

Abstract
Anaplasia may be identified in a subset of tumors with a presumed pilocytic astrocytoma (PA) component or piloid features, which may be associated with aggressive behavior, but the biologic basis of this change remains unclear. Fifty‐seven resections from 36 patients (23 M, 13 F, mean age 32 years, range 3–75) were included. A clinical diagnosis of NF1 was present in 8 (22%). Alternative lengthening of telomeres (ALT) was assessed by telomere‐specific FISH and/or CISH. A combination of immunohistochemistry, DNA sequencing and FISH were used to study BRAF,ATRX,CDKN2A/p16, mutant IDH1 p.R132H and H3‐K27M proteins. ALT was present in 25 (69%) cases and ATRX loss in 20 (57%), mostly in the expected association of ALT+/ATRX‐ (20/24, 83%) or ALT‐/ATRX+ (11/11, 100%). BRAF duplication was present in 8 (of 26) (31%). H3‐K27M was present in 5 of 32 (16%) cases, all with concurrent ATRX loss and ALT. ALT was also present in 9 (of 11) cases in the benign PA precursor, 7 of which also had ATRX loss in both the precursor and the anaplastic tumor. In a single pediatric case, ALT and ATRX loss developed in the anaplastic component only, and in another adult case, ALT was present in the PA‐A component only, but ATRX was not tested. Features associated with worse prognosis included subtotal resection, adult vs. pediatric, presence of a PA precursor preceding a diagnosis of anaplasia, necrosis, presence of ALT and ATRX expression loss. ALT and ATRX loss, as well as alterations involving the MAPK pathway, are frequent in PA with anaplasia at the time of development of anaplasia or in their precursors. Additionally, a small subset of PA with anaplasia have H3‐K27M mutations. These findings further support the concept that PA with anaplasia is a neoplasm with heterogeneous genetic features and alterations typical of both PA and diffuse gliomas.
Keywords: alternative lengthening of telomeres, ATRX, glioma, H3‐K27M, pilocytic astrocytoma
Introduction
Pilocytic astrocytoma (PA) represents the most frequent primary brain tumor in children, and accounts for 15.5% of CNS tumors in patients in the 0–19 age range in the United States 30. These tumors have a predilection for the cerebellum, but may also develop in the optic pathways, cerebrum, brainstem or spinal cord. PAs are well‐differentiated astrocytomas, characterized at the histologic level by bipolar astrocytes and a biphasic architecture in classic cases, including compact areas with Rosenthal fibers alternating with loose areas with myxoid stroma, microcysts and variable numbers of eosinophilic granular bodies. Glomeruloid microvasculature is frequent, but has no prognostic significance when compared with diffuse gliomas. Bland necrosis is another feature that may be present in up to 10% of cases, but is also not associated with a worse outcome11. In general, mitotic activity is low to altogether absent.
Our knowledge of the underlying biologic features of PA has expanded in the past decade (reviewed in 5). Numerous studies have demonstrated that a tandem duplication involving the kinase domain of BRAF and leading to a novel fusion (KIAA1549‐BRAF) is present in most sporadic PA 2, 10, 18, 33, 42, while NF1 gene inactivation is the key feature of tumors developing in NF1 patients 12. Molecular alterations in single genes that lead to MAPK pathway activation have emerged as a universal finding in PA, occurring in 100% of tumors through a variety of mechanisms 16. Other signaling pathways are active and participate in PA biology, particularly mTOR, which is being explored as a therapeutic target 15, 19.
PA is assigned a WHO grade I designation given its slow growth potential, long patient survival and potential for cure when totally resected. However, a small subset of PA develop anaplastic changes either de novo or in the setting of prior irradiation 8, 48. Grading criteria for these tumors remain to be defined 6, although working criteria on a prior series of 34 cases included brisk mitotic activity with or without necrosis 38. These findings were associated with a worse prognosis, akin to diffuse gliomas when compared to historic cohorts. The molecular alterations associated with PA with anaplasia are currently being identified. We have previously documented frequent PI3K/mTOR activation in these tumors, in addition to MAPK activation and deletions involving PTEN and CDKN2A 37. A recent study by Reinhardt et al. has identified a separate molecular methylation class corresponding to a subset of PA with anaplasia, which is also characterized by frequent CDKN2A and alpha thalassemia/mental retardation syndrome X‐linked (ATRX) alterations 36. In the current study, we explore the frequency of ATRX and H3‐K27M alterations, as well as the presence of alternative lengthening of telomeres (ALT) for the first time, in PA with anaplasia based on strict histopathologic criteria.
Materials and methods
Patients and Samples
Clinical and demographic characteristics, including NF1 status, were abstracted from retrospective chart review and consult letters. A total of 36 patients (23 M, 13 F) were included in the study. A total of 57 pathologic specimens were reviewed, including 36 primary resections of PA‐A, 16 biopsies representing PA preceding a diagnosis of anaplasia, and 5 for recurrences/second biopsies after anaplasia. The mean age of diagnosis of anaplasia was 32 years (range 3–75). Patients included 17 cases reported in 2 prior studies 37, 38 but with updated clinical follow‐up. Cerebellum/posterior fossa location was most common (n = 21), followed by supratentorial hemispheric (n = 7), supratentorial intraventricular (n = 3), tectum/pineal (n = 3), and spinal cord (n = 2). Demographics and basic clinical features of the cohort studied are presented in Table 1. Cases were included in the study if they satisfied strict histologic criteria for anaplasia as previously described 38. In brief, all tumors were characterized by brisk mitotic activity (at least 5 mitoses per 10 high‐power fields), as well as hypercellularity and moderate‐to‐severe cytologic atypia. Necrosis was recorded as present or absent. Anaplasia was designated as focal when present in a single low‐power (X20) field, otherwise it was interpreted as diffuse. To exclude the possibility of an infiltrating glioma, particularly glioblastoma IDH wildtype, specific morphologic features of a low grade, circumscribed PA precursor were required in addition to anaplasia, including compact piloid morphology, Rosenthal fibers and/or eosinophilic granular bodies. It must be noted that none of these features in isolation are specific for PA, since infiltrating gliomas may have any one of these. Tumors with overt tissue infiltration or high‐grade features in the precursor were excluded. Morphologic mimics of PA (eg, ganglioglioma, pleomorphic xanthoastrocytoma, rosette forming glioneuronal tumors) were also excluded. In addition, a group of 163 PA or low‐grade gliomas with PA features were studied as a control using telomere‐specific FISH or CISH, as well as ATRX and H3‐K27M immunohistochemistry, obtained from TMA (n = 116) or whole sections (n = 47) evaluated by one of us (FJR) using the same conditions as the study cases. All studies were performed following standard ethical guidelines under Institutional Review Board (IRB) approval.
Table 1.
