

Abstract
Control of synapse number and function in the developing central nervous system is critical to the formation of neural circuits. Astrocytes play a key role in this process by releasing factors that promote the formation of excitatory synapses. Astrocyte‐secreted thrombospondins (TSPs) induce the formation of structural synapses, which however remain post‐synaptically silent, suggesting that completion of early synaptogenesis may require a two‐step mechanism. Here, we show that the humoral innate immune molecule Pentraxin 3 (PTX3) is expressed in the developing rodent brain. PTX3 plays a key role in promoting functionally‐active CNS synapses, by increasing the surface levels and synaptic clustering of AMPA glutamate receptors. This process involves tumor necrosis factor‐induced protein 6 (TSG6), remodeling of the perineuronal network, and a β1‐integrin/ERK pathway. Furthermore, PTX3 activity is regulated by TSP1, which directly interacts with the N‐terminal region of PTX3. These data unveil a fundamental role of PTX3 in promoting the first wave of synaptogenesis, and show that interplay of TSP1 and PTX3 sets the proper balance between synaptic growth and synapse function in the developing brain.
Keywords: AMPARs, astrocyte, PTX3, synapse, thrombospondin
Subject Categories: Neuroscience
Introduction
During embryonic brain development, the innervation of cortical neurons by projecting axons is followed by an intense period of synapse formation. During the time window when synaptogenesis mostly occurs, a population of non‐neuronal cells, the astrocytes, begin to populate the cortex and start secreting factors, such as the proteins thrombospondins (TSPs; Christopherson et al, 2005; Eroglu et al, 2009; Kucukdereli et al, 2011; Thatipamula & Hossain, 2014) and hevin (Kucukdereli et al, 2011), that help promoting the formation of synapses. Astrocytes also potentiate presynaptic function by enhancing release probability and quantal content (Mauch et al, 2001) and increase postsynaptic activity through the release of factors which induce glutamate receptor localization and stabilization to the postsynaptic density (Blondel et al, 2000; Allen et al, 2012) and reviewed in Chung et al (2015).
Notably, astrocytes contribute to the establishment of synapses also by the production of extracellular matrix (ECM) molecules (Chung et al, 2015). Extracellular matrix provides a highly organized extracellular environment, localized to neuronal soma and dendrites and delineating synapses on neuronal surfaces (Frischknecht & Gundelfinger, 2012). The ECM acts as a passive diffusion barrier for cell surface molecules, including neurotransmitter receptors, and thus, it contributes to the definition of plasma membrane functional domains. In the past years, strong evidence has been provided that synaptic ECM (also defined as perineuronal network, PNN) heavily contributes to the regulation of neuronal plasticity (Dityatev & Schachner, 2003). Several ECM components, including tenascin‐R (Tnr), neurocan, versican, phosphacan, brevican, Crtl1, Bral2, and HAPLN3, are expressed in a glia‐dependent manner (Okuda et al, 2014; Dzyubenko et al, 2016), whereas aggrecan expression is neuron‐dependent, and hyaluronic acid (HA) synthesis is both neuron‐ and glia‐dependent (Dityatev & Fellin, 2008). Despite this extensive characterization, it is still unknown whether astrocytes or neurons, besides producing ECM components, also control ECM remodeling through the release of soluble factors. This would have a strong impact, given the crucial role of ECM organization in processes of synaptic function and plasticity.
Pentraxins are soluble pattern recognition molecules which critically contribute to the humoral arm of innate immunity. Pentraxin 3 (PTX3) is a prototypical member of the long pentraxin family characterized by a C‐terminal pentraxin like domain and a unique N‐terminal domain (Bottazzi et al, 2010; Garlanda et al, 2016). Macrophages, neutrophils, and dendritic cells produce and release PTX3 in response to inflammatory signals (e.g., IL‐1β and TNF‐α) and Toll‐like receptor activation (Alles et al, 1994; Doni et al, 2003; Jaillon et al, 2007; Bottazzi et al, 2010). Among the physiological functions attributed to pentraxins, which include recognition and binding of microbial moieties and complement components, PTX3 also interacts with extracellular matrix components, such as the key HA‐binding component tumor necrosis factor‐induced protein‐6 (TNFIP6 or TSG6) and inter‐alpha‐trypsin inhibitor (IαI), and participates to tissue remodeling (Salustri et al, 2004; Baranova et al, 2014; Doni et al, 2015). In particular, PTX3 is essential for female fertility, acting on the assembly of the extracellular matrix in the cumulus oophorus through the formation of multimolecular complexes that can cross‐link HA chains (Salustri et al, 2004). Of note, PTX3 is expressed in brain, where it is induced by primary inflammatory signals (Polentarutti et al, 2000), epilepsy (Ravizza et al, 2001), or stroke (Zanier et al, 2011; Rodriguez‐Grande et al, 2014).
Here, we discovered that astrocyte‐derived PTX3 induces functional synapse formation. In particular, PTX3 expression is developmentally regulated in the temporal window of synapse maturation, when it promotes the increase of synaptic AMPA receptors through a pathway directly involving ECM components and integrin β1.
Results
Astrocyte‐derived PTX3 increases excitatory neurotransmission
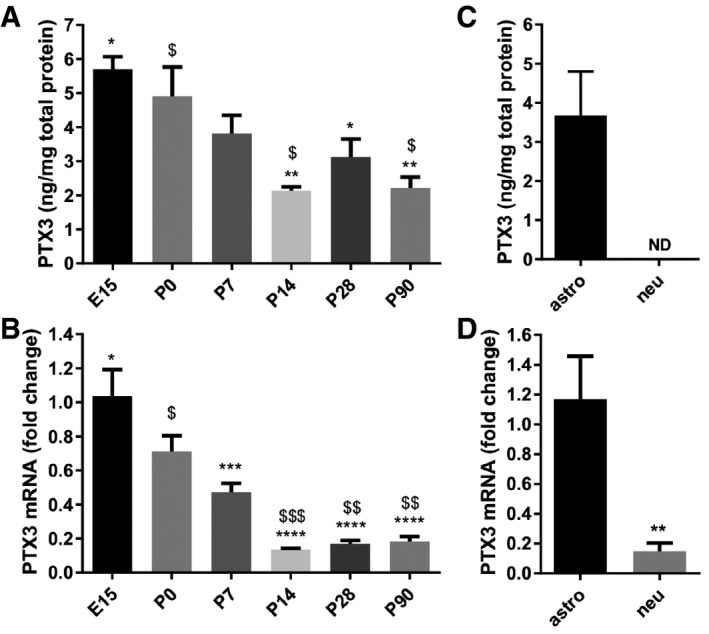
Pentraxin 3 was detected by both ELISA (Fig 1A) and qRT–PCR (Fig 1B) in the healthy mouse brain, in the absence of inflammatory stimuli. The specificity of the ELISA assay used to quantify murine PTX3 (Fig EV1A) and the amplification efficiency of the qRT–PCR for the analysis of PTX3 mRNA expression (Fig EV1B–D) have been tested. PTX3 expression is dynamically regulated during brain development, being higher in late embryonic and early postnatal brain, a time window that closely correlates with the period of synaptogenesis (Reemst et al, 2016). The presence of PTX3 in the medium of pure astrocytic, but not neuronal, cultures defines the glial origin of the molecule (Fig 1C). In addition, analysis of Ptx3 mRNA by qRT–PCR in the corresponding cellular lysates indicates that astrocytes, but not neurons, produce and release PTX3 (Fig 1D).
Figure 1. PTX3 expression is developmentally regulated and astrocyte dependent.
-
A, B(A) ELISA assay and (B) real‐time qPCR from brain lysates showing higher PTX3 expression in late embryonic and early postnatal brain, and lower in the adult brain. Data are presented as mean ± SEM. (A) ng PTX3/mg of total proteins, E15 = 5.702 ± 0.365, P0 = 4.904 ± 0.865, P7 = 3.818 ± 0.530, P14 = 2.140 ± 0.107, P28 = 3.125 ± 0.525, P90 = 2.212 ± 0.324. One‐way ANOVA followed by post hoc Tukey test: E15 vs. P14 **P = 0.001, E15 vs. P28 *P = 0.022, E15 vs. P90 **P = 0.002; P0 vs. P14 $ P = 0.013, P0 vs. P90 $ P = 0.016; four animals for each time point. (B) qPCR fold change normalized on E15, E15 = 1.037 ± 0.154, P0 = 0.711 ± 0.094, P7 = 0.472 ± 0.0524, P14 = 0.135 ± 0.007, P28 = 0.168 ± 0.020, P90 = 0.183 ± 0.029. One‐way ANOVA followed by post hoc Tukey test: E15 vs. P7 ***P = 0.0001, E15 vs. P14 ****P < 0.0001, E15 vs. P28 ****P < 0.0001, E15 vs. P90 ****P < 0.0001; P0 vs. P14 $$$ P = 0.0007, P0 vs. P28 $$ P = 0.001, P0 vs. P90 $$ P = 0.002; four animals for each time point.
-
C, DQuantitation of PTX3 levels in pure astrocyte and neuronal cultures. (C) ELISA assay performed on astrocyte and neuronal culture medium and (D) real‐time qPCR performed on astrocyte and neuronal lysates. Only astrocytes release PTX3 (ELISA, astro = 3.699 ± 1.129, neu = N.D., not detectable. Three independent astrocyte cultures evaluated. qPCR astro = 1.167 ± 0.288; neu = 0.148 ± 0.057; Mann–Whitney test **P = 0.0095. Two independent neuronal cultures evaluated. Data are presented as mean ± SEM.
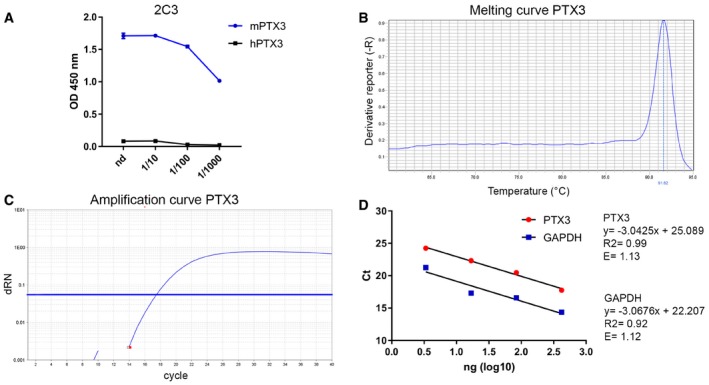
Figure EV1. Specificity of ELISA and RT–PCR assays.
-
AThe specificity of the PTX3 ELISA was tested using different dilutions of 2C3 antibody to measure immobilized murine and human PTX3. Purified recombinant murine and human PTX3 were immobilized in 96‐well ELISA plates, and then, different dilutions of 2C3 were added. The graph shows dose–response of 2C3 on immobilized murine or human PTX3. Human PTX3 was not detected by 2C3 antibody.
-
B–DEvaluation of the amplification efficiency of real‐time RT–PCR assay designed for PTX3 expression in astrocyte cell cultures. (B, C) Melting curve and amplification plot of PTX3 RT–qPCR assay. (D) Standard curves of PTX3 and GAPDH, used as reference mRNA, obtained using fivefold serial dilutions of the cDNA (420, 84, 16.8, 3.36 ng). The threshold cycle (C t) values (y‐axis) are plotted against log10 values of cDNA input amounts (x‐axis). The graphs are parallel lines and the calculated efficiencies (E) are, respectively, of 1.13 and 1.12 from a y‐slope of −3.04 and −3.07 and a correlation coefficient (R 2) > 0.9.
The developmentally regulated expression of PTX3 in a time window coinciding with synaptogenesis suggests a role for the protein in synapse formation and maturation, as already described for other synaptogenic, astrocyte‐derived molecules (Eroglu et al, 2009; Pyka et al, 2011). To investigate this possibility, recombinant PTX3 (1 μg/ml) was applied to 14DIV hippocampal neurons maintained in the presence of the anti‐mitotic agent arabinoside C (AraC), to avoid astrocyte contamination, and synapse formation was assessed 48 h later by electrophysiological recording of miniature synaptic activity and confocal microscopy. Application of PTX3 significantly increased the frequency and amplitude of glutamatergic miniature synaptic events (Fig 2A–C). This increase occurred in the absence of changes in the density of glutamatergic synapses (Fig EV2A and B), size of pre‐ and postsynaptic puncta (Fig EV2C), density of dendritic spines (Fig EV2D and E), and synaptic protein expression levels (Fig EV2F). These data indicate that PTX3 alters synaptic basal transmission without affecting the number of synapses—not even if chronically applied, i.e., from DIV 7 to DIV 14 (Fig EV2G–I). Similar to controls, PTX3‐treated neurons showed paired‐pulse facilitation in response to EPSCs elicited by two closely spaced action potentials (50 ms interval), although the EPSC2/EPSC1 ratio was slightly decreased (EPSC2/EPSC1 = 1.502± 0.051 N = 13 for Ctr and EPSC2/EPSC1 = 1.277 ± 0.065 N = 14 for PTX3. Student's t‐test, P = 0.012. Data are expressed as mean ± SEM), suggesting that a presynaptic effect is unlikely. Also, PTX3 did not affect the GABAergic inhibitory synapse number (Fig EV3A–C) nor their function (Fig EV3D–F), indicating that its action is specific for the excitatory neurotransmission.
Figure 2. PTX3 increases excitatory glutamatergic neurotransmission by promoting AMPA receptors insertion at the synapse.
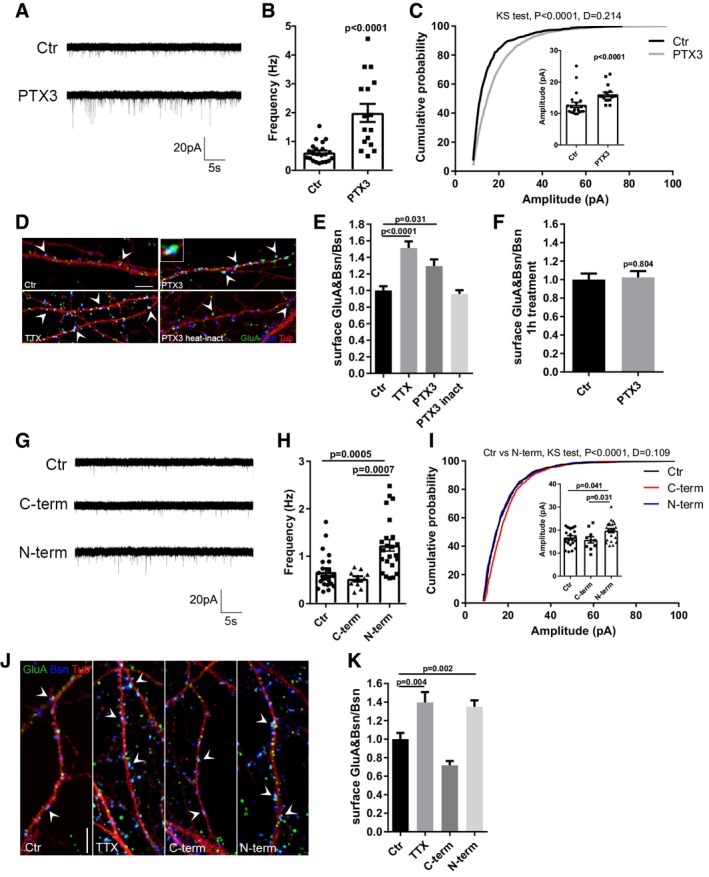
- Representative mEPSC traces recorded from control and PTX3‐treated (1 μg/ml; 48 h) neurons.
- mEPSC frequency quantitation (Hz, Ctr = 0.618 ± 0.069; PTX3 = 1.991 ± 0.313; number of neurons: Ctr = 22, PTX3 = 16; three independent experiments, Mann–Whitney test, data are presented as a distribution plus mean ± SEM).
- mEPSC amplitude quantitation and cumulative probability distribution of mEPSC amplitudes (pA, Ctr = 12.76 ± 0.813; PTX3 = 16.07 ± 0.709. Number of neurons: Ctr = 22, PTX3 = 16; three independent experiments, Mann–Whitney test. Data are presented as a distribution plus mean ± SEM). Cumulative probability distributions are analyzed by Kolmogorov–Smirnov test.
- Representative images showing 14DIV neurons stained for surface AMPAR (GluA, green), the presynaptic protein Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Inset: Example of surface synaptic AMPARs cluster (GluA&Bsn). Scale bar: 5 μm.
