the Orbitrap Fusion Lumos using CID MS2 acquisition of peak pairs for peptide sequence identification and HCD SPS-MS3 acquisition for accurate 24-plex quantification via DiLeu reporter ions.
The labeling scheme for cPILOT necessitates that peptides contain a lysine residue for isobaric tag labeling and quantification, and quantification by MS3 requires that selected fragment ions contain the tag to produce reporter ions in MS3 spectra. Digestion by Lys-C, as opposed to trypsin, would ensure that all peptides contain lysine for isobaric tagging and quantification, and in a typical isobaric tagging method, both b and y ions would contain the tag and generate reporter ions in MS3 spectra.10 However, peptides are dimethylated at their N-termini in the cPILOT workflow, so only y ions are viable for quantification by MS3 for both Lys-C and trypsin. Additionally, compared to trypsin, Lys-C generates peptides with longer sequence lengths that are less successfully identified using CID fragmentation, resulting in fewer numbers of identified peptides and proteins.16,24,25 While tryptic peptides with arginine at their C-termini do not contribute quantitative information, their analysis improves protein identification confidence. Trypsin was used in this work based on its greater proteome coverage compared to Lys-C.
Quantification by hybrid labeling strategies necessitates that both light and heavy precursors be acquired by MS/MS to achieve full multiplexing, and MS3 acquisition is required to mitigate coisolation interference effects that compromise the accuracy of reporter ion quantification. To evaluate the impact of these aspects of the DiLeu cPILOT acquisition strategy on proteomic depth, rate of quantification, and quantitative precision, we prepared light (L) and heavy (H) dimethylated yeast protein extract digests labeled with 12-plex DiLeu and combined them in unity across all channels to create an “L” sample, “H” sample, and a pooled “LH” sample at equal overall peptide concentration. Following SCX cleanup, each sample was analyzed by LC-MS/MS using an MS2-only acquisition method and an SPS-MS3 acquisition method on the Orbitrap Fusion Lumos. In terms of acquired MS2 scans, the duty cycle efficiencies of the two methods differed by about 20%; for every ten HCD FT-MS2 scans (RP 60K) acquired by the MS2 method, the SPS-MS3 method acquired eight CID IT-MS2 scans (turbo scan). The numbers of proteins, peptides, and PSMs identified (reported as total identifications and identifications with lysine residues) and quantified in each sample are summarized in Figure S1. The average numbers of proteins, unique peptide sequences, and PSMs identified in the three samples by the MS3 method were 74%, 64%, and 55%, respectively, of the MS2 method. As anticipated, the less efficient duty cycle of the MS3 method reduced proteomic depth compared to the MS2 method, and redundant sampling of peak pairs in the LH sample resulted in fewer protein and peptide identifications compared to the L or H samples. Overall, the rate of quantification of PSMs, calculated by dividing the number of quantified PSMs by the number of PSMs containing a DiLeu-labeled lysine residue, for the MS2 and MS3 methods was 97% and 88%, respectively. Despite this high success rate, just 50% and 40% of the total quantified peptides were quantified by both partners for the MS2 and MS3 methods, respectively, indicating that precursor partners were sometimes either not selected or selected at low signal levels. The median relative abundances and average signal-to-noise ratio of reporter ion intensities of all quantified PSMs for the MS2 and MS3 acquisitions of the LH sample are plotted against each other in Figure S2. Reporter ion abundances closely match the expected unity mixing ratios within 5% across all channels for both the MS2 and MS3 methods, with respective average coefficients of variation (CV) of 9.8% and 16.5%. Comparing the pooled LH sample to the individually acquired L and H samples reveals the impact of choosing 24-plex cPILOT over two 12-plex experiments; the pooled LH sample achieved approximately 65%, 57%, and 55% of the combined number of proteins, peptides, and PSMs, respectively, identified by separate L and H sample runs (MS3 method). The numbers of proteins, unique peptides, and PSMs quantified across all 24 channels in the pooled LH sample were 80%, 52%, and 54%, respectively, of the L and H samples acquired as separate runs. Thus, compared to two 12-plex experiments, halving the instrument run-time with a single 24-plex experiment yields just over half the number of identifications and quantifications of peptides and PSMs while achieving two-thirds of the protein identifications and 80% of the protein quantifications.
