

Abstract
Co-inhibitory molecules, such as PD-1, CTLA-4, 2B4 and BTLA, are an important new family of mediators in the pathophysiology of severe bacterial and/or fungal infection, as well as the combined insults of shock and sepsis. Further, the expression of these molecules may serve as indicators of the immune status of the septic individual. Using PD-1:PD-L as an example, we discuss in this review how such checkpoint molecules may affect the host response to infection by mediating the balance between effective immune defense and immune-mediated tissue injury. Additionally, we explore how the upregulation of PD-1 and/or PD-L1 expression on not only adaptive immune cells (e.g. T cells) but also on innate immune cells (e.g. macrophages, monocytes and neutrophils) as well as non-immune cells during sepsis and/or shock contributes to functional alterations, often with detrimental sequelae.
Keywords: Infection, Immunosuppression, Immunomodulation, PD-1, PD-L1, Innate Immunity
Summary Sentence:
Review of checkpoint proteins’ contributions to immunological signaling in shock and sepsis.
INTRODUCTION:
Sepsis
Sepsis remains a serious clinical problem worldwide and in the United States, with an annual incidence in the U.S. estimated at 1.5 million (1). Sepsis is the leading cause of death in non-cardiac Intensive Care Units, with 250,000 deaths each year in the United States (1). Additionally, it ranks as one of the most expensive conditions treated in U.S. hospitals, with costs exceeding twenty billion dollars annually. A concerning factor is that the incidence of sepsis is increasing, with current levels giving rise to more than triple the mortality compared to the 1970’s (2–5). Despite its concerning prevalence within the healthcare system, sepsis and related terminology remain difficult to define due to the heterogeneity and complexity of sepsis pathobiology. In 2016, the definition for sepsis was updated by the Third International Consensus Definitions for Sepsis and Septic Shock. Today sepsis is defined as a life-threatening organ dysfunction caused by a dysregulated host response to infection (6).
Sepsis is a complex clinical syndrome that is thought to develop when the initial host response against infection and/or injured tissue becomes inappropriately amplified, then dysregulated. Ultimately this results in a harmful host response. The homeostasis between eliminating invading pathogens and protecting tissue health is disrupted; multisystem organ failure (MSOF) ensues. Much of sepsis related mortality and long-term morbidity is driven by organ dysfunction and failure. On the cellular level, disruption of the immune system’s balanced response to infectious challenge and/or tissue injury causes circulating immune cells to influx in abnormal proportions into distal organs in the absence of a clear nidus of infection and/or overt tissue damage (6–8). All organs are susceptible, including the pulmonary, hepatic, renal and gastrointestinal organ systems.
SIRS/CARS
The central hypothesis that has driven the search for novel therapeutics is that sepsis induces an overwhelming Systemic Inflammatory Response Syndrome (SIRS). Sequelae include significant bystander cell/tissue injury within the host. Although the term SIRS has been largely overtaken by the modified definition encompassing an expanded pathophysiologic perspective of sepsis, the underlying principle of SIRS has historically influenced the framework with which sepsis has been researched (6, 9, 10). Unfortunately, however, while numerous treatments that were based on anti-inflammatory or anti-coagulant concepts showed promise in the experimental setting, they have all failed to provide a survival benefit in randomized human clinical trials (11, 12). And while one can argue the nature and quality of the clinical trials designed to test these anti-inflammatory approaches, it remains critical to continue to work towards a better understanding of the complex pathology of the septic state in order to develop a truly effective therapy.
The term Compensatory Anti-inflammatory Response Syndrome (CARS) was coined by Roger Bone to describe the immune suppression that occurs after sepsis (13). It arose from the initial descriptions of the development of immune suppression associated with severe shock/trauma/sepsis and with reports dating back to as early as the 1960’s. The syndrome is characterized by the immune system undergoing reduced activation with limited efficacy; in its most severe form it has even been referred to as immunoparalysis (14–20). Historically, however, this facet of the severely injured or septic patient has not been considered as a significant therapeutic target.
Characteristics of septic immune suppression/CARS include cutaneous anergy as manifested by delayed type hypersensitivity, leukopenia as determined by decreased lymphocyte count on a patient’s CBC (complete blood count) with differential, and persistence of or failure to resolve an infection. The mechanisms proposed include decreased human leukocyte antigen-D related (HLA-DR), lymphocyte reprogramming (movement from an Th1/M1 immune cell phenotype to a more Th2/M2 phenotype), the induction of programmed cell death/apoptosis, increased expression of anti-inflammatory mediators (prostaglandin E, IL-10, steroid hormones), and finally increased expression of cell-associated co-inhibitory receptors and ligands (such as PD-1/PD-L1, CTLA-4) (19–23).
CENTRAL QUESTION:
Out of this history grew the principal question which we undertake to review here: does modulating aspects of developing immune suppression improve the capacity of the critically-ill patient to ward off end-organ and tissue injury that thus far has been commensurate with the septic state? Specifically, what roles do such co-inhibitory/checkpoint protein signaling cascades play in this process of immunomodulation, and how does this affect the balance of innate and/or adaptive immunity?
OBJECTIVES:
From our central question, we derived four objectives for this discussion, as follows:
To provide a succinct overview of membrane co-stimulatory/co-inhibitory molecules and their role in classic immunity;
To discuss how sepsis and sepsis plus hypovolemic shock affect the expression of co-inhibitory molecules such as PD-1 and its ligands;
To describe the data that speak to the patho-physiological contributions of PD-1:PD-Ls to the morbidity/mortality seen in models of sepsis and/or shock (hemorrhage); and
To consider possible adaptive and innate immune mechanisms by which PD-1:PD-L1 interactions may drive morbidity.
DISCUSSION:
1. The role of checkpoint proteins in classic antigen-driven immunity.
Antigen presentation is mediated by the antigen presenting cell (APC), with examples including innate immune cells such as professional phagocytes, B cells, or dendritic cells, as well as non-classic immune cells in some cases, such as endothelial cells [EC]. The process is incited by a bacterial challenge or tissue injury, during which time components of the pathogen or damaged cell are taken up by the APC. The products of these lytic processes are antigenic peptides that are associated with the HLA/MHC molecule (Figure 1, Signal 1). A CD4+ or CD8+ T cell receptor binds to the APC’s HLA/MHC complex as well as the checkpoint molecule concomitant with the T cell receptor’s binding of the APC’s antigen-containing HLA/MHC molecule (Figure 1, Signal 2). If the predominant co-receptors on the APC are co-stimulatory molecules/receptors, such as CD28, ICOS, or CD40, this licenses the T cells that see presented antigen to respond to this connection by activation or differentiation. This transformation is a driving force behind what has been characterized as a Th1-driven response into an activated effector T cell (cell-mediated immunity) response. Alternatively, if the APC expresses a preponderance of co-inhibitory molecules (i.e. checkpoint proteins), the CD4+ or CD8+ T lymphocytes are not licensed to respond (i.e. becoming anergic and potentially apoptotic). It should be noted that many of these co-inhibitory cell surface receptor molecules actively signal --the simple over-expression of these receptors, or their cell surface ligands in some cases (e.g. the most overt example being the over-expression of some of these co-inhibitory ligands on tumor cells), is sufficient to directly (via check point protein cell signaling and/or the indirect stimulation immune suppressive mediators, like IL-4, IL-10 and IL-13) prompt an anergic state (Th1 to Th2 and/or M1 to M2 phenotypic shift) among cells within such an environment (24). That such shifting occurs in the setting of the experimental animal and septic patient has been documented by several labs (19, 23). Ultimately, these receptors and their ligands are often first regarded as toleragens (25, 26).
Figure 1.
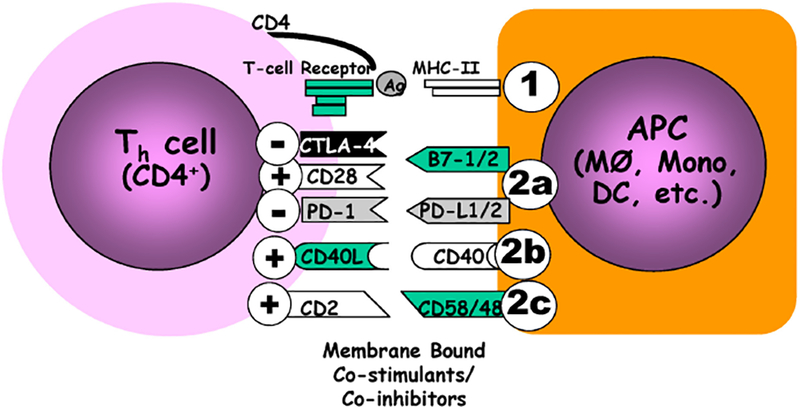
Antigen presentation is typically a two-signal process, in which antigens derived from a foreign pathogenic source (and/or at times tissue components/debris) are processed (commonly in a lytic fashion) by an APC, i.e., macrophage (MØ), dendritic cell (DC), monocyte (Mono), for formal association with the an HLA/mouse MHC II receptor and presentation/exposure to the appropriate T cell receptor expressing lymphocyte (CD4+ T helper cell)(This is signal one; ①.).However, for formal T cell activation/differentiation to proceed, the APC must not only provide a 2nd co-stimulatory (+) signal (Signal 2; ②) that licenses T cell differentiation, but this must overcome and/or suppress concomitant co-inhibitory (−) signals that are often expressed by the APC (but not exclusively by them). Of note, there are three loosely-termed families of these costimulatory/co-inhibitory molecules, as broken down by protein structure: (2a) the B7:CD28 superfamily, (2b) the TNF:TNFRs that lack death receptor domain, and (2c) the CD2 superfamily & select integrins.
Checkpoint proteins are not limited to solely the APC to T cell interaction. Communication among monocytes/macrophages/dendritic cells with epithelial/endothelial/tumor cells works via this mechanism (Figure 2).
Figure 2.