- Quantification of the surface synaptic AMPARs (GluA&Bsn) normalized to the total number of Bsn shows a statistically significant increase after TTX or PTX3 exposure (Ctr = 1 ± 0.051, TTX = 1.512 ± 0.080, PTX3 = 1.294 ± 0.081, PTX3 heat‐inactivated = 0.959 ± 0.044; number of fields examined: 27, 36, 37, 22, 19, respectively; one‐way ANOVA, P < 0.0001, followed by post hoc Tukey test for multiple comparison as indicated in figure; three independent experiments, data are presented as normalized mean values ± SEM).
- One hour of PTX3 administration is not sufficient to elicit an increase in synaptic GluA content (Ctr: 1 ± 0.065, PTX3: 1.023 ± 0.068; number of fields: Ctr = 26, PTX3 = 26; unpaired t‐test, three independent experiments, data are presented as normalized mean values ± SEM).
- Representative mEPSC traces recorded from control, C‐term PTX3 fragment‐treated, or N‐term PTX3 fragment‐treated cultures.
- mEPSC frequency quantitation (Hz, Ctr = 0.668 ± 0.078; C‐term = 0.523 ± 0.056; N‐term = 1.228 ± 0.121. Number of neurons: Ctr = 23, C‐term = 10, N‐term = 23; three independent experiments. Kruskal–Wallis test, P < 0.0001 followed by Dunn's test as indicated, data are presented as a distribution plus mean ± SEM).
- mEPSC amplitude quantitation (pA, Ctr = 16.81 ± 0.78; C‐term = 15.79 ± 1.37; N‐term = 19.78 ± 0.87. Number of neurons: Ctr = 23, C‐term = 10, N‐term = 23; three independent experiments. One‐way ANOVA, P = 0.013 followed by post hoc Tukey test as indicated in figure, data are presented as a distribution plus mean ± SEM) and cumulative probability distribution of mEPSC amplitudes (Kolmogorov–Smirnov test).
- Representative images of 14DIV neurons stained for surface AMPARs (GluA, green), the presynaptic protein Bassoon (blue) and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
- Quantification of the surface synaptic AMPARs normalized to the total number of Bsn shows an increase upon N‐terminal peptide application, but not upon C‐terminal peptide exposure. TTX is used as positive control (Ctr = 1.000 ± 0.066, TTX = 1.395 ± 0.113, C‐term = 0.717 ± 0.047, N‐term = 1.349 ± 0.069. Number of fields examined: 55, 31, 18, 54, respectively; one‐way ANOVA, P < 0.0001 followed by post hoc Tukey test as indicated in figure; at least three independent experiments, data are presented as normalized mean values ± SEM).
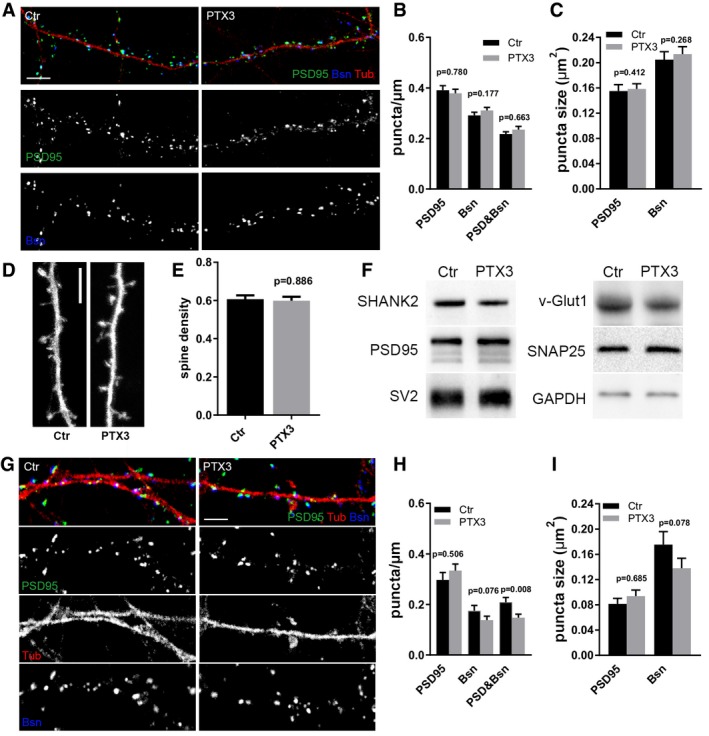
Figure EV2. PTX3 does not affect synapse number and organization.
-
ARepresentative images of 14DIV control and PTX3‐treated cultures stained for the presynaptic marker Bassoon (blue), the postsynaptic protein PSD95 (green), and the microtubule protein tubulin (red). Scale bar: 5 μm.
-
BQuantification of synaptic density shows no differences either for postsynaptic marker (PSD95/μm), or for presynaptic marker (Bsn/μm) or as a total number of synapses (PSD95&Bsn/μm) in control and PTX3‐treated cultures (PSD95/μm, Ctr = 0.391 ± 0.018; PTX3 = 0.379 ± 0.016; Bsn/μm, Ctr = 0.291 ± 0.012, PTX3 = 0.311 ± 0.012; PSD95&Bsn/μm, Ctr = 0.218 ± 0.009, PTX3 = 0.235 ± 0.013. Number of dendrites: 109 Ctr, 107 PTX3, Mann–Whitney test; five independent experiments, data are presented as mean ± SEM).
-
CQuantitative analysis of the mean size of PSD95 and Bsn puncta shows no differences in control or PTX3‐treated neurons (in μm2, PSD95: Ctr = 0.155 ± 0.010; PTX3 = 0.158 ± 0.008; Bsn: Ctr = 0.205 ± 0.013, PTX3 = 0.214 ± 0.012; Number of dendrites: 134 ctr, 124 PTX3, Mann–Whitney test; five independent experiments, data are presented as mean ± SEM).
-
D, E(D) Representative images of GFP‐expressing dendritic branches of control and PTX3‐treated neurons and (E) quantification of dendritic spine density, i.e., number of spines per μm of parent dendrite (Ctr = 0.607 ± 0.020, PTX3 = 0.598 ± 0.021, number of examined dendrites: 73 and 71 respectively; Mann–Whitney test; three independent experiments, data are presented as mean ± SEM). Scale bar: 5 μm.
-
FWestern blotting analysis of major synaptic proteins on lysates from control or PTX3‐treated cultures. GAPDH was used as reference marker.
-
G–IChronic PTX3 administration does not increase neither synaptic density nor synaptic puncta size. (G) Representative images of 14DIV control and PTX3‐treated cultures stained for the presynaptic marker Bassoon (blue), and the postsynaptic protein PSD95 (green) and tubulin (red). Scale bar: 5 μm. (H) Quantification of synaptic density parameters (PSD95/μm; Bsn/μm; PSD95&Bsn/μm) in control and PTX3‐treated neurons (PSD95/μm, Ctr = 0.297 ± 0.029; PTX3 = 0.334 ± 0.026; Bsn/μm, Ctr = 0.261 ± 0.021, PTX3 = 0.221 ± 0.020; PSD95&Bsn/μm, Ctr = 0.209 ± 0.019, PTX3 = 0.148 ± 0.014. Number of dendrites: Ctr = 20, PTX3 = 40, Mann–Whitney test; three independent experiments; data represented as mean ± SEM). (I) Analysis of the mean size of PSD95 and Bsn puncta shows no differences in control or PTX3‐treated neurons (in μm2, PSD95: Ctr = 0.081 ± 0.009; PTX3 = 0.094 ± 0.010; Bsn: Ctr = 0.175 ± 0.021, PTX3 = 0.138 ± 0.016; number of dendrites: Ctr = 22, PTX3 = 48, Mann–Whitney test; three independent experiments, data represented as mean ± SEM).
Source data are available online for this figure.
Figure EV3. PTX3 does not modify structure and function of inhibitory synapses.
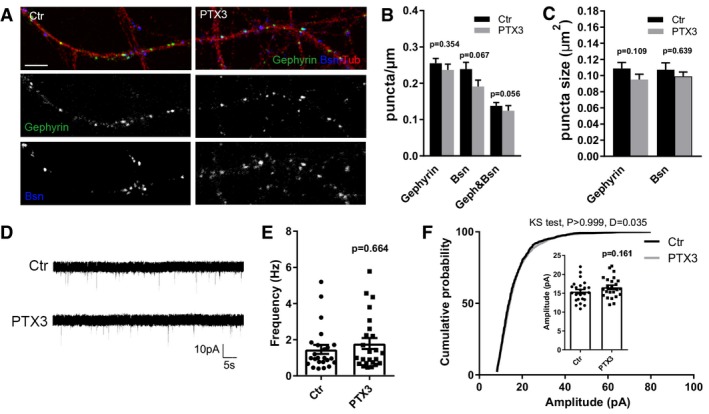
- Representative images of 14DIV control and PTX3‐treated cultures stained for the presynaptic marker bassoon (blue), and the postsynaptic inhibitory protein gephyrin (green) and the microtubule protein tubulin (red). Scale bar: 5 μm.
- Quantification of synaptic density showing no differences either for postsynaptic marker (gephyrin) or for presynaptic marker (Bsn) or as a total number of synapses (geph&Bsn) in control or PTX3‐treated neurons (gephyrin/μm, Ctr = 0.255 ± 0.014, PTX3 = 0.237 ± 0.016; Bsn/μm, Ctr = 0.239 ± 0.018, PTX3 = 0.191 ± 0.018; gephyrin&Bsn/μm, ctr = 0.138 ± 0.010, PTX3 = 0.125 ± 0.014. Number of dendrites: Ctr = 71, PTX3 = 56; Mann–Whitney test. Three independent experiments, data are presented as mean ± SEM).
- Quantitative analysis of the mean size of gephyrin and bassoon puncta showing no differences in control and PTX3‐treated neurons (in μm2, gephyrin: Ctr = 0.109 ± 0.008; PTX3 = 0.095 ± 0.006; Bsn: Ctr = 0.107 ± 0.008, PTX3 = 0.099 ± 0.005, number of dendrites: Ctr = 53, PTX3 = 41; Mann–Whitney test; three independent experiments, data are presented as mean ± SEM).
- Representative traces of mIPSCs recorded from control and PTX3‐treated neurons.
- mIPSC frequency quantitation (Hz, Ctr = 1.464 ± 0.250; PTX3 = 1.792 ± 0.306. Number of neurons: Ctr = 24, PTX3 = 24; five independent experiments. Mann–Whitney test, data are presented as a distribution plus mean ± SEM).
- Cumulative probability plot and (inset) average of mIPSC amplitude (pA, Ctr = 15.43 ± 0.572; PTX3 = 16.58 ± 0.565. Number of neurons: Ctr = 24, PTX3 = 24; five independent experiments. Unpaired t‐test, data are presented as a distribution, mean ± SEM and cumulative probability distribution of amplitudes analyzed with Kolmogorov–Smirnov test).
PTX3 promotes AMPA receptors recruitment at the synapse
The enhanced excitatory synaptic activity could result from an increased number of AMPA‐type glutamate receptors (AMPARs) inserted at the synaptic level (O'Brien et al, 1998; Chater & Goda, 2014). To investigate this possibility, surface AMPARs were labeled by live staining with an antibody specifically recognizing the extracellular domain of GluA receptors (here on called GluA) 48 h after culture exposure to PTX3. Neuronal cultures were then examined by confocal microscopy upon staining for Bassoon, to identify the presynaptic active zones, and for tubulin, to visualize the neuronal processes. Given that the number of synapses does not differ between vehicle‐treated and PTX3‐treated cultures (Fig EV2A and B), Bassoon was used as a reference marker. Quantitation of the percentage of juxtaposed Bassoon and surface GluA puncta relative to the total presynaptic sites (surface GluA&Bsn/Bsn) revealed that exposure of hippocampal neurons to PTX3 increases the amount of surface AMPARs at the synapse (Fig 2D and inset, and Fig 2E). Exposure to heat‐inactivated recombinant PTX3 (1 μg/ml) did not change the amount of GluA receptors, whereas application of TTX (1 μM, 24 h), which silences neuronal network activity and induces AMPAR insertion at the synapse (Wierenga et al, 2006; Turrigiano, 2008), increased, as expected, the amount of surface AMPAR (Fig 2D and E). Notably, no effect was observed within 1 h of stimulation with PTX3 (Fig 2F), suggesting that PTX3‐induced AMPAR insertion at the synapse may involve long‐term signaling cascades.
In order to investigate whether the PTX3‐dependent AMPAR insertion occurs specifically at synaptic sites, we quantified the total surface levels of GluA subunits. No changes in the total expression levels of GluA subunits (GluA1, GluA2/3, and GluA4) were induced by PTX3 as shown by quantitative Western blotting analysis (Fig EV4A). Also, the levels of surface GluA determined by biotinylation followed by Western blotting were not affected by PTX3 (Fig EV4B). Consistently, whole‐cell patch‐clamp quantitation of the total AMPAR current density upon agonist application revealed no differences in total AMPA current density before or after PTX3 application (Fig EV4C and D). These results indicate that PTX3 specifically regulates excitatory neurotransmission by selectively promoting postsynaptic AMPAR clustering.
Figure EV4. PTX3 does not affect the total amount of surface AMPARs.
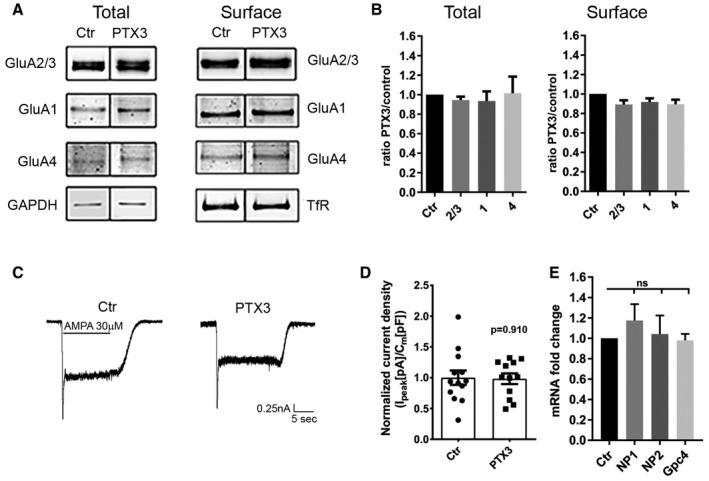
-
AWestern blotting analysis of total and surface (biotinylated) GluA subunits of control and PTX3‐treated neurons as indicated showing no differences either for the total levels of expression or for the global surface levels of the different GluA subunits.
-
BQuantification of total receptors levels was normalized to GAPDH in each sample, whereas surface GluA subunits were normalized to surface transferrin receptor, TfR, in each sample. Data are shown as ratio PTX3 over control for each GluA subunit (total: Ctr = 1 ± 0; GluA2/3 = 0.943 ± 0.036, GluA1 = 0.93 ± 0.098, GluA4 = 1.014 ± 0.17. Number of samples: 7, 6, 7, 7 respectively; Kruskal–Wallis test, P = 0.472. Surface: Ctr = 1 ± 0; GluA2/3 = 0.89 ± 0.041, GluA1 = 0.92 ± 0.038, GluA4 = 0.89 ± 0.047; number of samples: 6, 6, 6, 6, respectively. Kruskal–Wallis test, P = 0.112; 3 independent experiments, normalized values, mean ± SEM).
-
C, D(C) Representative traces and (D) quantitative analysis of total current density evoked by application of 30 μM AMPA in control and PTX3‐treated cultures (Ctr = 1 ± 0.117; PTX3 = 0.983 ± 0.87; Student's t‐test; number of neurons: Ctr = 13, PTX3 = 12; 3 independent experiments, data are presented as distribution plus normalized mean ± SEM).
-
EPTX3 does not affect neuronal pentraxins and glypican 4 expression. qRT–PCR analysis of mRNA levels for NP1, NP2, and glypican 4 (Gpc4) performed in pure neuronal cultures treated with PTX3 (1 μg/ml for 48 h). qPCR fold change normalized on control: Ctr = 1.000 ± 0.000; NP1 = 1.174 ± 0.16; NP2 = 1.041 ± 0.182; Gpc4 = 0.980 ± 0.062. Wilcoxon test: Ctr vs. NP1: P = 0.312, Ctr vs. NP2: P = 0.688, Ctr vs. Gpc4: P > 0.999. Graphs show mean ± SEM. At least 5 independent experiments.
Source data are available online for this figure.