To further evaluate the quantitative performance achievable by the DiLeu cPILOT strategy, we prepared dimethylated yeast digests, labeled them with 12-plex DiLeu, and combined them in unity (1) and 16:8:4:2:1:10:10:1:2:4:8:16 (16) ratios between channels. The light and heavy samples were combined in equal amounts to create two pooled samples, “L1:H1” and “L16:H16”, at equal overall peptide concentration. Samples were fractionated by SCX into five fractions with the aim of partially counteracting the increase in complexity resulting from mass difference labeling. Each set of samples was analyzed on the Fusion Lumos using the SPS-MS3 acquisition method, and targeted MS2 selection of both precursor partners was enabled. The numbers of proteins, peptides, and PSMs identified and quantified in the L1:H1 sample, along with their distribution as either shared or exclusive to light or heavy, are summarized in Figure 2A. Out of 1953 proteins, 1351 were identified with at least one quantifiable peptide containing lysine, and of those, 1232 (91%) were quantified with complete light or heavy 12-plex data; 1121 (83%) were quantified across all 24 channels. This high rate of fully quantified proteins may aid in justifying the trade-off of identification numbers for highly multiplexed quantification, particularly for large-scale applications in which high sample throughput and quantification may be preferred over deep proteome coverage. For unique peptides, 5344 out of 10 064 contained lysine, and of those, 4605 (86%) were quantified with complete light or heavy 12-plex data; 2629 (49%) were quantified across all 24 channels. The median relative abundances of reporter ion intensities of all quantified PSMs, plotted in Figure 2B,C, closely match the expected mixing ratios within ±4.1% and ±7.4% across all channels for L1:H1 and L16:H16, respectively, with average CVs of 18% for both, showing that the quantitative performance of this cPILOT approach is in line with traditional isobaric tag approaches. Example spectra of peptide dimethyl-DSILEVLK-DiLeu detected in the L1:H1 sample are shown in Figure S3.
Figure 2.
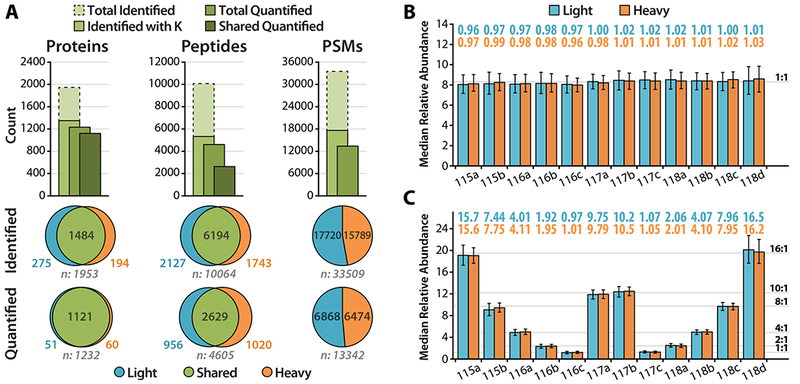
(A) The numbers of proteins, unique peptides, and PSMs identified and quantified in the labeled yeast digest sample L1:H1, along with their distributions as either shared or exclusive to light or heavy, are summarized. The median relative abundances of quantified PSMs are plotted for labeled yeast digest samples (B) L1:H1 and (C) L16:H16. Error bars mark the interquartile range.
Complex samples suffer distortion of reporter ion abundances measured in MS2 spectra due to ubiquitous coisolation of near-isobaric contaminants along with target precursors during MS/MS acquisition.9 In hyperplexed samples, the presence of light and heavy precursor peaks for every peptide exacerbates this phenomenon due to interfering heavy-labeled peptides that overlap with other light-labeled peptides and vice versa, making SPS-MS3 acquisition even more imperative for accurate quantification. However, coisolation interference effects are not made apparent with the L1:H1 and L16:H16 samples due to the like mixing ratios for every channel for both light and heavy, so we prepared and analyzed L1:H16 and L16:H1 samples to better evaluate the efficacy of SPS-MS3 in eliminating reporter ion abundance distortion. These mixtures replicate the two-proteome model used to impart discretely measurable precursor coisolation interference and induce ratio compression in previous reports.10–12 By comparing the quantitative results from these samples to the L1:H1 and L16:H16 samples, the impact of coisolation interference on reporter ion abundances becomes apparent; the measured reporter ion abundances of the L sample are distorted in the direction of the abundances of the H sample and vice versa (Figure S4). We reason that this may be due to interfering precursors in hyperplexed samples (two proteomes, light- and heavy-labeled) having greater intensity compared to those in typical samples (one proteome), leading to high-intensity contaminating fragments that are more likely to be selected by SPS-MS3 alongside fragments from the target precursor. We note that the mass range of 400–1200 m/z considered for SPS-MS3 in our acquisition method excluded the y1 fragment ion (K-DiLeu) at 292 m/z, as it is shared by the target precursor and any coisolated precursors and is a sure source of ratio distortion. Additionally, limiting the number of SPS-MS3 notches has been reported previously to reduce interference effects.12 The acquisition parameters for MS1 and MS2 precursor isolation widths (0.5 and 2.0 Da, respectively) and number of SPS-MS3 notches (four) used for the quantification experiments were chosen on the basis of this previous study12 and preliminary experiments (data not shown) in an effort to further mitigate interference effects. Though reporter ion abundance distortions were evident when induced, SPS-MS3 acquisition made the overall impact fairly modest; the median abundances remained within ±8.4% and ±15% of the expected values for the unity mixtures and differing ratio mixtures, respectively, with a single channel deviating by +24%. Furthermore, we demonstrate with these reverse and forward experiments that consistent results can be obtained with the DiLeu cPILOT approach across independent sample workups.
