

Abstract
Recently developed carbonyl-reactive aminoxy tandem mass tag (aminoxyTMT) reagents enable multiplexed characterization and quantitative comparison of structurally complex glycans between different biological samples. Compared to some previously reported isotopic labeling strategies for glycans, the use of the aminoxyTMT method features a simple labeling procedure, excellent labeling efficiency, and reduced spectral complexity at the MS1 level. Presence of the tertiary amine functionality in the reporter region of the aminoxyTMT labels leads to increased ionization efficiency of the labeled glycans thus improving electrospray ionization (ESI)-mass spectrometry (MS) detection sensitivity. The use of the labeling reagent also makes electrophoretic separation of the labeled neutral and acidic glycans feasible. In this work, we characterized the ESI and collision induced dissociation (CID) behavior of the aminoxyTMT-labeled neutral and sialylated glycans. For the high-mannose N-glycans and small sialylated oligosaccharides, CID fragmentation of [M + Na + H]2+ provides the most informative MS2 spectra for both quantitative and qualitative analysis. For complex N-glycans, MS3 of the protonated Y1(H) ion can be used for relative quantification without interference from the HexNAc fragments. Online capillary electrophoresis (CE)-ESI-MS/MS analyses of multiplexed aminoxyTMT-labeled human milk oligosaccharides (HMOs) and different types of N-glycans released from glycoprotein standards were demonstrated. Improved resolution and quantification accuracy of the labeled HMO isomers was achieved by coupling CE with traveling wave ion mobility (TWIM)-CID-MS/MS. N-Glycans released from human serum protein digests were labeled with six-plex aminoxyTMT and subjected to CE-ESI-MS/pseudo-MS3 analysis, which demonstrated the potential utility of this glycan relative quantification platform for more complex biological samples.
Graphical Abstract
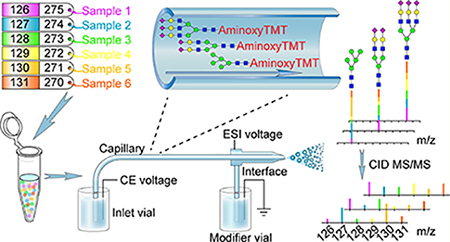
Glycosylation of proteins is one of the most important post-translational modifications. Studies have shown that the glycan moieties on the glycoproteins play critical roles in structural modulation and function as specific binding ligands for endogenous receptors or exogenous agents in many biological processes such as protein trafficking, cell−cell signaling, cellular adhesion.1–4 Changes in glycomic profiles have been linked to various diseases 5–9 including immunological disorders, cancer, and cardiovascular problems. These implications urge researchers to develop cutting-edge bioanalytical platforms for quantitative analysis of glycans which would facilitate the elucidation of the diverse biological roles of glycans and their roles in human diseases.10
Quantitative analysis of native glycans remains challenging due to high complexity and diversity of glycan structures,11 the difficulty of synthesizing glycan standards, and the relatively low response in both optical and mass spectrometric detection methods. Several fluorescence detection methods based on reducing-end labeling strategies have been developed12,13 and are routinely applied in conjunction with liquid chromatography (LC)14 or capillary electrophoresis (CE) separation.15–17 Reductive amination is the most common method used to attach the glycans with chromophores, such as 2-aminobenzamide (2-AB),18 2-aminopyridine (2-PA),19 2-aminobenzoic acid (2-AA),20 and 1-aminopyrene-3,6,8-trisulfonic acid (APTS).21 Although high separation efficiency and sensitive detection could be achieved with LC or CE combined with fluorescence or laser-induced fluorescence detection,8,12,22 structural identification is largely dependent on the availability of standards, sequential enzymatic digestion, and highly reproducible LC retention times or CE migration times.
With the recent advances in mass spectrometry, a variety of MS-based glycan quantification strategies have been developed, which also allow more confident assignment of glycan compositions based on mass-to-charge ratios of the intact glycans and fragment ions generated by different types of activation methods.23–26 These include both chemical and metabolic labeling approaches.24 Metabolic incorporation of 15 N into N- and O-linked glycans for quantitative glycomics has been demonstrated by Orlando et al., which involves the use of amide-15N-Gln in the cell medium to provide the only source of nitrogen for biosynthesis of amino-sugars.27 While metabolic labeling is relatively more expensive and restricted to the investigation of limited types of biological samples, chemical labeling strategies can be applied to glycan samples from different sources. MS1 precursor ion intensity-based relative quantification is realized by labeling glycans with “heavy” and “light” pairs of reagents. These strategies include permethylation of glycan samples with d0- and d3-CH3I,28 or 12CH3I and 13CH3I,29 enzymatic release of glycans in H216O and H218O,30 reductive amination with normal and isotopically encoded tags such as d0- and d4-PA,31 d0 and d4 2-AA,32 2-12[C6]-AA and 2-13[C6]-AA,33 12[C6] aniline, and 13[C6]-aniline,34 or even tetra-plex isotopic tags,35 formation of glycan hydrazones with 12[C6] and 13[C6] 4-phenethylbenzohydrazide,36 or d0 and d5 Girard’s reagent P.37 Incorporation of stable isotopes through permethylation can lead to variable mass shift between the differentially labeled glycans, depending on the total number of −OH, −NH, and −COOH groups in one glycan and thus complicating the spectral interpretation. In contrast, isotopic labeling at the reducing end of the glycans results in constant mass shift between the heavy and light labeled pairs; however, spectral interference caused by overlapping of the isotopes of the differentially labeled glycans is observed if the mass difference induced by the isotopic labeling is not large enough.