Clinicopathologic and molecular features of pilocytic astrocytoma with anaplasia (PA‐A)
| CASE | Age | Sex | Location | NF1 clinical status | History of Radiation | Temporal PA precursor | Extent of Resection | Necrosis | ALT | ATRX IHC | IDH1 (R132H) | p53 IHC | BRAF Duplication | BRAF (V600E) | H3‐K27M | p16 IHC | CDKN2A (FISH or NGS) | Other | Treatment for precursor | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 17 | M | Cerebellum | Sporadic | No | Yes | STR | Yes | POSITIVE | Lost | Negative* | Strongly positive | Negative | Negative* | Negative* | observation | Dead | |||
| 2 | 68 | M | fourth ventricle | Sporadic | No | No | STR | Yes | POSITIVE | Lost | Negative | negative | Negative | Negative | Negative | loss | No mutation | NF1 p.E595Nfs*14, c.1783_1784delGA; ATRX p.T1239fsN*10, c.3715dupA | Dead | |
| 3 | 13 | F | Cerebellum | Sporadic | No | No | GTR | Yes | POSITIVE | Lost | Negative* | negative | Alive | |||||||
| 4 | 12 | F | cerebellum | NF1 | No | No | Yes | NEGATIVE | Preserved | Negative* | focal | Negative* | Negative* | Partial loss | Dead | |||||
| 5 | 22 | M | Pineal | NF1 | No | No | STR | No | POSITIVE | Lost | Negative* | Negative* | Dead | |||||||
| 6 | 74 | F | cerebellum | Sporadic | Yes | Yes | POSITIVE | partial | Negative* | Strongly positive | Unknown | Dead | ||||||||
| 7 | 40 | F | Lateral ventricle | Sporadic | Yes | Yes | STR | Yes | POSITIVE | partial | Negative* | focal | Negative | Negative | Negative* | loss | Irradiation | Alive | ||
| 8 | 31 | M | Spinal cord | Sporadic | Yes | Yes | Yes | POSITIVE | Lost | Negative* | Strongly positive | Positive | Negative* | Negative* | loss | Irradiation | Alive | |||
| 9 | 53 | F | Cerebellum | Sporadic | Yes | Yes | Yes | POSITIVE | Lost | Negative* | Strongly positive | Positive | Negative | positive | No mutation | H3F3A p.K27M, ATRX p. R1739X, TP53 I255S | irradiation +temozolomide | Alive | ||
| 10 | 66 | M | Cerebellum | Sporadic | Yes | Yes | No | POSITIVE | Preserved | Negative* | focal | Negative* | Negative* | irradiation +chemo | Alive | |||||
| 11 | 12 | M | 3rd ventricle | Sporadic | No | No | STR | Yes | NEGATIVE | Preserved | Negative* | focal | Negative | Negative* | Negative* | loss | Alive | |||
| 12 | 3 | M | L hemisphere | Sporadic | No | No | GTR | Yes | NEGATIVE | Preserved | Negative* | Negative | Negative* | Negative* | Partial loss | Alive | ||||
| 13 | 4 | M | Posterior fossa | Sporadic | No | No | STR | Yes | POSITIVE | Lost | Negative* | Strongly positive | Negative | Negative* | positive | Partial loss | No mutation | H3F3A p.K27M, KRAS p.Q61H, TP53 p.C275F, ATRX p.E1767, ATRX p.R2085 | Dead | |
| 14 | 22 | F | Cerebellum | Sporadic | yes | Yes | Yes | POSITIVE | lost | Negative* | focal | Positive | Negative* | Negative* | loss | irradiation +chemo | Alive | |||
| 15 | 24 | M | Cerebellum | NF1 | No | Yes | GTR | No | POSITIVE | lost | Negative* | focal | Negative | Negative | Negative* | No mutation | NF1 mutation, STAG2 p.R1207T mutation, ATRX p.Q219fs deletion, LOH | observation | Alive | |
| 16 | 36 | M | Cerebellum | Sporadic | No | Yes | GTR | Yes | POSITIVE | lost | Negative* | focal | Positive | Negative* | observation | Alive | ||||
| 17 | 5 | M | Cerebellum | Sporadic | No | GTR | No | NEGATIVE | Preserved | Negative* | Positive | Negative* | Partial loss | no deletion | Alive | |||||
| 18 | 36 | F | cerebellum | Sporadic | No | No | No | POSITIVE | lost | Negative* | Negative | Negative* | Positive | Partial loss | no deletion | Alive | ||||
| 19 | 46 | F | R temporal lobe | NF1 | No | No | GTR | No | NEGATIVE | Preserved | Negative* | Negative | Negative* | Negative* | preserved | Dead | ||||
| 20 | 75 | M | L temporal lobe | Sporadic | No | No | GTR | No | NEGATIVE | Preserved | Negative* | Negative | Negative | Negative* | partial loss | no deletion | Dead | |||
| 21 | 29 | M | cerebellum | NF1 | No | No | STR | No | POSITIVE | Lost | Negative* | Negative | Negative* | Negative* | homozygous deletion | observation | Dead | |||
| 22 | 10 | M | 3rd ventricle | Sporadic | No | No | STR | No | POSITIVE | Lost | Negative* | Negative | Negative* | Positive | No deletion | Dead | ||||
| 23 | 70 | M | cerebellum | Sporadic | No | No | Yes | NEGATIVE | Preserved | Negative* | Negative | Negative* | Negative* | homozygous deletion | Dead | |||||
| 24 | 28 | M | cerebellum | Sporadic | Yes | Yes | Yes | POSITIVE | Negative* | Negative* | Negative* | Irradiation | Dead | |||||||
| 25 | 11 | M | R frontal lobe | Sporadic | No | No | GTR | Yes | NEGATIVE | Preserved | Negative* | Negative | Negative | Negative* | No deletion | Alive | ||||
| 26 | 11 | F | R occipital lobe | NF1 | No | No | GTR | No | NEGATIVE | Preserved | Alive | |||||||||
| 27 | 14 | M | cerebellum | Sporadic | No | No | Yes | NEGATIVE | Preserved | Negative* | Negative* | Dead | ||||||||
| 28 | 20 | M | cerebellum | Sporadic | No | No | Yes | POSITIVE | lost | Negative* | Positive | Negative* | Negative* | No deletion | Dead | |||||
| 29 | 27 | M | cerebellum | Sporadic | Yes | Yes | GTR | Yes | POSITIVE | Lost | Negative* | Positive | Negative* | Negative* | Homozygous deletion | Irradiation | Dead | |||
| 30 | 39 | F | cerebellum | Sporadic | Yes | Yes | Yes | POSITIVE | Lost | Negative* | Positive | Negative* | Negative* | No deletion | Irradiation | Dead | ||||
| 31 | 46 | M | L parietal lobe | NF1 | No | Yes | STR | Yes | POSITIVE | Lost | Negative* | Negative* | Negative* | observation | Dead | |||||
| 32 | 73 | F | tectum | Sporadic | No | Yes | STR | Yes | POSITIVE | Preserved | Negative* | Negative | Negative | Negative* | No deletion | observation | Dead | |||
| 33 | 51 | M | spinal cord | Sporadic | No | No | STR | No | POSITIVE | partial | Negative* | Negative | Negative* | Positive | No deletion or mutation | H3F3A p.K27M, ATRX pS856del, NF1 p.L2398M, AKT1 p.R206S, BRAF p.L64I, TSC1 p.G389 fs | Dead | |||
| 34 | 18 | M | L parietal lobe | NF1 | No | Yes | NEGATIVE | Preserved | focal | Negative* | loss | Alive | ||||||||
| 35 | 25 | F | Tectum | Sporadic | Yes | Yes | No | POSITIVE | Preserved | Negative* | focal | Negative | Negative | Negative | No mutation | NF1 p.R816*, NF1 p.A2099D, H3F3A p.A115G | Irradiation | Alive | ||
| 36 | 20 | F | R cerebellum | Sporadic | No | No | STR | Yes | POSITIVE | Preserved | Negative* | Strongly positive | Negative | BRAF p.D594G | Negative | loss | No mutation | NF1 p.Y1545* NF1 p.K1440* | Alive |
NF1 = neurofibromatosis type 1, STR = subtotal resection, GTR = Gross total resection, PA = pilocytic astrocytoma, ALT = alternative lenghtening of telomeres, IHC = immunohistochemistry.
*Evaluated by immunohistochemistry.
Telomere‐specific FISH and CISH
ALT status was interpreted using previously published criteria, and was characterized by the presence of distinct large signals, in FISH or CISH. Cases were considered ALT‐positive when foci defined as >10‐fold brighter (in telomere FISH) or larger (in telomere CISH) compared to the average signal of telomeres in stromal cells was present in >1% of cancer nuclei 13, 14, 39. Additional details are given in our prior publications. ALT status was tested by FISH only in 23 cases, CISH only in 4 cases and by both methods in 9 cases. Whole slides (n = 23) or tissue microarrays (n = 13) were used.
Telomere‐specific FISH was performed as previously described 13, 14, 39. In brief, deparaffinized slides were hydrated, steamed for 25 minutes in citrate buffer (Vector Laboratories), dehydrated and hybridized with a Cy3‐labeled peptide nucleic acid (PNA) probe complementary to the mammalian telomere repeat sequence [(N‐terminus to C‐terminus) CCCTAACCCTAACCCTAA]. As a positive control for hybridization efficiency, an Alexa Fluor‐488–labeled PNA probe specific to human centromeric DNA repeats (ATTCGTTGGAAACGGGA; CENP‐B–binding sequence) was included in the hybridization solution. Following post‐hybridization washes, the slides were counterstained with DAPI.