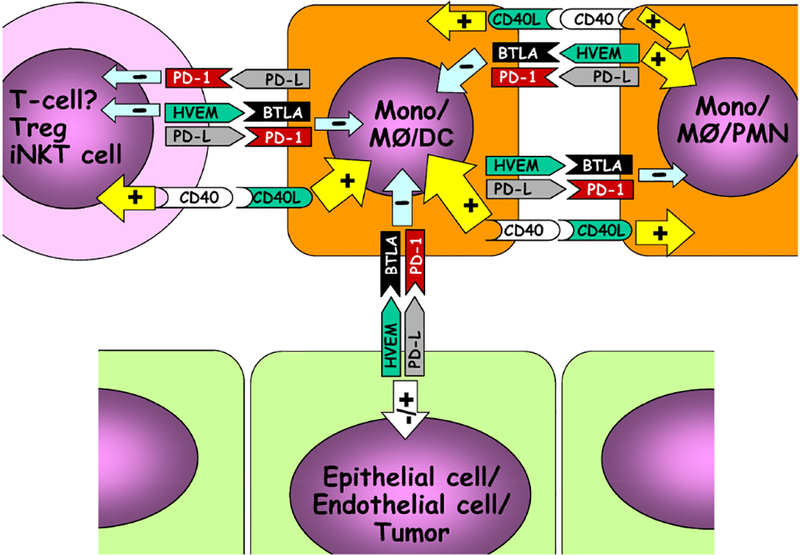
While co-inhibitors (a.k.a., checkpoint proteins)/co-stimulants are best appreciated for their role in stimulating or inhibiting the activation/differentiation of the CD4+ T helper cell, these same cell-surface co-inhibitors/co-stimulants appear to have potentially unique roles in cell:cell interactions between not only various leukocyte sub-sets, but with non-immune cells within tissue. Positive (+), stimulatory activity reported; negative (−), inhibitory activity reported.
(a). Programmed cell death receptor-1 (PD-1):
Programmed Cell Death Receptor (PD)-1, with pseudonyms including Pcdc1 and CD279, is a Type I transmembrane glycoprotein-Ig (IgV) superfamily member, containing an immunoreceptor tyrosine-based inhibition motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) for intracellular signaling. PD-1 participates across a spectrum of immune responses relative to many other B7:CD28 superfamily members (27–29). Most observations indicate that ligation of PD-1 recruits phosphatases Src homology region 2 domain-containing phosphatase (SHP)-1 and/or SHP-2, prompting an inhibition of PI3K pathway signaling resulting typically from CD28/CD3/immunoreceptor tyrosine-based activation motif (ITAM) activation (30–34) (Figure 3).
Figure 3.
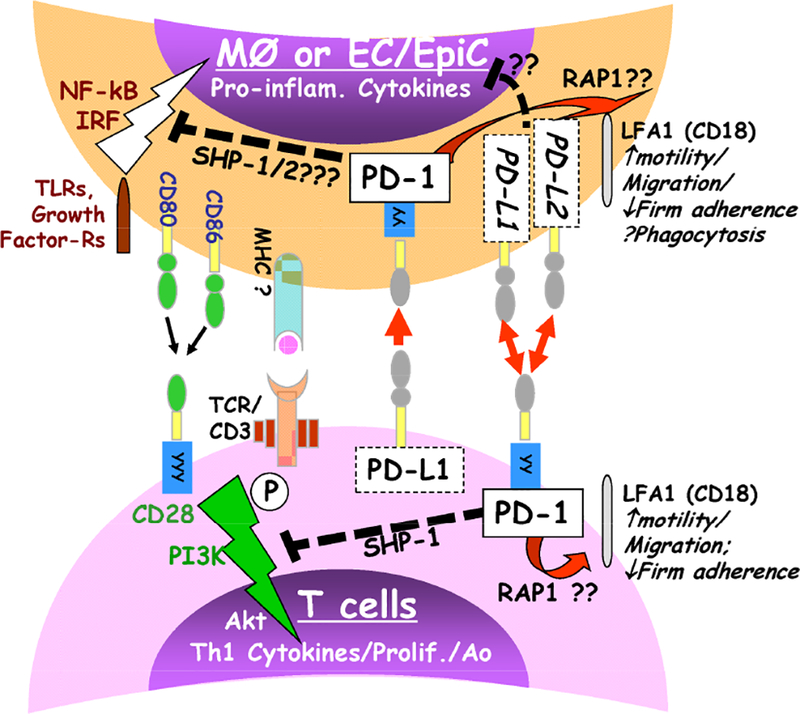
Overview of proposed PD-1 & PD-L1/L2 intra-cellular signaling between T cells and/or macrophage/monocytes, among others (e.g. PMN, DC and/or EC/EpiCs, which may express PD-1 and/or PD-Ls). The activation pathway is indicated by a ‘thunder bolt’ symbol, while suppressive effects are denoted with a dashed line.
Programmed Cell Death Receptor Ligand-1 (PD-L1), also known as B7-H1 or CD274, is considered the primary ligand of PD-1. Importantly, it is ubiquitously expressed on not only immune, but also a wide variety of non-immune tissues and organs (35–37). Alternatively, PDL2 is more restrictively expressed on APCs and immune cells (38). Like PD-1, these ligands are both type I transmembrane glycoprotein receptors (IgC in addition to IgV). Unlike PD-1, they have neither an ITIM nor ITSM motifs. However, there are reports that intra-cellular signaling may still be achieved via these PD-1 ligands themselves, perhaps from surrogate ligation of B7.1/CD80 (26, 39).
(b). B and T Lymphocyte Attenuator (BTLA) and Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4):
B and T Lymphocyte Attenuator (BTLA) is considered a co-inhibitory receptor, related in molecular structure to PD-1; it acts as an inhibitor of B cell and CD4+ T cell function, as its name describes (40). It is induced on anergic CD4+ T cells and is associated with attenuated pro-survival signaling (41, 42).
Cytotoxic T Lymphocyte-Antigen-4 (CTLA-4) is another inhibitory regulator of T cell activation and proliferation. CTLA-4 competes with CD28 (a potent co-stimulatory molecule/receptor) for binding to CD80 and CD86 on APCs, thus, preventing T cell activation (43, 44). CTLA-4 plays a prominent role in maintaining T cell responses which is evident in the phenotype of CTLA-4−/− mouse strain where CTLA-4 deficiency results in death from lymphoproliferative disorders (45, 46)
2. Checkpoint proteins and their ligands during sepsis and shock.
Studies from several laboratories initially utilizing experimental models of sepsis, and subsequently models complexed with aspects of severe shock or secondary infectious challenge, have shown that several of these families of cell-surface molecules have an impact on survival. This points to potential roles in pathological processes driving sepsis. Studies by Huang et al. (47) showed that mice in which the gene for PD-1 was deleted exhibited a marked reduction in mortality in response to CLP-induced polymicrobial septic challenge. From an experimental perspective, the role of PD-1:PD-L ligation in driving these morbid effects was further corroborated by two independent investigations utilizing antibodies against PD-1 (33), and its primary ligand PD-L1 (48), where both demonstrated that CLP morbidity and mortality could be significantly reduced by such antibody post-treatment. Importantly, observational clinical studies have shown that critically ill patients who developed severe septic shock plus secondary nosocomial infections expressed markedly levels of PD-1 and PD-L on various leukocyte subsets (49). A similar observation was made on severely injured patients, where the expression of high PD-1 blood leukocyte levels was correlated with worsening physiological dysfunction (50). Furthermore, to the extent that these are simply the aberrations of the CLP model as applied in adult male mice, the importance of PD-1 on throughout the age spectrum has been demonstrated (51, 52). Work by Young et al. (51), using a cecal slurry model, documented that PD-1 gene deficiency also confers a survival advantage in this model of polymicrobial sepsis in neonatal mice. Several studies have also shown a clear role for PD-1 in modulating the geriatric immune response to sepsis (53, 54).
Using a similar experimental approach, Shubin et al. (55) subsequently demonstrated that CD4+ T cells’ BTLA expression among ICU patients was associated with sepsis and development of subsequent nosocomial infections. When the interaction between BTLA and its primary ligand herpes virus entry mediator (HVEM) is limited by genetic deletion of BTLA, murine survival improves in the setting of septic challenge (56). Among adult septic patients and septic adult mice, BTLA and HVEM are both elevated on myeloid and lymphoid cell populations (55, 56).
Unsinger et al. (57) have also shown that in response to septic challenge, expression of CTLA-4 on CD4, CD8, and regulatory T cells is increased in the murine CLP model. Furthermore, antibody neutralization of CTLA signaling capacity with anti-CTLA-4 antibody treatment after the onset of CLP led to a decrease in sepsis-induced apoptosis, better antigen recall responsiveness, and improved survival (58). In studies looking at the role of CTLA-4 in sepsis-induced immunosuppression, blockade of CTLA-4 in conjunction with PD-1 also improved survival and reversed sepsis-induced immunosuppression as measured by reduced lymphocyte apoptosis in a sequential sepsis challenge model where CLP was followed by a secondary fungal insult (59). Recent studies by Chen et al. (60) investigating the co-inhibitory molecule 2B4 indicate it may also markedly suppress CLP-induced complications. Together these results point to pathologically important roles for these co-inhibitory/checkpoint proteins and their signaling as underpinning the development of septic morbidity.
(a). From PD-1 to its Ligands:
Similar to the survival analysis undertaken for PD-1−/− mice, a survival analysis of PD-L1 gene deficient mice after sepsis demonstrated comparable results(61). In this respect, elevated PD-L1 and PD-L2 expression has been documented in pulmonary epithelia from patients who died of sepsis (62). These investigators found that not only the expression of PD-1, but also the expression of its two ligands PD-L1 and PD-L2, are associated with poor outcome in sepsis and shock. Other groups have also reported the expression of the ligands for PD-1, namely PD-L1 and PD-L2, as being elevated on a variety of non-professional immune cells/tissue beds/tumor cells (63–65). As such, the over-expression of these toleragenic checkpoint protein ligands represents a novel, oft-overlooked, interface between non-professional immune cells/tissue beds/organs that may speak not only to how local immune, but organ function might be regulated directly orindirectly through their ligation (Figure 2 and Figure 4). So, what evidence is there for these ligands of co-inhibitors having an effect on immune dysfunction in sepsis and shock?
Figure 4.
