

Abstract
The transcription factor, T‐bet, regulates type 1 inflammatory responses against a range of infections. Here, we demonstrate a previously unaddressed role of T‐bet, to influenza virus and bacterial superinfection. Interestingly, we found that T‐bet deficiency did not adversely affect the efficacy of viral clearance or recovery compared to wild‐type hosts. Instead, increased infiltration of neutrophils and production of Th17 cytokines (IL‐17 and IL‐22), in lungs of influenza virus‐infected T‐bet−/− mice, were correlated with survival advantage against subsequent infection by Streptococcus pneumoniae. Neutralization of IL‐17, but not IL‐22, in T‐bet−/− mice increased pulmonary bacterial load, concomitant with decreased neutrophil infiltration and reduced survival of T‐bet−/− mice. IL‐17 production by CD8+, CD4+ and γδ T cell types was identified to contribute to this protection against bacterial superinfection. We further showed that neutrophil depletion in T‐bet−/− lungs increased pulmonary bacterial burden. These results thus indicate that despite the loss of T‐bet, immune defences required for influenza viral clearance are fully functional, which in turn enhances protective type 17 immune responses against lethal bacterial superinfections.
Keywords: bacteria, influenza, Streptococcus pneumonia, T‐bet
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Influenza viruses are respiratory intracellular pathogens that cause significant morbidity and mortality in human populations. Although vaccines and antiviral drugs have been developed against influenza viruses, rapid evolution and host adaptation of the virus underlie its constant threat for pandemic formation and debilitating disease (Taubenberger & Kash, 2010), notably from recent H5N1 and H7N9 strains. In individuals infected by influenza viruses, the eventual recovery depends on an efficient immune response spanning the innate and adaptive immune systems (Braciale et al, 2012; Iwasaki & Pillai, 2014), suggesting the robustness of host defences against influenza viruses. Conversely, overactivation of immune responses causes many influenza related deaths, indicating the need for proper balance in immune regulation during infection (Taubenberger & Morens, 2008; Newton et al, 2016). Hence, understanding and targeting of host immune responses during influenza virus infections is an important facet in tackling influenza pneumonia.
Flu infections are frequently complicated by secondary bacterial infections, significantly increasing the risk of severe pneumonia (Metersky et al, 2012; Chertow & Memoli, 2013; McCullers, 2014). In fact, bacterial complicated influenza infections accounted for nearly all the deaths in the 1918 flu pandemic (Morens et al, 2008) and up to 55% of deaths from the 2009 H1N1 pandemic (Rice et al, 2012; Centers for Disease Control and Prevention (CDC), 2009; Mauad et al, 2010). Recent studies have identified interactions between host, virus and bacteria as a main cause of heightened host susceptibility (Sun & Metzger, 2008; Shahangian et al, 2009; Ghoneim et al, 2013; Cao et al, 2014; Ellis et al, 2015). Based on these insights, the identification and manipulation of factors central to viral–bacterial lethality will formulate a viable treatment strategy against post‐influenza bacterial superinfection.
The transcription factor T‐bet is a central regulator of type 1 immune responses. Its functions are hitherto known to be mediated through the expression of cytokines IFNγ and IL‐12, chemokines CCL3 and CCL4 as well as chemokine receptors such as CXCR3. Furthermore, the expression of T‐bet is involved in various cellular functions—the development of CD4+ Th1 cells (Szabo et al, 2002); suppression of Th2 and Th17 immunity (Hwang et al, 2005; Djuretic et al, 2007; Lazarevic et al, 2011); maturation and cytolytic activity of natural killer (NK) and CD8+ T cells (Sullivan et al, 2003; Townsend et al, 2004); formation of memory immune cells (Intlekofer et al, 2005, 2007; Joshi et al, 2007; Marshall et al, 2011; Wang et al, 2012); and regulation of IgG class switching (Peng et al, 2002; Liu et al, 2003). As a result, T‐bet is purportedly required for immune defence against many classes of pathogens. Indeed, the absence of T‐bet has rendered hosts more susceptible to primary infections by the parasite, Leishmania major (Szabo et al, 2002), and bacterial species such as Mycobacterium tuberculosis (Sullivan et al, 2005), Staphylococcus aureus (Hultgren et al, 2004), Salmonella typhimurium (Ravindran et al, 2005) and Francisiella tularensis (Melillo et al, 2014). However, the role of T‐bet in viral immunity is less well defined. Earlier studies demonstrated its requirement for control of Vaccina virus and herpesvirus infections (Matsui et al, 2005; Svensson et al, 2005; Rubtsova et al, 2013), while a recent study showed that loss of T‐bet did not affect eventual viral clearance from rhinovirus infection, but is instead involved in suppression of viral‐mediated allergic airway response (Glanville et al, 2016). Hence, the regulatory functions and role of T‐bet in viral infections and coinfections of pathogens remain to be addressed.
In influenza virus infections, T‐bet‐mediated functions in CD8+ T, CD4+ T, Treg and B cells have been described (Mayer et al, 2008; Bedoya et al, 2013; Dolfi et al, 2013; Dutta et al, 2013; Hua et al, 2013; Naradikian et al, 2016). Nevertheless, the outcome from deficiency of T‐bet on pathogenesis or survival on hosts infected with influenza virus and subsequent bacterial complications have, to our knowledge, not been investigated. Given the wide immunoregulatory effects of T‐bet on type 1 immunity, we unexpectedly found that T‐bet‐deficient mice could effectively clear influenza viruses and recover from sub‐lethal dose of infection. Furthermore, the loss of T‐bet led to increased neutrophil and eosinophil infiltration in the lungs during influenza virus infection with concomitant production of Th2 and Th17 cytokines. This viral‐induced inflammation was required for increased protection against secondary respiratory infection by Streptococcus pneumoniae, as neutralization of IL‐17 or neutrophil depletion led to increased susceptibility to secondary bacterial infection. These findings shed new perspectives on T‐bet under influenza virus infection and subsequent bacterial superinfections, with implications for modulation of T‐bet as a strategy to improve survival outcomes from post‐influenza bacterial pneumonia.