Telomere‐specific CISH was performed as previously described 39. Briefly, deparaffinized slides were hydrated, steamed for 25 minutes in citrate buffer, dehydrated and hybridized with a Cy3‐labeled PNA probe (described above). Sections were blocked against endogenous peroxidase activity with Dual Endogenous Enzyme‐Blocking Agent (Dako) for 10 minutes, incubated with a monoclonal anti‐Cy3/Cy5 antibody (ab52060, Abcam, 1:2500) for 1 h at room temperature, then incubated with an anti‐mouse secondary antibody (Leica Microsystems) for 30 minutes and detected with 3,30‐diaminobenzidine (Sigma‐Aldrich) after 10 minutes. Finally, sections were counterstained with hematoxylin, rehydrated and mounted.
BRAF and CDKN2A alterations
BRAF duplication or KIAA1549:BRAF fusion and CDKN2A status was retrospectively obtained from a prior study (n = 14) 37, as well as through diagnostic evaluation as part of the clinical workup through next‐generation sequencing (n = 2), or using a BRAF (7q34) rearrangement by FISH assay employing a 186‐kb red probe centromeric(3′) to the BRAF gene and two overlapping green probes telomeric (5′) to BRAF and spanning 335 kb (Empire Genomics).
In a subset of cases (n = 12), FISH for BRAF duplication was systematically performed as a laboratory developed test that is clinically validated in a CAP/CLIA certified laboratory at St Jude Children's Research Hospital. The design includes a target probe at the BRAF locus on 7q34 and a control Probe at 7p11.2. This design demonstrates a “doublet” pattern in the setting of the KIAA1549‐BRAF fusion which is presumed to be related to the tandem duplication of the BRAF gene during the generation of the fusion 18. The dual‐color duplication probe set for BRAF was derived from BAC clones RP11‐96I22 with 7p controls RP11‐251I15 and RP11‐746C13 (BACPAC Resources, Oakland, CA). Probes were labeled with either AlexaFluor‐488 or AlexaFluor‐555 fluorochromes and nuclei were counterstained with DAPI (200 ng/mL)(Vector Labs) for viewing on an Olympus BX51 fluorescence microscope equipped with a 100‐watt mercury lamp; FITC, Rhodamine and DAPI filters; 100X PlanApo (1.40) oil objective; and a Jai CV digital camera. Images were captured and processed using the Cytovision v7.3 software from Leica Biosystems (Richmond, IL).
CDKN2A homozygous deletion was tested using a commercially available locus‐specific probe (9p21, Abbott Molecular/Vysis) and centromere 9 (CEP9) control probe. Deletion was defined as a ratio of LSI to control probe of <0.8, and homozygous deletion as loss of both CDKN2A copies in at least 10% of neoplastic cells, with preserved centromere signals in the same neoplastic cells and both target and centromere signals in non‐neoplastic internal controls.
Immunohistochemistry
Immunohistochemical slides performed as part of the clinical evaluation were reviewed. In missing or equivocal cases, immunohistochemical studies were systematically performed using the following antibodies: ATRX (Rabbit polyclonal, 1:200 dilution, catalog# HPA001906 Sigma‐Aldrich), H3‐K27M (Rabbit polyclonal, Dilution 1:400, catalog# ABE419, Millipore Sigma), p16 (CINtec®, prediluted, catalog# 705‐4713, Roche), IDH1‐R132H (Clone H09, 1:100 dilution, catalog#,Dianova), p53 (clone BP53‐11, prediluted, catalog# 760‐2542, Ventana). Immunostaining was performed on automated instruments (BenchMark, Ventana Medical Systems, Tucson, AZ, USA). The immunohistochemical protocol included deparaffinization, hydration, antigen retrieval, primary antibody incubation, and detection and visualization as per manufacturer's instructions. Immunohistochemistry for DAXX (Rabbit polyclonal, 1:100 dilution, catalog# HPA008736, Atlas Antibodies) was performed manually. Sections were incubated with primary antibody for 2 h at room temperature followed by secondary antibody (Leica Microsystems) for 30 minutes and detected with 3,30‐diaminobenzidine (Sigma‐Aldrich) after 10 minutes. For ATRX and DAXX, preserved immunoreactivity in internal non‐neoplastic cell components was required for valid interpretation in all cases.
Targeted next‐generation sequencing
Next‐generation sequencing (NGS) studies were performed by various methods at referring institutions as part of the clinical workup (n = 4). In two additional H3‐K27M positive cases and one H3‐K27M negative case, NGS was performed at Johns Hopkins as previously described 32. Briefly, DNA libraries were prepared using Agilent SureSelect‐XT reagents (Agilent Technologies, Inc., Santa Clara, CA) with genomic regions of interest captured by means of an Agilent custom‐designed bait set covering the full coding regions of 644 cancer‐associated genes (exon 1 is poorly covered for some genes).
Statistics
Variables were described using proportions, ranges, means and medians as appropriate. Proportions were compared using Chi‐Square or Fisher's exact tests as appropriate, while Student's t‐test or Wilcoxon rank sum was used to compare continuous variables between groups of interest. Survival rates were analyzed using Kaplan–Meier curves and the log‐rank test. Overall survival was calculated from the time of diagnosis of anaplasia to death. Recurrence/progression‐free survival was calculated from the time of diagnosis of anaplasia to the first evidence of tumor growth per clinical record, or time of death. Patients were jointly classified by the presence of necrosis and ALT status and by the presence of necrosis and ATRX expression. Cox proportional hazards regression was used to estimate the age‐adjusted relative hazard (HR) and 95% confidence interval (CI) of overall survival and of recurrence/progression‐free survival for each joint category, with necrosis‐free/ALT‐negative patients (or loss of ATRX expression) as the reference, and for the joint categories modeled as an ordinal variable to estimate a p‐trend. JMP v10 or SAS v9.4 software (SAS Institute) were used for statistical analyses. All analyses were two‐sided with p values <0.05 considered statistically significant.
Results
Alternative lengthening of telomeres and ATRX loss are frequent in pilocytic astrocytoma with anaplasia
Anaplastic changes were predominantly diffuse (n = 32) but focal in four cases. ALT was present in 25/36 (69%) cases and ATRX loss in 20/35 (57%), mostly in the expected pattern ALT+/ATRX‐ (20/24, 83%) or ALT‐/ATRX+ (11/11, 100%) (Figures 1, 2, 3, 4). ATRX loss was diffuse in 17 (of 20) cases and partial (ie, clearly involving a subset of tumor cells) in the 3 remaining cases. Discrepant results included ALT+/ATRX+ in four cases. DAXX immunohistochemistry was performed in two of these ALT+/ATRX+ cases and demonstrated preserved immunoreactivity in both. Eleven patients were strictly pediatric (<18 years old) at diagnosis with an age range 3–17 years. Four (36%) of these pediatric tumors had ALT and concurrent ATRX loss. In contrast, only 5/88 (5%) conventional PA or low‐grade gliomas with PA features had ALT, all of which represented recurrences or tumors that behaved aggressively, and only one of these had concurrent ATRX loss. Only 4/154 (2.5%) PA or low‐grade gliomas with PA features had ATRX expression loss (1 partial, 3 complete).
Figure 1.
