

Abstract
O-GlcNAcylation is a nutrient-responsive glycosylation that plays a pivotal role in transcriptional regulation. Human RNA polymerase II (Pol II) is extensively modified by O-linked N-acetylglucosamine (O-GlcNAc) on its unique C-terminal domain (CTD), which consists of 52 heptad repeats. One approach to understanding the function of glycosylated Pol II is to determine the mechanism of dynamic O-GlcNAcylation on the CTD. Here, we discovered that the Pol II CTD can be extensively O-GlcNAcylated in vitro and in cells. Efficient glycosylation requires a minimum of 20 heptad repeats of the CTD and more than half of the N-terminal domain of O-GlcNAc transferase (OGT). Under conditions of saturated sugar donor, we monitored the attachment of more than 20 residues of O-GlcNAc to the full-length CTD. Surprisingly, glycosylation on the periodic CTD follows a distributive mechanism, resulting in highly heterogeneous glycoforms. Our data suggest that initial O-GlcNAcylation can take place either on the proximal or on the distal region of the CTD, and subsequent glycosylation occurs similarly over the entire CTD with nonuniform distributions. Moreover, removal of O-GlcNAc from glycosylated CTD is also distributive and is independent of O-GlcNAcylation level. Our results suggest that O-GlcNAc cycling enzymes can employ a similar mechanism to react with other protein substrates on multiple sites. Distributive O-GlcNAcylation on Pol II provides another regulatory mechanism of transcription in response to fluctuating cellular conditions.
Graphical Abstract
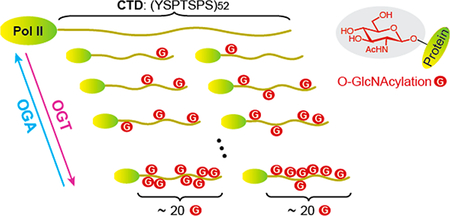
RNA polymerase II (Pol II) is the central machine of transcription in all eukaryotes. Unique to the largest subunit of polymerase (RPB1) is the C-terminal domain (CTD) that consists of 52 imperfect heptad repeats of Y1S2P3T4S5P6S7 (Figure 1A)1,2. This extended tail of Pol II is evolutionarily conserved in eukaryotes with the number of heptads varying from 26 in yeast to 52 in humans.3 A wide variety of posttranslational modifications (PTMs) have been found on the Pol II CTD.4 Most notably, phosphorylation is present from transcription initiation to termination.5,6 Extensive research over the past three decades has clearly demonstrated that specific patterns of phosphorylation on CTD spatially and temporally coordinate different functions of Pol II for the recruitment of enzymes and regulatory proteins that are involved in transcription and RNA processing.7,8 For example, phosphorylation of Ser5 on CTD heptads releases Pol II from the DNA promoter and initiates transcription, while phosphorylation of Ser2 switches Pol II to be elongation proficient.5
Figure 1.

Dynamic O-GlcNAcylation of the Pol II CTD. (A) The Pol II CTD consists of 52 imperfect heptad repeats of YSPTSPS, which can be dynamically O-GlcNAcylated (circled G). (B) Illustration showing that O-GlcNAcylation is catalyzed by OGT and removed by OGA.
Pol II can also be highly O-GlcNAcylated, as found in the transcription preinitiation step, but the specific function is still unclear.9,10 O-GlcNAcylation is the transfer of N-acetylglucosamine from uridine diphosphate N-acetyl glucosamine (UDPGlcNAc) to hydroxyl side chains of serine or threonine residues on thousands of intracellular proteins (Figure 1B).11–13 Like phosphorylation, O-GlcNAcylation is dynamic: installed by OGlcNAc transferase (OGT)14 and removed by O-GlcNAcase (OGA).15 The level of O-GlcNAcylation rapidly changes in response to nutrient and cellular stress to modulate gene expression and signal transduction.16,17 Early studies detected abundant O-GlcNAcylation on the Pol II CTD isolated from calf thymus.9 O-GlcNAc site mapping by manual Edman degradation had identified three CTD glycopeptides and suggested that glycosylation may occur on any of the four Ser/Thr residues of a heptad. More recently, extensive OGlcNAcylation was also detected on human Pol II.10 Using CTD mutants that replaced each of the four Ser/Thr residues of a heptad with alanine, Ser5 has been identified as a major glycosylation site.10 In the same report, pharmacological inhibition of OGT and OGA abolished transcription during the preinitiation step, suggesting that dynamic installation and removal of O-GlcNAc are essential for regulating tran-scription.10,16,18
To date, a number of potential roles of O-GlcNAcylated Pol II have been proposed, including directing Pol II to active promoters, facilitating the assembly of transcription preinitiation complex, and preventing premature association of Pol II elongation or mRNA processing factors.19 To understand the molecular basis of how glycosylated Pol II functions in transcription, a comprehensive mechanistic picture of O-GlcNAcylation along the entire Pol II CTD is crucial. While the study of O-GlcNAcylation on short synthetic CTD peptides was reported a decade ago,19 many fundamental questions regarding this modification in the context of the full-length CTD remain elusive. For example, how many residues of OGlcNAc can be added to the Pol II CTD, which contains more than 200 potential glycosylation sites? Is the O-GlcNAcylation mechanism on the repetitive CTD heptads processive (in which OGT glycosylates the CTD multiple times without dissociating) or distributive (one glycosylation per binding event)? Similarly, what mechanism does OGA utilize to remove O-GlcNAc from the glycosylated CTD? Different mechanisms are expected to yield differentially glycosylated Pol II and thus may lead to different functions. Here, we report biochemical and mass spectrometric studies of the mechanism of dynamic O-GlcNAcylation along the entire CTD of human Pol II to answer these questions and pave the way for a molecular understanding of the function of glycosylated Pol II in transcription.
EXPERIMENTAL PROCEDURES
In Vitro Transcription–Translation (IVTT) and MassTagging Assay.