Results
T‐bet−/− mice clear influenza virus infection with increased cellular infiltration into the lungs
To determine whether global loss of T‐bet had an impact on host susceptibility during influenza virus infection, we followed and compared the body weights of T‐bet−/− and wild‐type mice throughout the course of infection. Both wild‐type and T‐bet−/− mice survived sub‐lethal doses of the H1N1 PR8 mouse‐adapted influenza virus with no significant differences in percentage weight loss at all timepoints (Fig 1A). Similar to wild‐type mice, infected T‐bet−/− mice started to recover body weight after day 11 post‐infection and regained its original body weight by day 25. Weight loss and survival under lethal dose of influenza infection were also not significantly different between wild‐type and T‐bet−/− hosts (Fig EV1A and B). Survival and recovery of both wild‐type and T‐bet−/− mice under sub‐lethal infection were correlated with their ability to clear the virus by day 9 post‐infection, as measured by the amount of PR8 nucleoprotein (NP) mRNA in the lungs (Fig 1B). In contrast to wild‐type counterparts, histology of the T‐bet−/− lungs revealed notably higher cellular infiltration into the alveolar spaces at days 5, 9 and 15 post‐infection (Fig 1C), an observation which was further exemplified by increased numbers of CD45+ immune cells in the lung tissue and bronchoalveolar lavage (BAL; Fig 1D). Pulmonary injury was more severe at day 9 post‐infection in T‐bet−/− lungs. Nevertheless, the increased infiltration of immune cells and heightened inflammation did not cause increased cytotoxicity in T‐bet−/− lungs as reflected by the level of lactate dehydrogenase (LDH) in the BAL (Fig 1E). In fact, T‐bet−/− lungs displayed better recovery at day 15 post‐infection, with increased repair of alveolar walls and bronchiole epithelium and reduced pulmonary injury (Figs 1C and EV1C). To further assess tissue repair in T‐bet−/− lungs, podoplanin (PDPN), a glycoprotein expressed on alveolar type 1 epithelial cells and marker of lung injury and repair (Li et al, 2015; Zuo et al, 2015), was stained by Western blot (Figs 1F and EV1D) and immunohistochemistry (Fig EV1E). Days 15 and 25 post‐infected T‐bet−/− lungs displayed equal expression of podoplanin to wild‐type lungs, confirming that increased inflammation at earlier timepoints did not affect subsequent tissue recovery and repair. Decreased leakiness of T‐bet−/− lungs at day 25 post‐infection, as assessed by the concentration of total protein and albumin in the BAL (Fig 1G and H), further supports intact tissue homeostasis in T‐bet−/− mice. Together, these observations suggest that T‐bet deficiency does not affect host ability to clear influenza virus infections, and increased inflammation in T‐bet−/− lungs does not affect subsequent tissue repair and homeostasis.
Figure 1. T‐bet‐deficient lungs exhibit increased cellular infiltration and reduced leakiness following influenza virus infection.
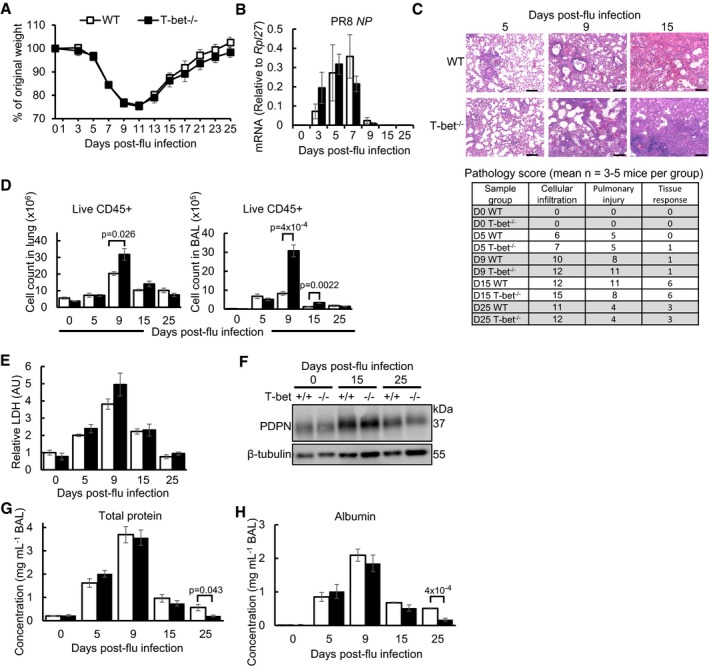
- Percentage body weight loss. See also Fig EV1A and B.
- Relative amount of PR8 viral NP mRNA, by qPCR.
- Histology of lungs by H&E staining with pathology scores. Scale bars = 200 μm. See also Fig EV1C.
- Absolute number of live CD45+ cells in whole lungs and BAL, by flow cytometry.
- Level of LDH in BAL, by LDH cytotoxicity assay.
- Expression of PDPN in lungs, by Western blot. See also Fig EV1D and E.
- Total protein in BAL, by Bradford assay.
- Concentration of albumin in BAL, by ELISA.
Figure EV1. T‐bet‐deficient mice exhibit comparable disease outcomes to wild‐type hosts, with increased infiltration of pulmonary granulocytes and lymphocytes, related to Fig 1 .
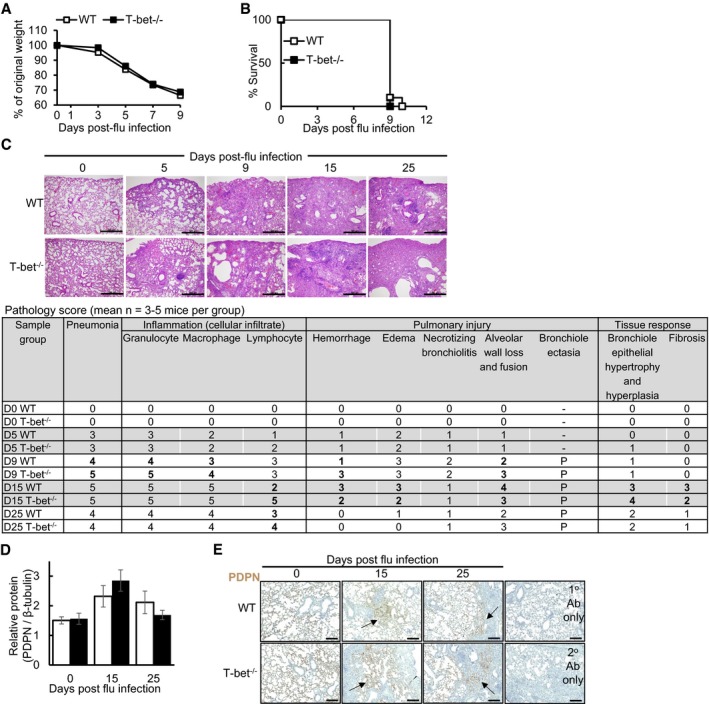
-
A, B(A) Weight loss and (B) survival of mice infected with lethal influenza virus dose (500 pfu per mice).
-
C–EMice were infected with sub‐lethal dose of influenza virus. (C) Histopathology of H&E‐stained lungs. Scale bars = 500 μm. Mean pathology scores from 3 to 5 H&E‐stained mouse lungs, with detailed assessment of inflammation, pulmonary injury and tissue responses in the lungs shown. Scores for each sub‐category range from 0 (least evident) to 5 (most evident). (D) Expression levels of PDPN in mouse lungs, assessed by densitometry of Western blots. (E) Immunohistochemistry staining of PDPN on mouse lungs. Arrow heads indicate tissue repair of damaged areas. To assess specificity of antibody staining, negative controls with no anti‐goat HRP secondary antibody (1° antibody only) or no anti‐PDPN antibody (2° antibody only) were processed simultaneously. Scale bars = 200 μm.