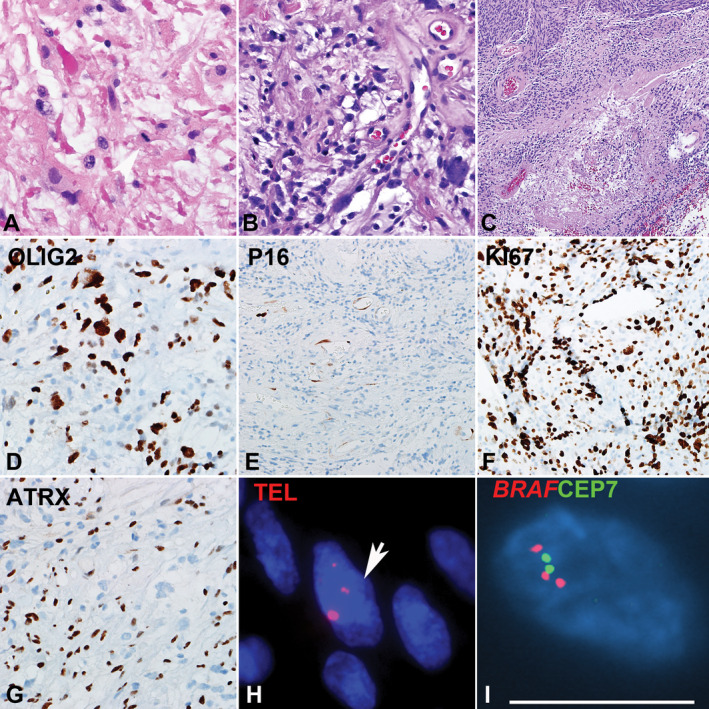
Pilocytic astrocytoma with anaplasia (PA‐A) with concurrent BRAF duplication and ALT. PA‐A developing in the spinal cord demonstrating Rosenthal fibers and frequent mitotic activity. A. Spinal cord PA‐A with well‐differentiated pilocytic areas, containing loose microcystic areas with eosinophilic granular bodies. B. Concurrent areas of anaplasia were characterized by hypercellularity, brisk mitotic activity and pseudopalisading necrosis. C. The immunoprofile included expression of OLIG2 D, p16 loss E, high ki67 labeling index F, and ATRX loss. G. Ultrabright telomere FISH signals consistent with ALT (arrow) H and BRAF duplication I were also present.
Figure 2.
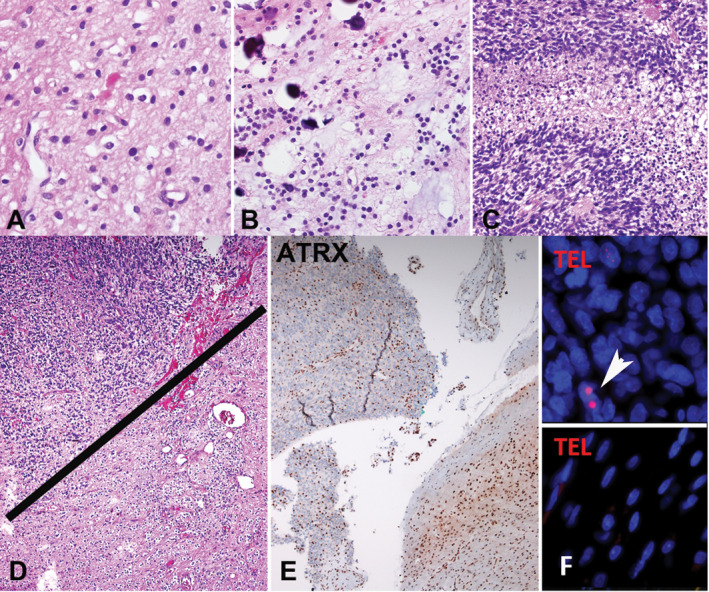
Pilocytic astrocytoma with anaplasia (PA‐A) with ATRX loss and ALT in the anaplastic component only (case 1). The tumor at first resection was hypocellular, composed of piloid areas with oligodendroglial‐like cells and Rosenthal fibers. A. The same component was present in a subsequent resection, in the absence of radiotherapy B, but in addition contained sharply demarcated hypercellular areas with brisk mitotic activity and pseudopallisading necrosis C consistent with spontaneous anaplastic transformation. Distinct anaplastic areas in upper fields D, contained ATRX loss E and ultrabright foci on telomere FISH indicating ALT (arrow) F, which were absent in the lower grade PA in lower fields D‐F, as well as in the PA precursor two years prior (not shown).
Figure 3.
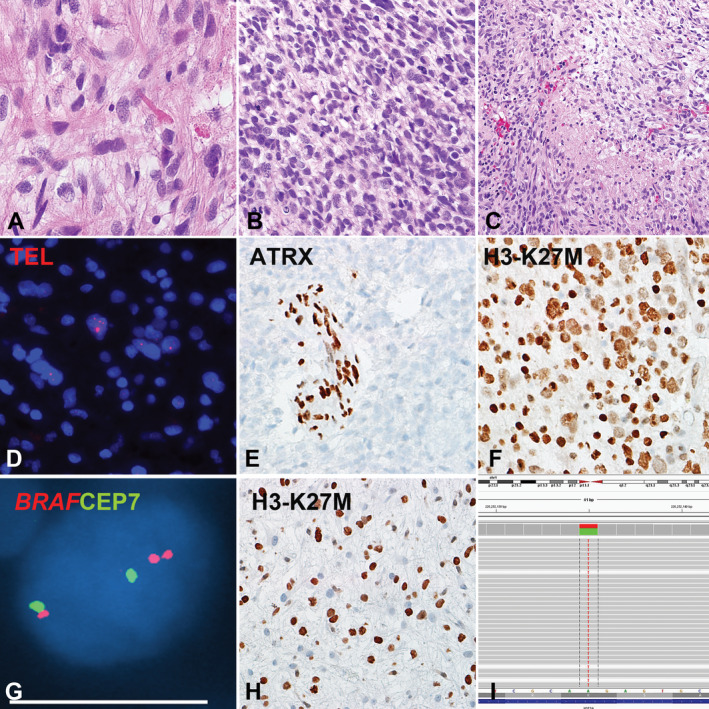
Pilocytic astrocytoma with anaplasia (PA‐A) with concurrent BRAF duplication, ALT and H3‐K27M mutation (Case 9). This PA‐A developed in the cerebellum of a 53‐year‐old woman during progression from a pre‐existing PA. The precursor and the most recent resection had compact piloid areas with Rosenthal fibers and eosinophilic granular bodies. A. The anaplastic component had brisk mitotic activity B and pseudopallisading necrosis. C. Molecular alterations detected included ultrabright telomere FISH signals indicating ALT D, ATRX loss E, expression of H3‐K27M mutant protein in almost 100% of neoplastic cells F and BRAF duplication G. H3‐K27M expression H and ATRX loss (not shown) were also present in a precursor 2 years prior, although the latter appeared to be present at a lower frequency in neoplastic cells. Next‐generation sequencing detected a AAG‐>ATG missense mutation in H3F3A at codon 27 Lysine K that was changed to Methionine M in all three H3‐K27M immunohistochemistry positive cases tested I.
Figure 4.
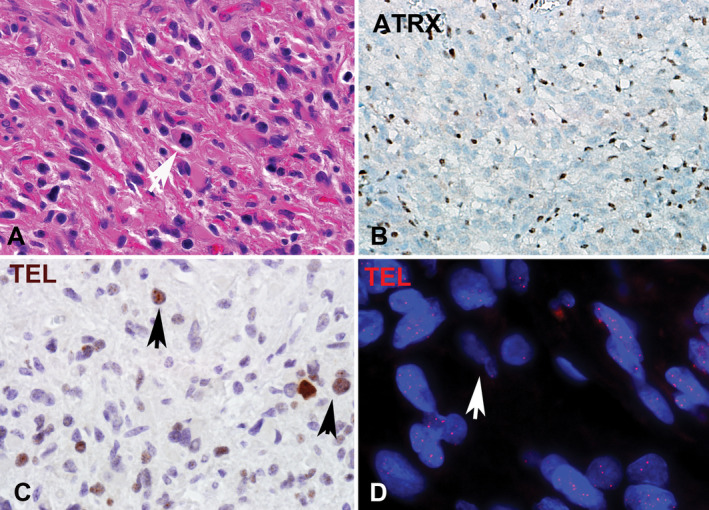
NF1‐associated pilocytic astrocytoma with anaplasia (PA‐A). PA‐A developing in a patient with NF1 A and loss of ATRX expression. B. ALT‐positive NF1‐associated PA‐A demonstrating large brown immunopositive telomeric foci through chromogenic in situ hybridization in >1% of cells corresponding to the ultrabright foci identified by telomere FISH indicative of ALT. C. ALT‐negative PA‐A had a tendency to have longer telomeres (brighter signals by telomere FISH) compared to internal non‐neoplastic controls (arrow) (Case 4, Telomere FISH) D.