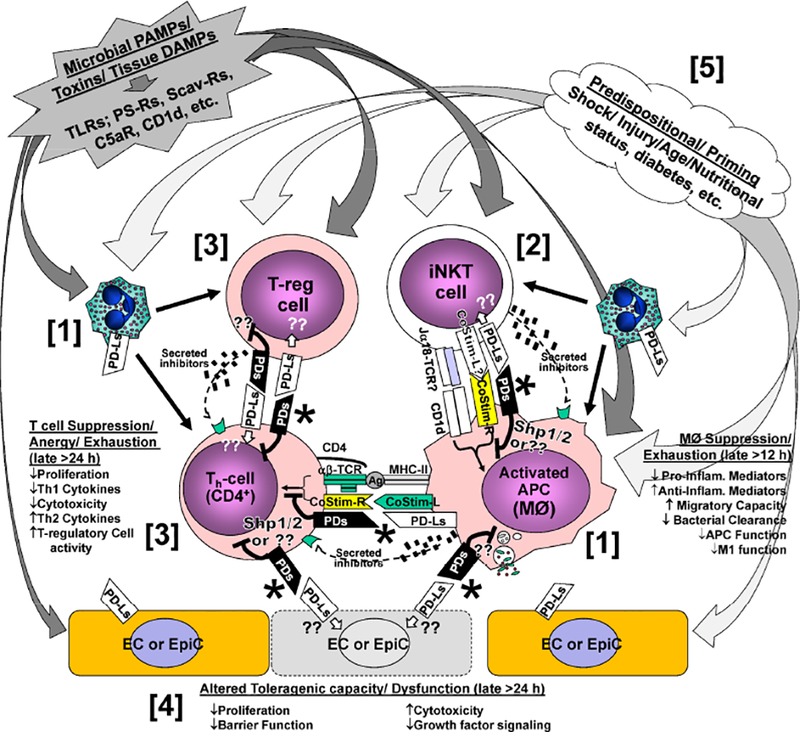
Hypothetical points at which Programmed Cell Death Receptor (PD) family members, i.e., PD-1, BTLA and their ligands (PD-Ls, HVEM) interact to alter iNKT cell activation in response to the diverse signal(s) derived from tissue injury and infectious microbial challenge. This leads to: [1] suppression of phagocyte clearance of the septic challenge; PMN and macrophage dysfunction (via direct/indirect effects of PD-1/PD-L1 ligation [*]); [2] overzealous activation of innate immune cells via overt iNKT cell activation through CD1d-Ag stimulation; [3] immune suppression of classic CD4 Th1 cell actions via anergy/chronic stimulation by Treg cells and/or iNKT-cells; and/or [4] through the induction of altered ECs/EpiCs capacity or dysfunction (hence a link to organ injury and dysfunction). Finally, [5] factors such as prior injury/shock and subjects age, background, nutritional status, prior health, etc., may serve to alter PD-family member expression, contributing to a state of priming/ pre-disposition/ innate immune memory.
3. Immune Dysfunction in Sepsis.
(a). Adaptive Immunity
(i). Classic T cell Immune Dysfunction:
Studies of sepsis with PD-1 gene-deficient mice and anti-PD-1 antibody treatments point to PD-1’s impact on adaptive immune responsiveness. These changes include the restoration of the delayed type hypersensitivity response and a reduction of sepsis-induced CD4+/CD8+ T as well as B lymphocyte cell death (47, 57, 61). In this regard, Chen et al. (60) have recently reported that the expression of the co-inhibitory molecule 2B4 (CD244, SLAM4), which was initially reported to be upregulated on activated natural killer cells and CD8+ T cells (but not in experimental models of sepsis), is markedly upregulated on the splenic CD4+ T cells of not only CLP mice, but also on human patients with severe sepsis. Further, these CD4+2B4+ cells appear to upregulate PD-1 and CTLA-4, but inhibition of 2B4 activity is non-redundant to either PD-1 or CTLA-4. Importantly, gene deficiency of 2B4 preserves CD4 cell Th1 functions and cell survival (reduced septic CD4 T cell loss), while providing a survival benefit to CLP mice (59).
Since many co-inhibitory receptors, e.g., PD-1, BTLA, CD47 and CTLA-4, express an ITIM and/or ITSM, it is thought that the suppression of classic CD4+/ CD8+ T cell function is a result of the recruitment of the phosphatase, SHP-1 and/or SHP-2 (27, 30, 66). These SHPs then antagonize not only T cell receptor driven, but other pathways that drive the activation of the Akt/ PI3K pathway, which underpin the immune-suppressed’exhausted’ T cell phenotype seen with PD-1 in conditions such as cancer and viral infection (27, 30, 66). In this respect, studies by Bandyopadhyay et al. (67) have documented that, at least in T lymphocytes isolated from severely injured patients, which they have previously reported as expressing a number of co-inhibitory molecules as well as exhibiting T cell anergy/ ‘exhaustion’ (21, 68), indicate that these cells appeared hyper-responsive to expressing the activated form of the phosphatase SHP-1 (Figure 3). Importantly, Huang et al. (47, 61) also showed that macrophages derived from septic mice had a markedly increased capacity to produce the immune suppressive/ anti-inflammatory cytokine IL-10 and this, more so than many of the mediators examined, could be attenuated by the loss of either PD-1 or PD-L1 gene expression. This implies that these cells shifted from an M1 to an M2 macrophage phenotype, and this change also has the capacity to support an antigen-independent induction of immune suppressive/Th2/anergic T cell state.
Intriguingly, an alternative signaling mechanism has been put forward for PD-1 related co-inhibitors, such as CTLA-4, involving interactions with integrins and various components of the cell cytoskeletal machinery (69–71) or the recruitment of alternative receptors like CD80, CD24 versus TLR4 by Siglec10 (72, 73) (Figure 3). Much, however, remains to be understood about sepsis-induced changes in T cell co-inhibitor molecule signaling and how it shapes the septic cellular phenotype and function.
(ii). Regulatory Lymphocytes:
invariant Natural Killer T-cell (iNKT-cell):
The innate regulatory lymphocyte populations are emerging as key regulators of the immune and inflammatory response a variety of infectious insults. Among these sub-populations, the iNKT-cell has been shown to play some of the most central roles in immune response to a wide variety of infections and septic events. iNKT-cells are capable of both a Th1 and a Th2 response and interact with a diverse array of immune cells serving to both positively and negatively regulate the immune and inflammatory response iNKT-cells are capable of trafficking both within the liver in response to a sterile injury (74), in and out the lung in response to an infection (75). As well as distally to a septic source in the abdomen (76). iNKT-cells are themselves regulated by co-inhibitory/checkpoint proteins, most notably PD-1. Initial activation of iNKT-cells is associated with increased PD-1 expression upon iNKT-cells (77) (Figure 4, Component [2]). Young et al. (76) showed that direct ligation of PD-1 with its ligand PD-L1 is essential to trafficking and migration of iNKT-cells and PD-1 is essential to regulation of chemokine receptor expression upon iNKT-cells. Conversely, repeated stimulation of PD-1 upon iNKT-cells has been shown to induce anergy (78) a mechanism that is believed to have developed to protect against an over-exuberant iNKT-cell driven immune response. In similar fashion, PD-1:PD-L blockade has been shown to prevent the induction of iNKT-cell anergy (79). Although PD-1 has been the most explored co-inhibitory pathway in iNKT-cell responses to stimuli, several other co-inhibitory molecules have been described. CD28 and OX40 have been described as indispensable for full activation of iNKT-cells, and glucocorticoid-induced TNF receptor (GITR) plays a key negative regulatory role in antigen-induced activation of iNKT-cells (80). Akin to CD8+ T-cells, the negative checkpoint regulator 2B4 has also been shown to play a key role in suppressing an excessive iNKT-cells response. Ahmad et al demonstrated that among patient with HIV infection, levels of 2B4 expression was significantly upregulated upon both CD3+ and HIV-specific CD8+ T-cells as well upon and iNKT-cells. Levels of 2B4 expression correlated with degree of suppression of intra-cellular production of IFN-gamma within iNKT-cells. Importantly, treatment with anti-retroviral therapy among these HIV patients was noted to lead to a decline of 2B4 levels upon CD8+ T-cells, but not among iNKT-cells (81).
CD4+CD25+ Regulatory T Cells (Tregs):
A regulatory/anti-inflammatory role for T regulatory cells (Tregs) has been identified in several models of lung diseases: allergy/asthma, pneumocystis and tuberculosis (82–84). However, lymphocytes, especially the small population of Tregs that reside in the lung, have garnered little attention relative to the role of these cells in the pathophysiology of ARDS, let alone indirect (i)ARDS. Using a well-established model of direct lung injury (IT instillation of lipopolysaccharide [LPS]), D’Alessio et al. (85) were the first to demonstrate that Tregs accumulate in the bronchoalveolar lavage fluid (BALF) of mice. In addition, they identified the presence of Tregs in patients with ARDS suggesting a role for Tregs in the resolution of ARDS. Based on these results, Aggarwal et al. (86) documented that adoptive transfer of Tregs to lymphocyte-deficient recombinase-activating gene-1-deficient (Rag-1−/−) mice restored them to a normal pattern of resolution after IT LPS-induced direct ALI. Furthermore, a recent study demonstrated that transplantation of human umbilical cord mesenchymal stem cells ameliorated ARDS by restoring the diminished levels of alveolar CD4+CD25+Foxp3+Tregs via a re-balancing of levels of anti- and pro-inflammatory factors in a direct ARDS mouse model (87). More recently, a study by Ehrentraut et al. has indicated that CD73-dependent adenosine generating Tregs could promote the resolution of LPS-induced direct ARDS (88). However, none of these studies illustrated a specific role of Tregs in iARDS. Using a two-hit model of Hem-CLP, Venet et al. observed that CD4+CD25+Foxp3+ cells appeared to be recruited to the lung in iARDS and that the Foxp3 as well as IL-10 gene expression in these cells were increased during this process (89). Most importantly, Venet et al. also showed that down-regulation of the Tregs’ function using Foxp3 targeting siRNA, administered 12 hours before the induction of experimental iARDS, echoed lung injury obtained in CD4-gene deficient as well as anti-CD4 antibody depleted mice; this was consistently associated with a reduction in lung IL-10 levels (89). However, how the activation of this small sub-population of lymphocytes regulated iARDS in this study was only partially elucidated.