Originating from the tandem mass tags (TMT) that have been widely applied in quantitative proteomics,38–40 several multiplex isobaric tags, which are usually composed of a reporter group and an aldehyde reactive group linked by a mass balance group, have been developed for high-throughput relative quantification of glycans based on reporter ion intensities generated by MS/MS. Compared to isotopic labeling, the MS1 spectra of isobarically labeled glycans are simplified because the differentially labeled glycans have the same mass. The isobaric aldehyde reactive tags (iARTs) were derived from the deuterium isobaric amine-reactive tags (DiARTs) by modifying the amino reactive group to a primary amine group for reductive amination.41 Two versions of glycanspecific carbonyl-reactive TMT reagents with hydrazide and alkoxyamine reactive functionalities, which form hydrazones or oximes with reducing sugars, respectively, were evaluated by Hahne et al.42 One of these tags, the aminoxyTMT that employs the alkoxyamine functional group, has recently become commercially available. The previous study indicated that under MALDI conditions, mainly the monosodium adduct ions of the aminoxyTMT-labeled glycans were observed, and the low intensity of the reporter ions generated by post-source decay (PSD) compromised the accuracy and dynamic range of relative quantification.42 However, the performance of the aminoxy-TMT labeled glycans in ESI mode, CID fragmentation of the multiply charged precursor ions, as well as the reporter ion intensity based quantification by ESI-CID MS/MS remain unexplored. In this work, we characterized the aminoxyTMTlabeled different types of glycans using an ESI-quadrupole (Q)time-of-flight (TOF) instrument and found that the CID MS/ MS of multiply charged aminoxyTMT-labeled glycan generated sufficient reporter ions for accurate quantification.
Another aspect that has not been explored previously is the coupling of microscale separation platforms with ESI-MS detection for analysis of multiplex aminoxyTMT-labeled glycans, which is critical for improving coverage of complex samples with limited amounts. The tertiary amine group in the aminoxyTMT makes the labeled glycan positively charged in acidic buffers and thus provides the possibility of electrophoretic separation of the aminoxyTMT-labeled glycans. As an attractive separation technique alternative to hydrophilic interaction chromatography (HILIC) and porous graphitized carbon (PGC) liquid chromatography, CE offers advantages for separation of polar molecules including low-cost of column material, very small amount of sample consumption, fast separation speed, and high resolving power. Accompanied by efforts toward developing robust and sensitive CE-ESI-MS over the past decades,43,44 more and more researchers have incorporated this technique in their toolboxes for analysis of oligosaccharides in biological samples and glycans from different types of glycoconjugates,22,45–48 such as human milk oligosaccharides,49 N-glycans from therapeutic recombinant glycoproteins,50–53 N-glycans released from serum,54,55 and oligosaccharides derived from glycosphingolipids.56 Most of these CE-ESI-MS studies involve separation of native glycans carrying negatively charged functionalities50,54,55,57 or APTSlabeled glycans,49,51–53,55,57,58 which have to be coupled with negative ESI detection that is inherently less sensitive and more susceptible to corona discharge compared to positive ESI.59–61 In contrast, the aminoxyTMT labeling promotes sensitivity in positive ESI of the labeled glycans by introducing hydrophobic and basic groups. Here, we demonstrate, for the first time, online coupling of CE with ESI-CID-MS/MS for the analysis of multiplexed aminoxyTMT-labeled human milk oligosaccharide (HMO) standards, different types of N-glycans released from glycoprotein standards and human serum proteins. In addition, CE coupled with traveling wave ion mobility (TWIM)-MS/MS was explored and proven to be effective in improving the resolution of small isomeric glycans and quantification accuracy. These results show significant promise in applying the multiplex aminoxyTMT labeling technique coupled with CEESI-MS/MS for high-throughput characterization of glycans in more complicated biological systems.
EXPERIMENTAL SECTION
For information regarding chemicals and materials, N-glycan release and aminoxyTMT labeling of glycans, direct infusion ESI-MS/MS and ESI-TWIM-MS, and CE-ESI-MS setup, see the Supporting Information.
MS Parameters for Online CE-ESI-MS/MS and CE-ESIIM-MS/MS Analysis.
For CE-MS/MS analysis of three-plex aminoxyTMT-labeled N-glycans released from RNase B, a full MS scan over the range of m/z 400−1500 was acquired to generate the inclusion list for the subsequent data-dependent analysis (DDA). Top 2 DDA with an inclusion list of all the [M + H + Na]2+ and [M + 2H + K]3+ precursor ion m/z was performed in a separate CE-MS run using parameters as follows: the MS survey scan range was acquired over the range of m/z 400−1400 Da, scan duration was 1 s; 2+ and 3+ charged ions with intensity above a threshold of 1500 were selected for MS/MS scans; the precursor ion isolation window was ~3 m/z, the MS/MS scan was acquired over the range of m/z 100−1900, and the scan duration was 1.5 s. The trap collision cell voltage profile was set as the following equation: CE (V) = 25 + ((m/z)−400) × 0.055. After one MS/MS scan, the current precursor was then excluded from the rest of the run.
For CE-ESI-ion mobility (IM)-MS/MS analysis of differentially labeled HMO standards, the traveling wave velocity was set as 700 m/s and wave height 40 V. Alternating full MS scans and targeted MS/MS scans of m/z 661.9 were acquired during the CE separation. The CID MS/MS was performed in the transfer collision cell with a collision energy of 50 V.