BRAF duplications/fusions were identified in 8 (of 26)(31%) cases tested, including one case tested by a rearrangement probe set with ATRX loss and ALT. BRAF p.V600E was not encountered in 31 cases tested. However, rare mutations involving the MAPK pathway were identified in three cases: one case had a BRAF p.D594G, NF1 p.Y1545, NF1 p.K1440 mutations and concurrent ALT, but demonstrated no ATRX loss or ATRX mutation by NGS. One case had BRAF p.L64I and NF1 p.L2398M mutations as well as ALT and ATRX p.S856del, and another case had a KRAS p.Q61H and two ATRX mutations (Table 1).
CDKN2A homozygous deletions were identified in three (of 18; 17%) cases tested by FISH (n = 12), NGS (n = 6) or by both FISH and NGS (n = 1). P16 protein loss was present in 13 (of 14) cases tested, being complete in 7 and partial in 6 cases. Strong p53 immunolabeling, defined as strong (3+) intensity present in >50% of neoplastic cells, was identified in 6 (of 17; 35%), while all cases tested for mutant IDH1 (R132H) by immunohistochemistry (n = 32) were negative. Eight cases studied by sequencing showed no IDH1 or IDH2 hotspot mutations. Immunophenotypic and molecular alterations are summarized in Table 2.
Table 2.
Summary of immunophenotypic and molecular features of pilocytic astrocytomas with anaplasia (PA‐A)
| Alteration | PA‐A | PA precursor |
|---|---|---|
| ALT | 25/36 (69%) | 9/14 |
| ATRX (IHC) loss | 20/35 (57%) | 7/11 |
| IDH1 (R132H) | 0/32 (0%) | NA |
| BRAF duplication | 8/26 (31%) | 8/14 |
| BRAF (V600E) | 0/31 (0%) * | NA |
| NF1 status | 12/36 (33%) † | 3/19 |
| H3‐K27M | 5/32 (16%) | 2/14 |
| CDKN2A | 3/18 (17%) ‡ | NA |
| P53 (strong IHC) | 6/17 (35%) | NA |
ALT = alternative lengthening of telomeres, NF1 = neurofibromatosis type 1.
A single case had an activating mutation in BRAF (non V600E) by sequencing (BRAF p.D594G).
A total of 8 patients satisfied clinical criteria for NF1 while four additional patients demonstrated NF1 mutations in their tumors by sequencing studies.
A total of 3 patients had homozygous deletion as tested by FISH (n = 12), NGS (n = 6), or both (n = 1).
Histologic precursors to pilocytic astrocytoma with anaplasia
PA precursors for PA‐A (ie, pathologically confirmed PAs preceding a diagnosis in PA‐A) were present in 15 cases, with an interval for anaplastic progression ranging from 5 to 307 months (mean 67 months). In the remaining 21 cases, anaplastic changes were present at first diagnosis. Treatments for the precursor PA tumors included observation (n = 5), irradiation only (n = 6) and irradiation and chemotherapy (n = 3). Treatment information was unknown in one case.
ALT and/or ATRX expression was also tested in the low‐grade/PA component in 14 cases where the PA precursor component was available for testing, including samples preceding a diagnosis of PA‐A (n = 6) or present in the same specimen as the anaplastic component (n = 8). ALT was present in 9/14 and ATRX loss in 7/11.
When looking at ALT‐positive PA‐A, ALT was also present in nine (of 11) PA precursors, seven of which also demonstrated ATRX loss in both the precursor and the anaplastic tumor. In a single case (case 1), ALT and ATRX loss developed in the anaplastic component only, and were absent in the PA precursor two years prior and the persistent benign PA component present concurrently with the anaplastic areas (Figure 2). In another case (case 24), ALT was present in the PA‐A, but not in the concurrent low‐grade precursor, although ATRX was not tested. In no cases was ALT present in the low‐grade PA precursor only.
A subset of pilocytic astrocytoma with anaplasia have H3‐K27M mutations
H3‐K27M was present in 5 of 32 (16%) cases tested (Table 1). This subgroup included three males and two females, with a median age of 36 (range 4–53). Anatomic locations of the H3‐K27M mutant cases included the posterior fossa/cerebellum (n = 3), the spinal cord (n = 1), and the third ventricle (n = 1). All demonstrated ATRX loss and ALT, and 1 (of 5) had a concurrent BRAF duplication (Figure 3). This patient also had tissue available from a low‐grade pilocytic precursor resected two years prior (case 9). ATRX loss and the H3‐K27M mutation were identified in this precursor, as well, although the proportion of H3‐K27M positive cells was lower (Figure 3). In one additional H3‐K27M positive case, the alteration was also present in the concurrent presumed PA precursor. Another case (case 35) had a H3F3A p.A115G mutation. In contrast only 1/145 (0.6%) conventional PA or low‐grade gliomas with PA features was H3‐K27M positive. Interestingly, this was a recurrent tumor.
In three of these five PA‐A with available material, NGS studies confirmed H3F3A p.K27M and ATRX mutations, as well as other alterations (Table 1). Three (of 5) patients with H3‐K27M mutations died 4, 25 and 55 months after diagnosis. The two remaining patients are alive without evidence of disease at 6 and 39 months after diagnosis.
NF1‐associated pilocytic astrocytoma with anaplasia
A clinical diagnosis of NF1 was present in eight cases (22%), four (50%) of which were ALT‐positive and five (63%) displayed ATRX loss (Figure 4). Anatomic locations included the cerebellum (n = 3), hemispheres (n = 4) and pineal region (n = 1). Four additional patients not satisfying clinical criteria for NF1 had NF1 mutations identified by NGS, including the single case with BRAF p.D594G. All relevant alterations tested in PA‐A and precusors, are summarized by case and group in Figure 5.
Figure 5.

Relevant clinicopathologic features of pilocytic astrocytoma with anaplasia (PA‐A). Relevant demographic, phenotypic and genetic features of pilocytic astrocytoma with anaplasia (PA‐A) are summarized in A. Relevant features for precursors are summarized in B.
Pilocytic astrocytoma with anaplasia are associated with a poor outcome
Clinical follow‐up was available in 31 patients. Of this cohort, 19 patients died during the follow‐up period, with a median overall survival of 13 months (range 2–55 months) after the diagnosis of anaplasia. Recurrence/progression was documented in 21 patients, with a median recurrence/progression‐free survival of 9.8 months. Interval to recurrence/progression in all cases from the time of the primary pathologic diagnosis (whether anaplasia was present or not) was 48 months (range 2–317). Clinicopathologic and molecular features associated with a worse overall survival included subtotal vs. gross total resection (P = 0.01), adult vs. pediatric patients (P = 0.03), presence of a PA precursor preceding PA‐A vs. de novo anaplastic changes (P = 0.02), presence of necrosis (P = 0.009), presence of ALT (P = 0.03) and ATRX loss (P = 0.04) (Figure 6, Table 3). There was a trend for worse overall survival in patients with a history of irradiation (P = 0.06) that did not reach statistical significance. Features not associated with overall survival included BRAF duplication/fusion, presence of H3‐K27M mutant protein, clinical NF1 diagnosis, supratentorial vs. infratentorial location and sex (P > 0.05). Clinicopathologic features associated with a decreased recurrence/progression‐free survival included subtotal vs. gross total resection (P = 0.0005), ALT+ vs. ALT tumors (P = 0.02), ATRX loss (P = 0.03) and necrosis (P = 0.02) (Figure 6). Features not associated with recurrence/progression‐free survival included BRAF duplication/fusion, presence of H3‐K27M mutant protein, clinical NF1 diagnosis, supratentorial vs. infratentorial location, presence of a PA precursor preceding a diagnosis of PA‐A vs. de novo anaplastic changes, adult vs. pediatric patients, prior irradiation and sex (P > 0.05). Finally, compared to patients with necrosis‐free/ALT‐negative tumors, patients with necrosis‐free/ALT‐positive tumors had no difference in survival (HR = 0.97; 95% CI: 0.17–5.42). In contrast, patients with necrotic/ALT‐negative tumors had a nonsignificant higher risk of death (HR = 1.95; 95% CI: 0.32–11.83); whereas, patients with necrotic/ALT‐positive tumors had a significantly higher risk of death (HR = 6.52; 95% CI: 1.28–33.19; P‐trend = 0.005). Results were similar when recurrence/progression‐free survival was modeled as the outcome, and when ATRX expression, instead of ALT status, was used in the joint categories. Given the small sample size and therefore imprecision of these estimates, findings should be interpreted with caution. Nonetheless, these findings strongly support further study of ALT and ATRX expression in this neoplasm.