With respect to the role of the checkpoint protein PD-1, Tang et al. (90) demonstrated a significant role for PD-1 in regulating the Tregs response to iARDS. Adoptive transfer of Tregs derived from WT naïve mice were capable of suppressing iARDS, including decreased pulmonary neutrophil influx. However, these beneficial effects upon the severity of lung injury were not evident when PD-1−/− derived Tregs were transferred into recipient WT mice during the resuscitation period of Hem or 24 hours before CLP via tail vein injection. Furthermore, the expression of CTLA-4, another co-inhibitory regulator, was also affected by the presence of PD-1, wherein the expression of CTLA-4 upon CD4+Foxp3+ T-cells was increased following transfer of Tregs from WT, whereas no increase in CTLA-4 expression was noted in mice receiving PD-1−/− Tregs. (Figure 4, Component [3]). Subsequently, Tang et al. expanded the significance of PD-1:PD-L1 ligation on Tregs on mediating lung-protective effects by documenting that AT transfer of WT donor Tregs into PD-L1 gene deficient recipient mice did not confer protection against lung injury/inflammation. Further, the ligation PD-1:PD-L1 in this system also appears to preferentially signal through SHP-1 as opposed to SHP-2, similar to that reported above for septic patient T cells (91, 92).
La et al. also noted the suppressive effect of PD-1 upon Tregs in a model of hydatidosis, a parasitic infection (93). They noted that the percentage of CD4+CD25+PD-1+ cells correlated with duration of infection and size of abscess. The authors speculated that the suppressive effect of PD-1+ expression upon Tregs was essential for infection growth and avoidance of the immune surveillance. These findings have been correlated clinically, wherein Liu et al. noted PD-1 expression was increased on Tregs in patients with sepsis when compared with healthy controls. Most significantly, levels of PD-1 expression upon Tregs were lowest among patients who subsequently survived the septic event, again implying the negative regulatory effect of PD-1 upon Tregs may significantly contribute to sepsis related mortality (94).
(b). Innate Immunity
(i). Macrophages:
One of the more interesting observations made by Huang et al. is that early following septic challenge/CLP (as defined as 12-hours post-CLP or less), changes in PD-1 expression on CD4+ T cells, CD8+ T cells or B cells are not yet evident (typically these are not seen until at 24 hours post CLP) (61). Further, dendritic cells did not express PD-1 at this time point. However, Huang et al. observed that peritoneal leukocytes, as defined by F4/80+ cells harvested from peritoneal lavage, demonstrated increased PD-1 and PD-L1 expression (61), at both protein and message levels, after as little as 12-hour post-CLP. This effect was sustained at the 24-hour and 48-hour time points. Mouse blood monocytes and Kupffer cells have also been, subsequently, demonstrated to exhibit increased PD-1 expression in response to CLP (47, 95). This upregulation of PD-1, from a naïve state of nearly undetectable levels, was also a little surprising as a number of groups had indicated that tissue macrophage did not express elevated levels of PD-1 typically. However, as most of this work was conducted with naïve cells’ response to LPS as a stimulant, unsurprisingly this was not detected. To elaborate, LPS, unlike TNF-α or interferon-γ, was not reported to be a strong inducer of PD-1 expression (49). In fact, Guignant et al. (49) noted that monocytes derived from patients with severe sepsis express both PD-1 and PD-L1 out to 3–5 days post diagnosis of sepsis.
These initial studies by Huang et al. also showed that following CLP, there was a decline in both phagocytic capacity (which occurs with both opsonized and non-opsonized targets) and stimulant-induced cytokine production among macrophages (47). Additionally, random migratory capacity (measured both in rate and direction) and cell spreading (following surface adherence) increased markedly after sepsis among WT mice. Importantly, these phenomena were equivalent to findings among sham control samples in the setting of PD-1 gene deficiency (96). These latter changes in migratory capacity, cell spreading and motility, appear to be partly due to PD-1-mediated changes in cytoskeletal alpha-actinin and F-actin aggregation capabilities. Proposed mechanisms include PD-1 association with CD11b and changes in PD-1-mediated upstream suppression of phosphatase and Rap-1-dependent migration. Ultimately, septic PD-1−/− macrophages exhibit less dysfunctional migration compared to their septic WT counterparts.
With this stated, it is unknown whether macrophages and monocytes expressing PD-1 under the stress of sepsis and/or severe shock/injury are the same cells that express PD-L1 or whether they are unique sub-populations. Does it affect macrophage M1 to M2 polarization?Does paracrine or even autocrine signaling occur for PD-1: PD-L1 expressed on these macrophages? Further, while the study by Ayala et al. suggests that phosphatase signaling, as reported in classical T helper cells and Tregs, appears to be involved in some of the changes seen in these septic macrophages, the exactnature of this signaling remains to be elucidated (Figure 3).
(ii). Neutrophils:
Neutrophils (PMNs) are proposed to have an important role in inducing iARDS. When recruited to a site of infection, they exert a variety of beneficial functions critical to clearance of the invading pathogen (97). However, it is also thought that the recruitment of activated PMNs may be harmful when these functions are directed at otherwise normal host tissue (bystander injury) (97, 98). In this regard, Ayala et al. (99) and Lomas et al. (100) reported on PMNs that were isolated from donor animals after hemorrhage (a form of PMN priming) (Figure 4, Component [5]), and were then adoptively transferred into naïve animals. They found that when these naïve animals were then subjected to a septic challenge, the recipient lungs became inflamed. In this setting, PMN in vivo ‘priming’ was not only associated with an increase in respiratory burst capacity in vitro, but also with a decrease in PMN apoptosis (Ao) (99, 100); a finding confirmed by others in experimental iARDS (101). Jimenez et al. (98) found a significant decline in PMN AO in patients with SIRS and the plasma from these individuals suppressed AO of PMNs derived from healthy controls (98). These findings and others (102–104) support the hypothesis that while delayed AO in an infectious environment aids PMN mediated pathogen killing, in a pathogen-free setting delay in AO contributes to inflammation/SIRS and organ failure/injury (98). With that stated, the mechanisms underpinning these effects in PMN have not been fully clarified.
As mentioned earlier, while it has been documented that neutrophils can express the co-inhibitory ligand PD-L1 (105), until recently little was known about how disease states may alter its expression, and what might be the pathological significance of such changes. In this regard, using the CLP model of sepsis Huang et al. (105) initially documented a trend toward increased frequency of PD-L1 (B7-H1)+ cells in the mouse blood PMN population following CLP, though this was not statistically significant due to wide variance. Upon closer inspection of the data, however, expression of PD-L1+ staining appeared to segregate into distinct PD-L1high and PDL1low sub-populations. Further it was found that if mice exhibited PD-L1high expressing PMNs in their peripheral blood at 24 hours post-CLP this correlated not only with higher levels of systemic pro- and anti-inflammatory cytokine/ chemokine levels (CCL2, IL-6, CXCL2, KC, TNF-α, and IL-10), but with lethal outcome as well. Subsequently, Wang et al. (106) showed that PMNs from CLP mice and patients diagnosed with severe sepsis exhibited a substantial rise in their PD-L1 expression, correlating with increased morbidity in the septic patients. Additionally, these cells, via direct contact, were capable of inducing a marked increase in lymphocytic cell death/dysfunction (another common feature of the septic animal/patient) as well as altering the migrational functionality of the cell (a dysfunction also noted by Young et al. (107) in septic mouse iNKT cells) (22). More recently, Patera et al. (53) have further found that peripheral blood PMNs from septic patients that expressed high levels of PD-L1 exhibited a decline in their capacity to phagocytize bacteria, a reduction of CD168 expression and a poor production of TNF-α in response to stimuli. Utilizing neutralizing antibodies directed against either PD-1 or PD-L1, to treat these septic patients’ PMNs ex vivo, the investigators also found that the decline in PMN bacterial phagocytic function could be markedly reversed. Together this illustrates that via direct ligation of PD-L1, or via indirect ligation of PD-1 and its downstream signaling, it appears possible that sepsis can drive altered PMN function as well as set up the PMNs to serve as a potent toleragenic cell population through PD-1:PD-L1 ligation.
3. End-organ sequelae of cellular immune disharmony
(a). End-organ Injury – the Gut
In the intestine, the epithelial lining serves as an important barrier to prevent the absorption of toxins, antigens, and microorganisms across the intestinal wall. Intestinal barrier dysfunction and increased intestinal permeability are morbid states that often accompany developing sepsis. This developing epithelial cell barrier dysfunction has been thought to contribute to the development of the multiple organ dysfunction syndrome (MODS) during sepsis (108–110).
Studies have shown that human gastric epithelial cells constitutively express basal levels of PD-L1, but when exposed to Helicobacter pylori this expression significantly increases (111). PD-L1 mRNA was also detected in colonic epithelial cells from healthy individuals and PD-L1 protein expression has been reported on the surface of colonic epithelial cells from patients with inflammatory bowel disease (IBD). Interestingly, blockade of PD-L1/B7-H1 ligation has been shown to suppress the development of experimental chronic intestinal inflammation (63–65), which implies that this ligand could be involved in mediating aspects of gastrointestinal dysfunction seen in septic or traumatic pathologies. In this vein, Huang et al. (61, 112) showed that jejunal villus injury was evident on histologic analysis as well splenic apoptosis in CLP mice – two findings which were ameliorated upon deletion of either PD-1 or its primary ligand PD-L1 (B7-H1) gene. However, a patho-mechanistic basis for these findings in the gut were not further explored in those studies.
In this respect, Wu et al. have recently shown that PD-L1 protein expression increases in small intestine and colon after CLP (113). This corresponds to changes in PD-L1 expression on colonic tissue sections of septic patients (113). Additionally, a time course demonstrated that intestinal epithelial cells maximally expressed PD-L1 at 24 hours after septic challenge – a finding also markedly increased across all sham time-points (113). Intestinal epithelial cell barrier integrity was markedly attenuated after sepsis in those mice with PD-L1 deficiency (113). But what is the possible basis for the improvement of gut barrier function? Wu et al. further demonstrated that an absence of PD-L1 leads to decreased expression of cytokines/chemokines, such as TNF-α, MCP-1, IL-6 and IL-10, in the ileum following CLP (113). Absence of PD-L1 also allows for a preservation of the levels of tight junction proteins as measured by western blot for ZO-1, Occludin and Claudin-1. To the extent that these findings are a result of PD-L1 ligation, it was observed by Wu et al. that antibody blockade of PD-L1 ligation produced a partial, yet significant, restoration of tight junction protein expression on the epithelial cell line that had lost such expression when subjected to inflammatory stimuli, as encountered in septic animals and septic patients (113). Taken together, these data support the concept that blockade of PD-1’s primary ligand PD-L1, and not just PD-1 itself, has a restorative effect on gut histology and function in the face of experimental septic challenge (Figure 4, Component [4]).