Pol II-WT and Pol II-delCTD plasmids were gifts from B. Blencowe (Addgene plasmids 35175 and 35176, respectively).21 The 35S-radiolabeled Pol II protein was produced from the plasmid following the user manual of the TNT Quick Coupled Transcription/Translation System (Promega). Purified ncOGT (1.2 μM), UDP-GlcNAz (200 μM), and OGA inhibitor PUGNAc (50 μM) were added to the reaction mixture and incubated at 37 °C for 60 min. Subsequently, azadibenzocyclooctyne-PEG 5 kDa (or ADIBO-PEG, 400 μM) was added to the mixture and incubated at 21 °C for an additional 1 h. Samples were mixed with SDS loading dye, heated at 60 °C for 5 min, and separated on a 4% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel. The gel was dried for 2 h in a gel dryer (Bio-Rad), exposed to a storage phosphor screen (GE Healthcare) overnight, and imaged with a Storm scanner (Amersham Biosciences). For O-GlcNAcylation of a mixture of Pol II-WT and SUMO-CTD52, each 35S-radiolabeled protein was individually generated before they were mixed together to react with ncOGT and UDP-GlcNAz following the same protocol described above. Products were separated on a 6% SDS–PAGE gel. A control reaction without the addition of DNA was conducted to measure any background incorporation of labeled amino acids.
Cell Culture, Metabolic Labeling with Ac4GlcNAz, and Western Blot.
Human HeLa S3 cells were cultured in DMEM containing 10% FBS at 37 °C and 5% CO2 to reach 60–70% confluence. Ac4GlcNAz (200 μM)22 was added to treat cells for 12 h before OGA inhibitor was added (Thiamet-G, 1 μM) and the mixture incubated for an additional 4 h. Cells were washed with PBS and harvested by centrifugation. Cell pellets were first lysed in cytoplasmic extraction buffer containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, and 10 mM KCl with a Dounce homogenizer. The cytoplasmic fraction was removed by centrifugation at 11000g for 20 min at 4 °C. The nuclear pellets were resuspended in nuclear extraction buffer [20 mM HEPES (pH 7.9), 25% (v/v) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, and 0.2 mM EDTA] and incubated at 4 °C for 1 h. Cell debris was removed by centrifugation at 16000g for 10 min at 4 °C. Nuclear proteins (30 μg) were incubated in reaction buffer [10 mM Tris (pH 8.0) and 150 mM NaCl] in the presence or absence of ADIBO-PEG (1 mM) at 21 °C for 1 h. Samples were separated on a 4% SDS–PAGE gel and detected by Western blot with an antibody that is specific for the Pol II Nterminal domain (sc-899, Santa Cruz) or the Pol II CTD (ab817, Abcam).
In Vitro O-GlcNAcylation Reaction.
To test the requirement of tetratricopeptide repeat (TPR) length for O-GlcNAcylation on CTD20, purified His-SUMO-CTD20 (10 μM) was preincubated in reaction buffer [10 mM Tris (pH 8.0), 75 mM NaCl, 1 mM THP, 500 units/mL CIP alkaline phosphatase, and 30 mM MgCl2] at 37 °C for 30 min to remove potential phosphorylation and reduce product inhibition in the following O-GlcNAcylation process. The UDP-GlcNAc mixture (2.88:97.12 UDP-[14C]GlcNAc:UDPGlcNAc ratio, total concentration of 3.34 mM, specific activity of 7.2 mCi/mmol) and OGT protein (ncOGT, OGT6.5, OGT5.5, or OGT4.5, final concentration of 4 μM) were added to the reaction mixtures and incubated at 37 °C for 14 h. The samples were mixed with SDS loading dye and boiled. Each sample was then split into 5 and 15 μL and analyzed on a 10% SDS–PAGE gel. The first half of the gel loaded with 5 μL of each sample was stained by Coomassie Blue to confirm the amount of loaded proteins. The second half of the gel loaded with 15 μL of each sample was dried, exposed to a storage phosphor screen overnight, and imaged with a Storm scanner. Intensities of protein bands on a Coomassie Blue-stained gel and the 14C signal on an exposed gel were integrated by ImageJ (version 1.45s, National Institutes of Health). To minimize exposure variations between different 14C gels from three independent experiments, the 14C signal of each CTD band was divided by the total 14C signal of all CTDs on the same gel to calculate their 14C percentages, which should be proportional to their O-GlcNAcylation levels. These percentages were divided by normalized concentrations of CTD20. For this experiment, the activity of ncOGT was set as 100%, and the activities of other truncated OGTs were normalized to ncOGT.
To test the requirement of CTD length for efficient O-GlcNAcylation, the same amount of CTD heptads of each purified protein (29.7 μM His-SUMO-CTD7, 20.8 μM His-SUMO-CTD10, 13.9 μM His-SUMO-CTD15, 10.4 μM His-SUMO-CTD20, or 4.0 μM His-SUMO-CTD52) was preincubated in reaction buffer [10 mM Tris (pH 8.0), 75 mM NaCl, 1 mM THP, 500 units/mL CIP alkaline phosphatase, and 30 mM MgCl2] at 37 °C for 30 min. The UDP-GlcNAc mixture (2.88:97.12 UDP-[14C]GlcNAc:UDP-GlcNAc ratio, total concentration of 3.34 mM, specific activity of 7.2 mCi/mmol) and ncOGT (final concentration of 4 μM) were added to the reaction mixtures and incubated at 37 °C for 14 h. The rest of steps were the same as described above. The percentages of the 14C signal were calculated similarly and divided by normalized total numbers of CTD heptads to evaluate the O-GlcNAcylation level on each heptad from different CTD constructs. In this experiment, the O-GlcNAc level on CTD52 was set as 100%, and the O-GlcNAc levels of other short CTD peptides were normalized to CTD52. This experiment was conducted in triplicate.
Intact Protein Mass Spectrometry (MS).
To determine the O-GlcNAcylation mechanism on CTD52, purified His-SUMO-CTD52 (10 μM) was preincubated in reaction buffer [10 mM Tris (pH 8.0), 75 mM NaCl, 1 mM THP, 500 units/ mL CIP alkaline phosphatase, and 30 mM MgCl2] at 37 °C for 30 min. UDP-GlcNAc (10 mM) and ncOGT (5 μM) were added to the reaction mixtures and incubated at 37 °C for different periods of time. O-GlcNAcylated CTD52 was bufferexchanged into 25 mM ammonium bicarbonate (pH 7.8) with a Zeba spin column (Thermo Scientific). Similar O-GlcNAcylation with an increased ratio of His-SUMO-CTD52 (50 μM) to ncOGT (5 μM) of 10:1 was conducted at 37 °C for 3 h to confirm the distributive mechanism. To analyze the large fragment of the CTD (2–30 repeats), proteins were digested with sequencing-grade trypsin [1:50 (w/w), Promega] overnight. A 5% formic acid/10% acetonitrile mixture was added to the intact and digested CTD samples for LC–MS analysis.