Alternating full MS scans and pseudo-MS3 scans were applied for CE-MS/MS analysis of aminoxyTMT labeled N-glycans released from fetuin and human serum protein digests. The cone voltage was set to 30 V for 0.5 s of full MS scan in the range of m/z 400−1700 and switched to 100 V for 0.5 s of the pseudo-MS3 scan during CE separation. For the pseudo-MS3 scan, the Y1(H) ion at m/z 523.3 generated in the ion source was selected by the quadrupole and fragmented in the trap collision cell with a collision energy of 40 V and the TOF scan range was m/z 100−150.
RESULTS AND DISCUSSION
CID MS/MS fragmentation of aminoxyTMT-labeled glycan standards and relative quantification of isobarically labeled glucose oligomers (see the Supporting Information and Figures S1 and S2)
CE-ESI-MS Analysis of aminoxyTMT Labeled HighMannose N-Glycans.
For improving the glycan coverage and resolving potentially existing isomeric species, a unique platform that couples the microscale separation technique with MS detection is usually required for analysis of complex samples with small volume. CE-ESI-MS is a promising tool for analysis of aminoxyTMT labeled glycans because the modification introduces a tertiary amine group at the reducing end, making electrophoretic separation of neutral glycans feasible in an acidic environment. When the tertiary amine on the reporter ion group is attached by a proton under an acidic environment, the labeled carbohydrate molecule will possess overall positive charge and migrate toward the cathode end of the capillary under an electric field.
CE separation of three-plex aminoxyTMT-labeled N-glycans released from RNase B (1:1:1 ratio) was conducted using methanol/water/formic acid (v/v/v) 50:49:1 as a background electrolyte (BGE). Figure 1 shows the extracted ion electropherograms (EIEs) of the [M + H + Na]2+ ions of the (Man)5−9(GlcNAc)2. The five labeled high-mannose N-glycans migrated out of the separation capillary in the order of increasing number of monosaccharide residues. The total amount of glycans loaded onto the column corresponded to the glycans released from approximately 0.2 μg of RNase B. By pooling together the differentially labeled glycans, ion intensities of both MS1 and MS2 scans were boosted because the isobarically labeled glycans were detected at the same precursor m/z, and most fragment ions (except the reporter ions) that originated from different samples were also detected at the same m/z.
Figure 1.

Extracted ion electropherograms (EIEs) of [M + H + Na]2+ ions of aminoxyTMT-labeled N-glycans released from a glycoprotein RNase B. BGE, 50:49:1 (v/v/v) MeOH/H2O/formic acid; modifier solution, 50:49.8:0.2 (v/v/v) MeOH/H2O/formic acid. The full MS scan was acquired from m/z 400−1500.
Similar to the glucose oligomers, the ESI generated [M + Na + H]2+ precursor ions of the high-mannose N-glycans produce abundant reporter ions for quantification and diverse fragment ions for structural inference. The CID MS/MS spectrum of the [(Man)5(GlcNAc)2-TMT + Na + H]2+ ion (m/z 779.8) is shown in Figure 2 as an example. At a trap collision cell voltage of 46 V, the reporter ions dominated the spectrum, and abundant fragment ions associated with the N-glycan structure were present as well. Except Y1(H) ion (m/z 523.3) and several doubly charged Y(Na+H) ions, most fragment ions were observed as sodium adducts. Sodiated Y(Na) ions (m/z 1396.6, 1234.5, 1072.5, 910.4) and doubly charged Y(Na+H) ions (m/z 698.8, 617.7) were formed by consecutive loss of mannose residues. B2(Na), B3(Na), B4(Na) ions (m/z 509.1, 833.2, 1036.3), as well as some B/Y(Na) ions (m/z 874.2, 671.2, 550.1, 347.1) produced from internal cleavage formed by different routes were observed. C3(Na) and C3−2H (Na) (m/z 851.2, 849.2) ions, and C4(Na) and C4−2H(Na) ions (m/z 1054.3, 1052.3) were formed by cleavage of the two β1–4 bonds of the chitobiose core, respectively. Cross-ring cleavages of the GlcNAc at the reducing terminus produced 3,5A4(Na) (m/z 907.2), 2,4A5(Na) (m/ z 1096.3), 0,2A5(Na) (m/z 1156.3), and internal fragment ion 0,2A5 /Y3β(Na) or 0,2A5 /Y4α(Na) (m/z 944.3). The intensity of the [M-reporter ion-CO + H + Na]+ ion was also prominent.
Figure 2.

CID MS/MS spectrum of [(Man)5(GlcNAc)2-aminoxyTMT + Na + H]2+ (m/z 779.8) acquired by data-dependent analysis during online CE separation. Domon and Costello nomenclature65 is used for annotation of fragment ions in this and the following figures.
In addition to the [M + Na + H]2+ precursor ion, the [M + 2H]2+ and [M + 2H + K]3+ precursor ions were also abundant in the MS1 scan and could be subjected to CID MS/MS during the online data-dependent analysis. Figure S3 in the Supporting Information shows the CID MS/MS spectra of [(Man)6(GlcNAc)2-TMT + 2H]2+, [(Man)6(GlcNAc)2-TMT + 2H + K]3+, and [(Man)6(GlcNAc)2-TMT + Na + H]2+ acquired during CE-MS separation. Although the trap collision energy profile was optimized for [M + Na + H]2+ precursor ions, [M + 2H]2+ and [M + 2H + K]3+ ions of the high-mannose type N-glycans also had decent reporter ion intensity despite the lack of many fragment ions in the high mass range. Table 1 summarizes the reporter ion intensity ratios calculated from the sum of the reporter ion intensities generated from CID MS/MS of [M + Na + H]2+, [M + 2H]2+, [M + 2H + K]3+ precursor ions and the error calculated in this way was within 17% for the five high-mannose N-glycans.