Increased infiltration of granulocytes and B cells into lungs of T‐bet−/− mice during influenza virus infection
To date, studies on infection of T‐bet−/− mice with various pathogens have mostly reported some form of susceptibility to infection, which is congruent with the central role of T‐bet in regulating type 1 immune responses. Recently, it was reported that T‐bet‐deficient Th1 cells display an aberrant amplification of type I interferon (IFN) response (Iwata et al, 2017). To determine whether T‐bet−/− mice were able to survive influenza infection due to increased antiviral responses, we quantified the expression of type I IFNs (IFNα and IFNβ) at the mRNA and protein levels in the lungs and BAL, respectively, at days 5 and 9 post‐influenza infection (Fig EV2A and B). T‐bet deficiency did not result in a notable change in expression of these cytokines. Furthermore, mRNA expression of a number of interferon response genes (ISGs) was equally expressed in wild‐type and T‐bet−/− lungs at day 5 post‐influenza infection (Fig EV2A) where viral load is at its peak (Fig 1B). In fact, expression of Isg15, Ifit3, Oasl1 and Irf7 was reduced at day 9 post‐influenza infection in T‐bet−/− lungs relative to wild‐type lungs. Hence, assessment of global antiviral responses did not reveal elevated type I IFN responses which would have conferred additional protective immunity against influenza in T‐bet−/− mice. To further understand immune responses of T‐bet deficiency in clearing influenza viruses and identify specific immune cells infiltrating T‐bet−/− lungs, we performed flow cytometry of single cells from influenza virus‐infected lungs. Strikingly, T‐bet−/− lungs had increased numbers and frequencies of neutrophils and eosinophils on days 9 and 15 post‐influenza infection (Fig 2A and B) while wild‐type mice lungs displayed a decline in these cell types. These results imply dysregulated immune responses or compensatory mechanisms for combating influenza virus infection in T‐bet−/− mice. Despite their increase in the earlier phase of infection, the numbers of neutrophils and eosinophils were reduced to basal levels by day 25 post‐infection in both wild‐type and T‐bet−/− lungs. No difference in numbers of monocytes was detected.
Figure EV2. Analysis of wild‐type and T‐bet−/− immune response in the lungs following influenza virus infection, related to Fig 2 .
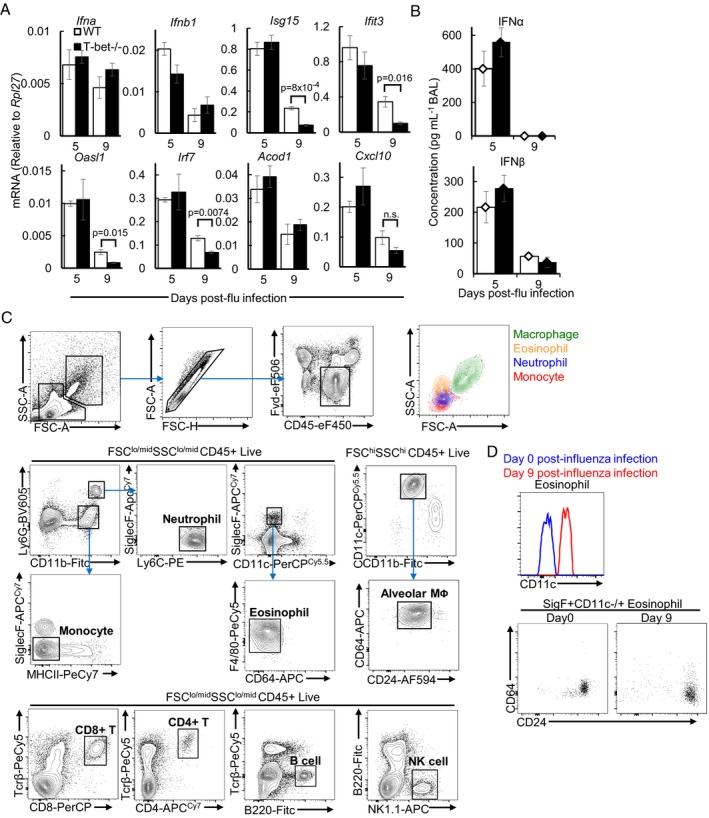
-
A, BMice were infected with sub‐lethal dose of influenza virus. (A) Relative expression of type I IFNs and antiviral mRNA in lungs of mice, as assessed by qPCR. (B) Protein concentrations of type I IFN in BAL, as assessed by ProcartaPlex assay.
-
CRepresentative flow cytometry gating strategy used for identification of immune cells in the lungs. Back‐gating of myeloid cells in the FSC‐SSC plot is shown in the top right‐hand corner.
-
DIncreased CD11c expression in eosinophils from infected lungs compared to uninfected lungs. Identification of eosinophils was further confirmed with CD64−CD24hi phenotype.
Figure 2. Increased infiltration of neutrophils, eosinophils and B cells in T‐bet‐deficient lungs infected with influenza virus.
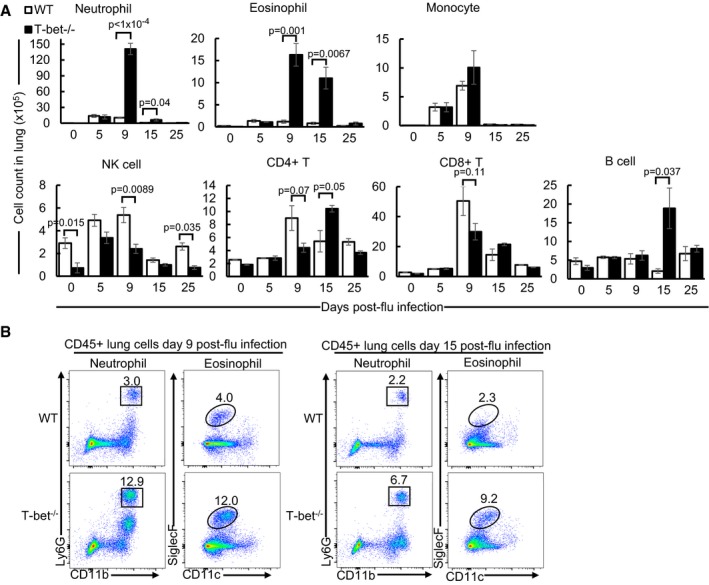
- Absolute cell counts from lungs of sub‐lethally influenza virus‐infected mice following analysis by flow cytometry. After gating out alveolar macrophages by forward and side scatter, immune cells are defined as follows: neutrophil (Ly6G+CD11b+SiglecF−Ly6Cint), eosinophil (SiglecF+CD11c−/+F4/80−CD64−), monocyte (Ly6G−CD11b+MHCII−SiglecF−), natural killer cell (NK, B220−/intNK1.1+), B cell (B220hiNK1.1−), CD8+ T cell (TCRβ+CD8+), CD4+ T cell (TCRβ+CD4+). See also Fig EV2C and D.