Pentraxin 3 is a large 340‐kDa protein, predominantly assembled in a multimeric complex of 8 protomers (45 kDa) linked by inter‐chain disulfide bonds (Bottazzi et al, 1997; Inforzato et al, 2008). The C‐ and N‐terminal protein domains of PTX3 play distinct and specific roles (for a review, see Bottazzi et al, 2010). The N‐terminal domain binds FGF2, inter‐α‐inhibitor (IαI), TNF‐α‐induced protein 6 (TNFAIP6 or TSG‐6; Scarchilli et al, 2007; Leali et al, 2009), the C‐terminal pentraxin domain binds C1q and P‐selectin (Bottazzi et al, 1997; Nauta et al, 2003; Deban et al, 2010), whereas both domains have been implicated in the interaction of PTX3 with complement factor H (FH), a major soluble inhibitor of the complement system (Deban et al, 2008). To identify the region of PTX3 responsible for postsynaptic potentiation, neuronal cultures were exposed to equimolar concentrations of either C‐ or N‐terminal fragments of PTX3 (see Materials and Methods; Scarchilli et al, 2007). mEPSC frequency and amplitude were evaluated by patch‐clamp recording, while the surface synaptic GluA receptor clustering was assessed by confocal microscopy. Differently from neuronal pentraxins (NPTXs), which promote GluA clustering through their C‐terminal pentraxin domain (O'Brien et al, 1999, 2002; Xu et al, 2003; Sia et al, 2007; Lee et al, 2017), the N‐terminal but not the C‐terminal fragment of PTX3 elicited a significant increase of mEPSC frequency (Fig 2G and H) and a slight, although significant, increase in amplitude (Fig 2I), along with the enhancement of the synaptic surface GluA (Fig 2J and K). The lower efficacy of the N‐terminal PTX3 fragment, as compared to the full‐length PTX3 protein, in potentiating mEPSC frequency and amplitude (compare panel 2H with 2B, and 2I with 2C), is in line with the lower molecular stability of PTX3 fragment in the absence of the protein C‐terminal domain (Bottazzi et al, 2010). Furthermore, no significant change of NP1, NP2, and glypican 4 mRNA levels has been detected in neuronal cultures treated with PTX3 (1 μg/ml, 48 h; Fig EV4E) indicating that PTX3 does not affect NPTXs and glypican 4 expression in neurons.
PTX3‐dependent AMPAR clustering requires an intact perineural network
We next aimed at investigating the mechanisms by which PTX3, through its N‐terminal domain, regulates the clustering of GluA receptors. Indeed, the N‐terminal domain of PTX3 has been found to be fundamental for a proper organization of the HA‐rich ECM in the cumulus oophorus through the interaction with the key HA‐binding components TSG6 and IαI (Salustri et al, 2004; Scarchilli et al, 2007; Baranova et al, 2014). Brain ECM, also called perineural network (PNN), plays a crucial role in synapse development as well as in AMPAR stability and mobility (Dityatev et al, 2007, 2010). IαI and TSG‐6 are present in the CNS (Bertling et al, 2016; Chen et al, 2016), opening the possibility that the PTX3‐dependent regulation of GluA content at the synapse may involve PNN remodeling processes. To examine whether PTX3 affects PNN organization, the synaptic distribution of the HA‐ligand aggrecan, a main component of the PNN, also known as chondroitin sulfate proteoglycan 1, was analyzed. An increase of aggrecan co‐localization with PSD95 and bassoon, assessed by quantitation of both integrated density and mean intensity of aggrecan staining, was detected in neuronal cultures upon PTX3 exposure (Fig 3A–C). No changes were observed in the total area of aggrecan signal, thus suggesting that PTX3 induces a remodeling rather than a variation in the amount of PNN (Fig 3A and D).
Figure 3. An intact perineural network is necessary for GluA recruitment to the postsynaptic membrane.
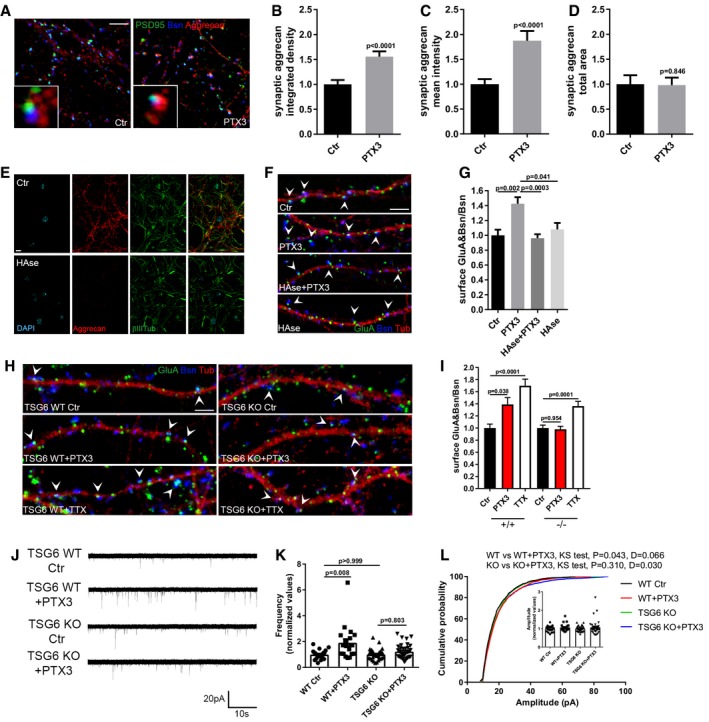
-
ALow and high magnification images of control and PTX3‐treated neurons stained for the PNN main component, aggrecan (red), the synaptic proteins PSD95 (green), and Bsn (blue). Scale bar: 5 μm.
-
B–DPTX3 application induces a remodeling of the PNN in culture, as assessed by the increase mean intensity and integrated density value of the synapse‐co‐localizing aggrecan signal, whereas no difference in the total area of aggrecan is evident (integrated density: Ctr = 1 ± 0.103, PTX3 = 1.874 ± 0.197; mean intensity: Ctr = 1.000 ± 0.088, PTX3 = 1.556 ± 0.107; total area: Ctr = 1 ± 0.179, PTX3 = 0.982 ± 0.149. Number of fields examined: 26 Ctr, 22 PTX3; Mann–Whitney test; three independent experiments, data are presented as normalized mean values ± SEM).
-
EOvernight treatment with hyaluronidase destroys PNN as shown by immunofluorescence for aggrecan (red), DAPI (cyan), and βIII tubulin (green) and confocal analysis. Scale bar: 20 μm.
-
FRepresentative images showing 14DIV neurons stained for surface AMPARs (GluA, green), the presynaptic protein Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
-
GHAse treatment blocks PTX3‐induced synaptic surface AMPA receptors clustering (Ctr = 1.000 ± 0.075, PTX3 = 1.425 ± 0.088, PTX3+HAse = 0.961 ± 0.053, HAse = 1.080 ± 0.087. Number of fields examined: 37, 23, 53, 28, respectively; Kruskal–Wallis test, P = 0.0004 followed by post hoc Tukey test as indicated in figure; three independent experiments, data are presented as normalized mean value ± SEM).
-
HRepresentative images showing 14DIV WT and TSG6 KO neurons stained for surface AMPARs (GluA, green), the presynaptic protein Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
-
ISynaptic surface GluA quantitation showing no effect of PTX3 treatment in TSG6 KO cultures. On the contrary WT cultures (from littermates) display increased surface GluA&Bsn/Bsn upon PTX3 treatment. A significant enhancement of surface GluA receptors was induced by TTX in both TSG6 KO and WT cultures (WT = 1 ± 0.06; WT+PTX3 = 1.389 ± 0.113; WT+TTX = 1.698 ± 0.109; Number of fields examined: 40, 39, 32 respectively; Kruskal–Wallis test P < 0.0001 followed by post hoc Dunn's test. TSG6 KO = 1.000 ± 0.047; TSG6 KO+PTX3 = 0.979 ± 0.048; TSG6 KO + TTX = 1.363 ± 0.077. Number of fields examined: 54, 57, 29 respectively; one‐way ANOVA, P < 0.0001 followed by post hoc Tukey test as indicated in figure. n = 3 independent experiments, data are presented as normalized mean values ± SEM).
-
JRepresentative mEPSC traces recorded from WT and TSG6 KO littermates cultures treated or not with PTX3 (1 μg/ml for 48 h).
-
KOn the contrary of WT cultures, mEPSC frequency quantitation shows no increase in frequency in TSG6 KO cultures when treated with PTX3 (normalized frequency, WT Ctr: 1.000 ± 0.074; Wt + PTX3 = 1.912 ± 0.289; TSG6 KO Ctr = 1 ± 0.079; TSG6 KO + PTX3 = 1.226 ± 0.096; 22, 20, 30, 24 cells, respectively, 6 TSG6 KO mice and 4 WT littermates. One‐way ANOVA test P = 0.002 followed by post hoc Dunn's test, data are presented as normalized mean ± SEM plus the distribution).
-
MCumulative probability plot of mEPSC amplitude and average mEPSC amplitude quantitation (inset) showing no difference in amplitude (WT Ctr: 1.000 ± 0.034, Wt + PTX3 = 1.069 ± 0.046, TSG6 KO Ctr = 1.000 ± 0.028; TSG6 KO + PTX3 = 1.074 ± 0.67; 22, 20, 30, 24 cells, respectively, 6 TSG6 KO mice and 4 WT littermates. One‐way ANOVA test P = 0.817 followed by post hoc Dunn's test, data are presented as normalized mean ± SEM plus the distribution. Cumulative distribution is analyzed by Kolmogorov–Smirnov test).
To investigate whether the synaptic GluA clustering induced by PTX3 resulted from a modulation of PNN architecture, neuronal cultures were exposed overnight (i.e., 14–16 h) to hyaluronidase (HAse, 8 U/ml), which destroys the extracellular matrix, as demonstrated by the loss of aggrecan signal (Fig 3E). Notably, HAse prevented the PTX3‐induced increase of GluA receptors at the synapse (Fig 3F and G). These data demonstrate that an intact PNN is necessary for PTX3‐mediated regulation of GluA content at the synapse.
To directly demonstrate that PTX3 action occurred through PNN remodeling, neurons genetically lacking the key HA‐binding component and PTX3 interactor, TSG6 (TSG6 KO) (Fulop et al, 2003; Salustri et al, 2004), and their WT littermates controls were exposed to PTX3 (1 μg/ml for 48 h) and subsequently analyzed by confocal microscopy and patch‐clamp electrophysiology. PTX3 application to TSG6 KO neurons failed to increase surface synaptic AMPARs, while being effective in WT cultures (Fig 3H and I). Of note, TSG6 KO cultured neurons exposed to 1 μM TTX for 24 h displayed increased synaptic GluA content (Fig 3H and I), thus excluding that lack of TSG6 induces per se a more general impairment of AMPAR trafficking. Finally, consistently with the lack of surface AMPAR increment, electrophysiological recordings of miniature excitatory activity revealed a PTX3‐dependent increase of mEPSC frequency and amplitude in WT but not TSG6 KO littermates cultures (1 μg/ml PTX3 for 48 h; Fig 3J–L). Altogether, these data further demonstrate that PTX3 regulates synaptic AMPARs through the remodeling of ECM surrounding excitatory synapses and identify TSG6 as a key molecular factor involved in this process.
β1‐Integrin and ERK1/2 mediate PTX3‐induced recruitment of AMPA receptors at the synapse
To define the molecular mechanisms through which the PTX3‐induced remodeling of PNN results in the recruitment of AMPARs at the synapse, we investigated the possible involvement of integrins, heterodimeric transmembrane receptors for ECM proteins, which play a crucial role in regulating synaptic transmission and plasticity. Some integrin subunits, such as β3, are enriched at synapses (Pinkstaff et al, 1999; Chavis & Westbrook, 2001; Chan et al, 2003; Shi & Ethell, 2006) and have been involved in GluA2‐containing AMPAR trafficking (Cingolani et al, 2008; Pozo et al, 2012). Of note, incubation of neurons with a specific anti‐integrin β1‐monoclonal antibody (αCD29) completely prevented the PTX3‐dependent increase of synaptic GluA content (Fig 4A and B), indicating that β1‐containing integrins are involved in the process of AMPARs recruitment induced by PTX3. To define whether integrins control AMPAR trafficking either directly or through downstream signaling pathways (Pozo et al, 2012; Park & Goda, 2016), we focused on the possible involvement of ERK1/2. In line with an increased levels of phospho‐ERK1/2 in neurons upon PTX3 application (Fig 4C), blockade of ERK1/2 phosphorylation through the specific MEK1 inhibitor, PD98059, completely prevented the PTX3‐induced increase of mEPSC frequency and amplitude (Fig 4D–F) as well as the synaptic AMPARs recruitment (Fig 4G and H). These experiments indicate that β1 integrin and ERK1/2 pathways are both involved in the PTX3‐mediated GluA clustering; however, they do not help to define whether they are linked or act as independent pathways in this process. To investigate this issue, we evaluated the effect of the concomitant blockade of β1‐integrin and ERK1/2 activation on the postsynaptic receptor clustering induced by PTX3 application. The results indicated that the simultaneous inhibition of β1 and ERK1/2 signaling pathway by co‐incubation with αCD29 and PD does not induce additive effects with respect to single αCD29 or PD applications (Fig 4I). Indeed, the inhibition of each of the two pathways individually produced an almost complete blockade of the PTX3‐induced effect. These data demonstrate that PTX3 induces postsynaptic AMPAR recruitment by promoting PNN remodeling through the involvement of β1‐containing integrin‐ERK1/2 signaling pathways.
Figure 4. PTX3 exerts its effects on AMPA receptors through β1‐integrin and MAPK activation.
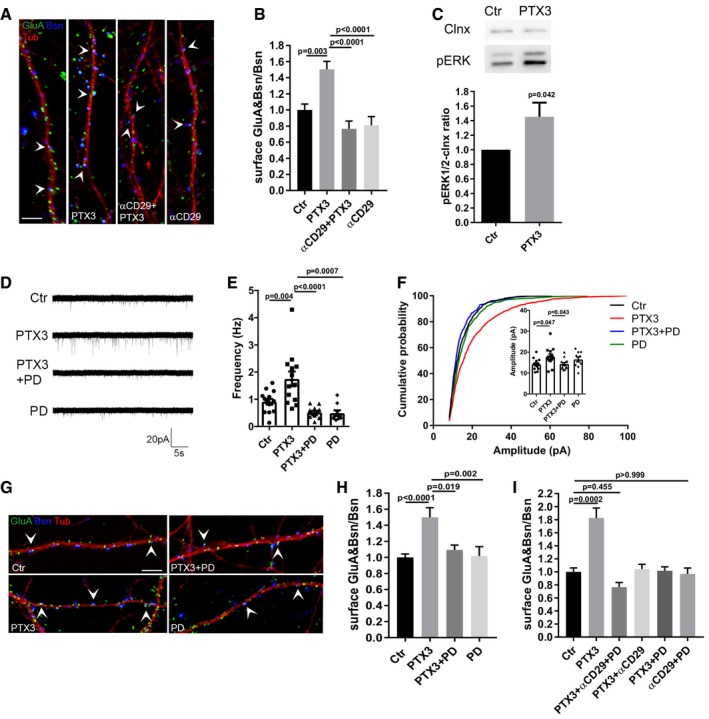
- Representative images showing 14DIV neurons stained for surface AMPARs (GluA, green), the presynaptic protein Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
- Blocking β1‐integrins activity by using the specific anti‐β1 integrin monoclonal antibody prevents the PTX3‐induced postsynaptic AMPAR recruitment (Ctr = 1.000 ± 0.074, PTX3 = 1.504 ± 0.098, αCD29+PTX3 = 0.766 ± 0.097, αCD29 = 0.810 ± 0.108. Number of fields examined: 37, 33, 23, 24 respectively; one‐way ANOVA, P < 0.0001 followed by post hoc Tukey test; three independent experiments, data are presented as normalized mean values ± SEM).
- Western blotting analysis of p‐ERK levels on lysates from control and PTX3‐treated neurons upon 30‐min stimulation (Ctr = 1 ± 0; PTX3 = 1.453 ± 0.195, six independent experiments, unpaired t‐test, data are presented normalized on control and as mean ± SEM).
- Representative mEPSC traces recorded from the indicated experimental conditions.
- mEPSC frequency quantitation showing that pre‐incubation with PD98059 (30 μM) completely prevents the PTX3‐dependent increase of mEPSC frequency (Hz, Ctr = 0.911 ± 0.104; PTX3 = 1.748 ± 0.273; PTX3 + PD = 0.518 ± 0.055; PD = 0.491 ± 0.104. Number of neurons: Ctr = 14, PTX3 = 14, PTX3 + PD = 13; PD = 9; three independent experiments. One‐way ANOVA, P < 0.0001 followed by post hoc Tukey test as indicated in figure. Data are presented as a distribution plus mean ± SEM).