Table 1.
Reporter Ion Intensity Ratios of the Three-Plex aminoxyTMT-Labeled N-Glycans Released from RNase Ba
| glycan composition | precursor m/z | I128/I130/I131b | CV (%, I130/I128)c | CV (%, I131/I128)c |
|---|---|---|---|---|
| (Man)5(GlcNAc)2 | 779.83(Na+H), 768.85(2H) | 1:1.12:0.89 | 11.1 | 1.8 |
| (Man)6(GlcNAc)2 | 860.87(Na+H), 849.88(2H), 579.56(2H+K) | 1:1.11:0.88 | 3.0 | 7.8 |
| (Man)7(GlcNAc)2 | 941.88(Na+H), 930.91(2H), 633.58(2H+K) | 1:1.11:0.91 | 3.8 | 10.6 |
| (Man)8(GlcNAc)2 | 1022.92(Na+H), 1011.93(2H), 687.56(2H+K) | 1:1.12:0.91 | 2.5 | 0.3 |
| (Man)9(GlcNAc)2 | 1103.95(Na+H), 1092.96(2H), 741.62(2H+K) | 1:1.17:0.96 | 6.9 | 5.4 |
N-Glycans released from RNase B was labeled with aminoxyTMT6 −128, aminoxyTMT6 −130, and aminoxyTMT6 −131 in a 1:1:1 ratio. The ratios were calculated from CID MS/MS spectra acquired from CE separation coupled with online data-dependent analysis.
Reporter ion intensities from MS/MS scans of precursor ions belonging to the same glycan composition are summed for the calculation of the ratios.
CVs are calculated from three technical replicates of CE-ESI-MS/MS analysis.
CE-ESI-TWIM-CID MS/MS Analysis of aminoxyTMT Labeled Acidic Oligosaccharides.
Oligosaccharides containing acidic monosaccharide residues such as NeuAc and NeuGc become amphoteric compounds after being labeled with aminoxyTMT reagents. To study the CE separation behavior of aminoxyTMT labeled acidic glycans, we selected four human milk oligosaccharides (sialyllacto-N-tetraose a, b, c and DSLNT) and subjected them to isobaric labeling and CEESI-MS analysis. With methanol/water/formic acid (v/v/v) 50:49:1 as BGE, the aminoxyTMT labeled DSLNT could be separated from the sialyllacto-N-tetraoses because of its lower pKa and larger size; however, only two CE peaks corresponding to the three pentasaccharide isomers were observed. By further investigation, it was confirmed that the labeled sialyllacto-N-tetraose b (LSTb) and sialyllacto-N-tetraose c (LSTc) coeluted at 34.6 min, followed by sialyllacto-N-tetraose a (LSTa) eluting at 38.1 min (Figure 3A). If the comigrated isomeric compound ions were subjected to CID MS/MS simultaneously, the observed reporter ion intensities would only reflect the total amounts of the coeluted isomers instead of the relative quantity of each individual component in the samples.
Figure 3.

CE-ESI-TWIM-MS/MS analysis of aminxoyTMT6-128 and aminoxyTMT6-131 differentially labeled LSTa (128/131, 1:4), LSTb (128/ 131, 1:1), LSTc (128/131, 3:1) and DSLNT mixture. (A) EIEs of [LST-aminoxyTMT + H + Na]2+ m/z 661.9 (top) and [DLST-aminoxyTMT + H + 2Na]3+ m/z 545.9 (bottom). The peak at 47.3 min in the top panel corresponds to the in-source fragmentation ion Y4α(Na + H) or Y3β(Na + H) of labeled DSLNT. (B) Arrival time distributions of [LST-aminoxyTMT + H + Na]2+ (m/z 661.9) at a CE migration time of 34.6 min (top), 38 min (middle), and 47.3 min (bottom). (C) Reporter ion ratios of aminoxyTMT-labeled LSTb (top), LSTc (middle), LSTa (bottom) obtained by CEIM-targeted CID MS/MS. The precursor ion [LST-aminoxyTMT + H + Na]2+ (m/z 661.9) was isolated by the quadrupole, separated in the ion mobility drift cell and then subjected to CID MS/MS in the transfer collision cell. CE conditions were as described in Figure 1. Alternating IM-full MS scan and IM-targeted MS/MS scan of m/z 661.9 were acquired during CE separation. TWIM conditions: trap bias voltage = 48 V, helium cell dc = 35 V, wave velocity = 700 m/s, and wave height = 40 V. The transfer cell collision energy was set to 50 V for targeted MS/MS.