(b). End-organ Injury – the Liver
Liver Sinusoidal Endothelial Cells (LSECs) are located within the hepatic sinusoid. They have a characteristically discontinuous basement membrane; this fenestration facilitates exchange of material with adjacent hepatocytes. A well-known phenotypic marker of the LSEC population is CD146. The LSECs can also act as antigen presenting cells (as alluded to earlier) as marked by their expression of CD80, CD86, MHC I and MHC II. They are also considered to have roles in protecting hepatocytes from infection and maintaining the liver’s immunotolerant state.
These characteristics are in contrast to the vascular endothelial cells which have a continuous basement membrane, express CD31, secrete inflammatory mediators such as IL-6, and directly interact with the intra-luminal leukocytes during an inflammatory state by expressing selectins (114–116).
Hutchins et al. (117) explored the LSEC population’s response to experimental septic challenge in relation to PD-L1 expression and found that indeed its increased expression appears to contribute to LSEC dysfunction and liver injury in response to CLP (Figure 4, Component [4]). Indices of dysfunction include: increased apoptosis, decreased VEGF-R2 expression, decreased barrier function, and decreased proliferative capacity – all of these parameters of injury were diminished in the setting of PD-L1 gene deletion/loss (116). Additionally, depletion of Kupffer cells (on which increased levels of PD-1 were expressed) reversed septic mouse LSEC rise in PD-L1 (95, 117). Zhu et al. (118) also investigated liver damage after CLP, finding that in vivo anti-PD-L1 antibody treatment reduced the onset of transaminitis otherwise experienced in the sham-treated wildtype mice.
(c). End-organ Injury – the Lung
Using an experimental model of sequential hemorrhagic shock (Hem) followed by CLP, which is reported to more consistently produce a condition clinically resembling moderate acute respiratory distress syndrome (ARDS), pulmonary pathophysiology has been studied. This was chosen over the model of standalone CLP because the combination of insults consistently produced moderate or worse acute pulmonary injury in the setting of a low lethality rate at 24 hours (99, 119).
Among wildtype mice after Hem-CLP, the P:F ratio (arterial P02:Fi02) decreases, suggesting loss of pulmonary compliance (99, 120). Among mice who were either PD-1−/− or PD-L1−/−, indices of lung inflammation (e.g. IL-6, MIP-2, MPO) and injury (pulmonary edema, protein leak) were mitigated following the same Hem-CLP challenge (99, 121). Histologic analysis has also revealed increased leukocyte presence and cellular apoptosis in lung tissue following Hem-CLP, a finding attenuated by PD-1 gene deletion (122). As a clinical correlate, PD-1 expression on circulating T cells of ICU patients corresponded with ARDS and poor survival (122).
Returning to the membrane-bound checkpoint proteins: by what mechanism are PD-1 and PD-L1 protecting the lung against septic injury? The answer may lie in endothelial cell regulation through the Angiopoietin/Tie2 pathway. Angiopoietin (Ang)-1 is constitutively released by surrounding pericytes surrounding vascular ECs (123). The tyrosine receptor Tie2, which is expressed on vascular ECs, is stimulated by the binding of Ang-1. This interaction results in the synthesis of a cascade of anti-inflammatory/pro-survival proteins, as well as the down-regulation of ICAM-1 expression, thus leading to diminished leukocyte recruitment.
Ang-2 has the opposite effects, causing downstream Rho kinase expression with subsequent de-stabilization of cell-cell junctions and exaggerated vascular permeability (124, 125). Ang-2 is elevated in the experimental model for indirect ARDS mentioned earlier, as well in the blood of critically ill patients with ARDS (124). Direct EC:neutrophil interactions contribute significantly to EC Ang-2 release (124). Suppression of Ang-2 (using either in vivo siRNA or blocking Ab) significantly decreases inflammatory lung injury, neutrophil influx, and lung as well as plasma IL-6 and TNF-α (124, 126, 127). Does this mean that alteration of the expression of PD-1 and/or PD-L1 affects the Ang-1:Ang-2 regulatory axis during the development of iARDS in mice? Yes, as gleaned from the experiments of Lomas-Neira et al. (Lomas-Neira, J.L., Monaghan, S.F., Huang, X., Ayala A. 2017. Novel role for PD-1:PD-L1 as mediator of neutrophil:endothelial interactions in pathogenesis of indirect ARDS in mice. Front Immunol. [Submitted]). Gene deletion of either PD-1 or PD-L1 suppresses the rise in lung Ang-2, but not Ang-1 following Hem-CLP. Similarly, phosphorylated Tie-2 in the lung is diminished after lung injury, but PD-1 or PD-L1 gene deletion restores the Tie-2 to baseline levels (122). Synthesis of this data suggests a role for PD-1:PD-L1 in endothelial cell interaction and septic dysfunction.
Of note, soluble PD-1 (sPD-1), a solubilized form of the cell-surface receptor PD-1 that appears to be synthesized and released as a product of alternative splicing, is increased in patients with ARDS and in Hem-CLP mice compared to sham controls (128). Furthermore, Monaghan et al. documented that the sPD-1 that was released also appears to be biologically functional (128). This was demonstrated by the reduced capacity of splenocytes to respond to stimuli following ex vivo murine serum co-culture with sPD-1 following Hem-CLP (an experimental model for ARDS, as discussed) (128). Questions regarding soluble PD-1 remain, specifically: what is the overall significance of sPD-1, not only as a potential biomarker, but also as a contributor to sepsis/ARDS pathophysiology? The larger question is: what is the significance of the changes in alternative splicing itself that not only lead to sPD-1, but many other soluble protein isoforms seen in response to stress? Further investigations are underway to delineate these unknowns.
(d). The Interface of Endothelium with Epithelium
The lung, similar to the gut (with its epithelial digestive system interacting with endothelium) and the liver (with sinusoidal tissue interacting with robust trees of vascular endothelium), has an interface of these two cellular lining entities, namely the endothelium and epithelium. To localize the PD-L1 expression within the lung, expression of CD31 as an endothelial marker and EpCAM as an epithelial marker were utilized – shock/sepsis conditions caused increased PD-L1 expressions on both of these cell types (Xu, S. et al. Blockade of PD-L1 attenuates shock/sepsis-induced iARDS in mice through targeting endothelial cells but not epithelial cells. [Manuscript in preparation]). Intra-tracheal (IT) delivery of naked siRNA to target pulmonary epithelial cells and intravenous (IV) delivery of liposomal-encapsulated siRNA to target vascular endothelial cells were undertaken (129, 130). The results support the concept that PD-L1 expression on the endothelial cell, but not on the pulmonary epithelial cell, contributes to shock/sepsis induced iARDS as produced in mice (129–131). In ex vivo studies, PD-L1 gene deficiency also renders the lung endothelial cell monolayer more resilient against tight junction loss following IFN-γ/TNF-α stimulation (Figure 4, Component [4]).
CONCLUSION:
The Future of Checkpoint Proteins in Sepsis
Expression of PD-1 and its ligands (as seen with gene deficient mice) appears to contribute to the morbidity and mortality seen in not only acute models of lethal septic challenge and hemorrhage/sepsis, but also in patients with septic shock. Additionally, inhibition of PD-1:PD-L signaling, when provided in a delayed (post-treatment) fashion in a low mortality model of chronic sepsis and/or fungal challenge, also appears protective. The effects of these co-inhibitors are not restricted to APC:lymphocyte communication associated with the antigen presentation process, but also phagocyte:phagocyte, phagocyte:lymphocyte (non-APC) and non-immune:immune cell interactions. These co-inhibitors (alone or together) and/or the downstream signaling process may represent novel diagnostic markers and/or potential therapeutic targets.
Finally, these investigative endeavors have potential to be highly clinically relevant and impactful. As evidenced by the body of work in cancer research, immunomodulatory therapy has been effective against several types of malignancies in clinical trials (132–135). Anti-PD-1 blocking antibody (Keytruda [pembrolizumab] by Merck and Opdivo [Nivolumab] by Bristol-Myers Squibb) are FDA-approved for treatment of a variety of metastatic cancers (136). Other anti-PD-1 and anti-PD-L1 antibodies (alone or with anti-CTLA-4) are in Phase I, II & III clinical trials for cancer patients. From malignancy back to sepsis, a phase I clinical trial has been initiated for anti-PD-1 monoclonal antibody in patients with severe sepsis and/or septic shock (137).
In conclusion, checkpoint proteins such as PD-1 and PD-L1, extensively considered here, appear to be important mediators in the pathophysiology of severe infection as well as the combined insults of shock and sepsis. PD-1 expression, either in membrane-bound or soluble form, may serve as and indicator of the immune status of the individual afflicted with sepsis or shock. PD-1 may influence the outcome of bacterial infection by influencing the balance between effective antimicrobial immune defense and immune-mediated tissue damage. Upregulated PD-1 expression on innate immune cells as well as on T cells during sepsis and/or shock appears to play a role in their functional decline. The future of immune signaling pathways as regulated by checkpoint proteins may lie in clinical therapy targeted towards unleashing the potential not only of the adaptive, but of the innate immune system.
Cover Letter Statement:
All authors herein affirm that we concur with contents of this manuscript submitted for publication. The materials being submitted are not under consideration for publication anywhere else.
ACKNOWLEDGEMENTS:
This work was supported by grants R01 GM046354–20 & R35 GM118097 (A.A.), P20 GM103652 (J.L.N.), K08 GM110495 (D.S.H.), and T32 HL065085 (B.M.B) from the National Institute of Health / National Institute of General Medical Sciences; C. James Carrico Faculty Research Fellowship award-Amer. Coll. Surg. (S.F.M.) as well as “Armand D. Versaci” Research Scholar in Surgical Sciences Fellowship and Surgical Infection Society Foundation-Basic & Translational Research Fellowship awards (E.A.F.).