- Inset: average mEPSC amplitude quantitation (pA), Ctr = 14.19 ± 0.70; PTX3 = 19.09 ± 1.71; PTX3+PD = 14.31 ± 0.70, PD = 16.51 ± 1.17. Number of neurons: Ctr = 14, PTX3 = 14, PTX3+PD = 13; PD = 9; three independent experiments. Kruskal–Wallis test, P = 0.015 followed by Dunn's test as indicated in figure, data are presented as a distribution plus mean ± SEM. Cumulative probability plot of mEPSC amplitudes is analyzed with Kolmogorov–Smirnov test: Ctr vs. PTX3: P < 0.0001, D = 0.183, PTX3 vs. PTX3 + PD: P < 0.0001, D = 0.251, PTX3 vs. PD: P < 0.0001, D = 0.157.
- Representative images showing 14DIV neurons stained for surface AMPARs (GluA, green), the presynaptic protein Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
- Quantification of the surface synaptic AMPARs normalized to the total number of Bsn [(GluA&Bsn)/Bsn] shows a statistically significant increase after PTX3 exposure, while pre‐incubation with PD98059 prevents the PTX3‐dependent increase (Ctr = 1 ± 0.042, PTX3 = 1.499 ± 0.121, PTX3 + PD = 1.093 ± 0.060, PD = 1.017 ± 0.117; number of fields examined: 49, 15, 15, 20, respectively; one‐way ANOVA, P = 0.0002 followed by post hoc Tukey test as indicated in figure; at least three independent experiments, data are presented as normalized mean values ± SEM).
- Quantification of surface synaptic GluA upon PTX3 application and simultaneous inhibition of β1‐integrin and ERK1/2 signaling pathways reveals no additive effects with respect to single αCD29 or PD applications (Ctr = 1 ± 0.061; PTX3 = 1.828 ± 0.151; αCD29 + PD + PTX3 = 0.766 ± 0.070; PTX3 + αCD29 = 1.043 ± 0.073; PTX3 + PD = 1.016 ± 0.061; PD = 0.969 ± 0.090; number of fields examined: 29, 31, 30, 23, 14, 33, respectively; one‐way ANOVA, P < 0.0001 followed by post hoc Dunn's test as indicated in figure; at least three independent experiments, data are presented as normalized mean values ± SEM).
Source data are available online for this figure.
PTX3 activity is inhibited by thrombospondin‐1 through direct interaction
Given the temporal coincidence of the expression patterns of PTX3 (our study) and TSPs (Christopherson et al, 2005; Eroglu et al, 2009) during brain development, we aimed at investigating the possible interaction between PTX3 and TSPs in controlling synaptogenesis and inducing the functional switch of postsynaptically silent excitatory synapses. TSP1 and PTX3 are modular proteins that act by establishing molecular and functional interactions with a variety of different ligands. TSPs in particular are classified into two main groups: group A, including TSP1 and TSP2 (see Fig 5C for a schematic representation of the multi‐modular organization of TSP1), and group B, including TSP3, 4, and 5. The major differences between the two groups are the presence of von Willebrand type C domain (vWC) and the type I thrombospondin repeats in the group A members and of four rather than three EGF‐like repeats in group B members (Carlson et al, 2008). We found that PTX3 directly interacts with TSP1 and TSP2, but not TSP4 (Fig 5A), mainly through the PTX3 N‐terminal domain (Fig 5B). To map the region of TSP1 recognized by PTX3, proteolytic fragments and recombinant proteins comprising different thrombospondin modules were tested. A scheme is reported in Fig 5C.
Figure 5. PTX3 activity is inhibited by thrombospondin‐1 through direct interaction.
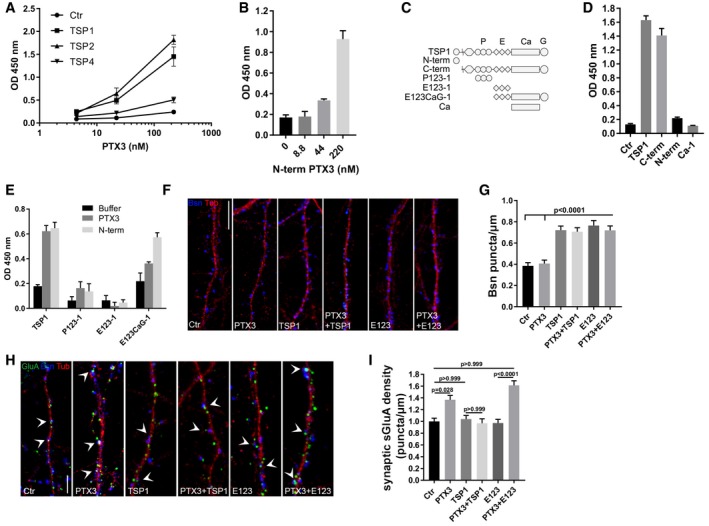
- PTX3 binds TSP1 and TSP2 but not TSP4. Different amounts of human recombinant PTX3 were incubated in microplate wells coated with purified human TSP1 or recombinant TSP2 and TSP4. Binding is reported as absorbance at 450 nm (mean ± SD). Data are from one experiment out of three performed.
- N‐terminal PTX3 binds TSP1. Binding of PTX3 N‐term domain was performed on TSP1 immobilized on plastic wells. Data are reported as absorbance at 450 nm (mean ± SD) and are representative of one out of two experiments performed.
- Schematic representation of TSP1 monomer and the TSP1 proteolytic fragments (N‐term, C‐term) and recombinant domains (P123‐1, E123‐1, E123CaG‐1, Ca‐1) used in the study.
- PTX3 binds the C‐term proteolytic fragment of TSP1. TSP1 and its fragments C‐term, N‐term, and Ca‐1 (type III repeats) were immobilized in plastic wells (5 μg/ml) and binding of PTX3 is reported as absorbance at 450 nm (mean ± SD). Data refers to one out of two experiments performed with similar results.
- PTX3 and its N‐terminal domain bind TSP1 C‐terminal globular domain. 50 nM of P123‐1 (type I “properdin” repeats), E123‐1 (type II EGF repeats), E123CaG‐1 (type II repeats plus type III repeats and globular C‐terminus), and TSP1 were immobilized in plastic well. Binding with PTX3 or N‐terminal domain (both at 220 nM) was analyzed. Data are reported as absorbance at 450 nm (mean ± SD) and refer to one out of two experiments performed with similar results.
- Representative images of 14DIV control and PTX3‐treated cultures stained for the presynaptic marker bassoon (blue) and the microtubule protein tubulin (red). Scale bar: 10 μm.
- Quantification of synaptic density (Bsn/μm) in the different experimental conditions (Ctr = 0.385 ± 0.029, PTX3 = 0.407 ± 0.032, TSP1 = 0.721 ± 0.039, PTX3+TSP1 = 0.707 ± 0.037, E123 = 0.764 ± 0.047, PTX3+E123 = 0.719 ± 0.041. Number of fields examined: 86, 80, 75, 72, 46, 55 respectively; Kruskal–Wallis test, P < 0.0001 followed by Dunn's test as indicated in figure; at least three independent experiments, data are presented as mean values ± SEM).
- Representative images showing 14DIV neurons stained for surface AMPAR (GluA, green), the presynaptic protein Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
- Quantification of the synaptic surface GluA density (number of sGluA puncta/μm) in the different tested conditions showing that GluA density increases in neuronal cultures treated with PTX3 or E123 + PTX3 but not in cultures treated with the full‐length TSP1 + PTX3 (Ctr = 1.000 ± 0.055; PTX3 = 1.367 ± 0.074; TSP1 = 1.04 ± 0.061; PTX3 + TSP1 = 0.968 ± 0.076; E123 = 0.971 ± 0.063; PTX3 + E123 = 1.613 ± 0.077. Number of dendrites examined: 103, 80, 88, 57, 51, 62, respectively; Kruskal–Wallis test, P < 0.0001 followed by Dunn's test as indicated in figure; at least three independent experiments, data are presented as normalized mean values ± SEM).
We found that PTX3 did not interact with constructs comprising the isolated N‐terminal heparin‐binding domain, type I repeats (P123), EGF‐like type II repeats (E123 domain; Fig 5E), or type III repeats Ca‐1 (Fig 5D). Notably, E123 binds the gabapentin receptor, α2δ‐1, also known as the neuronal thrombospondin receptor, and is responsible for the synaptogenic activity of the protein (Eroglu et al, 2009). Although PTX3 did not interact with E123, both intact PTX3 and the PTX3 N‐terminal domain bound a larger TSP1 construct containing E123, the type III repeats, and C‐terminal globular domain (E123CaG‐1, Fig 5D and E). The specific interaction between PTX3 and E123CaG‐1 but not P123‐1 and E123‐1 was also observed when PTX3 was immobilized in plastic wells and biotin‐labeled TSP1 constructs were added (64% fold increase of OD 450 nm for E123CaG‐1 over control buffer: no increase compared to control buffer for the other molecules). Overall, these results indicate that elements within the intricate structure formed by the TSP1 EGF and type III repeats and C‐terminal globular domain (Carlson et al, 2008) interact with the N‐terminal domain of PTX3. Although E123‐1 does not directly bind PTX3, additional studies are needed to investigate a potential indirect involvement of the EGF modules in PTX3 recognition by TSP1.
To investigate the functional consequences of PTX3 and TSP1 interaction, neurons were exposed to (i) full‐length PTX3 (1 μg/ml corresponding to 0.0238 μM, 48 h) and TSP1 [5 μg/ml corresponding to 0.0384 μM, 48 h; as described in Christopherson et al (2005)], either alone or in combination; (ii) the synaptogenic domain of TSP1, E123, which does not interact with PTX3 (Fig 5E), either alone or together with the full‐length PTX3. The synapse number and the synaptic content of GluA were then evaluated by confocal analysis. The results showed that TSP1, or its E123 domain, are able to promote synapse formation when applied either alone or in combination with PTX3 (Fig 5F and G). Since TSP1 or E123 induces an increase in synapse number and therefore Bsn puncta, the analysis of synaptic GluA content was performed—in this specific case—by evaluating the density of synaptic surface GluA puncta, instead than measuring the (GluA&Bsn/Bsn) value. The results (Fig 5H and I) showed that (i) full‐length TSP1 or E123 domain of TSP1 did not affect per se the density of GluA puncta; (ii) co‐incubation of PTX3 with full‐length TSP1 did not elicit any increase of density of GluA puncta; (iii) whereas co‐incubation of PTX3 with E123 domain of TSP1 resulted in significant enhancement of the density of GluA puncta. These data indicate that the ability of PTX3 to increase GluA puncta density is inhibited by co‐incubation with full‐length TSP1, which is able to interact with PTX3 (Fig 5E), but not with the E123 domain of TSP1, which is not able to bind PTX3 (Fig 5E). The inhibitory effect of TSP1 on PTX3‐mediated synaptic GluA increase was confirmed by the analysis of mEPSC frequency and amplitude showing lack of effect of PTX3 upon co‐incubation with TSP1 (Fig EV5). Conversely, E123 per se slightly increased mEPSC frequency and amplitude (Fig EV5), possibly because of the E123 ability to bind and activate the α2δ1 subunit of voltage‐gated calcium channels on presynaptic compartment (Field et al, 2006; Eroglu et al, 2009) and to stimulate presynaptic release (Hoppa et al, 2012). Altogether, these data demonstrate that TSP1 acts as a negative regulator of PTX3 activity.
Figure EV5. Full‐length TSP1 exerts a negative regulation of PTX3 activity.
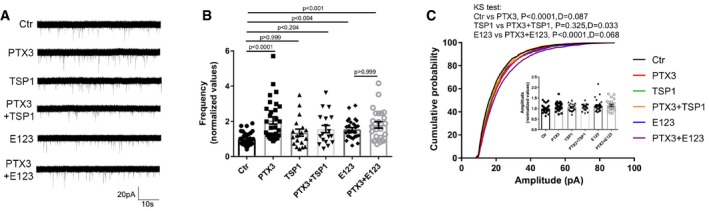
- Examples of mEPSC recordings in the indicated conditions.
- mEPSC frequency quantification showing that co‐incubation of PTX3 and full‐length TSP1 did not result in increased miniature frequency. Of note application of E123 alone induced per se a statistically significant increase of mEPSC frequency, whereas no difference was detectable upon co‐incubation of E123 fragment +PTX3 with respect to E123 alone (normalized average frequency: Ctr = 1.000 ± 0.065; PTX3 = 2.055 ± 0.209; TSP1 = 1.358 ± 0.202; PTX3+TSP1 = 1.562 ± 0.212; E123 = 1.548 ± 0.100; PTX3+E123 = 1.803 ± 0.183. Number of recorded cells: 33, 30, 18, 18, 29, 24, respectively, from at least 3 different culture preparations. Kruskal–Wallis test P < 0.0001 followed by post hoc Dunn's test. Data are presented normalized mean ± SEM along with distribution of values).
- Cumulative probability plot of mEPSCs amplitudes showing that there is a significant shift in the distribution by the Kolmogorov–Smirnov test in Ctr vs. PTX3 and E123 vs. E123 + PTX3 but not in TSP1 vs. TSP1 + PTX3 further supporting the evidence that full‐length TSP1, but not E123 fragment, inhibits PTX3 activity. Inset: No statistically significant difference was instead detectable in the average amplitude (normalized average amplitude: Ctr = 1.000 ± 0.033; PTX3 = 1.088 ± 0.036; TSP1 = 1.06 ± 0.043; PTX3+TSP1 = 1.053 ± 0.038; E123 = 1.137 ± 0.051; PTX3 + E123 = 1.152 ± 0.052. Number of recorded cells: 33, 30, 18, 18, 29, 24, respectively, from at least three different culture preparations. Kruskal–Wallis test P = 0.172 followed by post hoc Dunn's test. Data are presented normalized mean ± SEM along with the distribution of values.).
Lack of endogenous PTX3 results in weaker excitatory synapses both in vitro and in vivo
The role of astrocytes and astrocyte‐derived factors in regulating synapse development and function is well established in literature (Liu et al, 1996; Pfrieger & Barres, 1997; Li et al, 1999; Ullian et al, 2001). ELISA quantitation of TSP1 and PTX3 levels in the supernatant of astrocyte cultures showed that TSP1 and PTX3 are secreted by astrocytes under basal conditions (0.6 ± 0.1 ng/ml and 2.0 ± 0.3 ng/ml, respectively, data are expressed as mean ± SEM; n = 3), with a PTX3 molar concentration exceeding that of TSP1 (48 pM PTX3 versus 5 pM TSP1). Also, in vivo—in P7 brain—the concentration of PTX3 (3.8 ± 0.5 ng/mg corresponding to 84 pMol, Fig 1A) exceeded that of TSP1 (0.77 ± 0.05 ng/mg corresponding to 6 pmol. n = 7 WT mice, mean ± SEM). Of note, the inhibitory effect of TSP1 to PTX3 was evident in neuronal cultures (Fig 5H–I) when TSP1 exceeded PTX3 (0.0384 mM and 0.0238 mM, respectively) suggesting that, under basal conditions, the amount of physiologically released PTX3 is sufficient to promote synaptic strength, even in the presence of TSP1.