To detect the mechanism of removal of O-GlcNAc from glycosylated CTD52, purified O-GlcNAcylated His-SUMOCTD52 (20 μM) was incubated with OGA (0.6 μM for a 1:33 ratio or 5 μM for a 1:4 ratio) in reaction buffer [10 mM Tris (pH 7.5) and 0.5 mM THP] at 37 °C for different periods of time. The samples were buffer-exchanged similarly as mentioned above and analyzed via LC–MS.
Mass measurements of the intact and large fragment of the CTD were performed with a Q-TOF Bruker Maxis 4G instrument (Bruker Daltonics) coupled with an ACQUITY UPLC sustem (Waters). Samples were loaded onto a 2.1 mm × 100 mm BEH C4 column (1.7 μm, 300 Å, Waters). The injection volume was 9 μL with a flow rate of 300 μL/min. The mobile phases consisted of 0.1% formic acid (solvent A) and 0.1% formic acid in 95% acetonitrile (solvent B). LC program: 5% B from 0 to 10 min, 5 to 60% B from 10 to 25 min, 60 to 90% B from 25 to 26 min, 90% B from 26 to 31 min, and 90 to 5% B from 31 to 32 min. MS analysis was conducted in positive mode with electrospray voltage of 3.8 kV. The end plate offset and nebulizer pressure were −500 V and 2.1 bar, respectively. The interface heater temperature was set at 220 °C with a dry gas flow rate of 10 L/min. Data were acquired using one full MS scan (m/z 200–2900) with a scan rate of 1 Hz. Funnel 1 RF and multiple RF were set to 400 eV for ion transfer. The ion energy of the quadrupole was 3 eV. The collision energy was 6 eV. The transfer time was 110 μs. The prepulse storage time was 10 μs. LC–MS data were processed and analyzed by Compass Data Analysis software (version 4.1, Bruker). The spectrum was generated over the LC peak. The most intense charge states from 33+ to 73+ were selected for deconvolution performed by a maximum entropy algorithm. The parameters of maximum entropy include a mass range of 20000–130000 Da, auto data point spacing, and a resolving power of 10000. The increased mass resulting from O-GlcNAcylation was determined by subtracting the original protein mass (51655.2 Da) from the deconvoluted protein mass.
In-Gel Trypsin Digestion and Nano LC–MS/MS.
O-GlcNAcylated CTD52 was separated on a 4 to 20% Mini-PROTEAN precast gel (Bio-Rad) and stained with Coomassie Blue. The protein bands with O-GlcNAcylated CTD52 were excised into small slices and subjected to in-gel digestion. Gel slices were washed with a 25 mM NH4HCO3/50% acetonitrile mixture and digested with sequencing-grade trypsin [1:50 (w/ w)] overnight. Peptides were extracted by 100% acetonitrile, lyophilized, and redissolved in 0.1% formic acid for LC–MS/ MS analysis. Peptide samples were analyzed on an Orbitrap QExactive instrument (Thermo Scientific) equipped with a nanoAcaquity UPLC system (Waters). Samples were loaded onto a 75 μm × 1.5 cm nanoACQUITY 1.7 μm BEH C18 column at a flow rate of 350 nL/min. Mobile phase A consisted of 0.1% formic acid, and mobile phase B was 0.1% formic acid in acetonitrile. A linear gradient of 3 to 35% B in 30 min and 35 to 95% B in 1 min, 95% B in 5 min, and 95 to 3% B in 10 min was employed throughout this study. Mass spectra from full scans were acquired in a data-dependent mode (m/z 200–2000). The resolution of the survey scan was set to 70000 at m/ z 400 with an automated gain control (AGC) value of 106. The top 10 most intense precursor ions were selected from the MS scan for the subsequent higher-energy collisional dissociation (HCD, normalized collision energy of 30 eV) MS/MS scan. A resolution of 17500 at m/z 200 and an AGC target at 10000 were set for fragment spectra. Two biological analyses were performed for each time point.
For data analysis, the peak list of each raw MS spectrum was generated by MSConvert (release 3.0.3671). The peptide identification was performed with Mascot version 2.4 (Matrix Science) against a composite target-decoy protein sequence database containing the Uniprot database (release 2014_10, subset human, 20265 protein entries) and His-SUMO-CTD52 sequence. The search criteria used in this study include trypsin specificity allowing up to two missed cleavages and variable modifications of HexNAc (S/T), carbamidomethyl (C), and oxidation (M). The precursor mass tolerance and the fragment ion tolerance were set at ±10 ppm and ±0.6 Da, respectively. Data from each LC–MS/MS run were searched individually. Peptides were considered identified if the Mascot score yielded a confidence limit of >95% based on the significance threshold (p < 0.05). Via application of the criteria, the false discovery rate was 2.2%. The spectra of glycosylated peptides were manually inspected.
Distraction Assay.
Purified CTD52 (15 μM) was incubated with ncOGT (5 μM) and UDP-GlcNAc (6 mM) at 37 °C for 60 min. The reaction mixture (40 μL) was transferred and mixed with a second batch of CTD52, and the incubation was continued for an additional 15 min. The final concentrations of the first and second batches of CTD52 were 10 and 23.3 μM, respectively. After the incubation, this sample was immediately acidified by 2% formic acid to quench the reaction. As a control, formic acid was added to the same 40 μL of the reaction mixture immediately following addition of the second batch of CTD52 without further incubation. Reaction products were desalted and analyzed using the same intact protein MS protocol as mentioned above.
RESULTS
The Pol II CTD Is Heavily Glycosylated.