- Flow cytometry plots of neutrophils and eosinophils in lungs at days 9 and 15 post‐sub‐lethal influenza virus infection. Numbers in plots indicate frequency of gated immune cells in each throughput.
We observed a reduction of natural killer (NK) cells in T‐bet−/− lungs compared to wild‐type lungs at days 0, 9 and 25 post‐influenza virus infection (Fig 2A), consistent with the role of T‐bet in NK homeostasis and maturation (Townsend et al, 2004). Nevertheless, pulmonary NK cells in T‐bet−/− mice at day 5 post‐infection were comparable to wild‐type lungs, suggesting intact recruitment of NKs during the early phase of infection. The numbers of CD4+ T and CD8+ T cells between wild‐type and T‐bet−/− lungs were largely similar, although a consistent trend of their reduced numbers in T‐bet−/− lungs (with P > 0.05) across independent experiments was observed at day 9 post‐infection (Fig 2A). We observed a 10‐fold increase of B cells in T‐bet−/− lungs relative to wild‐type lungs specifically at day 15 post‐influenza infection, suggesting a role for T‐bet in regulating proliferation or migration of B cells. Collectively, the results demonstrate that absence of T‐bet does not abrogate innate or adaptive lymphocyte responses in the lungs.
Overall, analysis of immune responses revealed increased granulocytes and B cells in influenza‐infected T‐bet−/− lungs, while concomitant intact type I IFN responses and recruitment of cytotoxic and helper lymphocytes likely contributed to the effective clearance of influenza virus.
T‐bet deficiency confers survival advantage to post‐influenza bacterial superinfection
Excessive pulmonary infiltration of neutrophils and eosinophils has been observed in influenza virus‐induced pneumonia and deaths (Tumpey et al, 2005; Taubenberger & Morens, 2008; Jeon et al, 2010; Larrañaga et al, 2016), which was taken to indicate lung impairment. However, our results thus far did not suggest increased susceptibility or damage to lung tissues despite heightened infiltration of neutrophils and eosinophils in T‐bet−/− lungs. We then asked how this increase in granulocytes in influenza‐infected T‐bet−/− hosts might respond under bacterial superinfection, a frequent complication following influenza infections.
To investigate responses of T‐bet−/− mice to post‐influenza bacterial superinfection compared to wild‐type hosts, mice were infected with a sub‐lethal dose of PR8 influenza virus, followed by administration of a sub‐lethal dose of S. pneumoniae serotype 19F, at day 7 post‐influenza infection (Fig 3A). Remarkably, T‐bet−/− mice displayed significantly enhanced survival following secondary bacterial infection, compared to 85% lethality of wild‐type mice (Fig 3B). Furthermore, bacterial load in T‐bet−/− lungs was lower than wild‐type lungs (Fig 3C). These survival advantages in T‐bet−/− mice were correlated to increased infiltration of neutrophils and eosinophils in the lungs at days 1 and 3 of secondary bacterial infection (Fig 3D), suggesting that they could be involved in immune defence. Compared to wild‐type lungs, the number of NK cells transiently increased in T‐bet−/− lungs 1 day after secondary bacterial infection, supporting enhanced trafficking or proliferation in response to bacterial challenge, while CD8+ T cells were reduced at day 3 post‐infection. Similar to the viral infection only controls, B cell numbers were increased in T‐bet−/− lungs following secondary bacterial infection, while CD4+ T cells are comparable to wild‐type lungs. Enhanced protection and immune cell responses against secondary bacterial infection under T‐bet deficiency were reproduced with a more virulent S. pneumoniae serotype (S3; Fig EV3B–D).
Figure 3. Improved resistance of T‐bet‐deficient mice against post‐influenza bacterial superinfection.

-
AModel of post‐influenza bacterial infection illustrating infection with sub‐lethal dose of influenza virus (25 pfu) followed by sub‐lethal dose of secondary bacterial challenge (Streptococcus pneumoniae serotype 19F, 2 × 105 cfu) 7 days later. Where appropriate, mice lungs were analysed at days 0, 1 and 3 post‐secondary bacterial infection, corresponding to days 7, 8 and 10 post‐influenza virus infection, respectively.
-
B–GMice were superinfected as described in (A). (B) Survival of mice following bacterial superinfection. See also Fig EV3B. (C) Pneumococcal cfu in BAL of mice. See also Fig EV3C. (D) Absolute immune cell counts in lungs, assessed by flow cytometry. See also Fig EV3D. (E) Pneumococcal cfu in BAL of mice 12 h after infection with 2 × 105 cfu of S. pneumoniae serotype 19F alone (−Flu) or following influenza virus infection (+Flu). See also Fig EV3C. (F) Pneumococcal cfu in BAL 36 h after infection of naïve mice with 2 × 105 cfu of S. pneumoniae serotype 19F alone. (G) Absolute neutrophil and eosinophil cell count in lungs infected as described in (E), assessed by flow cytometry.
Figure EV3. Improved immune protection in T‐bet‐deficient mice following influenza virus infection and bacterial superinfection with Streptococcus pneumoniae serotype S3, related to Figs 3, 4B and 5B.
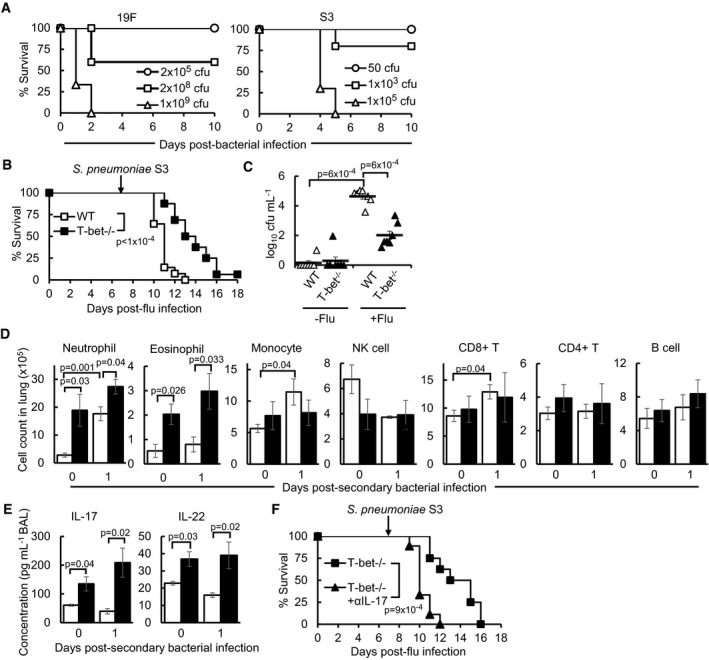
-
AWild‐type mice were infected with indicated doses of S. pneumoniae serotype 19F or S3 bacteria alone and assessed for survival.