To directly prove this possibility, synapse formation and function were examined in different experimental settings in which endogenous PTX3 activity was inhibited. First, endogenous PTX3 was functionally inhibited in the medium of WT neuron–astrocyte co‐cultures by using the N‐terminal‐specific monoclonal antibody (MNB4), previously characterized as a function‐blocking antibody (Scarchilli et al, 2007; Doni et al, 2015). MNB4‐treated cultures displayed a significant reduction in postsynaptic GluA clustering (Fig 6A and B). No effect was produced by the isotypic antibody. Second, a significantly lower amount of postsynaptic GluA content was detected in astrocyte–neuron co‐cultures established from PTX3 knockout mice (PTX3 KO) with respect to age‐matched WT cultures (Fig 6C and D), which occurred in the absence of changes in the density of excitatory glutamatergic synapses (number of PSD95/μm, WT = 0.416 ± 0.043; PTX3 KO = 0.492 ± 0.025; Bsn/μm, WT = 0.355 ± 0.037, PTX3 KO = 0.412 ± 0.031; number of PSD95&Bsn/μm, WT = 0.259 ± 0.028, PTX3 KO = 0.321 ± 0.021. Number of dendrites: 20 WT, 57 PTX3 KO from three independent experiments. Unpaired t‐test P = 0.123 P = 0.311; Mann–Whitney test P = 0.172, respectively; data are presented as mean ± SEM). Of note, the synaptic deficits of PTX3‐deficient neurons were recovered upon application of PTX3 N‐terminal domain, as shown by the increase in mEPSC frequency and amplitude (Fig 6E–G) and by the enhancement of synaptic surface GluA content (Fig 6H and I). Finally, we recorded mEPSCs from CA1 pyramidal neurons in acute hippocampal slices from PTX3 KO and WT littermates at P9, at the peak of endogenous PTX3 expression (Fig 1A) and the period of functional synapse maturation. Results showed that P9 PTX3 KO mice display weaker excitatory synapses in vivo, as shown by the lower mEPSC frequency (Fig 6J and K) and amplitude (Fig 6L and M). Of note, a slight increase of TSP1 expression, rather than a decrease, was detected in P9 PTX3 KO mice with respect to WT ruling out the possibility that decreased mEPSC activity in PTX3 KO slices may be due to lower TSP1 content (qPCR for TSP1: WT = 1.028 ± 0.09276; KO = 1.347 ± 0.09504; Mann–Whitney test, P = 0.040, n = 7 mice, mean ± SEM). Furthermore, lower mEPSC frequency and amplitude in PTX3 KO slices are detectable also at P30, when more mature synapses are present (Fig 6N–Q). These data indicate that lack of endogenous PTX3 results in defective synapse functioning, which apparently cannot be rescued by other astrocyte‐derived factors expressed at later developmental stages (Kucukdereli et al, 2011; Allen et al, 2012; Farhy‐Tselnicker et al, 2017). Collectively, these data demonstrate that either the functional inactivation or the genetic lack of PTX3 results in reduced postsynaptic GluA content and indicate that the endogenous activity of PTX3 is critical for synaptic formation in vivo and that it is not blocked by TSP1. Our finding further indicates that astrocyte‐derived PTX3 is crucial to promote the first wave of synaptogenesis in vivo and that the interplay of TSP1 and PTX3 sets the proper balance between synaptic growth and synapse activity during physiological early brain development.
Figure 6. Mice deficient in PTX3 have weaker excitatory synapses in vitro and in vivo .
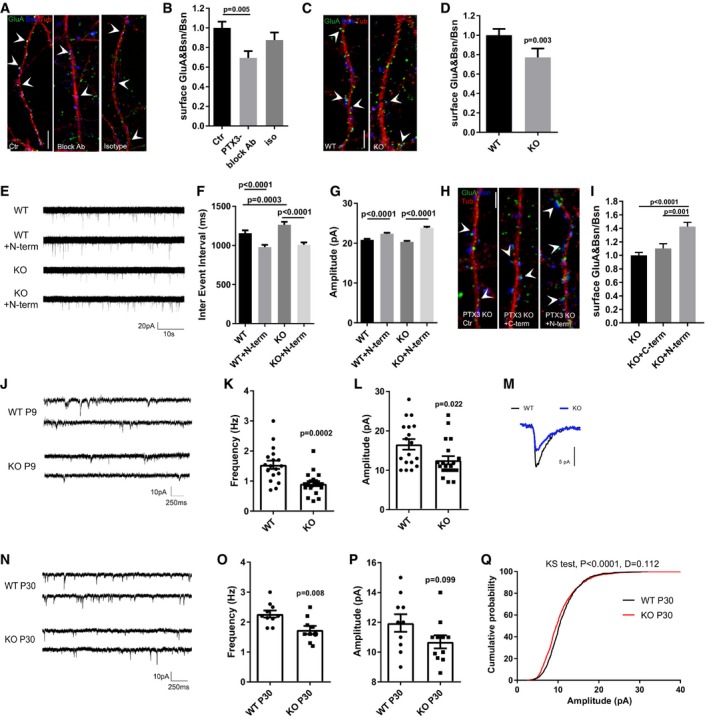
-
ARepresentative images of 14DIV neurons stained for surface AMPARs (GluA, green), Bassoon (blue), and tubulin (red) in the different tested conditions. Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
-
BQuantification of the surface synaptic AMPARs normalized to the total number of Bsn shows a decrease of the surface synaptic AMPAR clusters upon chronic application of the PTX3 blocking antibody to mixed cultures (Ctr = 1.000 ± 0.063, PTX3 block = 0.694 ± 0.069, isotype Ab = 0.876 ± 0.077. Number of fields examined: 34, 23, 19, respectively; one‐way ANOVA, P < 0.0001 followed by post hoc Tukey test as indicated in figure; three independent experiments, data are presented as normalized mean values ± SEM).
-
CRepresentative images of 14DIV WT and PTX3 KO neurons stained for surface AMPARs (GluA, green), Bassoon (blue), and tubulin (red). Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
-
DQuantification of the surface synaptic AMPARs normalized to the total number of Bsn shows a reduction in PTX3 KO cultures with respect to WT (WT = 1 ± 0.065, KO = 0.771 ± 0.0.92; number of fields examined: 36 and 42, respectively; Mann–Whitney test; number of animals 5 WT and 6 PTX3 KO, data are presented as normalized mean values ± SEM).
-
EExamples of mEPSCs recorded in the indicated experimental conditions.
-
FQuantitation of mEPSC inter‐event interval showing a rescue of mEPSC frequency in PTX3 KO cultures treated with the N‐terminal fragment of PTX3 (WT = 1158 ± 34.67; WT+N‐term = 979.1 ± 28.27; PTX3 KO = 1263 ± 38.99; PTX3 KO+N‐term = 1008 ± 30.14. Number of neurons: WT, Ctr = 25, WT+N‐term = 29; PTX3 KO, Ctr = 24; PTX3 KO+N‐term = 22; three independent experiments. Kruskal–Wallis test, P < 0.0001 followed by Dunn's test as indicated in figure, data are presented as mean ± SEM).
-
GQuantitation of mEPSC amplitude showing a rescue in PTX3 KO cultures treated with the N‐terminal fragment of PTX3 (pA, WT = 20.85 ± 0.249 WT+N‐term = 22.37 ± 0.234; PTX3 KO = 20.31 ± 0.264; PTX3 KO+N‐term = 23.8 ± 0.316. Number of neurons: WT, Ctr = 25, WT+N‐term = 29; PTX3 KO, Ctr = 24; PTX3 KO+N‐term = 22; 3 independent experiments. Kruskal–Wallis test, P < 0.0001 followed by Dunn's test as indicated in figure, data are presented as mean ± SEM).
-
HRepresentative images of 14DIV PTX3 KO neurons (Ctr, +N‐terminal PTX3, +C‐terminal PTX3) stained for surface AMPARs (GluA, green), Bassoon (blue), and tubulin (red). Arrowheads point to postsynaptic GluA clusters. Scale bar: 5 μm.
-
IQuantification of the surface synaptic AMPARs normalized to the total number of Bsn shows an increase in PTX3 KO treated with N‐terminal fragment of PTX3 but not with the C‐terminal fragment (PTX3 KO = 1.000 ± 0.042, PTX3 KO + C‐term = 1.103 ± 0.070, PTX3 KO + N‐term = 1.428 ± 0.061. Number of fields examined: 63, 38, 57 respectively; Kruskal–Wallis test, P < 0.0001 followed by Dunn's test; 3 independent experiments, data are presented as normalized mean values ± SEM).
-
J–L(J) Examples of mEPSCs recordings in WT and PTX3 KO (littermates) hippocampal slices at P8‐9 showing that frequency (K) and amplitude (L) are significantly decreased in KO neurons with respect to WT (Hz: WT = 1.54 ± 0.136; PTX3 KO = 0.912 ± 0.083; Mann–Whitney test. pA: WT = 16.57 ± 1.334; PTX3 KO = 12.55 ± 1.037; unpaired t‐test. Data are presented as a distribution plus mean ± SEM. WT: 18 cells, 4 mice; PTX3 KO: 20 cells, 5 mice).
-
MRepresentative superimposed average traces aligned by rise time.
-
NExamples of mEPSCs recordings of P30 WT and PTX3 KO hippocampal slices.
-
OQuantification of mEPSC frequency showing reduced frequency in PTX3 KO mice with respect to WT (Hz: WT = 2.27 ± 0.115; PTX3 KO = 1.74 ± 0.136. WT: 10 cells, 4 mice; PTX3 KO: 11 cells, 5 mice. Unpaired t‐test P = 0.008. Data are presented as a distribution plus mean ± SEM).
-
P, Q(P) No difference in the average amplitude is evident (pA: WT = 11.95 ± 0.589; PTX3 KO = 10.69 ± 0.438; unpaired t‐test P = 0.099. Data are presented as a distribution plus mean ± SEM. WT: 10 cells, 4 mice; PTX3 KO: 11 cells, 5 mice); however, the cumulative probability plot of amplitudes (Q) for mEPSCs in WT and PTX3 KO shows that there is a significant shift in the distribution by the Kolmogorov–Smirnov test (P < 0.0001, D = 0.112).
Discussion
Several cytokines, normally produced in the healthy brain, are now established to play critical roles in almost every aspect of neural development, including neurogenesis, migration, differentiation, synapse formation, plasticity, and responses to injury (Boulanger, 2009; Carpentier & Palmer, 2009; Deverman & Patterson, 2009). Our study introduces a new humoral innate immunity molecule in astrocyte secretome, PTX3, which adds to previous astrocyte‐derived immune molecules acting as modulators of brain development and functionality (Rivest, 2009; Garay & McAllister, 2010).
Pentraxin 3 is a molecule belonging to the humoral arm of innate immunity, involved in innate recognition and clearance of selected microbes and in modulating inflammatory responses. It has also been shown to contribute to ECM architecture and tissue remodeling, playing a non‐redundant role in female fertility and in tissue repair processes (Salustri et al, 2004; Doni et al, 2015). The results presented here show that PTX3 is developmentally regulated during brain development and plays a crucial role in promoting GluA clustering and functional maturation of synapses formed during the first wave of synaptogenesis. This occurs through a process involving the key PTX3 binding partner, TSG6, the remodeling of the perineural ECM and integrins β1 (Fig 7). Furthermore, we provide evidence that PTX3 interacts with TSP1 and 2 and that TSP1 acts as a negative regulator of PTX3 activity, since the ability of PTX3 to induce GluA clustering is blocked in the presence of exceeding amount of TSP1.
Figure 7. Cartoon depicting astrocyte‐derived PTX3 effects on PNN remodeling and on AMPA receptors at the synapse.
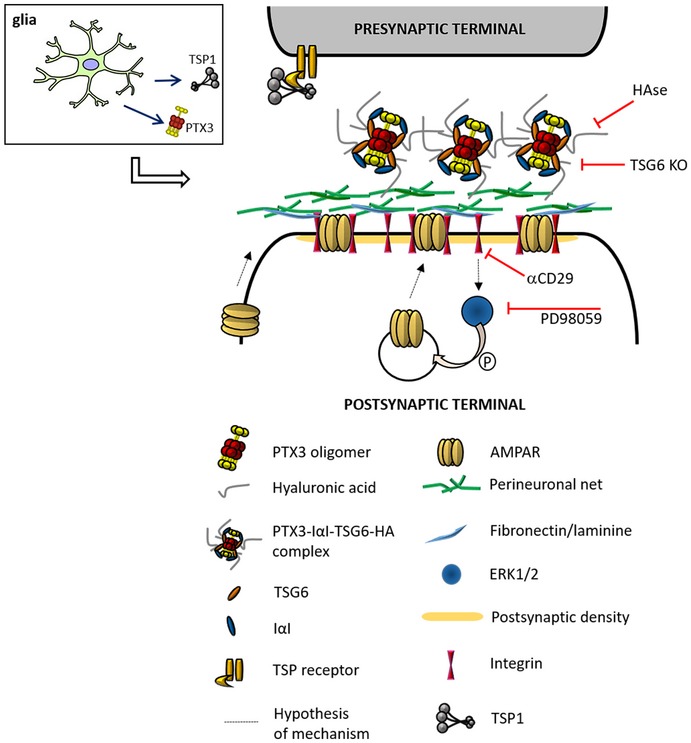
PTX3, released from astrocytes, binds to hyaluronic acid through TSG6 and activates β1‐integrin. β1‐integrin controls AMPAR trafficking through downstream MAP kinase activation. Indeed, the PTX3‐induced AMPAR enhancement is prevented by (1) genetic removal of the key HA‐binding factor, TSG6; (2) neuron exposure to hyaluronidase (HAse, which destroys the extracellular matrix); (3) inhibition of β1‐integrin activation; (4) pharmacological blockade of MAPK activation (PD98059).
The recruitment of AMPA receptors to the postsynaptic membrane at the nascent excitatory synapses is a critical process in synapse maturation, which allows glutamatergic transmission to become functional (Hall & Ghosh, 2008). During synapse maturation, PTX3 increases the numbers of functional synapses resulting in a net increase in the frequency of postsynaptic events. Indeed, following PTX3 modulation, a consistent change in both mEPSC frequency and synaptic surface GluA content is detectable. Of note, mEPSC amplitude changes are less robust, in line with literature data, showing that mEPSC frequency is the first parameter to be modulated by moderate increases of synaptic strength (Saglietti et al, 2007; Sun & Turrigiano, 2011; Sinnen et al, 2017).
Our data demonstrate that astrocytes, besides secreting ECM components and synaptogenic molecules, release PTX3 that acts on the remodeling of ECM and controls synaptic AMPA receptor clustering during synaptogenesis. PTX3, acting through its N‐terminal domain and via interaction with TSG6 and ECM molecules, contributes to β1 integrin‐dependent increase of AMPARs at the postsynaptic membrane. This process is relevant to the previously described role of another astrocyte‐derived immune molecule, TNF‐α, which induces the surface insertion of GluA1‐containing AMPARs, causing a rapid increase in the frequency of mEPSCs thus shifting neurons toward a more excitable state (Beattie et al, 2002). However, unlike PTX3, TNF‐α acts at mature synapses and operates on a fast time scale, causing a rapid (< 15 min) increase in the number synaptic AMPARs leading to synapse strengthening (Beattie et al, 2002; Leonoudakis et al, 2004).
Of note, PTX3 expression follows the developmental profile of thrombospondins, expressed by immature astrocytes only during the first week of postnatal development, which corresponds to the initiation of excitatory synapse formation. TSPs, which have a potent effect in increasing the number of synapses, completely fail in promoting synaptic activity. Indeed, thrombospondin‐induced synapses, although exhibiting normal postsynaptic densities containing PSD‐95, SAP‐102, and Homer and being endowed with NMDA receptors, completely lack AMPARs (Christopherson et al, 2005). Also, TSP1 has been shown to rapidly decrease postsynaptic surface AMPARs in cultured rat spinal cord neurons (Hennekinne et al, 2013). Early synaptogenesis appears therefore as a tightly regulated two‐step process, involving TSPs (first step) which provide the structural assembly of postsynaptically silent synapses and PTX3 (second step) which turns on newly TSP‐formed silent synapses, thus possibly representing the previously hypothesized “unidentified astrocyte signal [which] induces postsynaptic function by inserting functional AMPARs into postsynaptic sites” (Christopherson et al, 2005).
Notably, astrocytes control synapse formation and stabilization also at later developmental stages, when synaptic activity already starts driving the maturation of some synapses and the elimination of weaker ones (reviewed in Chung et al, 2015), by releasing distinct factors with temporal specificity. Among them, the astrocyte‐derived factors, Hevin (Kucukdereli et al, 2011) and glypican 4 (Allen et al, 2012), are expressed at later stages (second and third postnatal weeks of development) and could stabilize synaptic connections facilitating their maturation—glypican 4 by inducing NPTX1 (Farhy‐Tselnicker et al, 2017)—while TSPs and PTX3 would pioneer the first wave of synapse formation. Interestingly, the two proteins share the potential to orchestrate a pericellular interaction network. TSP1 is able to interact with components of the PNN (chondroitin sulfate proteoglycans including aggrecan; Eroglu et al, 2009; Resovi et al, 2014). Similarly, PTX3 has been shown to be fundamental for a proper organization of the HA‐rich ECM through the interaction with key HA‐binding components, such as IαI and TSG6, via different sites in the PTX3 N‐terminal domain (Salustri et al, 2004; Scarchilli et al, 2007; Baranova et al, 2014). Our findings that the PTX3‐mediated maturation of synapses requires an intact ECM and the presence of the HA‐binding TSG6 indicate that the remodeling of the perineural network is a key mechanism of PTX3 activity (Fig 7). Furthermore, our observation that neuronal exposure to PTX3 did not affect the expression of NP1, NP2, or glypican 4 (Fig EV4E) further supports the hypothesis that PTX3‐mediated GluA clustering and glypican 4/NPTX‐signaling (Farhy‐Tselnicker et al, 2017) are independent processes.