Figure 6.
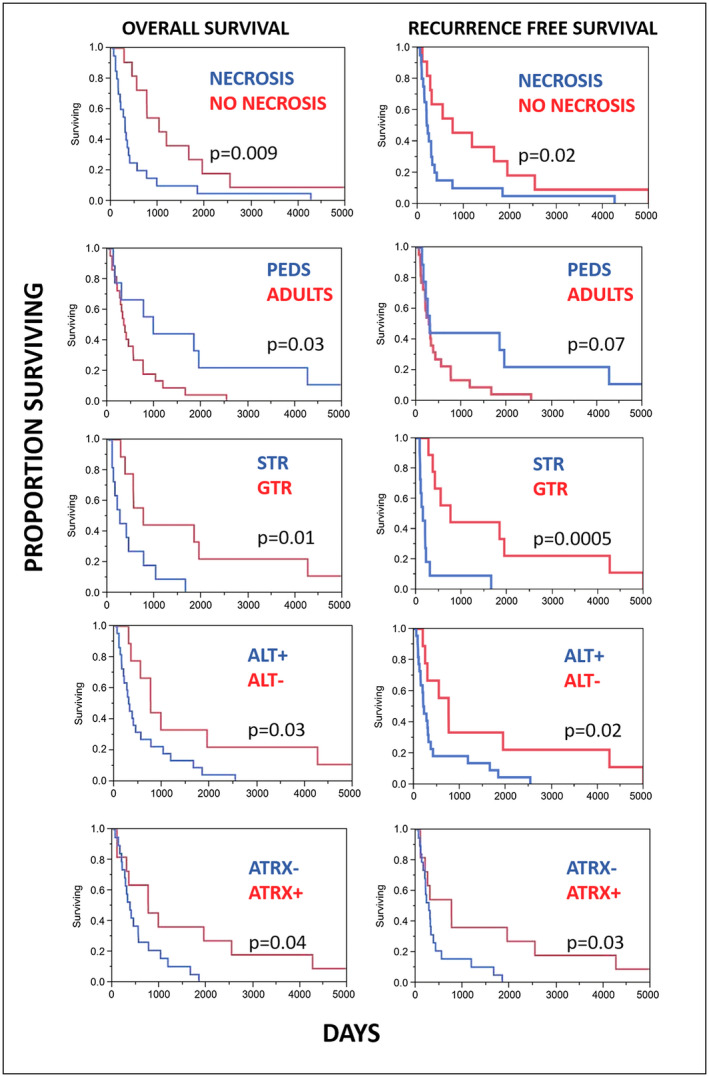
Factors associated with overall and recurrence/progression‐free survival in pilocytic astrocytoma with anaplasia (PA‐A). Features associated with a worse outcome included the presence of necrosis, adult age (vs. pediatric), extent of resection (subtotal resection, STR, vs. gross total resection, GTR), and presence of ALT (Kaplan Meier curves, Log‐rank test).
Table 3.
Clinicopathologic, molecular features and outcome in pilocytic astrocytoma with anaplasia (PA‐A)
| Comparison | N (%) | Overall survival in days | Progression/Recurrence‐free survival in days | P‐value1 |
|---|---|---|---|---|
| (mean ± Std Error) | (mean ± Std Error) | OS, PFS | ||
| Resection | ||||
| GTR | 10 (45%) | 1724 ± 587 | 1709 ± 591 | 0.01, 0.0005 |
| STR | 12 (55%) | 471 ± 148 | 293 ± 137 | |
| Age | ||||
| Adult | 11 (30%) | 565 ± 125 | 489 ± 128 | 0.03, 0.07 |
| Pediatric | 25 (70%) | 1703 ± 597 | 1560 ± 629 | |
| Sex | ||||
| Male | 23 (64%) | 863 ± 230 | 753 ± 238 | 0.99, 092 |
| Female | 13 (36%) | 954 ± 438 | 885 ± 443 | |
| Anatomic location | ||||
| Supratentorial | 12 (33%) | 1416 ± 612 | 1042 ± 540 | 0.15, 0.69 |
| Infratentorial | 24 (67%) | 683 ± 153 | 611 ± 154 | |
| Prior irradiation | ||||
| Yes | 9 (26%) | 312 ± 85 | 269 ± 57 | 0.06, 0.18 |
| No | 26 (74%) | 1141 ± 344 | 1047 ± 354 | |
| NF1 diagnosis | ||||
| Present | 8 (22%) | 1123 ± 562 | 842 ± 595 | 0.40, 0.97 |
| Absent | 28 (78%) | 817 ± 213 | 785 ± 215 | |
| PA precursor | ||||
| Preceding anaplasia | 15 (43%) | 442 ± 194 | 401 ± 196 | 0.02, 0.07 |
| De novo anaplasia | 20 (57%) | 1140 ± 321 | 1002 ± 335 | |
| Necrosis | ||||
| Present | 24 (67%) | 584 ± 213 | 516 ± 215 | 0.009, 0.02 |
| Absent | 12 (33%) | 1463 ± 409 | 1317 ± 439 | |
| Extent of anaplasia | ||||
| Focal | 4 (11%) | 1016 ± 924 | 534 ± 468 | 0.83, 0.34 |
| Diffuse | 32 (89%) | 887 ± 220 | 785 ± 226 | |
| ALT | ||||
| Positive | 25 (69%) | 587 ± 141 | 492 ± 143 | 0.03, 0.02 |
| Negative | 11 (31%) | 1650 ± 588 | 1553 ± 609 | |
| BRAF | ||||
| Duplication | 8 (31%) | 610 ± 270 | 588 ± 272 | 0.88, 0.87 |
| No duplication | 18 (69%) | 807 ± 273 | 720 ± 278 | |
| ATRX expression | ||||
| Loss | 20 (57%) | 558 ± 117 | 448 ± 119 | 0.04, 0.03 |
| Preserved | 15 (43%) | 1548 ± 515 | 1468 ± 529 | |
| H3‐K27M | ||||
| Present | 5 (16%) | 780 ± 291 | 669 ± 313 | 0.55, 0.80 |
| Absent | 27 (84%) | 684 ± 203 | 574 ± 206 | |
Log‐Rank Test.
Discussion
The concept of PA‐A historically has been controversial, but PA‐A is emerging as a brain tumor category of interest. Current evidence suggests a worse clinical outcome than conventional PA, but better than high‐grade astrocytomas, particularly those that are IDH wildtype 36, 38. Activation of the MAPK and PI3K/mTOR pathway, as well as CDKN2A alterations have been previously associated with anaplasia in PA 36, 37. More recently, the co‐occurrence of BRAF p.V600E and CDKN2A mutations have been associated with worse outcome in pediatric low‐grade glioma 23. Even though CDKN2A alterations were identified in our cohort, BRAF p.V600E was uniformly absent.