-
B–FWild‐type and T‐bet−/− mice were infected with sub‐lethal dose of influenza virus followed by sub‐lethal dose of secondary bacterial challenge (S. pneumoniae serotype S3, 50 cfu) 7 days later. (B) Survival of mice following post‐influenza bacterial infection. (C) Pneumococcal cfu in lung homogenates of mice infected with 50 cfu of S. pneumoniae serotype S3 alone or following influenza virus infection. (D) Absolute immune cell counts in lungs, as assessed by flow cytometry. (E) Concentrations of IL‐17 and IL‐22 in lung homogenates of mice, assessed by ELISA. (F) T‐bet−/− mice were administered αIL‐17 or IgG isotype control antibody every other day from day −2 post‐secondary bacterial infection and monitored for survival.
To determine whether mortality in wild‐type mice was due to viral‐mediated suppression of antibacterial defences through T‐bet, naïve or influenza‐infected mice were infected with equal doses of bacteria. Wild‐type mice infected with prior influenza virus suffered heavier bacterial burden compared to infection with bacteria alone, indicating lethality in viral–bacterial superinfection (Figs 3E and EV3C). While T‐bet−/− mice appeared to control bacteria more efficiently than wild‐type mice at 12 h post‐bacteria‐only infection (Fig 3E), both mice strains were able to clear bacteria by 36 h post‐infection (Fig 3F), indicating suppression of antibacterial defences by T‐bet during influenza and bacterial superinfections. Moreover, the numbers of neutrophils and eosinophils were significantly lower in the lungs of both bacteria‐only infected wild‐type and T‐bet−/− mice, compared to secondary infected T‐bet−/− lungs (Fig 3G). Together, these data demonstrate that enhanced inflammatory responses during primary influenza infection in T‐bet‐deficient lungs contribute to subsequent antibacterial defence and improved survival.
Increase in Th2 and Th17 cytokines in T‐bet−/− lungs are correlated to enhanced infiltration of neutrophils and eosinophils
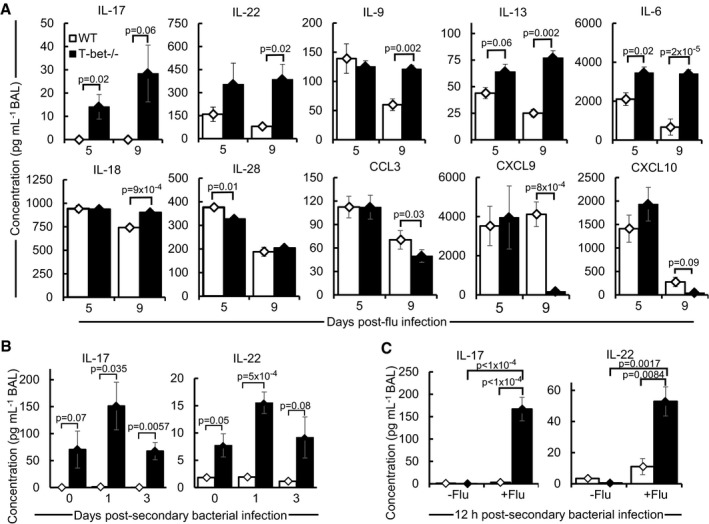
To further understand the underlying immune mechanisms in T‐bet−/− mice during the inflammatory phase of influenza virus infection, the BAL of days 5 and 9 post‐infection was screened for cytokines and chemokines using a protein bead array. Out of 24 analytes examined, significant differences in concentrations of 10 target proteins between wild‐type and T‐bet−/− lungs were observed (Fig 4A). Strikingly, Th2 cytokines, IL‐9 and IL‐13, as well as Th17 cytokines, IL‐17 and IL‐22, were significantly elevated in T‐bet−/− lungs (Figs 4A and EV4A), consistent with the role of T‐bet in repression of these immune responses (Hwang et al, 2005; Villarino et al, 2010; Lazarevic et al, 2011). Furthermore, IL‐13 and IL‐17 are known to drive pulmonary recruitment of eosinophils and neutrophils, respectively (Pope et al, 2001; Ye et al, 2001), which may explain the increased cellular infiltration of these cell types into T‐bet‐deficient lungs following infection. Secretion of pro‐inflammatory cytokines, IL‐6 and IL‐18, is increased in T‐bet‐deficient lungs (Fig 4A), suggesting possible compensatory inflammatory mechanisms perhaps in view of reduced antiviral cytokine IL‐28. Decrease in concentrations of T‐bet target protein, CCL3, was observed in T‐bet−/− lungs. IFNγ‐induced T cell chemoattractants like CXCL9 (MIG) and CXCL10 (IP‐10) were also reduced, which may provide an explanation for lowered numbers of pulmonary T cells in T‐bet−/− lungs (Figs 2A and 3D). Interestingly, no difference was observed in protein levels of the T‐bet target—IFNγ, as further confirmed by ELISA (Fig EV4B). Nevertheless, reduced expression of Ifng and Th1 cytokine Tnf mRNA was observed in T‐bet−/− lungs compared to wild‐type (Fig EV4C), implying post‐transcriptional regulation of Th1 effector cytokines. Overall, these mechanisms suggest a shift towards Th2 and Th17 immune responses in the absence of T‐bet during influenza virus infection, leading to increased infiltration of granulocytes.
Figure 4. Increased IL‐17 and IL‐22 production in T‐bet−/− lungs infected with influenza virus or superinfected with bacteria.
-
AConcentrations of cytokines or chemokines in the BAL of days 5 and 9 post‐sub‐lethal influenza virus infection as determined by protein bead array. See also Fig EV4A and B.
-
B, CMice were superinfected as described in Fig 3A. (B) Concentrations of IL‐17 and IL‐22 in BAL of mice, as determined by ELISA. See also Fig EV3E. (C) Concentrations of IL‐17 and IL‐22 in BAL of mice 12 h after infection with 2 × 105 cfu of Streptococcus pneumoniae serotype 19F alone (−Flu), or following superinfection (+Flu), assessed by ELISA.
Figure EV4. Analysis of cytokine expression in T‐bet−/− relative to wild‐type lungs following influenza virus infection, related to Fig 4 .
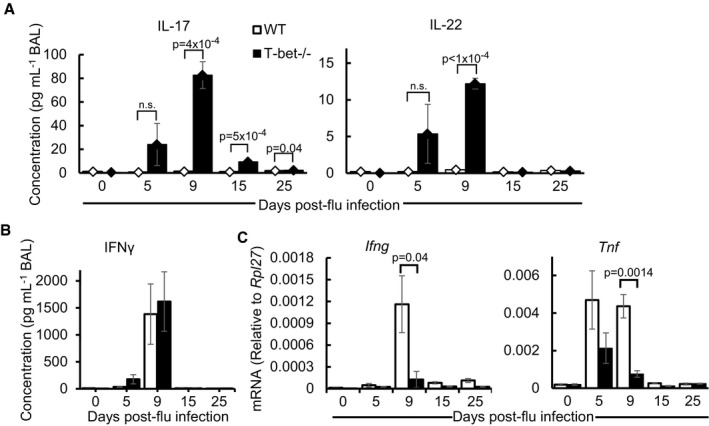
- Concentrations of IL‐17 and IL‐22 in BAL of mice determined by ELISA.