Besides TSP1, additional molecules with potential synaptogenic effects have been shown to bind PTX3, in particular C1q (Li et al, 2002; Yuzaki, 2017) and FGF2 (Terauchi et al, 2010). However, the involvement of these molecules in the PTX3‐dependent increase of AMPARs at the synapse is unlikely. Indeed, C1q binds the C‐term domain of PTX3 (Bottazzi et al, 1997; Nauta et al, 2003) which we demonstrated not to be involved in the described protein effect. Also, the binding of PTX3 to FGF2 is inhibited by TSG6 (Leali et al, 2012) which is instead necessary for the PTX3‐induced excitatory synapse maturation. TSP1 as well binds to TSG6, through the N‐terminal domain, and promotes TSG6 interaction with IαI and IαI association with hyaluronic acid (Kuznetsova et al, 2005). Both PTX3 and thrombospondins interact with the Link module, the hyaluronan‐recognizing domain present in TSG6 and in other ligands of hyaluronic acid including aggrecan. It could be hypothesized that the direct interaction with TSP1 might affect PTX3 binding to ECM ligands and ability to orchestrate PNN composition and organization, a requirement for PTX3 induction of functional synapses.
Astrocytes secrete both PTX3 and TSP1 and hence have the potential to enhance both the number and activity of synapses. Of note, PTX3 and TSP1 display a spatially and temporally overlapped expression also in human brain, being higher in the astrocytes of fetal cerebral cortex (http://web.stanford.edu/group/barres_lab/cgi-bin/geneSearchMariko.py?geneNameIn=PTX3; http://web.stanford.edu/group/barres_lab/cgi-bin/geneSearchMariko.py?geneNameIn=THBS1; Zhang et al, 2014). The functional and molecular interaction between the two molecules might represent an additional mechanism of control in the process of early synaptogenesis. Our findings indicate that TSP1 negatively modulates the “synaptogenic” action of PTX3, i.e., the ability of PTX3 to promote AMPARs insertion at the synapse. Conversely, the findings that α2δ‐1‐binding E123‐1 domain of TSP1 does not bind PTX3 and that E123‐1 activity is not inhibited by PTX3 indicate that PTX3 is not a negative regulatory factor for TSP1 binding to α2δ‐1.
These data suggest that a control mechanism may be in place, which, under excessive TSP1 concentrations, negatively controls AMPAR insertion and consequent synapse activation, thus avoiding excessive excitation. Of note, PTX3 is induced by primary inflammatory signals (Polentarutti et al, 2000) opening the possibility that increased levels of PTX3 upon prenatal or postnatal CNS infections may enhance brain vulnerability by adversely impacting the process of synapse formation. Indeed, changes of AMPARs content at the synapse have been shown to affect neuronal activity and to be linked to epilepsy (Rogawski & Donevan, 1999; Zhang et al, 2008; Bateup et al, 2013). Also, traumatic brain injuries—conditions characterized by PTX3 elevations in the brain parenchyma (Zanier et al, 2011; Rodriguez‐Grande et al, 2014)—are often followed by abnormal hyperexcitability, leading to acute seizures and epilepsy (Avramescu & Timofeev, 2008; Timofeev et al, 2010). It has to be noted, however, that also the expression levels of TSP1 and TSP2 are upregulated upon inflammation or injury (Risher & Eroglu, 2012), a process which may limit the excessive PTX3‐induced activation of TSP1‐induced silent synapses. The relative concentration of the two molecules could therefore be crucial to set the proper balance between synaptic growth and synapse function during physiological and pathological conditions. Under this perspective, the possibility that the interplay between TSPs and PTX3 might change in pathological conditions affecting brain vulnerability is worth being investigated.
Materials and Methods
Animals
Procedures involving animals handling and care were conformed to protocols approved by the Humanitas Clinical and Research Center (Rozzano, Milan, Italy) in compliance with national (4D.L. N.116, G.U., suppl. 40, 18‐2‐1992) and international law and policies (EEC Council Directive 2010/63/EU, OJ L 276/33, 22‐09‐2010; National Institutes of Health Guide for the Care and Use of Laboratory Animals, US National Research Council, 2011). All efforts were made to minimize the number of mice used and their suffering. Wild‐type pregnant mice were obtained from Charles River (Calco, Italy). PTX3‐deficient mice were generated as described (Garlanda et al, 2002). TSG6‐deficient mice come from Jackson Laboratories (C.129S6‐Tnfaip6tm1Cful/J, Strain 012903; Fulop et al, 2003). Animals were housed and bred in the SPF animal facility of Humanitas Clinical and Research Center in individually ventilated cages.
Cell cultures
Mouse hippocampal neurons were prepared from E18 wild‐type (WT) littermates from C57BL/6 mice as described by Fossati et al (2015) with slight modifications. Briefly, hippocampi were dissociated by treatment with trypsin (0.125% for 15 min at 37°C), followed by mechanical trituration. The dissociated cells were plated onto glass coverslips coated with poly‐L‐lysine at density of 200 cells/mm2. The cells were maintained in Neurobasal (Invitrogen, San Diego, CA) with B27 supplement and antibiotics, 2 mM glutamine, and 12.5 μM glutamate (neuronal medium). To obtain pure neuronal cultures, cytoarabinoside‐C (4 μM) has been added at DIV3.
Mouse astrocytes were prepared from P2 WT pups, and pure cultures of astrocytes (> 99.5%) were obtained by shaking flasks for 24 h at 37°C at days 2 and 6 after plating (Filipello et al, 2016). The cells were maintained in EMEM (Life Technologies, Carlsbad, CA, USA), 20% glucose, 1% Pen/Strep (Lonza, Basel, Switzerland) with 10% FBS (EuroClone, Milan, Italy).
DNA constructs, recombinant proteins, and cytokines
Recombinant human PTX3, and the PTX3 C‐terminal (C domain) and N‐terminal (N domain) fragments were purified under endotoxin‐free conditions by immunoaffinity, from the supernatants of stably transfected CHO cells as previously described (Bottazzi et al, 1997; Deban et al, 2010). Human thrombospondin‐1 (TSP1) was purified from thrombin‐stimulated human platelets (Taraboletti et al, 1990). Fragments were produced by digesting TSP1 with thrombin (20 U/ml) at 37°C, O/N. Digestion was stopped by 2 mM phenylmethylsulfonyl fluoride, and digestion products were separated by chromatography on heparin‐Sepharose (GE Healthcare Europe, Milano Italy; Margosio et al, 2003). Human TSP1 constructs E123CaG‐1, P123‐1, E123‐1, and Ca‐1 were prepared as secreted proteins from insect cells infected with recombinant baculoviruses as described (Margosio et al, 2008; Liu & Mosher, 2009; Liu et al, 2009). Recombinant human TSP‐2 and TSP‐4 used in some experiments were from R&D Systems (Minneapolis, MN, USA).
Rat monoclonal antibody anti‐PTX3 MNB4 (IgG2a) was obtained as previously described (Camozzi et al, 2006). The rat isotype control was from AbCam (IgG1; #RTK2071). Hyaluronidase (Sigma‐Aldrich, Milan, Italy) was used at 8 U/ml. PD98059 (Sigma‐Aldrich, Milan, Italy) was used 30 μM. CD29 blocking antibody (Biolegend, San Diego, CA, USA) was used 25 μg/ml. IL‐1β (Peprotech, rocky hill, NJ, USA) was used 100 ng/ml. Neuronal cultures were transfected with pEGFP‐C1 (Clontech, Palo Alto, CA) using Lipofectamine 2000 (Invitrogen) at 12 DIV and fixed at 14 DIV to evaluate dendritic spines.
Microtiter plate binding assays
Pentraxin 3 binding to TSP1 was performed as previously described (Deban et al, 2010). Briefly 96‐well plates (Nunc Maxisorb immunoplates, Roskilde, Denmark) were coated overnight with TSP1 (39–78 nM) in phosphate buffer (PBS++; contains 130 mg/l (1.2 mM) CaCl2 and 100 mg/l (1.4 mM) MgCl2; Lonza). After blocking of non‐specific sites with 0.5% dry milk in PBS++ (2 h at room temperature), plates are incubated with 100 μl of PTX3 (1.4–220 nM considering a molecular weight of 45 kDa for the PTX3 monomer) in PBS++ containing 0.05% Tween 20 (PBST). After washing, plates were first incubated with rabbit anti‐PTX3 polyclonal antibody (1:2,000) and then with anti‐rabbit‐IgG labeled with horseradish peroxidase (HRP: GE Healthcare, Pittsburgh, PA, USA). The chromogen substrate 3′,5,5′‐tetramethylbenzidine (TMB; 1 Step™ ULTRA TMB‐ELISA, Thermo Scientific, Rockford, IL, USA) was added and stopped with 2 N H2SO4 before reading absorbance at 450 nm. Binding to immobilized recombinant TSP‐2 and TSP‐4 and to the TSP1 fragments C‐term; N‐term; Ca‐1; P123‐1; E123‐1; and E123CaG‐1 all used at 50 nM, was performed following the same procedure. PTX3 N‐terminal domain (8.8–220 nM) was also tested in the same setting.
Immunocytochemical staining, image acquisition and analysis
Neuronal cultures were fixed with 4% paraformaldehyde + 4% sucrose, or with 100% cold methanol, depending on the markers. The following antibodies were used: rabbit anti‐tubulin (1:100; T3526 Sigma‐Aldrich, Milan, Italy), guinea pig anti‐Bassoon (1:300; 141004, Synaptic Systems, Goettingen, Germany), mouse anti‐PSD95 (1:400; 75‐028, UC Davis/NIH NeuroMab Facility, CA), mouse anti‐gephyrin (1:500; 147021, Synaptic Systems, Goettingen, Germany), mouse anti‐beta III tubulin (1:400; G712A, Promega Corporation, Madison, USA), rabbit anti‐tubulin (1:80; Sigma‐Aldrich, Milan, Italy), rabbit anti‐aggrecan (1:200; AB1031, Millipore, Billerica, MA, USA), DAPI (1:5,000, Thermo Fisher). Secondary antibodies were conjugated with Alexa‐488, Alexa‐555 or Alexa‐633 fluorophores (Invitrogen, San Diego, CA).
AMPA receptors live staining has been performed with a mouse anti‐GluA antibody (1:100; 182411, Synaptic Systems, Goettingen, Germany). Cells were stained for 5′ at 37°C, followed by three washes in KRH buffer [(in mM): 125 NaCl, 5 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 HEPES‐NaOH, 2 CaCl2, and 6 glucose at pH 7.4) before fixation with 4% paraformaldehyde + 4% sucrose.
Images were acquired using an Olympus FV1000 TIRF confocal microscope equipped with an UPLSAPO 60X OIL NA:1.35 Oil objective at a resolution 1,024 × 1,024. Confocal microscope settings were kept constant among the different conditions in each single experiment.
Analysis of synapse density and mean puncta size were performed using Fiji software (NIH, Bethesda, Maryland, USA) as described in Fossati et al (2015). Co‐localization of two or three selected markers was measured using the boolean function “AND” for the selected channels. The resulting image was binarized and used as a co‐localization mask to be subtracted to single channels. The number of the puncta resulting from co‐localization mask subtraction was measured for each marker. A co‐localization ratio was set as colocalizing puncta/total puncta number. For spine density evaluation, GFP‐positive protrusions with the following morphological characteristics were counted: (i) emerging from a parent dendrite; (ii) with a long neck and a visible small head (thin spines: length 41.2 μm, width 0.5 μm); (iii) well‐defined neck and a voluminous head (mushroom spines: length ≤ 1.2 μm, width 0.5 μm). At least three dendritic branches were analyzed for each neuron. The number of analyzed neurons is reported in each figure legend. At least three independent replications were performed for each experimental setting.
ELISA immunoassay
For ELISA assay from brain tissue, cortices were solubilized in lysis buffer (Tris–HCl 50 mM, Triton X‐100 0.1%, EDTA 2 mM, protease inhibitor cocktail) with Tissue Lyser III (Qiagen, Germany), centrifuged at 1,600 g for 15 min, and then, the supernatant was collected. For ELISA from neurons and astrocytes culture media, the supernatant was collected at the same moment for control or treated samples. To measure murine PTX3 levels, ELISA assay was performed as described in (Doni et al, 2015). The assay is based on two monoclonal antibodies generated by immunizing Ptx3−/− mice with recombinant murine PTX3 purified from transfected CHO. The antibodies 2C3 (also known as MnmE1) and 6B11 (also known as Mnme2) are both murine IgG1 and do not recognize human PTX3. Briefly, 96‐well ELISA plates (Nunc MaxiSorp, Thermo Fischer Scientific, Roskilde, Denmark) were coated with monoclonal antibody 2C3 anti‐mouse PTX3 in coating buffer (15 mM carbonate buffer pH 9.6) and incubated overnight at 4°C. After each step, plates were washed three times with washing buffer (PBS containing 1.17 mM CaCl2, 1.05 mM MgCl2 and 0.05% Tween 20, pH 7.00). Non‐specific binding sites were blocked with 5% dry milk in washing buffer. Standard (mouse recombinant PTX3, from 156 pg/ml to 10 ng/ml) or samples were added in duplicate and incubated for 2 h at 37°C. Then, plates were washed and the biotinylated monoclonal antibody 6B11 anti‐mouse PTX3 (25 ng/well) diluted in washing buffer was added. The plates were kept for 1 h at 37°C, washed, and incubated with streptavidin–horseradish peroxidase (Amersham, Milan, Italy). After 1‐h incubation at room temperature, the plates were washed extensively before the addition of 100 μl of tetramethylbenzidine substrate (Thermo Fischer Scientific, Rockford, IL, USA). The reaction was blocked with 2 N sulfuric acid. Absorbance was measured at 450 nm with an automatic ELISA reader. Mean PTX3 content was obtained converting Abs450 values to protein concentration using the standard curve with recombinant purified murine PTX3. Murine TSP1 in the culture supernatants or brain extracts was measured by ELISA (Cusabio, Hubei, China), following the manufacturer's instructions.
Real‐time RT–PCR
Total RNA was extracted using TRIzol™ reagent (Invitrogen™) following manufacturer's recommendations. RNA was purified using Direct‐zol™ RNA miniPrep kit (Zymo Research, Irvine, CA, USA). cDNA was synthesized using 300 ng of total RNA from neuronal cultures and 1 μg from brain tissue by reverse transcription using High Capacity cDNA Reverse Transcription Kit™ (Applied Biosystems™, Foster City, CA, USA). Relative quantitative real‐time PCR to evaluate the expression level of Ptx3 mRNA was performed using 80 ng of cDNA and 400 nM of specific primers in 20 μl while the amplification of np1, np2, tsp1, and glypican 4 was done using 30 ng of cDNA and 250 nM of specific primers in 15 μl of reaction. All qPCR assays were performed using the SensiFAST™ SYBR® low‐ROX kit (Bioline, Trento, Italy) on ViiA 7 Real‐Time PCR System (Applied Biosystems™, Foster City, CA, USA) with at least two technical replicates for each data point. The thermal cycling conditions were optimized based on primers’ sequences. Data were analyzed with the ΔΔCt method using GAPDH as reference gene and normalized as described in figure legends. Amplification specificity was confirmed by melting curve profile, and the efficiency of qPCR assays was verified by standard curves using serial dilutions of the cDNA. Primers used are the following: gapdh: forward 5′‐GCA AAG TGG AGA TTG TTG CCA T‐3′, reverse 5′‐CCT TGA CTG TGC CGT TGA ATT T‐3′; ptx3: forward 5′‐CGC TGT GCT GGA GGA ACT‐3′, reverse 5′‐ATT GCT GTT TCA CAA CCT G‐3′, tsp1 forward 5′‐GAA GCA ACA AGT GGT GTC AGT‐3′, reverse 5′‐ACA GTC TAT GTA GAG TTG AGC CC‐3′ (Liauw et al, 2008), np1 forward 5′‐CCC GCT TCA TCT GCA CTT C‐3′, reverse 5′‐TCA GCT CCC TGA TGG TCT CC‐3′, np2 forward 5′‐CGG AGC TGG AAG ATG AGA AG‐3′, reverse 5′‐GGC AGA TGG TAA AGG CGT A‐3′, gpc4: forward 5′‐GGC AGC TGG CAC TAG TTT G‐3′, reverse 5′‐AAC GGT GCT TGG GAG AGA G‐3′ (Gesta et al, 2006).