ABBREVIATIONS:
- Ao
Apoptosis
- Ab
Antibody
- Ag
Antigen
- Akt
Protein kinase B
- Ang-1
Angiopoietin-1
- Ang-2
Angiopoietin-2
- APC
Antigen Presenting Cell
- ARDS
Acute Respiratory Distress Syndrome
- AT
Adoptive Transfer
- BALF
Bronchoavleolar Lavage Fluid
- B7-H1
B7 Homolog 1
- BTLA
B and T Lymphocyte Attenuator
- CARS
Compensatory
- CBC
Complete Blood Count
- CCL
C-C motif Ligand
- CD
Cluster of Differentiation
- CLP
Cecal Ligation and Puncture
- CS
Cecal Slurry
- CTLA
Cytotoxic T Lymphocyte-Associated protein
- CXCL12
C-X-C motif chemokine 12
- DCs
Dendritic Cells
- EC
Endothelial Cell
- EpiCs
Epithelial Cells
- Foxp3
Forkhead Boxp3
- Hem-CLP
Hemorrhage-CLP
- HLA
Human Leukocyte Antigen
- HLA-DR
Human Leukocyte Antigen - D Related
- HVEM
Herpesvirus Entry Mediator
- iALI
indirect Acute Lung Injury
- IBD
Inflammatory Bowel Disease
- ICAM-1
Intercellular Adhesion Molecule 1
- IEC
Intestinal Epithelial Cells
- IFN-☐
Interferon-gamma
- IgC
Immunoglobulin-Constant
- IgV
Immunoglobulin-Variable
- IL
Interleukin
- IT
Intra-tracheal
- IV
Intravenous
- iNKT
cell invariant Natural Killer T cell
- ITIM
Immunoreceptor Tyrosine-based Inhibition Motif
- ITSM
Immunoreceptor Tyrosine-based Switch Motif
- KC
Keratinocyte Chemoattractant
- LPS
Lipopolysaccharide
- LSECs
Liver Sinusoidal Endothelial Cell
- MHC
Major Histocompatibility Complex
- MIP-2
Macrophage Inflammatory Protein-2
- MØ
Macrophage
- MODS
Multiple Organ Dysfunction Syndrome
- MOF
Multiple Organ Failure
- MPO
Myeloperoxidase
- NFkB
Nuclear Factor kappa-light-chain-enhancer of activated B cells
- PD-1
Programmed cell death receptor-1
- PD-L1
Programmed cell death receptor Ligand-1
- PD-L2
Programmed cell ligand-2
- PI3K
Phosphoinositide 3-kinase
- PMN
Polymorphonuclear leukocytes
- Rap-1
Repressor Activator Protein 1
- SHP
Src Homology region 2 domain-containing Phosphatase
- Siglec10
Sialic acid-binding Ig-like lectin 10
- SIRS
Systemic Inflammatory Response Syndrome
- SLAM4
Signaling Lymphocytic Activation Molecule 4
- sPD-1
soluble Programmed cell death receptor-1
- Tie2
Tyrosine kinase with Ig and Epidermal growth factor 2
- TLR4
Toll-Like Receptor 4
- TNF-α
Tumor Necrosis Factor-alpha
- TNFR
Tumor Necrosis Factor Receptor
- WT
Wildtype
- ZO-1
Zona Occludens-1
Footnotes
CONFLICT OF INTEREST DISCLOSURE:
The authors declare no conflict of interest.
Contributor Information
Eleanor A. Fallon, Email: efallon@lifespan.org.
Bethany M. Biron-Girard, Email: bethany_biron@brown.edu.
Chun-Shiang Chung, Email: cchung@lifespan.org.
Joanne Lomas-Neira, Email: jlomasneira@lifespan.org.
Daithi S. Heffernan, Email: dheffernan@brown.edu.
Sean F. Monaghan, Email: smonaghan@Lifespan.org.
REFERENCES:
- 1.2017. Data Reports | Sepsis | CDC. https://www.cdc.gov/sepsis/datareports/index.html
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. 2001. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303–10. [DOI] [PubMed] [Google Scholar]
- 3.Martin GS, Mannino DM, Eaton S, Moss M. 2003. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348:1546–54. [DOI] [PubMed] [Google Scholar]
- 4.van Ruler O, Schultz MJ, Reitsma JB, Gouma DJ, Boermeester MA. 2009. Has mortality from sepsis improved and what to expect from new treatment modalities: review of current insights. Surg Infect (Larchmt) 10:339–48. [DOI] [PubMed] [Google Scholar]
- 5.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. 2013. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 41:1167–74. [DOI] [PubMed] [Google Scholar]
- 6.Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, Rubenfeld G, Kahn JM, Shankar-Hari M, Singer M, Deutschman CS, Escobar GJ, Angus DC. 2016. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, Kochanek KD, Murphy SL, Tejada-Vera B. 2010. Deaths: final data for 2007. Natl Vital Stat Rep 58:1–19. [PubMed] [Google Scholar]
- 8.Rittirsch D, Flierl MA, Ward PA. 2008. Harmful molecular mechanisms in sepsis. Nat Rev Immunol 8:776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche J-D, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, Simpson SQ, Singer M, Thompson BT, Townsend SR, Van der Poll T, Vincent J-L, Wiersinga WJ, Zimmerman JL, Dellinger RP. 2017. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med 43:304–377. [DOI] [PubMed] [Google Scholar]
- 10.Osuchowski MF, Thiemermann C, Remick DG. 2017. Sepsis-3 on the Block: What Does It Mean for Preclinical Sepsis Modeling? Shock 47:658–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice TW, Bernard GR. 2005. Therapeutic intervention and targets for sepsis. Annu Rev Med 56:225–48. [DOI] [PubMed] [Google Scholar]
- 12.Deans KJ, Haley M, Natanson C, Eichacker PQ, Minneci PC. 2005. Novel therapies for sepsis: a review. J Trauma 58:867–74. [DOI] [PubMed] [Google Scholar]
- 13.Bone RC. 1996. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med 24:1125–8. [DOI] [PubMed] [Google Scholar]
- 14.MacLean LD, Meakins JL, Taguchi K, Duignan JP, Dhillon KS, Gordon J. 1975. Host resistance in sepsis and trauma. Ann Surg 182:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munster AM. 1976. Post-traumatic immunosuppression is due to activation of suppressor T cells. Lancet (London, England) 1:1329–30. [DOI] [PubMed] [Google Scholar]
- 16.Meakins JL, Pietsch JB, Bubenick O, Kelly R, Rode H, Gordon J, MacLean LD. 1977. Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg 186:241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller CL, Baker CC. 1979. Changes in lymphocyte activity after thermal injury. The role of suppressor cells. J Clin Invest 63:202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfe JH, Wu AV, O’Connor NE, Saporoschetz I, Mannick JA. 1982. Anergy, immunosuppressive serum, and impaired lymphocyte blastogenesis in burn patients. Arch Surg 117:1266–71. [DOI] [PubMed] [Google Scholar]
- 19.Ward NS, Casserly B, Ayala A. 2008. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med 29:617–25, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perl M, Chung C-S, Garber M, Huang X, Ayala A. 2006. Contribution of anti-inflammatory/immune suppressive processes to the pathology of sepsis. Front Biosci 11:272–99. [DOI] [PubMed] [Google Scholar]
- 21.Laudanski K, Miller-Graziano C, Xiao W, Mindrinos MN, Richards DR, De A, Moldawer LL, Maier RV, Bankey P, Baker HV, Brownstein BH, Cobb JP, Calvano SE, Davis RW, Tompkins RG. 2006. Cell-specific expression and pathway analyses reveal alterations in trauma-related human T cell and monocyte pathways. Proc Natl Acad Sci U S A 103:15564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wesche DE, Lomas-neira JL, Perl M, Chung C, Ayala A. 2005. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol 78. [DOI] [PubMed] [Google Scholar]
- 23.Hotchkiss RS, Opal S. 2010. Immunotherapy for sepsis--a new approach against an ancient foe. N Engl J Med 363:87–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. 2014. The new normal: Immunomodulatory agents against sepsis immune suppression. Trends Mol Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharpe AH. 2017. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol Rev 276:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. 2007. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 8:239–245. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Flies DB. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceeraz S, Nowak EC, Noelle RJ. 2013. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. NIH Public Access. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJR, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–4. [DOI] [PubMed] [Google Scholar]
- 30.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. 2008. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T, Kane LP, Ferris RL. 2015. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of t cells in the tumor microenvironment. Cancer Res 75:508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Folkl a Bienzle D. 2010. Structure and function of programmed death (PD) molecules. Vet Immunol Immunopathol 134:33–8. [DOI] [PubMed] [Google Scholar]
- 33.Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. 2010. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol 88:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. 2001. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A 98:13866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenwald RJ, Freeman GJ, Sharpe AH. 2005. The B7 family revisited. Annu Rev Immunol 23:515–48. [DOI] [PubMed] [Google Scholar]
- 36.Okazaki T, Honjo T. 2006. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 27:195–201. [DOI] [PubMed] [Google Scholar]
- 37.Chen L 2004. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol 4:336–47. [DOI] [PubMed] [Google Scholar]
- 38.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. 2001. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2:261–8. [DOI] [PubMed] [Google Scholar]
- 39.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. 2005. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25:9543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy KM, Nelson CA, Sedý JR. 2006. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol 6:671–81. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. 2003. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 4:670–9. [DOI] [PubMed] [Google Scholar]
- 42.Hurchla MA, Sedy JR, Gavrieli M, Gavrielli M, Drake CG, Murphy TL, Murphy KM. 2005. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J Immunol 174:3377–85. [DOI] [PubMed] [Google Scholar]
- 43.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. 1991. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 174:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. 1994. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1:405–13. [DOI] [PubMed] [Google Scholar]
- 45.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3:541–7. [DOI] [PubMed] [Google Scholar]
- 46.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. 1995. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270:985–8. [DOI] [PubMed] [Google Scholar]