Considering that the large size of Pol II (∼220 kDa) drastically limits the scope of experiments that can be used to monitor its dynamic O-GlcNAcylation, we decided to first examine if the Pol II CTD can be glycosylated. To avoid significant PTMs on Pol II that may interfere with O-GlcNAcylation, we freshly generated 35S-radiolabeled Pol II protein directly from cDNA using an in vitro transcription–translation (IVTT) assay (Figure 2A).23 Purified OGT and an azido analogue of UDP-GlcNAc (UDP-GlcNAz) were added to glycosylate Pol II in the mixture. Using click chemistry, O-GlcNAz was conjugated to an alkyne containing 5 kDa PEG-tag (ADIBO-PEG).23,24 Glycosylated Pol II was visualized as a smear at higher molecular weight via autoradiography (Figure 2B). A smear rather than a sharp band shift observed here could be due to the heterogeneous nature of glycosylated Pol II (see below). As a control, Pol II missing the entire CTD (Pol II-delCTD) remained unchanged after incubation with OGT (Figure 2B). This provides strong evidence suggesting that extensive O-GlcNAcylation is located on the CTD but not other regions of Pol II. To further test if the non-CTD part of Pol II affects O-GlcNAcylation on CTD, we compared glycosylation on the CTD of wild-type Pol II (Pol II-WT) and a construct containing the entire CTD fused to the C-terminus of SUMO protein (SUMO-CTD52). This fusion enhanced the stability and solubility of the CTD. In the same assay, SUMO-CTD52 can be O-GlcNAcylated like Pol IIWT as evidenced by the original band shifted to a smear at a higher molecular weight (Figure 2C). Importantly, both Pol IIWT and SUMO-CTD52 were as heavily glycosylated when mixed together as glycosylating them individually. This implies no strong preference for OGT to glycosylate CTD from either substrate; thus, CTD itself is a good substrate of OGT. We further tested if the results from the IVTT assay can faithfully report O-GlcNAcylation of Pol II in cells. We treated HeLa cells with an azido sugar analogue Ac4GlcNAz, which can be metabolically converted to UDP-GlcNAz for O-GlcNAcylation of proteins in cells.22 To detect the glycosylation of Pol II, nuclear extracts from these cells were “clicked” with ADIBOPEG followed by immunoblots with antibodies that are specific for Pol II. A PEG-shifted smear was observed (Figure 2D), which is consistent with the results from the IVTT assay and suggests that O-GlcNAcylated Pol II in cells is also a heterogeneous population.
Figure 2.

O-GlcNAcylation is located on the CTD of Pol II. (A) Schematic illustration of the IVTT experimental procedure. (B) Radiolabeled Pol II-WT or Pol II-delCTD protein generated from IVTT assays. Following incubation under the indicated conditions, O-GlcNAcylation was detected on Pol II-WT but not Pol II-delCTD by autoradiography. Abbreviations: Pol II-WT, wild-type RNA polymerase II; Pol II-delCTD, RNA polymerase II with the entire CTD region deleted; G-Pol II-WT, O-GlcNAcylated wild-type RNA polymerase. (C) IVTT experiment showing that SUMO-CTD52 can be glycosylated in a manner similar to that of Pol II-WT (lanes 1, 2, 5, and 6). For lanes 3 and 4, radiolabeled SUMO-CTD52 or Pol IIWT protein was generated individually before they were mixed together to react under the conditions as specified. Abbreviation: G-SUMO-CTD52, O-GlcNAcylated SUMO-CTD52. (D) O-GlcNAcylated Pol II was detected from Ac4GlcNAz-treated HeLa cells (200 μM, 16 h). The nuclear extracts from these cells were incubated in the presence or absence of ADIBO-PEG. Pol II was detected by anti-N-term Pol II (recognizing the Nterminal domain of Pol II) (top) or by anti-CTD (recognizing unmodified CTD heptads) (bottom). O-GlcNAcylated Pol II was visualized as a smear shift following its incubation with ADIBO-PEG. The loss of anti-CTD signal in the ADIBO-PEG sample could be due to the interference of “clicked” PEG with the recognition of unmodified CTD heptads by the anti-CTD antibody.
O-GlcNAcylation on CTD Requires Exceptionally Long Heptad Repeats and Maximum OGT TPRs.
Despite the abundant O-GlcNAcylation on Pol II CTD in vivo, previous in vitro O-GlcNAcylation on short synthetic CTD peptides (one to five heptads) was not observed.19 Increasing the length of the CTD peptide to 10 heptads (CTD10, 70 residues) detected on average only approximately one unit of O-GlcNAc per molecule of peptide, which corresponds to one of 40 potential glycosylation sites.19 Because OGT is known to glycosylate many short peptides,20,25 this report suggested a unique feature of OGT binding to CTD, but the minimal length of CTD for efficient O-GlcNAcylation remains to be determined. Intrigued by this report, we systematically tested the efficiency of O-GlcNAcylation on different lengths of CTD peptides. A series of SUMO-CTD constructs containing different numbers of heptads (SUMO-CTD7, −10, −15, −20, and −52) were recombinantly expressed and purified from E. coli. The same total number of heptad repeats of each CTD was incubated with OGT and radiolabeled UDP-[14C]GlcNAc. O-GlcNAcylation was quantified by autoradiography and normalized to the total number of CTD heptads (Figure 3A). It is clear that SUMO-CTD7 was not O-GlcNAcylated, suggesting that neither SUMO nor CTD7 is a good substrate of OGT. Weak O-GlcNAcylation corresponding to 26% relative to SUMOCTD52 was detected on SUMO-CTD10. This is consistent with a previous report on substoichiometric O-GlcNAcylation on the synthetic CTD10 peptide.19 It also supports the idea that the SUMO tag does not interfere with O-GlcNAcylation on the CTD. For longer CTD peptides, the O-GlcNAcylation level increased along SUMO-CTD15 (40%), CTD20 (77%), and CTD52 (100%). SUMO-CTD52 was the best substrate for O-GlcNAcylation compared to its C-terminally truncated counterparts. Therefore, when the same number of CTD heptads is provided, OGT prefers to glycosylate long CTD substrates. The structure of human OGT contains N-terminal tetratricopeptide repeats (TPRs) that are usually involved in protein–protein interactions, and a C-terminal catalytic domain for glycosyl transfer.20,26 We next investigated the requirement of TPR length of OGT for CTD O-GlcNAcylation. Tested constructs included full-length ncOGT (the most abundant isoform of OGT in the nucleus),14,27,28 OGT6.5, OGT5.5, and OGT4.5,20 which share the same catalytic domain but contain 13.5, 6.5, 5.5, and 4.5 TPRs, respectively (Figure 3B). The ability of each purified OGT to glycosylate SUMO-CTD20 was tested with UDP-[14C]GlcNAc via autoradiography (Figure 3B). CTD20 instead of CTD52 was used here to avoid overlap with OGT4.5 on the gel. Compared to ncOGT, truncated constructs (OGT6.5, OGT5.5, and OGT4.5) severely diminished O-GlcNAcylation on CTD, and only 10–20% relative activities were detected. Taken together, efficient O-GlcNAcylation requires a minimum of 20 heptad repeats of CTD and more than half of the TPR region of ncOGT.