Cell culture electrophysiology
For mEPSC and mIPSC, whole‐cell recordings were performed in cultured neurons at 14 DIV using the following external solution (in mM): 125 NaCl, 5 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 HEPES‐NaOH, 2 CaCl2, and 6 glucose at pH 7.4. Miniature excitatory postsynaptic current (mEPSC) was recorded in the presence of bicuculline (Tocris) 20 mM, 2‐amino‐5‐phosphonovalerate (D‐APV) (Tocris) 50 mM, tetrodotoxin (Tocris) 1 mM using the following internal solution (in mM): 135 K gluconate, 5 KCl, 2 MgCl2, 10 HEPES, 1 EGTA, 2 ATP, 0.5 GTP at pH 7.4. Miniature inhibitory postsynaptic current (mIPSC) was recorded in the presence of 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione, CNQX (Tocris) 25 mM, APV 50 mM, tetrodotoxin 1 mM using the following internal solution (in mM): 110 KCl, 1 EGTA, 10 HEPES, 2 ATP, 0.5 GTP at pH 7.2. Both mEPSC and mIPSC were recorded from a holding potential of −70 mV using borosilicate electrodes with tip resistance of 4–6 MΏ. Access resistance (R a), membrane resistance (R m), and capacitance (C m) were estimated in voltage‐clamp configuration using a response to 10 mV hyperpolarization step. R a was left uncompensated and monitored at the end of each recording sweep; only cells in which Ra was below 15 MW and did not change more than 30% were considered for the analysis. Signals were amplified, using a Axopatch 200B (Axon Instruments, USA), sampled at 20 kHz and filtered at 5 kHz using DIGIDATA 1440 (Axon Instruments, USA). Data were stored with pCLAMP software (Molecular Device, USA), analyzed with Mini Analysis Software (Synaptosoft), and processed with Origin 8 Pro and GraphPad 6.
For total AMPA currents, voltage‐clamp whole‐cell recordings were obtained from cultured neurons on DIV 13‐DIV16 under visual guidance using fluorescence and transmitted light illumination.
Extracellular solution contained 125 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM HEPES sodium salt, 2 mM CaCl2, and 6 mM glucose. Patch pipettes (2.5–4.5 MΩ resistance) made from borosilicate glass (World Precision Instruments, Sarasota, FL, USA) were filled with a potassium gluconate‐based solution containing 10 mM KCl, 2 mM MgCl2, 10 mM HEPES sodium salt, 130 mM potassium gluconate, 1 mM ethylene glycol tetraacetic acid (EGTA), 4 mM Mg‐ATP, and 0.3 mM Tris‐GTP. Whole‐cell AMPA receptor‐mediated currents were measured by holding neurons at −70 mV in the presence of 1 μM TTX, 20 μM bicuculline, 20 μM nifedipine, and 20 μM AP5. 30 μM AMPA was applied locally using a Perfusion Fast‐Step System SF‐77B (Warner Instruments, Hamden CT, USA) during 5 s and then washed. Recordings were performed at room temperature in voltage‐clamp mode using an Axopatch 200b amplifier (Molecular Devices) and pClamp‐10 software (Axon Instruments). Series resistance ranged from 5 to 20 MΩ and was monitored for consistency during recordings. Cells with leak currents > 200 pA or Vm > −40 mV were excluded from the analysis. Signals were amplified, sampled at 10 kHz and filtered to 4 KHz, and acquired using pClamp 10 data acquisition program. Analyses were carried out using Clampfit‐10.6 software. Traces were low‐pass‐filtered at 1 KHz. Whole‐cell AMPAR‐mediated current currents were calculated as the maximum peak of the current divided by cell capacitance (current density).
Electrophysiology in brain acute slices
To obtain acute hippocampal slices, PTX3−/− and WT littermates aged 8–9 days were deeply anesthetized with isoflurane inhalation and decapitated. Brains were removed and placed in ice‐cold solution containing the following (in millimolar): 87 NaCl, 21 NaHCO3, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 2.5 KCl, 25 d‐glucose, and 7 sucrose, equilibrated with 95% O2 and 5% CO2 (pH 7.4). Coronal slices (300 μm thick) were cut with a VT1000S Vibratome (Leica Microsystems) from the hippocampus. Slices were incubated at room temperature for at least 1 h, in the same solution as above, before being transferred to the recording chamber. During experiments, slices were perfused at 2.0 ml/min with artificial cerebrospinal fluid (ACSF) containing the following (in millimolar): 135 NaCl, 21 NaHCO3, 0.6 CaCl2, 3 KCl, 1.25 NaH2PO4, 1.8 MgSO4, and 10 d‐glucose, aerated with 95% O2 and 5% CO2 (pH 7.4). Cells were examined with a BX51WI upright microscope (Olympus) equipped with a water immersion differential interference contrast (DIC) objective and an infrared (IR) camera (XM10r Olympus). Neurons were voltage (or current) clamped with a Multiclamp 700B patch‐clamp amplifier (Molecular Devices, Union City, CA) at room temperature. Low‐resistance micropipettes (2–3 MΩ) were pulled from borosilicate glass. The cell capacitance and series resistance were always compensated. Experiments in which series resistance did not remain below 10 MΩ (typically 5–8 MΩ) were discarded. Input resistance was generally close to 100–200 MΩ. Synaptic currents were low‐pass‐filtered at 2 kHz, sampled at 10 kHz using DIGIDATA 1440 (Axon Instruments, USA), and analyzed with the pClamp 10.6 data acquisition and analysis program (Molecular Devices). Recordings were made from hippocampal CA1 pyramidal neurons. For miniature excitatory postsynaptic current (mEPSC) recordings, 1 μM TTX, 20 μM bicuculline, and 50 μM AP5‐(Tocris) were added to the ACSF. Pipettes contained (in millimolar): 135 K+‐gluconate, 1 EGTA, 10 HEPES, 2 MgCl2, 4 Mg‐ATP, and 0.3 Tris‐GTP (pH 7.4).
Western blot
14DIV hippocampal cultures were lysed in lysis buffer with 5% SDS and analyzed by Western blotting using mouse anti‐phospho ERK1/2 (1:1,000, M8159, Sigma‐Aldrich, Milan, Italy), rabbit anti‐calnexin (1:2,000, c4731, Sigma‐Aldrich, Milan, Italy), rabbit anti‐SHANK2 (1:2,000; 162202, Synaptic Systems, Goettingen, Germany), mouse anti‐PSD95 (1:10,000; UC Davis/NIH NeuroMab Facility, CA), rabbit anti‐SV2a (1:2,000; 119002, Synaptic Systems, Goettingen, Germany), guinea pig anti‐vGlut1 (1:1,000; 135304, Synaptic Systems, Goettingen, Germany), mouse anti‐SNAP‐25 (1:100,000; SMI81 Sternberger Monoclonals, Baltimore MD), rabbit anti‐GAPDH (1:4,000, 247002, Synaptic Systems, Goettingen, Germany). Membranes were washed and incubated for 1 h at room temperature with the secondary antibody anti‐mouse or anti‐rabbit HRP conjugated (Thermo Fisher, Waltham, MA, USA). Blots were scanned using a ChemiDoc‐MP system (Bio‐Rad, Segrate, Italy). Optical densities were measured using the Fiji software (NIH, Bethesda, Maryland, USA) with local background subtracted as described Fossati et al (2015). For each sample, calnexin was used as a loading control.
Biotinylation assay
Control or PTX3 treated neuronal cultures from DIV 14 were washed twice with phosphate buffer saline containing 0.3 mM CaCl2 and 1 mM MgCl2 (PBS++) and then biotinylated twice using 0.5 mg/ml of sulfo‐NHS‐SS‐Biotin (Thermo Scientific) dissolved in PBS++ for 10 min at 4°C with gentle agitation. The labeled neurons were then incubated for 10 min with 50 mM glycine in PBS++ to quench free biotin. After glycine incubation, the neurons were washed a 4°C with ice‐cold PBS++. The neurons were then incubated with the lysis buffer [Tris–HCl pH 8.9, 0.1 mM PMSF, 1% SDS, and phosphatase inhibitor mixture 1 (diluted 1:100) and mixture 2 (Sigma)] and passed 10 times through needles of different sizes (from 18G to 25G, BD Microlance, USA) and then centrifuged (20,000 g for 20 min at room temperature). Protein content of the supernatants (total receptors) was evaluated by using BCA protein assay (Pierce Chemical, Rockford, IL). Equal amounts of protein (150–600 μg) were incubated with streptavidin beads (Sigma‐Aldrich, USA) to isolate the biotinylated proteins overnight at room temperature. The resins were then washed by centrifugation and the proteins eluted from the streptavidin beads (surface receptors) using Laemmli sample buffer and subjected to SDS–PAGE followed by Western blotting. Briefly, the samples from control or PTX3 treated neurons were diluted 1:1 (v/v) with Laemmli buffer, separated by means of SDS–polyacrylamide gel electrophoresis using 7.5% acrylamide, and electrophoretically transferred to nitrocellulose membranes with 0.45‐μm pores (Schleicher and Schull ll, Dassel, Germany). In each lane of control or PTX3‐treated samples, 1% of the total lysate and 3.5% of the biotinylated samples were loaded for the analysis with anti‐GluA2/3 Abs; 3% of the total lysates and 10.5% of the biotinylated samples were loaded for the analysis with anti‐GluA1 and GluA4 Abs. The blots were blocked overnight in 4% non‐fat milk in Tris‐buffered saline, washed in a buffer containing 4% non‐fat milk and 0.3% Tween 20 in Tris‐buffered saline, and incubated for 2 h with the primary antibody at the following concentrations or dilutions : GluA1 and GluA2/3 (1 μg/ml); GluA4 (1:1,000; 182303, Synaptic System, Goettingen, Germany); and anti‐transferrin receptor (1:1,000; 13/6800, Life Technologies, Milan, Italy). Blots were then incubated for 1 h with the appropriate secondary antibody (anti‐rabbit Ly‐Cor IRDye800RD; anti‐mouse Ly‐Cor IRDye680RD). After another series of washes, the membranes were dried overnight in the dark at room temperature. The IR signal was measured using an Odyssey CLx—Infrared Imaging System. The signal intensity of the Western blot bands was quantified using iStudio software. The optical density ratio was calculated by taking the optical density of the control as 100%. The data are expressed as mean values ± SEM of four independent experiments using each antibody. We used affinity‐purified, subunit‐specific polyclonal antibodies (Abs), produced in rabbit against peptides derived from the C‐terminal (COOH), N‐terminal (NH) of mouse and AMPAR GluA1 and GluA2/3 subunits. The specificity of the affinity‐purified GluA1 and GluA2/3 abs was previously tested by Western blotting as reported in (Pistillo et al, 2016). All other reagents (PMSF, proteases inhibitors, chemicals) were from Sigma‐Aldrich.
Statistical analysis
Statistical analysis was performed using Prism6 (GraphPad); data are presented as mean ± SEM from the indicated number of experiments. After testing whether data were normally distributed or not, the appropriate statistical test, followed by specific multiple comparison post hoc tests, has been used as indicated in figure legends. Kolmogorov–Smirnov test was used to determine significance in cumulative distributions of mEPSC amplitudes. Differences were considered to be significant if P < 0.05 and are indicated by one asterisk; those at P < 0.01 are indicated by double asterisks; those at P < 0.001 are indicated by triple asterisks; those at P < 0.0001 are indicated by four asterisks.
Author contributions
GF, MM, and EM conceived the project and designed the study. GF, DP, GT, and EM designed experiments. GF conducted and analyzed experiments in neuronal cultures, and histological and biochemical experiments. AC performed and analyzed RT–PCR experiments and helped with in vitro experiments. DP, FM, and EG conducted and analyzed patch‐clamp experiments in neuronal cultures. RM conducted and analyzed patch‐clamp experiments in hippocampal slices. SV and BB performed PTX3 ELISA and PTX3‐TSP1 binding assay. GT performed TSP1 ELISA. MM and CG conducted biotinylation experiments in neuronal cultures. FF performed astrocyte purification; DSA, DFM, CG, and AM contributed unique reagents and transgenic mouse lines. MM and EM wrote the manuscript with input from all co‐authors. All authors read and commented on the manuscript. MM and EM procured funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Acknowledgments
The authors are grateful to Dr. E. Albani (Humanitas, Italy) for kindly providing Hase, Dr. Marina Sironi (Humanitas, Italy) for help with transgenic mice, Dr Maria Luisa Malosio (IN‐CNR, Milano), and Dr. A. Inforzato and L. Deban (Humanitas, Italy) for discussion and suggestions. We thank Monzino Foundation (Milano, Italy) for its generous gift of the Zeiss LSM800 confocal microscope to the section of Milan of the Institute of Neuroscience. GF was supported by Fondazione Umberto Veronesi. EM was supported by Fondazione Vodafone Italia, Progetto Bandiera Interomics 2015–2017 and Cariplo Rif. 2017‐0622. MM was supported by Ministero della Salute GR‐2011‐02347377, Cariplo 2015‐0594, Project “AMANDA” CUP_B42F16000440005 from Regione Lombardia and CNR Research Project on Aging. AM was supported by Cariplo Rif. 2015‐0564. DP was supported by Cariplo Rif. 2017‐0886.
The EMBO Journal (2019) 38: e99529
Contributor Information
Michela Matteoli, Email: michela.matteoli@hunimed.eu.
Elisabetta Menna, Email: e.menna@in.cnr.it.