Recent studies have shown promises of differentiation of isomeric carbohydrate as metal ion adducts by TWIM.62,63 Taking advantage of the TWIM capability of the Waters Synapt G2 Q-TOF instrument, we were able to further improve the resolution of aminoxyTMT-labeled isomeric HMO standards by integrating a second separation dimension, the ion mobility separation in the gas phase, with CE separation. Although both CE and ion mobility are electrophoretic based separation techniques, they could be orthogonal separation dimensions because the aqueous-phase mobility and gas-phase mobility of an analyte molecule are dependent on different parameters that affect “charge-to-shape ratio” of the molecule in a specific environment. In terms of an amphoteric compound during CE separation, the pKa values of the compound and pH of the BGE define the “average charge” carried by an ensemble of analyte molecules which comigrated in the aqueous phase and the “shape” of the molecule refers to the hydrated aqueous-phase conformation of the molecule in the buffer. After the analyte molecules are converted to gas-phase ions by ESI, the number of H+, Na+, K+, and other charge carriers attached to one analyte molecule defines the charges carried by the single analyte molecule ions. The “shape” of one analyte ion refers to the gas-phase conformation of a specific type of ion adduct of the analyte molecule. As different types of ion adducts of glycans are usually observed under positive ESI mode, we first compared the IM-MS separation of the [M + H + Na]2+, [M +H + K]2+, [M + 2Na]2+, [M + K + Na]2+ adducts of the three aminoxyTMT labeled isomeric pentasaccharides at a set of dc traveling wave velocity/wave height combinations by direct infusion experiments. Figure S4 in the Supporting Information shows the arrival time distributions of labeled LSTa, LSTb, and LSTc as different types of ion adducts at a wave velocity of 700 m/s and wave height of 40 V. Interestingly, the [M + H + Na]2+ adduct ion of aminoxyTMT labeled LSTc could be differentiated from those of LSTa and LSTb by TWIM, which provides complementary results to CE separation.
To demonstrate the coupling of CE with TWIM-CID-MS/ MS for improved relative quantification accuracy of the aminoxyTMT labeled isomeric oligosaccharides, LSTa, LSTb, and LSTc were differentially labeled with aminoxyTMT6-128 and aminoxyTMT6-131 in 1:4, 1:1, 3:1 ratios, respectively, and mixed together with aminoxyTMT labeled DSNLT, followed by CE-ESI-TWIM-CID-MS/MS analysis. With the optimized CE and IM-MS conditions, the coeluting aminoxyTMT labeled LSTb and LSTc at 34.6 min were resolved in the ion mobility drift cell, with the drift time of the [M + Na + H]2+ ion adducts at 4.35 and 4.83 ms, respectively (Figure 3B, top panel). It was also noticed that the in-source fragmentation products, the Y4α(H+Na) and Y3β(H+Na) ions of the labeled DSLNT, have the same arrival time distribution as the [M + Na + H]2+ ions of the earlier eluting aminoxyTMT labeled LSTa and LSTb (Figure 3B). After the two-dimensional separation, the three differentially labeled isomers were almost completely resolved and the [M + Na + H]2+ (m/z 661.9) precursor ions from each isomer were separately subjected to CID MS/MS in the transfer collision cell. The reporter ion intensities obtained at three different CE migration time/IM drift time combinations, shown in Figure 3C, represent the relative quantities of each individual pentasacchride isomer; the observed reporter ion ratios were close to the expected values because of the improved separation.
CE-ESI-Pseudo-MS3 Analysis of aminoxyTMT-Labeled Sialylated Complex N-Glycans.
Complex type N-glycans released from bovine fetuin, which contain terminal sialic acid residues, were also investigated by CID MS/MS and CE-MS after being labeled with aminoxyTMT reagents. By direct infusion CID MS/MS experiments, it was found that the reporter ion yield of the aminoxyTMT-labeled complex Nglycans are generally not as good as that of the high-mannose type N-glycans because the GlcNAc fragment ion, m/z 138.06, always dominates the CID MS/MS spectra. Representative MS/MS spectra of different ion adducts of the triantennary complex N-glycan (Hex)6(GlcNAc)5(NeuAc)3 is shown in Figure S5 in the Supporting Information. In order to overcome this limitation, we report a pseudo-MS3 based quantification method here for CE-MS applications on a Q-TOF-MS platform. Since acidic BGE and modifier are used for CE-MS and a tertiary amine group is incorporated into the aminoxyTMT reagent, abundant Y1(H) ions (m/z 523.3), which are common for all types of N-glycans labeled with aminoxyTMT reagents, can be easily generated in the ion source from the fully and partially protonated precursor ion adducts by raising the sample cone voltage and then subjected to CID MS/MS in the collision cell for production of reporter ions. In order to obtain both the mass of the intact glycan and decent reporter ion intensities during online separation, two scan events, a full MS scan at low cone voltage (30 V), and a targeted MS/MS scan of the Y1(H) ion at high cone voltage (100 V) were acquired in an alternating fashion during one CE-MS run.
The pseudo-MS3 method was applied to the aminoxyTMT6128,131 (1:1) labeled N-glycans released from bovine fetuin. As shown in Figure 4A, the labeled N-glycans from fetuin migrated in the order of neutral, monosialylated, disialylated, trisialylated, tetrasialylated, and pentasialylated glycans (total ion electropherogram (TIE) from full MS scans, top panel); the EIE of m/ z 523.3 (bottom panel) corresponding to the Y1(H) generated by in-source fragmentation at low cone voltage defined the migration zone of the aminoxyTMT labeled N-glycans. Figure 4B presents the EIEs of two reporter ions, m/z 128.1 and m/z 131.1, generated from targeted MS/MS scans of m/z 523.3 at high cone voltage (pseudo-MS3 scans). The migration peak profiles of the two ion channels were almost identical and the ion intensity ratios in the glycan migration zone were close to 1:1. Despite the fact that the pseudo-MS3 method is only ideal for highly efficient separation techniques coupled with MS and the generated reporter ions intensities have to be deconvoluted in the case of coeluting and partially resolved components, this issue can be alleviated if the real MS3 method, isolating a specific aminoxyTMT labeled precursor ion to produce the common Y1(H) and Y2(H) ions and submitting them to further fragmentation, is applied on an MS platform with a trap-type mass analyzer such as LTQ-Orbitrap.
Figure 4.