Figure 3.

O-GlcNAcylation on CTD requires exceptionally long heptad repeats and maximum OGT TPRs. (A) O-GlcNAcylation on a series of SUMO-CTDs containing different numbers of heptads (ranging from 7 to 52 repeats). The glycosylation level was quantified on the basis of [14C]GlcNAc signals via autoradiography and normalized to the same total number of CTD heptads. The data are presented as means ± the standard deviation (SD) (n = 3). (B) O-GlcNAcylation of SUMO-CTD20 by a series of OGTs (varying only in the TPR domain, 4.5, 5.5, 6.5, and 13.5 TPRs). The glycosylation level was quantified on the basis of [14C]GlcNAc signals via autoradiography and normalized to the same concentration of each OGT. The data are presented as means ± SD (n = 3).
O-GlcNAcylation on the CTD Follows a Distributive Mechanism.
Having validated that SUMO-CTD52 can be efficiently O-GlcNAcylated by ncOGT in vitro, we used this construct to examine the processivity of O-GlcNAcylation on CTD repeats by mass spectrometry (MS), which can decipher a complex mixture of CTD modifications. The periodic template structure of CTD allows for multisite O-GlcNAcylation, which can happen via a processive or distributive mechanism. In a processive mechanism, multiple O-GlcNAc modifications could occur during a single substrate binding event, and the substrate dissociates when it is maximally O-GlcNAcylated. In the presence of an excess of CTD substrate, we would expect to detect either unmodified or maximally glycosylated CTD. Alternatively, O-GlcNAcylation could take place via a distributive mechanism. In this case, only one O-GlcNAcylation occurs during a single enzyme–substrate binding event. After the CTD is glycosylated, OGT dissociates and then must rebind at a different site of O-GlcNAcylation on the same or a different molecule of the CTD. This mechanism will generate a mixture of similarly O-GlcNAcylated CTDs showing a timedependent increase in their glycosylation levels (Figure 4A). To test these mechanisms, we conducted high-resolution intact protein MS to monitor O-GlcNAcylation on SUMO-CTD52 after incubation with ncOGT and UDP-GlcNAc. MS accuracy was evaluated using unmodified SUMO-CTD52: deconvoluted MS showed a single peak at 51654.7 Da, matching closely its theoretical molecular weight of 51655.2 Da (Figure 4B, top). Interestingly, after reaction for only 5 min, the unmodified CTD became a mixture containing zero, one, and two residues of O-GlcNAc modifications (Figure 4B). This heterogeneous feature of glycosylated CTD was maintained at each tested time point from 5 min to 14 h. For example, 7–11 units of O-GlcNAc were detected after 1 h, and this number increased to 12–16 units after 3 h. At the longest time point of 14 h, 18–22 units of O-GlcNAc were detected. Further increasing the CTD:OGT molar ratio from 2:1 to 10:1 yielded a similar symmetric profile of glycosylated CTD, and no unmodified peptide was detected (Figure S1). These data support a distributive mechanism of O-GlcNAcylation on the Pol II CTD.
Figure 4.

Time-dependent analysis of intact protein MS of O-GlcNAcylated SUMO-CTD52. (A) Schematic illustration of different products expected from processive or distributive O-GlcNAcylation of the CTD: (---) early time point, (—) late time point, and (Ⓖ) O-GlcNAc. (B) Deconvoluted mass spectra of O-GlcNAcylation on SUMO-CTD52 at different time points (CTD:OGT molar ratio of 2:1). The numbers of O-GlcNAc residues are highlighted in red and bold.
Unambiguous discrimination between processive and distributive modes of action requires the demonstration of single or multiple dissociation events between the enzyme and its acceptor substrate throughout the reaction. This direct evidence will help rule out other possibilities during the catalytic processes, especially considering the CTD is an unusually long substrate of OGT and multisite interactions have been suggested.19 Inspired by the distraction assay for carbohydrate polymerases,29 we sought to use a similar assay to validate the distributive O-GlcNAcylation mechanism on the CTD. To avoid any impact from the SUMO tag, CTD52 without any tag (theoretical molecular weight of 39635.8 Da) was used in this experiment. In the first stage of the assay, the CTD was partially glycosylated by OGT, and then a second batch of the unmodified CTD was added to “distract” O-GlcNAcylation on the partially glycosylated CTD. Samples taken 0 and 15 min after the addition of the second batch of CTD were analyzed by intact protein MS (Figure S2). After the first stage, 5–8 units of O-GlcNAc residues were detected on the first batch of the CTD. Because of the severe ion suppression effect, the O-GlcNAcylated CTD ionized much more weakly than the unmodified CTD. Following the second stage of incubation, a processive mechanism would result in biased O-GlcNAcylation on the first batch versus the second batch of CTD due to the fact that newly added CTD cannot compete with the first batch for glycosylation. In contrast, two groups of the glycosylated CTD were detected. O-GlcNAcylation on the first batch of the CTD increased to 6–9 units, and the second batch was modified with 0–3 units of O-GlcNAc (Figure S2b). This result indicates that the unmodified CTD and the partially glycosylated CTD have equal access to the enzyme, providing direct evidence of the distributive mechanism of O-GlcNAcylation on the CTD.
O-GlcNAcylation Occurs over the Entire CTD.