Peptides with dimethylated N-termini exhibit unique collision-induced fragmentation patterns compared to their unmodified counterparts.26–29 The most notable is enhanced production of the immonium a1 ion through preferential fragmentation of the dimethylated N-terminal residue. We observed favorable fragmentation of side chain and dimethylamine moieties at the dimethylated N-terminal residue as well, in a residue-dependent manner, that yields abundant characteristic precursor neutral loss species, each with an intact isobaric tag on their C-terminal lysine residue. These neutral losses are summarized in Table S1. Example spectra are shown in Figure S5, and the distribution of N-terminal amino acid residues for unique peptide sequences is shown in Figure S6. For general analyses of dimethylated peptides, these neutral loss peaks could be disregarded without consequence, but for the SPS-MS3 method employed here, they are significant in that they can often be selected for the MS3 quantification scan. Common precursor neutral loss peaks resulting from loss of H2O and NH3 are excluded from MS3 selection to avoid coisolating contaminant precursors a second time, but side chain or dimethylamine losses relocate the target precursor to a unique lower m/z and make it available for selection. This phenomenon appears to be advantageous for reporter ion accumulation into the third MS stage, as the neutral loss serves to purify the target precursor from remaining contaminant precursor ions, and their generally high intensities yield abundant reporter ion signals in MS3 spectra. Additionally, peptides with hydrophobic residues at their N-termini produce dominant y(n−1) ions as the complementary ion to the immonium a1 ion. These are also good candidates for SPS-MS3, warranting inclusion of the high m/z range in the SPS parameters. While the effect of these unique fragmentation patterns seems to be beneficial to quantification by the DiLeu cPILOT strategy, the influence of coisolation interference effects between different light and heavy precursors on reporter ion abundances, as observed with the L1:H16 and L16:H1 samples, was still carried over into the MS3 stage.
CONCLUSION
By combining mass difference stable isotope dimethyl labeling and DiLeu isobaric tag labeling, we have demonstrated a straightforward method of doubling multiplexing to achieve 24-plex quantification in a single LC-MS/MS run. Through proof-of-principle quantification experiments performed on the Orbitrap Fusion Lumos using an SPS-MS3 method, we have shown high rates of quantification with high accuracy across a useful dynamic range. Notably, the DiLeu cPILOT approach provides a significant cost savings compared to previously reported hyperplexing strategies that leverage metabolic mass difference labeling paired with commercial multiplex isobaric tags, and the chemical labeling steps make the method broadly compatible with a diverse range of biological samples and research applications. Future implementations of the DiLeu cPILOT approach could include a third mass difference channel at +4 Da to achieve 36-plex quantification given samples with lower complexity or improved instrumentation equipped with even faster sampling rates. Potentially, an advanced acquisition mode that performs real-time peptide sequence identification could intelligently trigger MS3 acquisition only when necessary to improve duty cycle efficiency and increase proteomic coverage. Such real-time analysis could also be used to exclusively select y ions for SPS-MS3 acquisition to increase reporter ion signal intensities and rate of quantification. We conclude that DiLeu cPILOT offers a simple and cost-effective strategy for high-throughput quantitative proteomics analysis of many samples in a single MS experiment.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Adam R. Evans for his correspondence during preliminary experiments. This research was supported in part by NIH grants R01GM117191 (R.A.S.R.), R01DK071801, P41GM108538, R21AG05537701, and S10RR029531 and funding provided by the Office of the Vice Chancellor for Research and Graduate Education at UW–Madison. L.L. acknowledges a Vilas Distinguished Achievement Professorship and Janis Apinis Professorship with funding provided by the Wisconsin Alumni Research Foundation (WARF) and UW–Madison School of Pharmacy.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b01301.