- Concentration of IFNγ in the BAL, determined by ELISA.
- Relative mRNA expression of Ifng and Tnf in mouse lungs infected as assessed by qPCR.
Th17 cytokines, IL‐17 and IL‐22, were previously shown to be critical for defence against post‐influenza secondary bacterial infection (Kudva et al, 2011). We confirmed the increased production of these cytokines in the T‐bet−/− lungs following secondary bacterial infection (Figs 4B and EV3E). Infection with bacteria alone did not induce significant production of IL‐17 and IL‐22 relative to superinfected T‐bet−/− lungs (Fig 4C), indicating that prior virus infection is required for an elevated immune response in an imminent secondary bacterial infection. Hence, it is conceivable that the survival advantage of T‐bet−/− mice under post‐influenza bacterial infection is due to the induction of Th17‐mediated antibacterial defences.
Neutralization of IL‐17 and neutrophil depletion reduces immune defences against post‐influenza bacterial superinfection in T‐bet‐deficient mice
To specifically test whether IL‐17 and IL‐22 are responsible for the survival advantage of influenza‐infected T‐bet−/− mice against secondary bacterial infection, anti‐IL‐17 or anti‐IL‐22 monoclonal antibodies were administered to neutralize the effects of these two cytokines. Treatment with anti‐IL‐17 antibody, but not anti‐IL‐22 antibody resulted in increased bacterial load in T‐bet−/− lungs following post‐influenza bacterial superinfection (Fig 5A). Correspondingly, the survival of T‐bet−/− mice against secondary S. pneumoniae infection was also reduced following IL‐17 depletion (Figs 5B and EV3F). Concomitant with the role of IL‐17 in the recruitment of neutrophils (Ye et al, 2001; Miyamoto et al, 2003), neutralization of IL‐17 reduced the number of infiltrating neutrophils to T‐bet−/− lungs at days 1 and 3 post‐secondary bacterial infection (Fig 5C). Depletion of neutrophils through administration of anti‐Ly6G‐specific antibody (Fig EV5A) resulted in significantly heavier bacterial burden in T‐bet−/− lungs (Fig 5D), confirming direct neutrophil‐mediated protection. Reduced resistance against secondary bacterial infection upon IL‐17 or neutrophil depletion did not appear to be a result of the inability to clear prior influenza virus infection, as quantitation of PR8 NP in the lungs did not show increased viral load upon antibody administration (Fig EV5B and C).
Figure 5. Increased immunity against bacterial superinfection in T‐bet−/− mice is abrogated by neutralization of IL‐17 or neutrophil depletion.
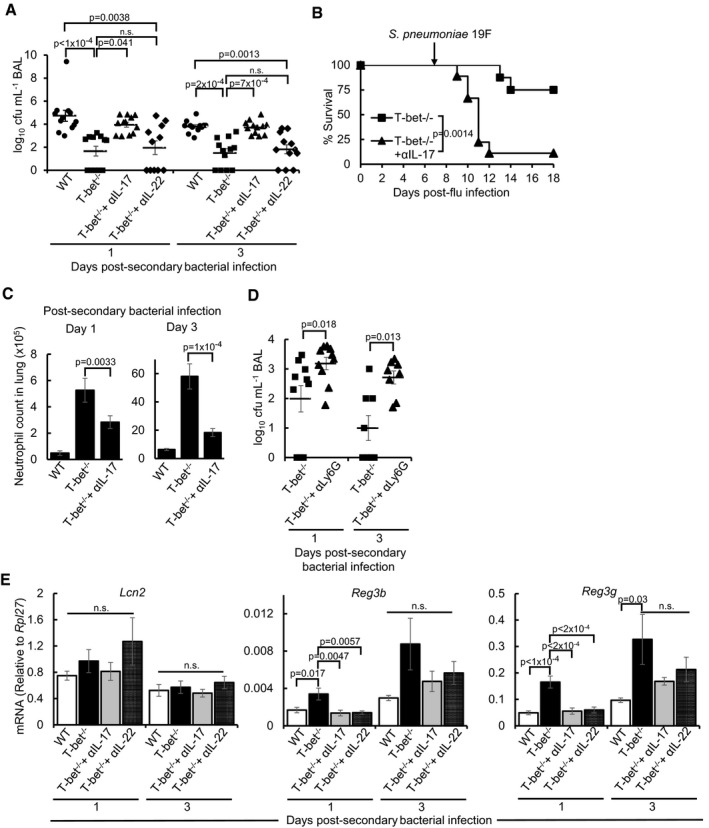
- Bacterial load as determined by enumeration of cfu in BAL following administration of αIL‐17 or αIL‐22 blocking antibody. Control wild‐type and T‐bet−/− groups were administered with IgG isotype control antibody.
- T‐bet−/− mice were administered αIL‐17 or IgG isotype control antibody every other day from day −2 post‐secondary bacterial infection and monitored for survival.
- Absolute neutrophil counts in the lungs following administration of αIL‐17 or IgG isotype control antibody, as determined by flow cytometry.
- Pneumococcal cfu in BAL following αLy6G antibody‐mediated neutrophil depletion. Control T‐bet−/− mice were administered with IgG isotype control antibody. See also Fig EV5A.
- mRNA expression of antimicrobial peptides in lungs following administration of αIL‐17, IL‐22 or IgG control antibody, as determined by qPCR.
Figure EV5. Neutrophil depletion and viral load determination following antibody treatment, related to Fig 5 .
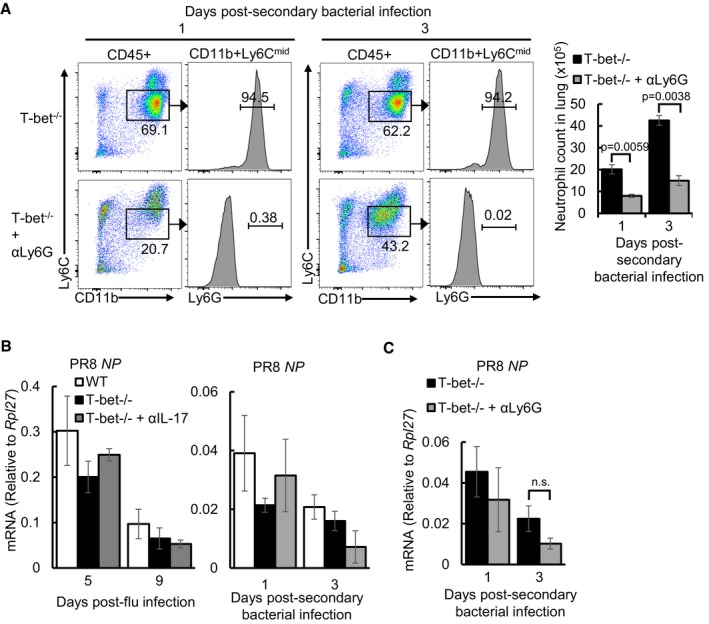
- Frequency and absolute numbers of pulmonary neutrophil depletion in T‐bet−/− lungs following αLy6G‐mediated depletion compared to IgG‐administered controls as assessed by flow cytometry. Numbers in flow cytometry plots indicate frequency of gated cells in each throughput.