CE-ESI-MS of aminoxyTMT6-128 and aminoxyTMT6-131 (1:1) labeled N-glycans released from bovine fetuin. (A) Total ion electropherogram (TIE) (top) and EIE of Y1(H) ion m/z 523.3 generated by in-source fragmentation. The cone voltage was set to 30 V for the full MS scan and the scan range was m/z 400−1700. (B) EIEs of reporter ions m/z 128.1 and m/z 131.1 generated by pseudo-MS3 scans. The cone voltage was set to 100 V, Y1(H) m/z 523.3 was isolated and fragmented in the trap collision cell with a collision energy of 40 V; the scan range was m/ z 100−150. (C) EIE of [Hex5GlcNAc4NeuAc2 + 3H]3+ (m/z 842.4). (D) EIE of [Hex6GlcNAc5NeuAc3 + 3H]3+ (m/z 1061.1).
Multiple isomeric structures associated with the same N-glycan composition occurred frequently in this sample. After examining and excluding the in-source fragmentation generated components, a total of 10 sialylated N-glycan compositions, containing at least 23 glycan structures, were putatively identified from the CE-MS experiments (Figure S6 in the Supporting Information). Two representative EIEs, corresponding to the [M + 3H]3+ ions of the biantennary complex N-glycan, (Hex)5(GlcNAc)4(NeuAc)2, and the triantennary complex N-glycan, (Hex)6(GlcNAc)5(NeuAc)3, respectively, are shown in Figure 4C,D. Four isomeric structures associated with the composition (Hex)5(GlcNAc)4(NeuAc)2 were resolved by CE and at least three isomeric structures associated with the composition (Hex)6(GlcNAc)5(NeuAc)3 were detected. Although efforts have been made to improve N-glycan isomer separation by employing TWIM as a second separation dimension, the limited resolution of TWIM, the more complicated tertiary structures of multiply charged large gasphase N-glycan ions and the multiple conformations generated from one isomer ion, are still barriers for the application of CEESI-IM-MS to N-glycan samples.
CE-ESI-Pseudo-MS3 Analysis of Six-Plex aminoxyTMT Labeled N-Glycans Released from Human Serum Proteins.
N-Glycans released from human serum proteins labeled with six-plex aminoxyTMT6-126, 127, 128, 129, 130, and 131 in a 1:2:1:1:1:2 ratio were used as a complex biological sample to assess the developed platform for glycan quantification. Initial data-dependent analysis was performed during online separation to inspect interfering ions in the reporter ion region. Figure S7 in the Supporting Information shows the MS/MS spectra of [M + 2H]2+, [M + 2H + K]3+, [M + H + Na]2+ ion adducts of the core-fucosylated complex N-glycan (Hex)3(GlcNAc)4Fuc. It was found that the reporter ion yield of neutral complex N-glycans was also low and the HexNAc fragment ion of [C6H8O2N]+ (m/z 126.06)42 and its isotope (m/z 127.06) could be just resolved from the two reporter ions at m/z 126.13 and m/z 127.13 by TOF-MS in resolution mode. However, the pseudo-MS3 method is still applicable for the core-fucosylated N-glycans since the Y1Y1δ(H) ion m/z 523.3 was also observed when the protonated precursor ions were activated. The TIE of the six-plex aminoxyTMT-labeled human serum N-glycans is shown in Figure 5. The hydrophilic peptides retained in the glycan fraction during sample purification migrated out of the capillary column first, followed by the aminoxyTMT-labeled neutral N-glycans, monosialylated, disialylated, trisialylated, and tetrasialylated N-glycans. A total of 46 N-glycan compositions were identified within 30 ppm accuracy with at least two precursor adduct ions belonging to the same composition. By manually excluding the in-source fragmentation peaks, 84 isomeric structures belonging to the 46 N-glycan compositions were putatively identified (Table S1 in the Supporting Information). The inset of Figure 5 shows the pseudo-MS3 spectra acquired during the migration time zones of the six-plex aminoxyTMT labeled (Hex)3(GlcNAc)4Fuc, (Hex)5(GlcNAc)4(NeuAc), (Hex)5(GlcNAc)4(NeuAc)2, and (Hex)6(GlcNAc)5(NeuAc)3, respectively, which were free from the spectral interference of the fragment ions of GlcNAc residues. The relative quantification ratios from these reporter ion intensities are summarized in Table S2 in the Supporting Information. As a “proof-of-principle” example, our preliminary study demonstrates the potential utility of the multiplex isobaric aminoxyTMT reagents in combination with the CE-ESI-MS platform for quantitative glycomics. Further optimization of this platform is needed to allow its robust application in the emerging field of clinical glycomics.64
Figure 5.

TIE of six-plex aminoxyTMT-labeled N-glycans released from human serum protein digests. Inset: four representative pseudo-MS3 spectra (MS/MS of Y1(H) ion m/z 523.3) integrated over 0.2 min periods marked with asterisks corresponding to the migration time zones of aminoxyTMTlabeled (Hex)3(GlcNAc)4Fuc, (Hex)5(GlcNAc)4(NeuAc), (Hex)5(GlcNAc)4(NeuAc)2, and (Hex)6(GlcNAc)5 (NeuAc)3, respectively.