We next characterized the order and distribution of O-GlcNAcylation along the entire CTD. Specifically, we are interested in the following questions. Where does the first O-GlcNAcylation take place on the CTD? Does O-GlcNAcylation on the CTD follow an ordered sequence from the N- to C-terminus? Is CTD O-GlcNAcylated uniformly on each heptad repeat? We sought to clarify the region-specific distribution of O-GlcNAcylation on the CTD using a combination of intact protein MS, trypsin digestion, and tandem LC–MS/MS approaches. Purified SUMO-CTD52 was incubated with ncOGT and UDP-GlcNAc for 0, 2, 10, 60, and 120 min. The sample from each time point was split into three fractions: one for intact protein MS to detect total O-GlcNAc modifications on the intact SUMOCTD52 protein; one for in-solution trypsin digestion followed by intact protein MS to detect total O-GlcNAc modifications on the highly repetitive CTD2–31 region (21 kDa), which is deficient with respect to trypsin digestion sites and also difficult to map O-GlcNAcylation to a specific CTD heptad; and the last fraction for in-gel trypsin digestion followed by LC–MS/ MS to detect time-dependent O-GlcNAc addition on the less conserved SUMO-CTD1 and distal CTD31–52 regions for relative comparison. Mascot searching of LC–MS/MS spectra of the third fraction against a customized database (containing the entire Uniprot human database and SUMO-CTD52 protein sequence) unambiguously matched to SUMO-CTD52 with 43% protein coverage. This corresponds to 94% of the tryptic CTD peptides in SUMO-CTD1 and CTD31–52 regions that would be detected under the MS setting. An example of sequence coverage is shown in Figure S3, and the total identified peptides are listed in the Supplementary Table. A summary of intact and tandem MS results is shown in Figure 5. As expected, no modification was detected at 0 min. After a brief incubation (2 min), we detected 0–3 units of O-GlcNAc modifications on the entire SUMO-CTD52 protein, which include 0–2 units on the proximal CTD2–31 region and 0–1 units each on CTD32–35, CTD36–38, and CTD43–45 peptides in the distal region. These data suggest that the first O-GlcNAcylation can take place on proximal or distal regions of the CTD. Consistent with this “random” pattern, continuously increased O-GlcNAcylation was observed over the entire CTD with prolonged incubation. The distribution of O-GlcNAc on each heptad appeared to be nonuniform because certain regions of CTD are more favorably O-GlcNAcylated (e.g., CTD2–31, CTD36–38, and CTD43–45), while other regions even containing similar potential glycosylation sites remain unmodified at the longest reaction time tested (e.g., CTD39–40) (Figure S3, Figure 5, and the Supplementary Table). Overall, these experiments revealed unexpected highly heterogeneous glycoforms of the CTD.
Figure 5.

Summary of intact protein MS and tandem MS/MS data from a time course showing that O-GlcNAcylation can take place over the entire CTD. The highly repetitive CTD2–30 region of SUMO-CTD52 is highlighted in dark gray, and less conserved CTD1 and CTD32–47 are highlighted in light gray. The numbers of O-GlcNAc residues are colored red. For example, “0–3” represent unmodified, singly, doubly, and triply O-GlcNAcylated CTD peptides, respectively. The peptides containing the same total number of O-GlcNAc modifications could vary in their modification sites, but our current study cannot unambiguously map O-GlcNAcylation sites on a particular CTD heptad repeat. Peptide CTD48–52 (white) was not detected (N). Results shown here could be reproduced from two biological analyses at each time point.
OGA Distributively Removes O-GlcNAc from the Glycosylated CTD.
With the establishment of distributive O-GlcNAcylation on CTD heptads, we further examined the mechanism of removal of O-GlcNAc from the glycosylated CTD in a reaction with OGA. We co-expressed SUMO-CTD52 and ncOGT in E. coli to generate and purify the glycosylated CTD. Following incubation with OGA at different time points, O-GlcNAc removal was monitored by intact protein MS. Processive and distributive mechanisms of deglycosylation are expected to yield distinct profiles of the CTD (Figure 6A), which can help to determine the mode of action. At 0 min, the deconvoluted mass spectrum showed a mixture of 5–15 units of O-GlcNAc on SUMO-CTD52, which peaked at 11 units (Figure 6B, top). After incubation for 30 min at a 1:33 OGA:glycosylated CTD ratio, O-GlcNAc was decreased to 4–14 units, and further reduced to 3–12 units after incubation for 1 h (Figure 6B). Deconvoluted mass spectra remained symmetric in a constrained range at various time points, and no unmodified CTD was detected. This observation indicates that OGA hydrolyzes O-GlcNAc distributively from periodic CTD heptads and displays no obvious preference for either more or less glycosylated substrates. To test if OGA can completely remove O-GlcNAc from the CTD, an increased ratio of OGA to glycosylated CTD of 1:4 was applied. After incubation for 30 min, only 0–6 units of O-GlcNAc were left on CTD, which dropped to 0–4 units after 2 h and decreased further to 0–2 units after 8 h (Figure S4). It is therefore conceivable that OGA is capable of completely removing O-GlcNAc from the Pol II CTD.
Figure 6.

Intact protein MS data of OGA-catalyzed deglycosylation of CTD along a time course. (A) Hypothetical schematic illustration of different products generated from the possibilities that OGA may preferentially remove O-GlcNAc from more or less glycosylated CTD during either distributive or processive deglycosylation (OGA’s concentration is lower than that of the glycosylated CTD). In each panel, the x-axis corresponds to the O-GlcNAcylation level of proteins and the y-axis corresponds to intensity. Dashed and solid curves represent deconvoluted intact protein mass spectra of glycosylated CTD at early and late reaction time points, respectively. (B) Intact protein mass spectra of deglycosylation of SUMO-CTD52 at different time points (OGA:CTD molar ratio of 1:33). The numbers of O-GlcNAc residues are highlighted in red and bold.
DISCUSSION AND CONCLUSIONS
Distributive O-GlcNAcylation on the Pol II CTD.