- Relative amounts of PR8 viral NP mRNA following αIL‐17 treatment from day 0 (virus only infection, left panel) or from day 5 (bacterial superinfection, right panel) post‐influenza virus infection. Subsequent doses were administered every other day. Wild‐type and control T‐bet−/− mice were given IgG isotype control antibody treatment.
- Relative amount of PR8 viral NP mRNA following αLy6G or IgG isotype control antibody treatment as assessed by qPCR.
Th17 responses are known to mediate antibacterial defences through inducing antimicrobial peptides (Liang et al, 2006). Compared to wild‐type lungs, the expression of Reg3b and Reg3g antimicrobial peptides, but not lipocalin 2 (Lcn2), was increased in T‐bet−/− lungs at day 1 post‐secondary bacterial infection (Fig 5E). Expression of Reg3g remained elevated in T‐bet−/− lungs up to day 3 post‐infection (Fig 5E). Neutralization of IL‐17 and IL‐22 reduced expression of Reg3b and Reg3g in T‐bet−/− lungs at day 1 post‐secondary bacterial infection. Therefore, it appears that IL‐17‐ and IL‐22‐mediated expression of antimicrobial peptides is involved only in the early phase of post‐influenza bacterial infection, while IL‐17‐mediated neutrophil recruitment is a major factor conferring protection to T‐bet−/− mice.
IL‐17‐producing CD8+, CD4+ and γδ T cells contribute to protection during post‐influenza bacterial superinfection in T‐bet‐deficient mice
To determine the cell types responsible for type‐17‐mediated immune protection, intracellular staining of IFNγ and IL‐17 on days 0, 1 and 3 post‐secondary bacterial‐infected lung lymphocytes was performed. The data indicated an elevated frequency of IL‐17‐producing cells and a reduction in IFNγ‐producing cells in T‐bet−/− lungs (Fig 6A), suggesting a shift from type 1 to type 17 immune responses due to the loss of T‐bet, as also observed by others (Lazarevic et al, 2011). Further assessment of individual cell types revealed that up to 35% of total CD8+, CD4+ and γδ T cells in T‐bet‐deficient lungs accounted for more than 75% of all IL‐17‐producing cells (Fig 6B and C), while the proportion of IL‐17‐producing NK cells was negligible. Amongst IL‐17+ cells in T‐bet−/− lungs, γδ T cells form the majority in proportion and absolute numbers prior to bacterial superinfection, while CD8+ T cells constitute the highest frequency and numbers by day 3 post‐secondary bacterial infection (Fig 6C). Hence, we conclude that IL‐17‐protection in T‐bet−/− lungs against secondary bacterial infection is mediated through IL‐17‐producing T cells, notably through CD8+ and γδ T cells.
Figure 6. CD8+, CD4+ and γδ T cells are major sources of IL‐17 in influenza and secondary bacterial superinfected T‐bet−/− lungs.
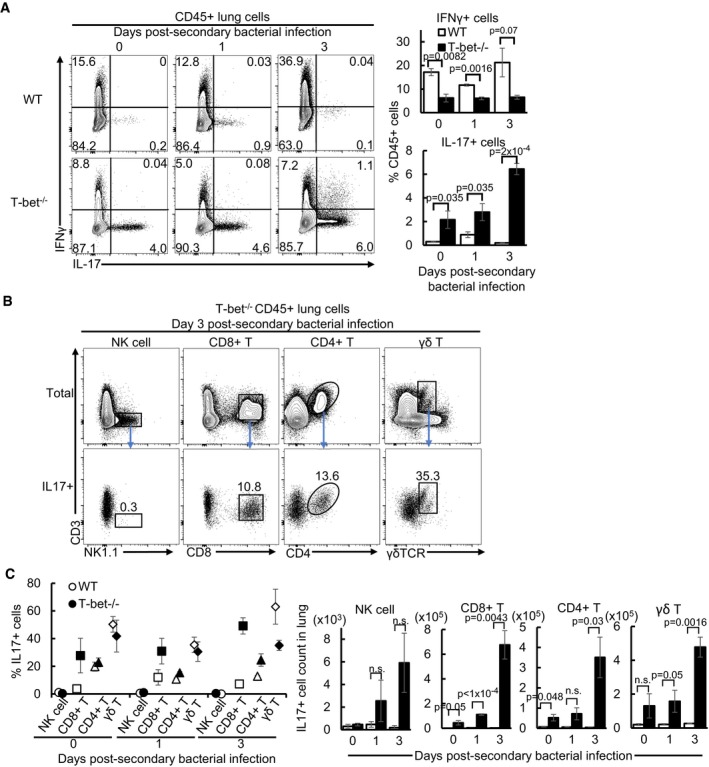
- Frequency of IFNγ+ and IL‐17+ immune cells following intracellular cytokine staining and flow cytometry.
- Representative flow cytometry plots of total and IL‐17+ lymphocyte subsets on day 3 post‐secondary bacterial‐infected lungs following intracellular cytokine staining. Numbers in plots indicate the frequency of each IL‐17+ cell subset over total numbers of respective cell types.
- Frequency over total IL‐17+ cells and absolute numbers of each IL‐17 producing lymphocyte subset on days 0, 1 and 3 post‐secondary bacterial superinfected lungs as assessed by flow cytometry of intracellular cytokine stained cells.
Taken together, our results support a mechanism of tripartite host–viral–bacterial interaction whereby induction of IL‐17 and IL‐22 in T‐bet−/− hosts during primary influenza virus infection establishes an inflammatory environment characterized by elevated neutrophils and antimicrobial peptide expression in the lungs. These inflammatory defences are suppressed in wild‐type hosts, but primes T‐bet‐deficient hosts against lethality from subsequent bacterial infections (Fig 7).
Figure 7. Summary of immune responses and effects of T‐bet deficiency following influenza virus and bacterial superinfection.