CONCLUSIONS
Combining the isobaric labeling strategy and CE-ESI-MS/MS, aminoxyTMT-labeled neutral and acidic glycans can be efficiently separated in acidic background electrolytes and quantified with CID MS/MS or pseudo-MS3 methods (targeted MS/MS of Y1(H) ion). CE coupled with TWIM-CID MS/MS was demonstrated to be efficient in resolving human milk oligosaccharide isomers and improving the relative quantification accuracy. The labeled sialylated isomeric N-glycans can also be resolved by CE separation in normal polarity. For the high-mannose type N-glycans, online data-dependent analysis of the precursor ion adducts [M + H + Na]2+, [M + 2H]2+, and [M + K + 2H]3+ could give rise to high-intensity reporter ions that can be used for accurate relative quantification between different samples, and CID MS/MS of [M + H + Na]2+ ions generate the most useful tandem mass spectra in terms of abundant reporter ions and rich fragment ions for structural inference. For the complex type of N-glycans, MS3 based quantification method could be applied to generate reporter ions and avoid interference from the fragment ions of HexNAc residues. Future work will extend the application of these aminoxyTMT tags to more complex biological systems for high-throughput glycan biomarker discovery.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank Professor David D. Y. Chen at Department of Chemistry at the University of British Columbia for generously providing the CE-ESI-MS interface unit. Our thanks also go to Gary Girdaukas and staff members of the machine shop of Department of Chemistry at the University of Wisconsin-Madison for their assistance with the CE and ESI source instrumentation. Erin Gemperline in the Li Research Group is acknowledged for her critical reading of the revised manuscript and providing helpful comments. This work was supported by grants from NIH Grants R01DK071801, R56DK071801, and S10RR029531. L.L. acknowledges an H.I. Romnes Faculty Fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Additional experimental information and results and discussion. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.5b01835.
REFERENCES
- (1).Biological Roles of Glycans, Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratories Press: Cold Spring Harbor, NY, 2009. [Google Scholar]
- (2).Moremen KW; Tiemeyer M; Nairn AV Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dwek RA Chem. Rev 1996, 96, 683–720. [DOI] [PubMed] [Google Scholar]
- (4).Defaus S; Gupta P; Andreu D; Gutierrez-Gallego R Analyst 2014, 139, 2944–2967. [DOI] [PubMed] [Google Scholar]
- (5).Jankovic MJ Med. Biochem. 2011, 30, 213–223. [Google Scholar]
- (6).Taniguchi N Mol. Cell Proteomics 2008, 7, 626–627. [PubMed] [Google Scholar]
- (7).An HJ; Kronewitter SR; de Leoz MLA; Lebrilla CB Curr. Opin. Chem. Biol. 2009, 13, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Alley WR; Mann BF; Novotny MV Chem. Rev. 2013, 113, 2668–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Arnold JN; Saldova R; Hamid UMA; Rudd PM Proteomics 2008, 8, 3284–3293. [DOI] [PubMed] [Google Scholar]
- (10).Hart GW; Copeland RJ Cell 2010, 143, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Marino K; Bones J; Kattla JJ; Rudd PM Nat. Chem. Biol. 2010, 6, 713–723. [DOI] [PubMed] [Google Scholar]
- (12).Ruhaak LR; Zauner G; Huhn C; Bruggink C; Deelder AM; Wuhrer M Anal. Bioanal. Chem. 2010, 397, 3457–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Harvey DJ J. Chromatogr. B 2011, 879, 1196–1225. [DOI] [PubMed] [Google Scholar]
- (14).Anumula KR Anal. Biochem. 2006, 350, 1–23. [DOI] [PubMed] [Google Scholar]
- (15).El Rassi Z Electrophoresis 1999, 20, 3134–3144. [DOI] [PubMed] [Google Scholar]
- (16).El Rassi Z Electrophoresis 1997, 18, 2400–2407. [DOI] [PubMed] [Google Scholar]
- (17).Suzuki S; Honda S Electrophoresis 1998, 19, 2539–2560. [DOI] [PubMed] [Google Scholar]
- (18).Bigge JC; Patel TP; Bruce JA; Goulding PN; Charles SM; Parekh RB Anal. Biochem. 1995, 230, 229–238. [DOI] [PubMed] [Google Scholar]
- (19).Hase S; Hara S; Matsushima YJ Biochem. 1979, 85, 217–220. [DOI] [PubMed] [Google Scholar]
- (20).Anumula KR Anal. Biochem. 1994, 220, 275–283. [DOI] [PubMed] [Google Scholar]
- (21).Chen FT; Evangelista RA Anal. Biochem. 1995, 230, 273–280. [DOI] [PubMed] [Google Scholar]
- (22).Mechref Y; Novotny MV Mass Spectrom. Rev. 2009, 28, 207–222. [DOI] [PubMed] [Google Scholar]