Prior studies have demonstrated a strong association of alterations in ATRX or death domain‐associated protein (DAXX) genes with the alternative lengthening of telomeres (ALT) phenotype in several cancer subtypes, particularly gliomas, pancreatic neuroendocrine tumors and certain sarcomas 13. However, in ALT‐positive gliomas, alterations involve predominantly ATRX. ATRX and DAXX are chromatin remodeling proteins that work at repetitive regions, including telomeres, to incorporate H3.3 into heterochromatin. ATRX mutations and ALT are also associated with specific molecular subgroups of brain tumors, particularly a subset of pediatric glioblastoma 41 and IDH mutant diffuse astrocytomas 3, 28. Specifically, previous studies have demonstrated an almost perfect concordance between ATRX mutations in high‐grade gliomas with ALT 13, 28. The vast majority of ATRX mutations occurring in association with ALT are inactivating and associated with nuclear protein loss as demonstrated by immunohistochemistry, which serves as a useful surrogate marker. ATRX protein loss is specific to neoplastic cells, since internal non‐neoplastic brain components preserve ATRX immunoreactivity within tumors. More recently, we demonstrated that ATRX loss and ALT are frequent features of diffuse and high‐grade astrocytomas developing in patients with NF1 39. This is of particular interest, since approximately 25% of PA‐A develop in patients with a clinical diagnosis of NF1 38. Interestingly, in our study, additional tumors demonstrated NF1 gene mutations in the absence of the absence of clinical NF1, and when combining these groups, the frequency of NF1 altered tumors reaches 33%.
The current study expands the glioma subgroups that are dependent on ALT for telomere maintenance for the first time to also include PA‐A. We identified ALT in 69% and ATRX protein loss in 55% of PA‐A. ATRX mutations were also identified in 45% in the study of Reinhardt et al., where a distinct molecular class was discovered 36, and referred to as anaplastic astrocytoma with piloid features. Other similarities with this study included an absence of IDH mutations. In the study by Reinhardt et al. the most frequent alteration was CDKN2A deletion (80%). We found homozygous deletions in CDKN2A in 20% of cases tested, but more cases with p16 loss by immunohistochemistry, suggesting inactivation in 67%. The lower frequency of CDKN2A deletions in our study may be in part explained by differences in methods, since most cases were tested for CDKN2A deletion by FISH rather than by higher resolution techniques such as next‐generation sequencing. The frequency of p16 loss by immunohistochemistry was higher in our cohort, and p16 loss by immunohistochemistry has been found to be associated with shorter overall survival in PA 35. However, loss of p16 expression by IHC is not the most optimal marker to assess CDKN2A inactivation, since p16 expression is very low in non‐neoplastic cells and it is difficult to confidently determine loss of expression in tumor cells.
In our study, the concordance between ALT and ATRX expression was somewhat lower than expected based on our prior experience and the literature. Several possibilities may explain these observations. First, in some instances loss of ATRX function may occur (eg, point mutations) while preserving protein levels and thereby leading to a normal immunohistochemistry result. Additionally, it is possible that alterations in DAXX or other heretofore unidentified ALT suppressors may occur in these cases. We were able to stain for DAXX in two ALT+ cases and expression was preserved in both. Additionally, no DAXX mutations were identified in the study by Reinhardt et al., suggesting that other genetic hits may explain anaplasia in PA with ALT lacking ATRX alterations.
The presence of H3‐K27M mutations defines the diffuse midline glioma H3‐K27M mutant, a WHO grade IV category that has been included in the 2016 WHO Classification update 26. This alteration is frequent in diffuse gliomas that develop in midline anatomical structures, particularly brainstem and spinal cord and are in general associated with poor clinical outcome irrespective of histologic grade 20, 44, 46, 49. However, recent studies have identified these mutations in other histologic subtypes, where the outcome may be relatively more favorable 29, 40. These include circumscribed gliomas that may also have other known drivers including BRAF p.V600E or NF1 mutations 34. Additionally, recent studies suggest that H3‐K27M may develop subclonally, presumably during tumor progression rather than an initiating event 24.Given these observations, in a recent statement cIMPACT‐NOW (the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy—Not Official WHO) recommends that the category of diffuse midline glioma, H3‐K27M mutant, be only applied to tumors that share all those properties encompassing its name (ie, they are diffuse, midline, glial and have the H3‐K27M mutation), excluding circumscribed gliomas and others 25.
In the current study, we found H3‐K27M mutant protein in five cases, which is a higher frequency compared with the study of Reinhardt et al, where H3‐K27M mutant protein was detected in 1 (of 47) cases 36. Of interest, all of the H3‐K27M mutant cases in our study also had ALT and ATRX loss, which was confirmed by sequencing in the three cases undergoing further testing. This is intriguing, since ATRX mutations are most often associated with H3F3A p.G34 mutations 9, 22, occurring in most of the latter, but in only 15–20% of H3‐K27M mutant tumors 9, 20. This suggests that ALT is an important biologic feature of PA‐A, irrespective of the underlying oncogenic drivers (eg, BRAF, NF1 or H3‐K27M). Interestingly, we encountered a single case with BRAF duplication and H3‐K27M. Concurrent BRAF p.V600E and H3‐K27M mutations have been rarely reported 31, 44, but BRAF duplications must be even less frequent. It must be noted that prior studies have not found either ATRX/H3‐K27M alterations or ALT in conventional PA, with only rare exceptions 9, 14, 29. One case in our study had a H3F3A p.A115G mutation, which has been identified rarely in squamous cell carcinoma of the upper aerodigestive tract 27 (https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=3930660), but not in brain tumors to our knowledge, and therefore its clinical significance in glioma is unclear.
The current study expands our knowledge regarding the molecular alterations associated with PA‐A. However, it is still unclear what is the precise sequence of events leading to anaplasia in these tumors. The mechanisms are likely heterogeneous, but alterations in MAPK pathway components or H3 histone (H3‐K27M) appear to be early drivers. The majority of MAPK pathway alterations involve NF1 loss or BRAF activation, as well as FGFR1 in some instances 1, 36. In most instances, ATRX alterations and ALT appear to develop early, but at least in two cases in our current study they were identified only in the anaplastic component suggesting that they play an important role in anaplastic change. One important conclusion of our study is that ALT, H3‐K27M and ATRX loss may identify PAs that are most likely to behave aggressively or develop anaplastic change, since these alterations were present in most precursors tested, when present in PA‐A. Furthermore, although these alterations were rare in a control group, with only 5% of PA showing ALT and <1% H3‐K27M, these tumors were either recurrent or clinically aggressive PA.
ATRX loss appears to have numerous effects and leads to substantial epigenomic changes that have cell context‐dependent oncogenic effects 7. However, not all tumors demonstrate ATRX alterations and ALT, and therefore other mediators of anaplasia remain to be discovered. Additionally, DNA methylation‐based classification is emerging as a powerful tool to classify brain tumors including rare entities 4. In our study, genetic and phenotypic features associated with PA‐A were present in most concurrent or earlier conventional PA precursors, so it is possible that they also share methylation profiles. However, methylation studies in the future could play a role in identifying early those rare PA that undergo anaplasia and PA‐A associated changes secondarily during the process of malignant progression, as our index case (case 1 and Figure 2).
Our current study confirms prior observations that PA‐A is a malignant neoplasm with a high potential for recurrence and death 36, 38. Features that were associated with a worse outcome in our study included subtotal resection (vs. complete), adult (>17 years) age at diagnosis of anaplasia, the presence of a PA precursor preceding an PA‐A diagnosis, necrosis during histologic examination, ALT‐positive and ATRX loss of expression. Some of these associations are expected in part, since PA is predominantly a surgical disease and the extent of resection historically is one of the most important prognostic factors. PA‐A is also more frequent in adult patients, and PA developing in adult patients tend to be more aggressive at the biologic and clinical levels 17, 45, 47. The association of a PA precursor preceding anaplasia with a worse outcome is intriguing and could be explained by several factors. It is possible that prior surgery or irradiation have an impact of outcome. Alternatively, a PA precursor not satisfying histologic criteria for anaplasia may still be more biologically aggressive, and affect overall prognosis starting at the time of diagnosis.
A novel feature of our study is that we found ALT and ATRX loss to be associated with worse overall and recurrence/progression‐free survival. This suggests that ALT‐positive/ATRX‐negative PA‐A is a distinct category with important prognostic implications. Given the association of ALT with different gliomas subsets, its specific role in prognosis appears to be context‐dependent, since it is also a feature of IDH‐mutant astrocytomas with ATRX loss, which have a better prognosis than IDH wildtype tumors. Interestingly, primary pancreatic neuroendocrine tumors with ALT and ATRX/DAXX loss are associated with a worse clinical outcome 21, 43.