Two PTM-specific forms of Pol II have been identified (either O-GlcNAcylation or phosphorylation).9 In contrast to the wellstudied phosphorylated Pol II, the specific function of O-GlcNAcylated Pol II remains largely unknown, partially because of the challenge of determining its O-GlcNAcylation pattern in vivo. Here, we study the mechanism of O-GlcNAcylation on Pol II as an approach to assess the distribution of O-GlcNAc residues on this large polymerase. Using an IVTT assay, we detected that O-GlcNAcylation of human Pol II is mainly located on the repetitive CTD region. This observation is in excellent agreement with the Pol II isolated from calf thymus.9 In addition, we found that removal of the polymerase core domain did not affect glycosylation on the CTD, implying that the CTD itself is a substrate of OGT. This result is in line with several pieces of reported evidence. First, crystal structures of yeast Pol II30–32 show a flexible transition linker (∼80 residues) between the polymerase core and the CTD, protruding from the enzyme and forming a tail-like extension (CTD is not visible due to mobility). Second, the entire CTD of yeast Pol II can be detached and reattached to other transcription factors in the assembly of the preinitiation complex, and the transcription function can be restored in vitro.33 On the basis of the evidence described above, we focused on O-GlcNAcylation of the CTD instead of the entire Pol II protein for the following MS and biochemical analysis.
To determine the structural features of the CTD and OGT needed for efficient O-GlcNAcylation, we first quantified the glycosylation level of a series of recombinant CTD peptides containing different heptad repeats. CTD7–15 peptides were poor substrates of OGT, and even peptide CTD20 (140 residues) achieved only 77% glycosylation on each repeat compared to that of CTD52. Because the increasing length of the CTD is unlikely to acquire a stable secondary structure,34 these results imply that O-GlcNAcylation of the CTD relies on extensive contacts with OGT to gain sufficient binding. We next tested the glycosylation level of the CTD with truncated TPRs of OGT. Removal of half or more of the TPR repeats from ncOGT significantly reduced its activity toward CTD20, which agrees with a previous report on synthetic peptide CTD10.19 Because OGT4.5 was shown to be fully competent with respect to glycosylating short peptides (e.g., CKII peptide with 14 residues),20,25,35 its deficiency on glycosylating CTD20 suggests that OGT binds to the CTD in a different mode involving multisite interactions through its TPR domain.
With our optimized O-GlcNAcylation assay and different types of MS analysis, we detected that >20 residues of O-GlcNAc were distributively added onto the CTD, resulting in a complex mixture of differentially glycosylated CTD in both proximal and distal regions. The heterogeneous nature of O-GlcNAcylated Pol II was detected from in vitro assays, and it is consistent with the PEG-shifted smear observed for glycosylated Pol II in IVTT assays and in metabolically labeled cells. We noted that the smear from metabolically labeled cells did not shift as high as that from the IVTT assay. This could be due to a lower rate of incorporation of O-GlcNAz to the Pol II CTD in the presence of endogenous UDP-GlcNAc in cells. While other chemoenzymatic assays may be able to detect higher levels of O-GlcNAcylated Pol II in cells, we employed this strategy for more consistent comparison with the IVTT results. With a large excess of UDP-GlcNAc supplied in vitro, even at the longest time point tested, the O-GlcNAcylation on CTD seems to be still unsaturated. Additional cellular factors for accelerated O-GlcNAcylation will require further investigation. It is possible that a minimal number of O-GlcNAc modifications on the Pol II CTD is required to achieve appropriate regulation of transcription in cells. While the O-GlcNAcylation level increased with longer incubations, the narrow symmetric profile of deconvoluted mass spectra remained, and no unmodified CTD was detected after the initial incubation. Symmetric mass spectra and a gradually slowed reaction rate argue against a consecutive mechanism, in which the first O-GlcNAcylation on a CTD heptad makes the subsequent glycosylation more favorable because of an increased binding affinity. A further distraction assay demonstrated that the unmodified CTD and the partially glycosylated CTD have equal access to the enzyme. Taken together, these results established a distributive mechanism for O-GlcNAcylation of the repetitive CTD. Furthermore, our experiments uncovered the fact that OGA also utilizes a distributive mechanism to remove O-GlcNAc from the glycosylated CTD. We envision that these O-GlcNAc cycling enzymes could employ a similar distributive mechanism to react with other substrate proteins on multiple sites. Compared to a processive mechanism, which usually includes one major ratelimiting event, forming the enzyme–substrate complex, a distributive mechanism relying on multiple binding events would be more sensitive to small changes in the concentrations of active enzymes and substrates. Therefore, distributive O-GlcNAcylation allows a more efficient response to acute stimuli. O-GlcNAcylation of Pol II provides another regulatory mechanism of transcription in response to the fluctuation of the nutritional and metabolic status of the cell.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Suzanne Walker for generously providing plasmids of ncOGT, OGT4.5, OGT5.5, and OGT6.5. Pol IIWT and Pol II-delCTD plasmids were gifts from B. Blencowe (Addgene plasmids 35175 and 35176, respectively). We thank Dr. Richard Hsung for advice and support on the synthesis of sugar analogues. We also thank Drs. Lingjun Li and Weiping Tang for comments on the manuscript.
Funding
This research was supported by University of Wisconsin— Madison startup funds, a Vilas Research Investigator Award from the William F. Vilas Trust Estate, and NIH Shared Instrument Program Grant S10 RR029531 for funding the MS instrument.
ABBREVIATIONS
- Pol II
RNA polymerase II
- CTD
C-terminal domain
- OGlcNAc
β-N-acetylglucosamine
- OGT
O-GlcNAc transferase
- OGA
O-GlcNAcase
- PTM
posttranslational modification
- IVTT
in vitro transcription–translation
- Ac4GlcNAz
Nazidoacetylglucosamine-tetraacylated
- TPR
tetratricopeptide repeat
- Pol II-WT
wild-type RNA polymerase II
- Pol IIdelCTD
RNA polymerase II with the entire CTD region deleted
- G-Pol II-WT
O-GlcNAcylated wild-type RNA polymerase II
- G-SUMO-CTD52
O-GlcNAcylated SUMO-CTD52
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b01280.