- 47.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung C-S, Ayala A. 2009. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci 106:6303–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, Wan X, Deng X, Cai Z. 2010. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care 14:R220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, Malcus C, Chéron A, Allaouchiche B, Gueyffier F, Ayala A, Monneret G, Venet F. 2011. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care 15:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monaghan SF, Thakkar RK, Tran ML, Huang X, Cioffi WG, Ayala A, Heffernan DS. 2012. Programmed death 1 expression as a marker for immune and physiological dysfunction in the critically ill surgical patient. Shock 38:117–22. [DOI] [PubMed] [Google Scholar]
- 51.Young WA, Fallon EA, Heffernan DS, Efron PA, Cioffi WG, Ayala A. 2017. Improved survival after induction of sepsis by cecal slurry in PD-1 knockout murine neonates. Surgery 161:1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fallon EA, Chun TT, Young WA, Gray C, Ayala A, Heffernan DS. 2017. Program Cell Death Receptor-1-Mediated Invariant Natural Killer T-Cell Control of Peritoneal Macrophage Modulates Survival in Neonatal Sepsis. Front Immunol 8:1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. 2016. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol 100:1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue S, Suzuki-Utsunomiya K, Okada Y, Taira T, Iida Y, Miura N, Tsuji T, Yamagiwa T, Morita S, Chiba T, Sato T, Inokuchi S. 2013. Reduction of immunocompetent T cells followed by prolonged lymphopenia in severe sepsis in the elderly. Crit Care Med 41:810–9. [DOI] [PubMed] [Google Scholar]
- 55.Shubin NJ, Monaghan SF, Heffernan DS, Chung C-S, Ayala A. 2013. B and T lymphocyte attenuator expression on CD4+ T-cells associates with sepsis and subsequent infections in ICU patients. Crit Care 17:R276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shubin NJ, Chung CS, Heffernan DS, Irwin LR, Monaghan SF, Ayala A. 2012. BTLA expression contributes to septic morbidity and mortality by inducing innate inflammatory cell dysfunction. J Leukoc Biol 92:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. 2010. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol 184:6766–6772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. 2011. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock 36:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang KC, Burnham C-A, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, Unsinger J, Korman AJ, Green JM, Hotchkiss RS. 2013. Blockade of the negative costimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care 17:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen C-W, Mittal R, Klingensmith NJ, Burd EM, Terhorst C, Martin GS, Coopersmith CM, Ford ML. 2017. Cutting Edge: 2B4-Mediated Coinhibition of CD4(+) T Cells Underlies Mortality in Experimental Sepsis. J Immunol 199:1961–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang X, Chen Y, Chung C-S, Yuan Z, Monaghan SF, Wang F, Ayala A. 2014. Identification of B7-H1 as a Novel Mediator of the Innate Immune/Proinflammatory Response as well as a Possible Myeloid Cell Prognostic Biomarker in Sepsis. J Immunol 192:1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306:2594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakazawa A, Dotan I, Brimnes J, Allez M, Shao L, Tsushima F, Azuma M, Mayer L. 2004. The expression and function of costimulatory molecules B7H and B7-H1 on colonic epithelial cells. Gastroenterology 126:1347–1357. [DOI] [PubMed] [Google Scholar]
- 64.Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, Fukushima T, Akiba H, Yagita H, Okumura K, Machida U, Iwai H, Azuma M, Chen L, Watanabe M. 2003. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol 171:4156–63. [DOI] [PubMed] [Google Scholar]
- 65.Wu Y-Y, Lin C-W, Cheng K-S, Lin C, Wang Y-M, Lin I-T, Chou Y-H, Hsu P-N. 2010. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol 161:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Riley JL. 2009. PD-1 signaling in primary T cells. Immunol Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bandyopadhyay G, Bandyopadhyay S, Bankey PE, Miller-Graziano CL. 2014. Elevated postinjury thrombospondin 1-CD47 triggering aids differentiation of patients’ defective inflammatory CD1a+dendritic cells. J Leukoc Biol 96:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bandyopadhyay G, De A, Laudanski K, Li F, Lentz C, Bankey P, Miller-Graziano C. 2007. Negative signaling contributes to T-cell anergy in trauma patients. Crit Care Med 35:794–801. [DOI] [PubMed] [Google Scholar]
- 69.Schneider H, Mandelbrot DA, Greenwald RJ, Ng F, Lechler R, Sharpe AH, Rudd CE. 2002. Cutting edge: CTLA-4 (CD152) differentially regulates mitogen-activated protein kinases (extracellular signal-regulated kinase and c-Jun N-terminal kinase) in CD4+ T cells from receptor/ligand-deficient mice. J Immunol 169:3475–3479. [DOI] [PubMed] [Google Scholar]
- 70.Schneider H, Valk E, da Rocha Dias S, Wei B, Rudd CE. 2005. CTLA-4 up-regulation of lymphocyte function-associated antigen 1 adhesion and clustering as an alternate basis for coreceptor function. Proc Natl Acad Sci U S A 102:12861–12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider H 2006. Reversal of the TCR Stop Signal by CTLA-4. Science (80- ) 313:1972–1975. [DOI] [PubMed] [Google Scholar]
- 72.Chen G-Y, Tang J, Zheng P, Liu Y. 2009. CD24 and Siglec-10 Selectively Repress Tissue Damage-Induced Immune Responses. Science (80- ) 323:1722–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen G-Y, Chen X, King S, Cavassani KA, Cheng J, Zheng X, Cao H, Yu H, Qu J, Fang D, Wu W, Bai X-F, Liu J-Q, Woodiga SA, Chen C, Sun L, Hogaboam CM, Kunkel SL, Zheng P, Liu Y. 2011. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat Biotechnol 29:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liew PX, Kubes P. 2015. Intravital imaging - dynamic insights into natural killer T cell biology. Front Immunol 6:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thanabalasuriar A, Neupane AS, Wang J, Krummel MF, Kubes P. 2016. iNKT Cell Emigration out of the Lung Vasculature Requires Neutrophils and Monocyte-Derived Dendritic Cells in Inflammation. Cell Rep 16:3260–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Young JS, Heffernan DS, Chung C-S, Kettenmann ML, Young WA, Guillen VS, Cioffi WG, Ayala A. 2016. Effect of PD-1: PD-L1 in Invariant Natural Killer T-Cell Emigration and Chemotaxis Following Sepsis. Shock 45:534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. 2017. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res 208:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chang W-S, Kim J-Y, Kim Y-J, Kim Y-S, Lee J-M, Azuma M, Yagita H, Kang C-Y. 2008. Cutting edge: Programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol 181:6707–10. [DOI] [PubMed] [Google Scholar]
- 79.Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, Kumar V, Wu L, Kaer L Van. 2009. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol 182:2816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen S, Ndhlovu LC, Takahashi T, Takeda K, Ikarashi Y, Kikuchi T, Murata K, Pandolfi PP, Riccardi C, Ono M, Sugamura K, Ishii N. 2008. Co-inhibitory roles for glucocorticoid-induced TNF receptor in CD1d-dependent natural killer T cells. Eur J Immunol 38:2229–40. [DOI] [PubMed] [Google Scholar]
- 81.Ahmad F, Shankar EM, Yong YK, Tan HY, Ahrenstorf G, Jacobs R, Larsson M, Schmidt RE, Kamarulzaman A, Ansari AW. 2017. Negative Checkpoint Regulatory Molecule 2B4 (CD244) Upregulation Is Associated with Invariant Natural Killer T Cell Alterations and Human Immunodeficiency Virus Disease Progression. Front Immunol 8:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kearley J, Barker JE, Robinson DS, Lloyd CM. 2005. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4 + CD25 + regulatory T cells is interleukin 10 dependent. J Exp Med 202:1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McKinley L, Logar AJ, McAllister F, Zheng M, Steele C, Kolls JK. 2006. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J Immunol 177:6215–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, Bevan MJ, Urdahl KB. 2007. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med 204:2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, King LS. 2009. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest 119:2898–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aggarwal NR, D’Alessio FR, Tsushima K, Sidhaye VK, Cheadle C, Grigoryev DN, Barnes KC, King LS. 2010. Regulatory T cell-mediated resolution of lung injury: identification of potential target genes via expression profiling. Physiol Genomics 41:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun J, Han ZB, Liao W, Yang SG, Yang Z, Yu J, Meng L, Wu R, Han ZC. 2011. Intrapulmonary delivery of human umbilical cord mesenchymal stem cells attenuates acute lung injury by expanding CD4CD25+ forkhead Boxp3 (FOXP3) + regulatory T cells and balancing anti- and pro-inflammatory factors. Cell Physiol Biochem 27:587–596. [DOI] [PubMed] [Google Scholar]
- 88.Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, Riegel AK, Westrich JA, Colgan SP, Eltzschig HK. 2013. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J 27:2207–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Venet F, Chung C-S, Huang X, Lomas-Neira J, Chen Y, Ayala A. 2009. Lymphocytes in the development of lung inflammation: a role for regulatory CD4+ T cells in indirect pulmonary lung injury. J Immunol 183:3472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang L, Bai J, Chung C-S, Lomas-Neira J, Chen Y, Huang X, Ayala A. 2014. Active players in resolution of shock/sepsis induced indirect lung injury: immunomodulatory effects of Tregs and PD-1. J Leukoc Biol 96:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Heffernan DS, Monaghan SF, Thakkar RK, Tran ML, Chung C-S, Gregory SH, Cioffi WG, Ayala A. 2013. Inflammatory mechanisms in sepsis: elevated invariant natural killer T-cell numbers in mouse and their modulatory effect on macrophage function. Shock 40:122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang L, Bai J, Chung C-S, Lomas-Neira J, Chen Y, Huang X, Ayala A. 2015. Programmed cell death receptor ligand 1 modulates the regulatory T cells’ capacity to repress shock/sepsis-induced indirect acute lung injury by recruiting phosphatase SRC homology region 2 domain-containing phosphatase 1. Shock 43:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.La X, Zhang F, Li Y, Li J, Guo Y, Zhao H, Pang N, Ma X, Wen H, Fan H, Ding J. 2015. Upregulation of PD-1 on CD4+CD25+ T cells is associated with immunosuppression in liver of mice infected with Echinococcus multilocularis. Int Immunopharmacol 26:357–66. [DOI] [PubMed] [Google Scholar]