References
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486: 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alles VV, Bottazzi B, Peri G, Golay J, Introna M, Mantovani A (1994) Inducible expression of PTX3, a new member of the pentraxin family, in human mononuclear phagocytes. Blood 84: 3483–3493 [PubMed] [Google Scholar]
- Avramescu S, Timofeev I (2008) Synaptic strength modulation after cortical trauma: a role in epileptogenesis. J Neurosci 28: 6760–6772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova NS, Inforzato A, Briggs DC, Tilakaratna V, Enghild JJ, Thakar D, Milner CM, Day AJ, Richter RP (2014) Incorporation of pentraxin 3 into hyaluronan matrices is tightly regulated and promotes matrix cross‐linking. J Biol Chem 289: 30481–30498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Denefrio CL, Johnson CA, Saulnier JL, Sabatini BL (2013) Temporal dynamics of a homeostatic pathway controlling neural network activity. Front Mol Neurosci 6: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295: 2282–2285 [DOI] [PubMed] [Google Scholar]
- Bertling F, Bendix I, Drommelschmidt K, Wisniewski HG, Felderhoff‐Mueser U, Keller M, Prager S (2016) Tumor necrosis factor‐inducible gene 6 protein: a novel neuroprotective factor against inflammation‐induced developmental brain injury. Exp Neurol 279: 283–289 [DOI] [PubMed] [Google Scholar]
- Blondel O, Collin C, McCarran WJ, Zhu S, Zamostiano R, Gozes I, Brenneman DE, McKay RD (2000) A glia‐derived signal regulating neuronal differentiation. J Neurosci 20: 8012–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottazzi B, Vouret‐Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, Spreafico F, Pausa M, D'Ettorre C, Gianazza E, Tagliabue A, Salmona M, Tedesco F, Introna M, Mantovani A (1997) Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C‐reactive protein and serum amyloid P component. J Biol Chem 272: 32817–32823 [DOI] [PubMed] [Google Scholar]
- Bottazzi B, Doni A, Garlanda C, Mantovani A (2010) An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol 28: 157–183 [DOI] [PubMed] [Google Scholar]
- Boulanger LM (2009) Immune proteins in brain development and synaptic plasticity. Neuron 64: 93–109 [DOI] [PubMed] [Google Scholar]
- Camozzi M, Rusnati M, Bugatti A, Bottazzi B, Mantovani A, Bastone A, Inforzato A, Vincenti S, Bracci L, Mastroianni D, Presta M (2006) Identification of an antiangiogenic FGF2‐binding site in the N terminus of the soluble pattern recognition receptor PTX3. J Biol Chem 281: 22605–22613 [DOI] [PubMed] [Google Scholar]
- Carlson CB, Lawler J, Mosher DF (2008) Structures of thrombospondins. Cell Mol Life Sci 65: 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Palmer TD (2009) Immune influence on adult neural stem cell regulation and function. Neuron 64: 79–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Weeber EJ, Kurup S, Sweatt JD, Davis RL (2003) Integrin requirement for hippocampal synaptic plasticity and spatial memory. J Neurosci 23: 7107–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chater TE, Goda Y (2014) The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci 8: 401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Westbrook G (2001) Integrins mediate functional pre‐ and postsynaptic maturation at a hippocampal synapse. Nature 411: 317–321 [DOI] [PubMed] [Google Scholar]
- Chen X, Rivard L, Naqvi S, Nakada S, Padbury JF, Sanchez‐Esteban J, Stopa EG, Lim YP, Stonestreet BS (2016) Expression and localization of Inter‐alpha Inhibitors in rodent brain. Neuroscience 324: 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell 120: 421–433 [DOI] [PubMed] [Google Scholar]
- Chung WS, Allen NJ, Eroglu C (2015) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7: a020370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y (2008) Activity‐dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron 58: 749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, Jokiranta TS, Mantovani A, Meri S (2008) Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol 181: 8433–8440 [DOI] [PubMed] [Google Scholar]
- Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, Cuccovillo I, Bastone A, Gobbi M, Valentino S, Doni A, Garlanda C, Danese S, Salvatori G, Sassano M, Evangelista V, Rossi B, Zenaro E, Constantin G, Laudanna C et al (2010) Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol 11: 328–334 [DOI] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH (2009) Cytokines and CNS development. Neuron 64: 61–78 [DOI] [PubMed] [Google Scholar]
- Dityatev A, Schachner M (2003) Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci 4: 456–468 [DOI] [PubMed] [Google Scholar]
- Dityatev A, Bruckner G, Dityateva G, Grosche J, Kleene R, Schachner M (2007) Activity‐dependent formation and functions of chondroitin sulfate‐rich extracellular matrix of perineuronal nets. Dev Neurobiol 67: 570–588 [DOI] [PubMed] [Google Scholar]
- Dityatev A, Fellin T (2008) Extracellular matrix in plasticity and epileptogenesis. Neuron Glia Biol 4: 235–247 [DOI] [PubMed] [Google Scholar]
- Dityatev A, Schachner M, Sonderegger P (2010) The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat Rev Neurosci 11: 735–746 [DOI] [PubMed] [Google Scholar]
- Doni A, Peri G, Chieppa M, Allavena P, Pasqualini F, Vago L, Romani L, Garlanda C, Mantovani A (2003) Production of the soluble pattern recognition receptor PTX3 by myeloid, but not plasmacytoid, dendritic cells. Eur J Immunol 33: 2886–2893 [DOI] [PubMed] [Google Scholar]
- Doni A, Garlanda C, Mantovani A (2015) PTX3 orchestrates tissue repair. Oncotarget 6: 30435–30436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzyubenko E, Gottschling C, Faissner A (2016) Neuron‐glia interactions in neural plasticity: contributions of neural extracellular matrix and perineuronal nets. Neural Plast 2016: 5214961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor alpha2delta‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139: 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhy‐Tselnicker I, van Casteren ACM, Lee A, Chang VT, Aricescu AR, Allen NJ (2017) Astrocyte‐secreted glypican 4 regulates release of neuronal pentraxin 1 from axons to induce functional synapse formation. Neuron 96: 428–445.e413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, Bramwell S, Corradini L, England S, Winks J, Kinloch RA, Hendrich J, Dolphin AC, Webb T, Williams D (2006) Identification of the alpha2‐delta‐1 subunit of voltage‐dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci USA 103: 17537–17542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Pozzi D, Proietti M, Romagnani A, Mazzitelli S, Matteoli M, Verderio C, Grassi F (2016) Ectonucleotidase activity and immunosuppression in astrocyte‐CD4 T cell bidirectional signaling. Oncotarget 7: 5143–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossati G, Morini R, Corradini I, Antonucci F, Trepte P, Edry E, Sharma V, Papale A, Pozzi D, Defilippi P, Meier JC, Brambilla R, Turco E, Rosenblum K, Wanker EE, Ziv NE, Menna E, Matteoli M (2015) Reduced SNAP‐25 increases PSD‐95 mobility and impairs spine morphogenesis. Cell Death Differ 22: 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht R, Gundelfinger ED (2012) The brain's extracellular matrix and its role in synaptic plasticity. Adv Exp Med Biol 970: 153–171 [DOI] [PubMed] [Google Scholar]
- Fulop C, Szanto S, Mukhopadhyay D, Bardos T, Kamath RV, Rugg MS, Day AJ, Salustri A, Hascall VC, Glant TT, Mikecz K (2003) Impaired cumulus mucification and female sterility in tumor necrosis factor‐induced protein‐6 deficient mice. Development 130: 2253–2261 [DOI] [PubMed] [Google Scholar]
- Garay PA, McAllister AK (2010) Novel roles for immune molecules in neural development: implications for neurodevelopmental disorders. Front Synaptic Neurosci 2: 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, Nota R, Maccagno A, Riva F, Bottazzi B, Peri G, Doni A, Vago L, Botto M, De Santis R, Carminati P, Siracusa G, Altruda F, Vecchi A, Romani L, Mantovani A (2002) Non‐redundant role of the long pentraxin PTX3 in anti‐fungal innate immune response. Nature 420: 182–186 [DOI] [PubMed] [Google Scholar]
- Garlanda C, Jaillon S, Doni A, Bottazzi B, Mantovani A (2016) PTX3, a humoral pattern recognition molecule at the interface between microbe and matrix recognition. Curr Opin Immunol 38: 39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesta S, Bluher M, Yamamoto Y, Norris AW, Berndt J, Kralisch S, Boucher J, Lewis C, Kahn CR (2006) Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci USA 103: 6676–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BJ, Ghosh A (2008) Regulation of AMPA receptor recruitment at developing synapses. Trends Neurosci 31: 82–89 [DOI] [PubMed] [Google Scholar]
- Hennekinne L, Colasse S, Triller A, Renner M (2013) Differential control of thrombospondin over synaptic glycine and AMPA receptors in spinal cord neurons. J Neurosci 33: 11432–11439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA (2012) alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature 486: 122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inforzato A, Rivieccio V, Morreale AP, Bastone A, Salustri A, Scarchilli L, Verdoliva A, Vincenti S, Gallo G, Chiapparino C, Pacello L, Nucera E, Serlupi‐Crescenzi O, Day AJ, Bottazzi B, Mantovani A, De Santis R, Salvatori G (2008) Structural characterization of PTX3 disulfide bond network and its multimeric status in cumulus matrix organization. J Biol Chem 283: 10147–10161 [DOI] [PubMed] [Google Scholar]
- Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A, Moalli F, Garlanda C, Romani L, Gascan H, Bellocchio S, Bozza S, Cassatella MA, Jeannin P, Mantovani A (2007) The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med 204: 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C (2011) Control of excitatory CNS synaptogenesis by astrocyte‐secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA 108: E440–E449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova SA, Day AJ, Mahoney DJ, Rugg MS, Mosher DF, Roberts DD (2005) The N‐terminal module of thrombospondin‐1 interacts with the link domain of TSG‐6 and enhances its covalent association with the heavy chains of inter‐alpha‐trypsin inhibitor. J Biol Chem 280: 30899–30908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leali D, Alessi P, Coltrini D, Rusnati M, Zetta L, Presta M (2009) Fibroblast growth factor‐2 antagonist and antiangiogenic activity of long‐pentraxin 3‐derived synthetic peptides. Curr Pharm Des 15: 3577–3589 [DOI] [PubMed] [Google Scholar]
- Leali D, Inforzato A, Ronca R, Bianchi R, Belleri M, Coltrini D, Di Salle E, Sironi M, Norata GD, Bottazzi B, Garlanda C, Day AJ, Presta M (2012) Long pentraxin 3/tumor necrosis factor‐stimulated gene‐6 interaction: a biological rheostat for fibroblast growth factor 2‐mediated angiogenesis. Arterioscler Thromb Vasc Biol 32: 696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Wei M, Zhang C, Maxeiner S, Pak C, Calado Botelho S, Trotter J, Sterky FH, Sudhof TC (2017) Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J Neurosci 37: 1062–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Braithwaite SP, Beattie MS, Beattie EC (2004) TNFalpha‐induced AMPA‐receptor trafficking in CNS neurons: relevance to excitotoxicity? Neuron Glia Biol 1: 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Schaffner AE, Barker JL (1999) Astrocytes regulate the developmental appearance of GABAergic and glutamatergic postsynaptic currents in cultured embryonic rat spinal neurons. Eur J Neuorsci 11: 2537–2551 [DOI] [PubMed] [Google Scholar]
- Li AJ, Suzuki S, Suzuki M, Mizukoshi E, Imamura T (2002) Fibroblast growth factor‐2 increases functional excitatory synapses on hippocampal neurons. Eur J Neuorsci 16: 1313–1324 [DOI] [PubMed] [Google Scholar]
- Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman‐Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK (2008) Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab 28: 1722–1732 [DOI] [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Li YX, Dunlap V, Barker JL (1996) Upregulation of GABAA current by astrocytes in cultured embryonic rat hippocampal neurons. J Neurosci 16: 2912–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Mosher DF (2009) Interactions among stalk modules of thrombospondin‐1. J Biol Chem 284: 28563–28570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Annis DS, Mosher DF (2009) Interactions among the epidermal growth factor‐like modules of thrombospondin‐1. J Biol Chem 284: 22206–22212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margosio B, Marchetti D, Vergani V, Giavazzi R, Rusnati M, Presta M, Taraboletti G (2003) Thrombospondin 1 as a scavenger for matrix‐associated fibroblast growth factor 2. Blood 102: 4399–4406 [DOI] [PubMed] [Google Scholar]
- Margosio B, Rusnati M, Bonezzi K, Cordes BL, Annis DS, Urbinati C, Giavazzi R, Presta M, Ribatti D, Mosher DF, Taraboletti G (2008) Fibroblast growth factor‐2 binding to the thrombospondin‐1 type III repeats, a novel antiangiogenic domain. Int J Biochem Cell Biol 40: 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW (2001) CNS synaptogenesis promoted by glia‐derived cholesterol. Science 294: 1354–1357 [DOI] [PubMed] [Google Scholar]
- Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, Gingras AR, Tzima S, Vivanco F, Egido J, Tijsma O, Hack EC, Daha MR, Roos A (2003) Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol 33: 465–473 [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity‐dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067–1078 [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P (1999) Synaptic clustering of AMPA receptors by the extracellular immediate‐early gene product Narp. Neuron 23: 309–323 [DOI] [PubMed] [Google Scholar]
- O'Brien R, Xu D, Mi R, Tang X, Hopf C, Worley P (2002) Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci 22: 4487–4498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H, Tatsumi K, Morita S, Shibukawa Y, Korekane H, Horii‐Hayashi N, Wada Y, Taniguchi N, Wanaka A (2014) Chondroitin sulfate proteoglycan tenascin‐R regulates glutamate uptake by adult brain astrocytes. J Biol Chem 289: 2620–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YK, Goda Y (2016) Integrins in synapse regulation. Nat Rev Neurosci 17: 745–756 [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA (1997) Synaptic efficacy enhanced by glial cells in vitro . Science 277: 1684–1687 [DOI] [PubMed] [Google Scholar]
- Pinkstaff JK, Detterich J, Lynch G, Gall C (1999) Integrin subunit gene expression is regionally differentiated in adult brain. J Neurosci 19: 1541–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistillo F, Fasoli F, Moretti M, McClure‐Begley T, Zoli M, Marks MJ, Gotti C (2016) Chronic nicotine and withdrawal affect glutamatergic but not nicotinic receptor expression in the mesocorticolimbic pathway in a region‐specific manner. Pharmacol Res 103: 167–176 [DOI] [PubMed] [Google Scholar]
- Polentarutti N, Bottazzi B, Di Santo E, Blasi E, Agnello D, Ghezzi P, Introna M, Bartfai T, Richards G, Mantovani A (2000) Inducible expression of the long pentraxin PTX3 in the central nervous system. J Neuroimmunol 106: 87–94 [DOI] [PubMed] [Google Scholar]
- Pozo K, Cingolani LA, Bassani S, Laurent F, Passafaro M, Goda Y (2012) beta3 integrin interacts directly with GluA2 AMPA receptor subunit and regulates AMPA receptor expression in hippocampal neurons. Proc Natl Acad Sci USA 109: 1323–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyka M, Wetzel C, Aguado A, Geissler M, Hatt H, Faissner A (2011) Chondroitin sulfate proteoglycans regulate astrocyte‐dependent synaptogenesis and modulate synaptic activity in primary embryonic hippocampal neurons. Eur J Neurosci 33: 2187–2202 [DOI] [PubMed] [Google Scholar]
- Ravizza T, Moneta D, Bottazzi B, Peri G, Garlanda C, Hirsch E, Richards GJ, Mantovani A, Vezzani A (2001) Dynamic induction of the long pentraxin PTX3 in the CNS after limbic seizures: evidence for a protective role in seizure‐induced neurodegeneration. Neuroscience 105: 43–53 [DOI] [PubMed] [Google Scholar]
- Reemst K, Noctor SC, Lucassen PJ, Hol EM (2016) The indispensable roles of microglia and astrocytes during brain development. Front Hum Neurosci 10: 566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resovi A, Pinessi D, Chiorino G, Taraboletti G (2014) Current understanding of the thrombospondin‐1 interactome. Matrix Biol 37: 83–91 [DOI] [PubMed] [Google Scholar]
- Risher WC, Eroglu C (2012) Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol 31: 170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9: 429–439 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Grande B, Swana M, Nguyen L, Englezou P, Maysami S, Allan SM, Rothwell NJ, Garlanda C, Denes A, Pinteaux E (2014) The acute‐phase protein PTX3 is an essential mediator of glial scar formation and resolution of brain edema after ischemic injury. J Cereb Blood Flow Metab 34: 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Donevan SD (1999) AMPA receptors in epilepsy and as targets for antiepileptic drugs. Adv Neurol 79: 947–963 [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M (2007) Extracellular interactions between GluR2 and N‐cadherin in spine regulation. Neuron 54: 461–477 [DOI] [PubMed] [Google Scholar]
- Salustri A, Garlanda C, Hirsch E, De Acetis M, Maccagno A, Bottazzi B, Doni A, Bastone A, Mantovani G, Beck Peccoz P, Salvatori G, Mahoney DJ, Day AJ, Siracusa G, Romani L, Mantovani A (2004) PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development 131: 1577–1586 [DOI] [PubMed] [Google Scholar]
- Scarchilli L, Camaioni A, Bottazzi B, Negri V, Doni A, Deban L, Bastone A, Salvatori G, Mantovani A, Siracusa G, Salustri A (2007) PTX3 interacts with inter‐alpha‐trypsin inhibitor: implications for hyaluronan organization and cumulus oophorus expansion. J Biol Chem 282: 30161–30170 [DOI] [PubMed] [Google Scholar]
- Shi Y, Ethell IM (2006) Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin‐dependent protein kinase II‐mediated actin reorganization. J Neurosci 26: 1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia GM, Beique JC, Rumbaugh G, Cho R, Worley PF, Huganir RL (2007) Interaction of the N‐terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron 55: 87–102 [DOI] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Forte JS, Hiester BG, Crosby KC, Gibson ES, Dell'Acqua ML, Kennedy MJ (2017) Optogenetic control of synaptic composition and function. Neuron 93: 646–660.e645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Turrigiano GG (2011) PSD‐95 and PSD‐93 play critical but distinct roles in synaptic scaling up and down. J Neurosci 31: 6800–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboletti G, Roberts D, Liotta LA, Giavazzi R (1990) Platelet thrombospondin modulates endothelial cell adhesion, motility, and growth: a potential angiogenesis regulatory factor. J Cell Biol 111: 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A, Johnson‐Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H (2010) Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465: 783–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatipamula S, Hossain MA (2014) Critical role of extracellularly secreted neuronal pentraxin 1 in ischemic neuronal death. BMC Neurosci 15: 133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Bazhenov M, Avramescu S, Nita DA (2010) Posttraumatic epilepsy: the roles of synaptic plasticity. Neuroscientist 16: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG (2008) The self‐tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA (2001) Control of synapse number by glia. Science 291: 657–661 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Walsh MF, Turrigiano GG (2006) Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol 96: 2127–2133 [DOI] [PubMed] [Google Scholar]
- Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O'Brien RJ, Worley P (2003) Narp and NP1 form heterocomplexes that function in developmental and activity‐dependent synaptic plasticity. Neuron 39: 513–528 [DOI] [PubMed] [Google Scholar]
- Yuzaki M (2017) The C1q complement family of synaptic organizers: not just complementary. Curr Opin Neurobiol 45: 9–15 [DOI] [PubMed] [Google Scholar]
- Zanier ER, Brandi G, Peri G, Longhi L, Zoerle T, Tettamanti M, Garlanda C, Sigurta A, Valaperta S, Mantovani A, De Simoni MG, Stocchetti N (2011) Cerebrospinal fluid pentraxin 3 early after subarachnoid hemorrhage is associated with vasospasm. Intensive Care Med 37: 302–309 [DOI] [PubMed] [Google Scholar]
- Zhang L, Schessl J, Werner M, Bonnemann C, Xiong G, Mojsilovic‐Petrovic J, Zhou W, Cohen A, Seeburg P, Misawa H, Jayaram A, Personius K, Hollmann M, Sprengel R, Kalb R (2008) Role of GluR1 in activity‐dependent motor system development. J Neurosci 28: 9953–9968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 4