Construction of Plasmids and Expression and Purification of Proteins, intact protein MS of O-GlcNAcylated SUMO-CTD52 at a higher molar ratio (Figure S1), deconvoluted mass spectra from the distraction assay (Figure S2), sequence coverage and detected glycosylated SUMO-CTD52 from in-gel trypsin digestion of glycosylated SUMO-CTD52 after incubation for 60 min with ncOGT and UDP-GlcNAc (Figure S3), and intact protein MS showing that OGA almost completely removed O-GlcNAc from the glycosylated CTD (Figure S4) (PDF)
A list of unique peptides detected from in-gel trypsin digestion (Supplementary Table) (XLSX)
REFERENCES
- (1).Corden JL (2013) RNA polymerase II C-terminal domain: tethering transcription to transcript and template. Chem. Rev 113, 8423–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Eick D, and Geyer M (2013) The RNA polymerase II carboxyterminal domain (CTD) code. Chem. Rev 113, 8456–8490. [DOI] [PubMed] [Google Scholar]
- (3).Hsin JP, and Manley JL (2012) The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 26, 2119–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Napolitano G, Lania L, and Majello B (2014) RNA polymerase II CTD modifications: How many tales from a single tail. J. Cell. Physiol 229, 538–544. [DOI] [PubMed] [Google Scholar]
- (5).Heidemann M, Hintermair C, Voß K, and Eick D (2013) Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta, Gene Regul. Mech 1829, 55–62. [DOI] [PubMed] [Google Scholar]
- (6).Yogesha SD, Mayfield JE, and Zhang Y (2014) Cross-talk of phosphorylation and prolyl isomerization of the C-terminal domain of RNA polymerase II. Molecules 19, 1481–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Komarnitsky P, Cho EJ, and Buratowski S (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jeronimo C, Bataille AR, and Robert F (2013) The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem. Rev 113, 8491–8522. [DOI] [PubMed] [Google Scholar]
- (9).Kelly WG, Dahmus ME, and Hart GW (1993) RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J. Biol. Chem 268, 10416–10424. [PubMed] [Google Scholar]
- (10).Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, and Lewis BA (2012) Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J. Biol. Chem 287, 23549–23561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Torres CR, and Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem 259, 3308–3317. [PubMed] [Google Scholar]
- (12).Ma J, and Hart GW (2014) O-GlcNAc profiling: from proteins to proteomes. Clin. Proteomics 11, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Bond MR, and Hanover JA (2015) A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol 208, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kreppel LK, Blomberg MA, and Hart GW (1997) Dynamic glycosylation of nuclear and cytosolic proteins cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem 272, 9308–9315. [DOI] [PubMed] [Google Scholar]
- (15).Gao Y, Wells L, Comer FI, Parker GJ, and Hart GW (2001) Dynamic O-glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a neutral, cytosolic β-N-acetylglucosaminidase from human brain. J. Biol. Chem 276, 9838–9845. [DOI] [PubMed] [Google Scholar]
- (16).Lewis BA, and Hanover JA (2014) O-GlcNAc and the epigenetic regulation of gene expression. J. Biol. Chem 289, 34440–34448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hart GW, Slawson C, Ramirez-Correa G, and Lagerlof O (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem 80, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lewis BA (2013) O-GlcNAcylation at promoters, nutrient sensors, and transcriptional regulation. Biochim. Biophys. Acta, Gene Regul. Mech 1829, 1202–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Comer FI, and Hart GW (2001) Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry 40, 7845–7852. [DOI] [PubMed] [Google Scholar]
- (20).Lazarus MB, Nam Y, Jiang J, Sliz P, and Walker S (2011) Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rosonina E, and Blencowe BJ (2004) Analysis of the requirement for RNA polymerase II CTD heptapeptide repeats in premRNA splicing and 3′-end cleavage. RNA 10, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Boyce M, Carrico IS, Ganguli AS, Yu SH, Hangauer MJ, Hubbard SC, Kohler JJ, and Bertozzi CR (2011) Metabolic cross-talk allows labeling of O-linked β-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc. Natl. Acad. Sci. U. S. A 108, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ortiz-Meoz RF, Merbl Y, Kirschner MW, and Walker S (2014) Microarray discovery of new OGT substrates: the medulloblastoma oncogene OTX2 is O-GlcNAcylated. J. Am. Chem. Soc 136, 4845–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, and Hsieh-Wilson LC (2012) Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat. Chem. Biol 8, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pathak S, Alonso J, Schimpl M, Rafie K, Blair DE, Borodkin VS, Schüttelkopf AW, Albarbarawi O, and van Aalten DMF (2015) The active site of O-GlcNAc transferase imposes constraints on substrate sequence. Nat. Struct. Mol. Biol 22, 744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, and Conti E (2004) The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin α. Nat. Struct. Mol. Biol 11, 1001–1007. [DOI] [PubMed] [Google Scholar]
- (27).Lubas WA, Frank DW, Krause M, and Hanover JA (1997) O-linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J. Biol. Chem 272, 9316–9324. [DOI] [PubMed] [Google Scholar]
- (28).Hanover JA, Krause MW, and Love DC (2012) Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol 13, 312–321. [DOI] [PubMed] [Google Scholar]
- (29).Levengood MR, Splain RA, and Kiessling LL (2011) Monitoring processivity and length control of a carbohydrate polymerase. J. Am. Chem. Soc 133, 12758–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cramer P, Bushnell DA, and Kornberg RD (2001) Structural basis of transcription: RNA polymerase II at 2.8 Ångstrom resolution. Science 292, 1863–1876. [DOI] [PubMed] [Google Scholar]
- (31).Armache KJ, Mitterweger S, Meinhart A, and Cramer P (2005) Structures of complete RNA polymerase II and its subcomplex, Rpb4/7. J. Biol. Chem 280, 7131–7134. [DOI] [PubMed] [Google Scholar]
- (32).Spåhr H, Calero G, Bushnell DA, and Kornberg RD° (2009) Schizosacharomyces pombe RNA polymerase II at 3.6-Å resolution. Proc. Natl. Acad. Sci. U. S. A 106, 9185–9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Suh H, Hazelbaker DZ, Soares LM, and Buratowski S (2013) The C-terminal domain of Rpb1 functions on other RNA polymerase II subunits. Mol. Cell 51, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Meinhart A, Kamenski T, Hoeppner S, Baumli S, and Cramer P (2005) A structural perspective of CTD function. Genes Dev. 19, 1401–1415. [DOI] [PubMed] [Google Scholar]
- (35).Rodriguez AC, Yu SH, Li B, Zegzouti H, and Kohler JJ (2015) Enhanced transfer of a photocross-linking N-acetylglucosamine (GlcNAc) analog by an O-GlcNAc transferase mutant with converted substrate specificity. J. Biol. Chem 290, 22638–22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.