Supplemental methods and results; MS2 vs. SPS-MS3 acquisition; MS spectra; precursor coisolation interference model; CID MS2 spectra; fraction of N-terminal amino acid residues of unique peptide sequences; frequently observed CID neutral losses of dimethylated peptides (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chen X; Smith LM; Bradbury EM Anal. Chem. 2000, 72, 1134–1143. [DOI] [PubMed] [Google Scholar]
- (2).Zhu H; Pan S; Gu S; Bradbury EM; Chen X Rapid Commun. Mass Spectrom. 2002, 16, 2115–2123. [DOI] [PubMed] [Google Scholar]
- (3).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (4).Hsu JL; Huang SY; Chow NH; Chen SH Anal. Chem. 2003, 75, 6843–6852. [DOI] [PubMed] [Google Scholar]
- (5).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Anal. Chem. 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (6).Dayon L; Hainard A; Licker V; Turck N; Kuhn K; Hochstrasser DF; Burkhard PR; Sanchez JC Anal. Chem. 2008, 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- (7).Ross PL; Huang YN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin DJ Mol. Cell. Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- (8).Choe L; D’Ascenzo M; Relkin NR; Pappin D; Ross P; Williamson B; Guertin S; Pribil P; Lee KH Proteomics 2007, 7, 3651–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ow SY; Salim M; Noirel J; Evans C; Rehman I; Wright PC J. Proteome Res. 2009, 8, 5347–5355. [DOI] [PubMed] [Google Scholar]
- (10).Ting L; Rad R; Gygi SP; Haas W Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McAlister GC; Nusinow DP; Jedrychowski MP; Wühr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP Anal. Chem. 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Erickson BK; Jedrychowski MP; McAlister GC; Everley RA; Kunz R; Gygi SP Anal. Chem. 2015, 87, 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dephoure N; Gygi SP Sci. Signaling 2012, 5, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Everley RA; Kunz RC; McAllister FE; Gygi SP Anal. Chem. 2013, 85, 5340–5346. [DOI] [PubMed] [Google Scholar]
- (15).Robinson RAS; Evans AR Anal. Chem. 2012, 84, 4677–4686. [DOI] [PubMed] [Google Scholar]
- (16).Evans AR; Robinson RA S Proteomics 2013, 13, 3267–3272. [DOI] [PubMed] [Google Scholar]
- (17).Evans AR; Gu L; Guerrero R; Robinson RAS Proteomics: Clin. Appl. 2015, 9, 872–884. [DOI] [PubMed] [Google Scholar]
- (18).Gu L; Evans AR; Robinson RA S. J. Am. Soc. Mass Spectrom. 2015, 26, 615–630. [DOI] [PubMed] [Google Scholar]
- (19).Gu L; Robinson AS Analyst 2016, 141, 3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lau H-T; Suh HW; Golkowski M; Ong S-EJ Proteome Res. 2014, 13, 4164–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiang F; Ye H; Chen R; Fu Q; Li L Anal. Chem. 2010, 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Frost DC; Greer T; Li L Anal. Chem. 2015, 87, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Greer T; Lietz CB; Xiang F; Li LJ Am. Soc. Mass Spectrom. 2015, 26, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Swaney DL; Wenger CD; Coon JJ J. Proteome Res. 2010, 9, 1323–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dayon L; Sonderegger B; Kussmann MJ Proteome Res. 2012, 11, 5081–5089. [DOI] [PubMed] [Google Scholar]
- (26).Hsu J-L; Huang S-Y; Shiea J-T; Huang W-Y; Chen S-HJ Proteome Res. 2005, 4, 101–108. [DOI] [PubMed] [Google Scholar]
- (27).Fu Q; Li L Anal. Chem. 2005, 77, 7783–7795. [DOI] [PubMed] [Google Scholar]
- (28).Fu Q; Li L Rapid Commun. Mass Spectrom. 2006, 20, 553–562. [DOI] [PubMed] [Google Scholar]
- (29).Fu Q; Li LJ Am. Soc. Mass Spectrom. 2006, 17, 859–866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.