In summary, ALT, ATRX loss and alterations involving components of the MAPK pathway are frequent in PA‐A at the time of development of anaplasia or their precursors. Additionally, a small subset of PA‐A have H3‐K27M mutations. These findings further support the concept that PA‐A is a neoplasm with heterogeneous genetic features and putative drivers and combined alterations typical of PA and diffuse gliomas.
Acknowledgments
The authors thank the pathologists and clinicians who contributed to this study, particularly Drs. David Mirkin, Naomi Rance, Carlos Ribeiro, and Ali Saad.
This work was supported in part by Pilocytic/Pilomyxoid Fund, including Lauren's First and Goal, and the Stick it to Brain Tumors Annual Women's Ice Hockey Tournament (F.J.R.); the PLGA foundation (F.J.R), NIH grant 2T32CA009110‐39A1 (J.A.B), and NIH grant P30 CA006973 to the Sidney Kimmel Comprehensive Cancer Center (PI: W. Nelson).
References
- 1. Ballester LY, Penas‐Prado M, Leeds NE, Huse JT, Fuller GN (2018) FGFR1 tyrosine kinase domain duplication in pilocytic astrocytoma with anaplasia. Cold Spring Harb Mol Case Stud 4:a002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG (2008) Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol 67:878–887. [DOI] [PubMed] [Google Scholar]
- 3. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA et al (2015) Comprehensive, integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med 372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation‐based classification of central nervous system tumours. Nature 555:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collins VP, Jones DT, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collins VP, Tihan T, VandenBerg SR, Burger PC, Hawkins C, Jones DT et al (2016) Pilocytic astrocytoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavennee WK, Ellison DW, Figarella‐Branger D et al. (eds). IARC Press: Lyon. [Google Scholar]
- 7. Danussi C, Bose P, Parthasarathy PT, Silberman PC, Van Arnam JS, Vitucci M et al (2018) Atrx inactivation drives disease‐defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat Commun 9:1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dirks PB, Jay V, Becker LE, Drake JM, Humphreys RP, Hoffman HJ, Rutka JT (1994) Development of anaplastic changes in low‐grade astrocytomas of childhood. Neurosurgery 34:68–78. [PubMed] [Google Scholar]
- 9. Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Muhleisen H, Eckert F et al (2016) ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun 4:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW et al (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181. [DOI] [PubMed] [Google Scholar]
- 11. Giannini C, Scheithauer BW, Burger PC, Christensen MR, Wollan PC, Sebo TJ et al (1999) Cellular proliferation in pilocytic and diffuse astrocytomas. J Neuropathol Exp Neurol 58:46–53. [DOI] [PubMed] [Google Scholar]
- 12. Gutmann DH, McLellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS et al (2013) Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1‐associated pilocytic astrocytoma. Genome Res 23:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C et al (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E et al (2011) Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 179:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hutt‐Cabezas M, Karajannis MA, Zagzag D, Shah S, Horkayne‐Szakaly I, Rushing EJ et al (2013) Activation of mTORC1/mTORC2 signaling in pediatric low‐grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro Oncol 15:1604–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones DT, Ichimura K, Liu L, Pearson DM, Plant K, Collins VP (2006) Genomic analysis of pilocytic astrocytomas at 0.97 Mb resolution shows an increasing tendency toward chromosomal copy number change with age. J Neuropathol Exp Neurol 65:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH (2012) Pediatric glioma‐associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type‐specific and mTOR‐dependent manner. Genes Dev 26:2561–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khuong‐Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim JY, Brosnan‐Cashman JA, An S, Kim SJ, Song KB, Kim MS et al (2017) Alternative lengthening of telomeres in primary pancreatic neuroendocrine tumors is associated with aggressive clinical behavior and poor survival. Clin Cancer Res 23:1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V et al (2016) Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 131:137–146. [DOI] [PubMed] [Google Scholar]
- 23. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R et al (2017) Therapeutic and prognostic implications of BRAF V600E in pediatric low‐grade gliomas. J Clin Oncol 35:2934–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lopez GY, Oberheim Bush NA, Phillips JJ, Bouffard JP, Moshel YA, Jaeckle K et al (2017) Diffuse midline gliomas with subclonal H3F3A K27M mutation and mosaic H3.3 K27M mutant protein expression. Acta Neuropathol 134:961–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louis DN, Giannini C, Capper D, Paulus W, Figarella‐Branger D, Lopes MB et al (2018) cIMPACT‐NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M‐mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH‐mutant. Acta Neuropathol. [DOI] [PubMed] [Google Scholar]
- 26. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella‐Branger D et al (2016) WHO Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer: Lyon, France. [Google Scholar]
- 27. Martin D, Abba MC, Molinolo AA, Vitale‐Cross L, Wang Z, Zaida M et al (2014) The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget 5:8906–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nguyen DN, Heaphy CM, de Wilde RF, Orr BA, Odia Y, Eberhart CG et al (2013) Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high‐grade astrocytomas. Brain Pathol 23:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orillac C, Thomas C, Dastagirzada Y, Hidalgo ET, Golfinos JG, Zagzag D et al (2016) Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol Commun 4:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz‐Sloan JS (2016) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2009–2013. Neuro Oncol 18(Suppl. 5):v1–v75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pages M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D et al (2016) Co‐occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palsgrove DN, Brosnan‐Cashman JA, Giannini C, Raghunathan A, Jentoft M, Bettegowda C et al (2018) Subependymal giant cell astrocytoma ‐like astrocytoma: a neoplasm with a distinct phenotype and frequent neurofibromatosis type 1‐association. Mod Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N et al (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low‐grade astrocytomas. J Clin Invest 118:1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pratt D, Natarajan SK, Banda A, Giannini C, Vats P, Koschmann C et al (2018) Circumscribed/non‐diffuse histology confers a better prognosis in H3K27M‐mutant gliomas. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G et al (2011) BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res 17:3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DE et al (2018) Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B et al (2011) PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol 121:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodriguez FJ, Scheithauer BW, Burger PC, Jenkins S, Giannini C (2010) Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol 34:147–160. [DOI] [PubMed] [Google Scholar]
- 39. Rodriguez FJ, Vizcaino MA, Blakeley J, Heaphy CM (2016) Frequent alternative lengthening of telomeres and ATRX loss in adult NF1‐associated diffuse and high‐grade astrocytomas. Acta Neuropathol 132:761–763. [DOI] [PubMed] [Google Scholar]
- 40. Ryall S, Guzman M, Elbabaa SK, Luu B, Mack SC, Zapotocky M et al (2017) H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33:1047–1051. [DOI] [PubMed] [Google Scholar]
- 41. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. [DOI] [PubMed] [Google Scholar]
- 42. Sievert AJ, Jackson EM, Gai X, Hakonarson H, Judkins AR, Resnick AC et al (2009) Duplication of 7q34 in pediatric low‐grade astrocytomas detected by high‐density single‐nucleotide polymorphism‐based genotype arrays results in a novel BRAF fusion gene. Brain Pathol 19:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singhi AD, Liu TC, Roncaioli JL, Cao D, Zeh HJ, Zureikat AH et al (2017) Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin Cancer Res 23:600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJ, Perry A (2016) Diffuse midline gliomas with histone H3‐K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol 26:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stuer C, Vilz B, Majores M, Becker A, Schramm J, Simon M (2007) Frequent recurrence and progression in pilocytic astrocytoma in adults. Cancer 110:2799–2808. [DOI] [PubMed] [Google Scholar]
- 46. Sturm D, Witt H, Hovestadt V, Khuong‐Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. [DOI] [PubMed] [Google Scholar]
- 47. Theeler BJ, Ellezam B, Sadighi ZS, Mehta V, Tran MD, Adesina AM et al (2014) Adult pilocytic astrocytomas: clinical features and molecular analysis. Neuro Oncol 16:841–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tomlinson FH, Scheithauer BW, Hayostek CJ, Parisi JE, Meyer FB, Shaw EG et al (1994) The significance of atypia and histologic malignancy in pilocytic astrocytoma of the cerebellum: a clinicopathologic and flow cytometric study. J Child Neurol 9:301–310. [DOI] [PubMed] [Google Scholar]
- 49. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 44:251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]