- 94.Liu Q, An L, Qi Z, Zhao Y, Li C. 2017. Increased expression of programmed cell death-1 in regulatory T-cells of patients with severe sepsis and septic shock: an observational clinical study. Scand J Immunol. [DOI] [PubMed] [Google Scholar]
- 95.Wang F, Huang X, Chung C-S, Chen Y, Hutchins NA, Ayala A. 2016. Contribution of programmed cell death receptor (PD)-1 to Kupffer cell dysfunction in murine polymicrobial sepsis. Am J Physiol Gastrointest Liver Physiol 311:G237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ayala A, Elphick GF, Kim YS, Huang X, Carreira-Rosario A, Santos SC, Shubin NJ, Chen Y, Reichner J, Chung C-S. 2014. Sepsis-induced potentiation of peritoneal macrophage migration is mitigated by programmed cell death receptor-1 gene deficiency. J Innate Immun 6:325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perl M, Chung CS, Perl U, Biffl WL, Cioffi WG, Ayala A. 2007. Beneficial Versus Detrimental Effects of Neutrophils Are Determined by the Nature of the Insult. J Am Coll Surg 204:840–852. [DOI] [PubMed] [Google Scholar]
- 98.Jimenez MF, Watson RW, Parodo J, Evans D, Foster D, Steinberg M, Rotstein OD, Marshall JC. 1997. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg 132:1263–9–70. [DOI] [PubMed] [Google Scholar]
- 99.Ayala A, Chung C-S, Lomas JL, Song GY, Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH, Grutkoski PS. 2002. Shock-induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am J Pathol 161:2283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lomas JL, Chung C-S, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. 2003. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock 19:358–365. [DOI] [PubMed] [Google Scholar]
- 101.Parsey MV, Kaneko D, Shenkar R, Abraham E 1999. Neutrophil Apoptosis in the Lung after Hemorrhage or Endotoxemia : Apoptosis and Migration Are Independent of IL-1 ▯ 1. Clin Immunol 91:219–225. [DOI] [PubMed] [Google Scholar]
- 102.Chitnis D, Dickerson C, Munster AM, Winchurch RA. 1996. Inhibition of apoptosis in polymorphonuclear neutrophils from burn patients. J Leukoc Biol 59:835–839. [DOI] [PubMed] [Google Scholar]
- 103.Fanning N, Kell M, Shorten G, Kirwan W, Bouchier-Hayes D, Cotter T, Redmond H. 1999. Circulating granulocyte macrophage colony-stimulating factor in plasma of patients with the systemic inflammatory response syndrome delays neutrophil apoptosis through inhibition of spontaneous reactive oxygen species generation. Shock 11:167–74. [DOI] [PubMed] [Google Scholar]
- 104.Ayala A, Karr SM, Evans T a, Chaudry IH. 1997. Factors responsible for peritoneal granulocyte apoptosis during sepsis. J Surg Res 69:67–75. [DOI] [PubMed] [Google Scholar]
- 105.Huang X, Chen Y, Chung C-S, Yuan Z, Monaghan SF, Wang F, Ayala A. 2014. Identification of B7-H1 as a Novel Mediator of the Innate Immune/Proinflammatory Response as well as a Possible Myeloid Cell Prognostic Biomarker in Sepsis. J Immunol 192:1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang J-F, Li J-B, Zhao Y-J, Yi W-J, Bian J-J, Wan X-J, Zhu K-M, Deng X-M. 2015. Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression: an animal study and a prospective case-control study. Anesthesiology 122:852–63. [DOI] [PubMed] [Google Scholar]
- 107.Young JS, Heffernan DS, Chung C-S, Kettenmann ML, Young WA, Guillen VS, Cioffi WG, Ayala A. 2016. Effect of PD-1. SHOCK 45:534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fink MP, Delude RL. 2005. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit Care Clin 21:177–96. [DOI] [PubMed] [Google Scholar]
- 109.Abu Faddan NH, Sherif TMK, Mohammed OA, Nasif KA, El Gezawy EM. 2017. Intestinal barrier integrity and function in infants with cholestasis. Intest Res 15:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thuijls G, Derikx JPM, Grootjans J, Buurman WA, van Waardenburg DA. 2011. Intestinal barrier loss in sepsis. Netherlands J Crit Care. [Google Scholar]
- 111.Wu Y-Y, Lin C-W, Cheng K-S, Lin C, Wang Y-M, Lin I-T, Chou Y-H, Hsu P-N. 2010. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol 161:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung C-S, Ayala A. 2009. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A 106:6303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu Y, Chung C-S, Chen Y, Monaghan SF, Patel S, Huang X, Heffernan DS, Ayala A. 2016. A Novel Role for Programmed Cell Death Receptor Ligand-1 (PD-L1) in Sepsis-Induced Intestinal Dysfunction. Mol Med 22:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scudellari M 2009. The innate debate over HSCs. Nat Reports Stem Cells 1–2. [Google Scholar]
- 115.Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. 2009. Hematopoietic cytokines can instruct lineage choice. Science 325:217–8. [DOI] [PubMed] [Google Scholar]
- 116.Sarrazin S, Mossadegh-Keller N, Fukao T, Aziz A, Mourcin F, Vanhille L, Kelly Modis L, Kastner P, Chan S, Duprez E, Otto C, Sieweke MH. 2009. MafB restricts MCSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell 138:300–13. [DOI] [PubMed] [Google Scholar]
- 117.Hutchins NA, Chung C-S, Borgerding JN, Ayala CA, Ayala A. 2013. Kupffer cells protect liver sinusoidal endothelial cells from Fas-dependent apoptosis in sepsis by down-regulating gp130. Am J Pathol 182:742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu W, Bao R, Fan X, Tao T, Zhu J, Wang J, Li J, Bo L, Deng X. 2013. PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediators Inflamm 2013:361501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Iskander KN, Craciun FL, Stepien DM, Duffy ER, Kim J, Moitra R, Vaickus LJ, Osuchowski MF, Remick DG. 2013. Cecal Ligation and Puncture-Induced Murine Sepsis Does Not Cause Lung Injury*. Crit Care Med 41:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lomas-Neira JL, Heffernan DS, Ayala A, Monaghan SF. 2016. BLOCKADE OF ENDOTHELIAL GROWTH FACTOR, ANGIOPOIETIN-2, REDUCES INDICES OF ARDS AND MORTALITY IN MICE RESULTING FROM THE DUAL-INSULTS OF HEMORRHAGIC SHOCK AND SEPSIS. Shock 45:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Monaghan SF, Thakkar RK, Heffernan DS, Huang X, Chung C-S, Lomas-Neira J, Cioffi WG, Ayala A. 2012. Mechanisms of indirect acute lung injury: a novel role for the coinhibitory receptor, programmed death-1. Ann Surg 255:158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Monaghan SF, Thakkar RK, Heffernan DS, Huang X, Chung C-S, Lomas-Neira J, Cioffi WG, Ayala A. 2012. Mechanisms of indirect acute lung injury: a novel role for the coinhibitory receptor, programmed death-1. Ann Surg 255:158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. 1996. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87:1171–1180. [DOI] [PubMed] [Google Scholar]
- 124.Lomas-Neira J, Venet F, Chung C-S, Thakkar R, Heffernan D, Ayala A. 2014. Neutrophil-endothelial interactions mediate angiopoietin-2-associated pulmonary endothelial cell dysfunction in indirect acute lung injury in mice. Am J Respir Cell Mol Biol 50:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maisonpierre PC. 1997. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in vivo Angiogenesis. Science (80- ) 277:55–60. [DOI] [PubMed] [Google Scholar]
- 126.van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VWM, Groeneveld ABJ. 2008. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 63:903–9. [DOI] [PubMed] [Google Scholar]
- 127.Orfanos SE, Kotanidou A, Glynos C, Athanasiou C, Tsigkos S, Dimopoulou I, Sotiropoulou C, Zakynthinos S, Armaganidis A, Papapetropoulos A, Roussos C. 2007. Angiopoietin-2 is increased in severe sepsis: correlation with inflammatory mediators. Crit Care Med 35:199–206. [DOI] [PubMed] [Google Scholar]
- 128.Monaghan SF, Chung C-S, Chen Y, Lomas-Neira J, Fairbrother WG, Heffernan DS, Cioffi WG, Ayala A. 2016. Soluble programmed cell death receptor-1 (sPD-1): a potential biomarker with anti-inflammatory properties in human and experimental acute respiratory distress syndrome (ARDS). J Transl Med 14:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thakkar RK, Chung C-S, Chen Y, Monaghan SF, Lomas-Neira J, Heffernan DS, Cioffi WG, Ayala A. 2011. Local tissue expression of the cell death ligand, fas ligand, plays a central role in the development of extrapulmonary acute lung injury. Shock 36:138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lomas-Neira J, Perl M, Venet F, Chung C-S, Ayala A. 2012. The role and source of tumor necrosis factor-α in hemorrhage-induced priming for septic lung injury. Shock 37:611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Del Sorbo L, Costamagna A, Muraca G, Rotondo G, Civiletti F, Vizio B, Bosco O, Martin Conte EL, Frati G, Delsedime L, Lupia E, Fanelli V, Ranieri VM. 2016. Intratracheal Administration of Small Interfering RNA Targeting Fas Reduces Lung Ischemia-Reperfusion Injury. Crit Care Med 44:e604–13. [DOI] [PubMed] [Google Scholar]
- 132.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. 2012. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, Berry S, Chartash EK, Daud A, Fling SP, Friedlander PA, Kluger HM, Kohrt HE, Lundgren L, Margolin K, Mitchell A, Olencki T, Pardoll DM, Reddy SA, Shantha EM, Sharfman WH, Sharon E, Shemanski LR, Shinohara MM, Sunshine JC, Taube JM, Thompson JA, Townson SM, Yearley JH, Topalian SL, Cheever MA. 2016. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med 374:2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, Kelly K, Mega AE, Britten CD, Ravaud A, Mita AC, Safran H, Stinchcombe TE, Srdanov M, Gelb AB, Schlichting M, Chin K, Gulley JL. 2017. Avelumab, an Anti-Programmed Death-Ligand 1 Antibody, In Patients With Refractory Metastatic Urothelial Carcinoma: Results From a Multicenter, Phase Ib Study. J Clin Oncol 35:2117–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Center for Drug Evaluation & Research UF. 2017. Approved Drugs - FDA grants nivolumab accelerated approval for MSI-H or dMMR colorectal cancer. US Food Drug Adm. Center for Drug Evaluation and Research. [Google Scholar]
- 137.Squibb Bristol-Myers. A Study of Nivolumab Safety and Pharmacokinetics in Patients With Severe Sepsis or Septic Shock. - Full Text View - ClinicalTrials.gov