- (23).Zaia J Chem. Biol. 2008, 15, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mechref Y; Hu YL; Desantos-Garcia JL; Hussein A; Tang HX Mol. Cell Proteomics 2013, 12, 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kailemia MJ; Ruhaak LR; Lebrilla CB; Amster IJ Anal. Chem. 2014, 86, 196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Alley WR; Novotny MV Annu. Rev. Anal. Chem 2013, 6, 237–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Orlando R; Lim JM; Atwood JA; Angel PM; Fang M; Aoki K; Alvarez-Manilla G; Moremen KW; York WS; Tiemeyer M; Pierce M; Dalton S; Wells LJ Proteome Res. 2009, 8, 3816–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kang P; Mechref Y; Kyselova Z; Goetz JA; Novotny MV Anal. Chem. 2007, 79, 6064–6073. [DOI] [PubMed] [Google Scholar]
- (29).Aoki K; Perlman M; Lim JM; Cantu R; Wells L; Tiemeyer MJ Biol. Chem. 2007, 282, 9127–9142. [DOI] [PubMed] [Google Scholar]
- (30).Zhang W; Wang H; Tang HL; Yang PY Anal. Chem. 2011, 83, 4975–4981. [DOI] [PubMed] [Google Scholar]
- (31).Yuan J; Hashii N; Kawasaki N; Itoh S; Kawanishi T; Hayakawa TJ Chromatogr. A 2005, 1067, 145–152. [DOI] [PubMed] [Google Scholar]
- (32).Hitchcock AM; Costello CE; Zaia J Biochemistry 2006, 45, 2350–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Prien JM; Prater BD; Qin Q; Cockrill SL Anal. Chem. 2010, 82, 1498–1508. [DOI] [PubMed] [Google Scholar]
- (34).Lawrence R; Olson SK; Steele RE; Wang LC; Warrior R; Cummings RD; Esko JD J. Biol. Chem. 2008, 283, 33674–33684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bowman MJ; Zaia J Anal. Chem. 2010, 82, 3023–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Walker SH; Budhathoki-Uprety J; Novak BM; Muddiman DC Anal. Chem. 2011, 83, 6738–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wang C; Wu Z; Yuan J; Wang B; Zhang P; Zhang Y; Wang Z; Huang LJ Proteome Res. 2014, 13, 372–384. [DOI] [PubMed] [Google Scholar]
- (38).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Anal. Chem. 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (39).Xiang F; Ye H; Chen RB; Fu Q; Li LJ Anal. Chem. 2010, 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ross PL; Huang YLN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin DJ Mol. Cell Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- (41).Yang S; Yuan W; Yang WM; Zhou JY; Harlan R; Edwards J; Li SW; Zhang H Anal. Chem. 2013, 85, 8188–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hahne H; Neubert P; Kuhn K; Etienne C; Bomgarden R; Rogers JC; Kuster B Anal. Chem. 2012, 84, 3716–3724. [DOI] [PubMed] [Google Scholar]
- (43).Maxwell EJ; Chen DD Anal. Chim. Acta 2008, 627, 25–33. [DOI] [PubMed] [Google Scholar]
- (44).( Zhong X; Zhang Z; Jiang S; Li L Electrophoresis 2014, 35, 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mittermayr S; Bones J; Guttman A Anal. Chem. 2013, 85, 4228–4238. [DOI] [PubMed] [Google Scholar]
- (46).Zaia J Methods Mol. Biol. 2013, 984, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhao SS; Zhong X; Tie C; Chen DD Proteomics 2012, 12, 2991–3012. [DOI] [PubMed] [Google Scholar]
- (48).Mechref Y Electrophoresis 2011, 32, 3467–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Albrecht S; Schols HA; van den Heuvel EGHM; Voragen AGJ; Gruppen H Electrophoresis 2010, 31, 1264–1273. [DOI] [PubMed] [Google Scholar]
- (50).Jayo RG; Thaysen-Andersen M; Lindenburg PW; Haselberg R; Hankemeier T; Ramautar R; Chen DDY Anal. Chem. 2014, 86, 6479–6486. [DOI] [PubMed] [Google Scholar]
- (51).Liu Y; Salas-Solano O; Gennaro LA Anal. Chem. 2009, 81, 6823–6829. [DOI] [PubMed] [Google Scholar]
- (52).Gennaro LA; Salas-Solano O Anal. Chem. 2008, 80, 3838–3845. [DOI] [PubMed] [Google Scholar]
- (53).Bunz SC; Rapp E; Neususs C Anal. Chem. 2013, 85, 10218–10224. [DOI] [PubMed] [Google Scholar]
- (54).Liu X; Afonso L; Altman E; Johnson S; Brown L; Li J Proteomics 2008, 8, 2849–2857. [DOI] [PubMed] [Google Scholar]
- (55).Jayo RG; Li JJ; Chen DDY Anal. Chem. 2012, 84, 8756–8762. [DOI] [PubMed] [Google Scholar]
- (56).Ito E; Nakajima K; Waki H; Miseki K; Shimada T; Sato TA; Kakehi K; Suzuki M; Taniguchi N; Suzuki A Anal. Chem. 2013, 85, 7859–7865. [DOI] [PubMed] [Google Scholar]
- (57).Bunz SC; Cutillo F; Neususs C Anal. Bioanal. Chem. 2013, 405, 8277–8284. [DOI] [PubMed] [Google Scholar]
- (58).Maxwell EJ; Ratnayake C; Jayo R; Zhong X; Chen DDY Electrophoresis 2011, 32, 2161–2166. [DOI] [PubMed] [Google Scholar]
- (59).Hiraoka K; Kudaka I Rapid Commun. Mass Spectrom. 1992, 6, 265–268. [Google Scholar]
- (60).Cole RB; Harrata AK J. Am. Soc. Mass Spectrom. 1993, 4, 546–556. [DOI] [PubMed] [Google Scholar]
- (61).Yamashita M; Fenn JB J. Phys. Chem. 1984, 88, 4671–4675. [Google Scholar]
- (62).Huang Y; Dodds ED Anal. Chem. 2013, 85, 9728–9735. [DOI] [PubMed] [Google Scholar]
- (63).Pagel K; Harvey DJ Anal. Chem. 2013, 85, 5138–5145. [DOI] [PubMed] [Google Scholar]
- (64).Moh ES; Thaysen-Andersen M; Packer NH Proteomics Clin. Appl. 2015, 9, 368–382. [DOI] [PubMed] [Google Scholar]
- (65).Domon B; Costello CE Glycoconjugate J. 1988, 5, 397–409. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.