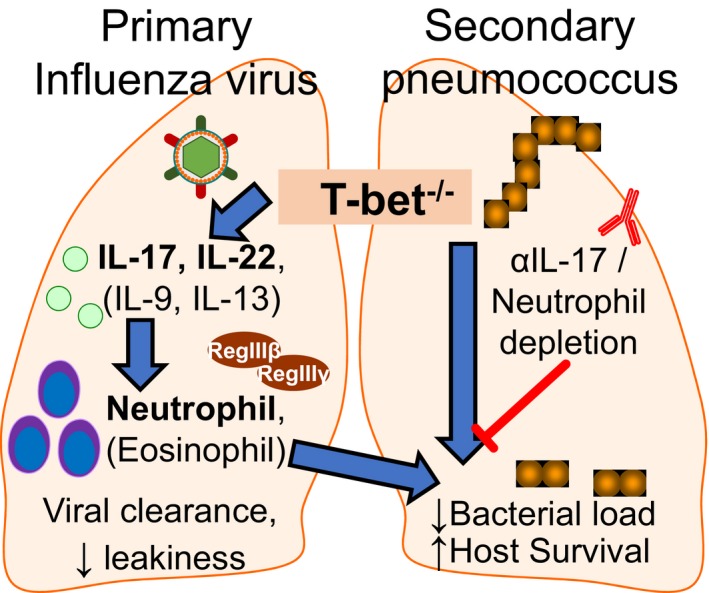
T‐bet‐deficient lungs infected with sub‐lethal dose of influenza virus upregulate Th2 and Th17 responses, leading to increased pulmonary infiltration of neutrophils and eosinophils. This altered inflammatory response did not compromise efficiency in viral clearance or eventual tissue homeostasis, and in fact reduced tissue leakiness following infection. Furthermore, the immune environment resulting from influenza‐infected T‐bet‐deficient lungs conferred immunity and survival advantage to pneumococcal superinfected hosts. Correspondingly, neutralization of IL‐17 or neutrophil depletion by antibody treatment in T‐bet−/− hosts reduced pulmonary neutrophil infiltration or antimicrobial peptide expression, reversing immunity against secondary bacterial infection.
Discussion
Type 1 immune responses are essential for defence against all classes of invading pathogens. As a central regulator of type 1 immunity, T‐bet has been implicated in infections by pathogenic bacteria and parasites affecting the lungs, skin, central nervous system and gut (reviewed by Lazarevic & Glimcher, 2011), wherein loss of function mostly resulted in greater pathogen burden. In viral infections, loss of T‐bet resulted in greater viral load (Matsui et al, 2005; Svensson et al, 2005; Rubtsova et al, 2013), attributable to reduced NK and CD8+ T cell cytolytic activity and dysregulated B cell responses. Beyond acute infections, T‐bet is also involved in secondary antiviral defences through mediating the formation of memory B and T cells (Joshi et al, 2007; Marshall et al, 2011; Knox et al, 2017). Nevertheless, a recent study found that the loss of T‐bet did not result in marked defects in the ability to clear acute respiratory syncytial virus (RSV) infection despite reduced cellular and humoral antiviral responses, but instead led to increased allergic responses (Glanville et al, 2016). Similarly, our study showed that infection with the influenza virus did not significantly affect viral loads, tissue homeostasis or survival in T‐bet‐deficient hosts. Strikingly, massive infiltration of neutrophils and eosinophils, accompanied by production of Th2 and Th17 cytokines, was observed in the inflammatory phase of influenza‐infected T‐bet‐deficient lungs. This altered inflammatory environment instead provided immunity and survival advantage against subsequent bacterial superinfection, while neutralization of IL‐17 or neutrophil depletion in T‐bet−/− mice decreased antibacterial immunity. Hence, contrary to the existing view that T‐bet is largely required for resistance against primary virus or bacterial infections, our results provide new evidence to the growing perspective that T‐bet loss need not eventually lead to greater susceptibility to infections.
T‐bet transcription factor is expressed in a wide variety of immune cells, including NK cells, NKT cells, B cells, CD4+ and CD8+ T cells, dendritic cells and γδ T cells (reviewed by Lazarevic et al, 2013). Its mechanism of function is mainly through expression of IFNγ and mediating maturation, cytotoxicity or IgG class switching of these cells. All these immune responses are crucial against viral infections, yet surprisingly, the loss of T‐bet did not result in marked deficiency in defence against the influenza virus. We show that the viral load is comparable in T‐bet‐deficient lungs and wild‐type counterparts throughout the course of virus infection, suggesting that the kinetics of viral clearance is not affected. Survival is also indifferent under high virus doses between wild‐type and T‐bet−/− mice. This can be explained by the largely intact immune responses in T‐bet−/− mice, with CD4+ T, CD8+ T and B cells still present in comparable numbers. Although NK cells were reduced in the resting lung, their numbers were unaffected at the early phase of virus infection, where they are known to be the predominant mediators of cytotoxicity against viral infection (Leung & Ada, 1981; Stein‐Streilein & Guffee, 1986). These mechanistic insights suggest that T‐bet is dispensable for the clearance of influenza virus, or there could be the presence of compensatory functions by other regulatory factors. Eomes, a T‐box transcription factor homologous to T‐bet and co‐expressed in CD8+ T and NK cells, could be one possible factor, as shown by earlier studies that demonstrated their complementary roles against virus infections (Intlekofer et al, 2005, 2008). Increased neutrophils and eosinophils in T‐bet−/− lungs may also play compensatory roles to other immune cells following influenza virus infection (Fujisawa, 2008; Tate et al, 2011; Hufford et al, 2012; Samarasinghe et al, 2017).
Further to cellular responses, antiviral gene expression and production of type I and II IFNs in the lungs were also similar, further supporting the robustness of immune responses in the context of sub‐lethal influenza infection. Our findings on T‐bet‐deficient mice support results from a number of related Th1 knockout studies that showed no defects in influenza resistance (Graham et al, 1993; Nguyen et al, 2000; Price et al, 2000).
The presence of IFNγ in the BAL in infected T‐bet−/− lungs at levels similar to that of wild‐type levels is particularly intriguing, considering the role of T‐bet in the production of IFNγ within several cell types (Szabo et al, 2002; Lugo‐Villarino et al, 2003; Chen et al, 2007; Klose et al, 2013; Barros‐Martins et al, 2016). Correspondingly, we also observed a reduction in mRNA expression of Ifng and frequency of IFNγ‐producing cells. Nevertheless, the unaffected levels of total secreted IFNγ in T‐bet‐deficient hosts during inflammation, as observed by us and in other studies (Burrell et al, 2008; Guo et al, 2009), suggest post‐translational control or expression based on specific immune environmental cues. This additional level of control on the physiological amounts of IFNγ warrants further investigation and may have important implications on aberrant type 1 responses.
Excessive neutrophils and eosinophils in the lungs following influenza virus infection has been linked to exacerbated lung injury and respiratory failure (Jeon et al, 2010; Narasaraju et al, 2011). In this study, pulmonary injury was assessed by histology to be more severe in day 9 post‐influenza‐infected T‐bet−/− lungs, correlating with significant increase in neutrophil and eosinophil numbers. Nevertheless, subsequent timepoint at day 15 post‐influenza virus infection showed reduced pulmonary injury and comparable efficacy in tissue repair. Moreover, reduced lung leakiness at day 25 was observed in T‐bet−/− lungs, supporting the presence of mechanisms that might ameliorate excessive inflammation or promote homeostasis. Previous studies have shown IL‐22 and IL‐6 cytokines to be essential in lung repair (Kumar et al, 2013; Pociask et al, 2013; Yang et al, 2017). While these two cytokines were observed to be elevated in T‐bet−/− lungs during day 9 post‐influenza infection, it is currently unclear whether this early increase in cytokines has contributed to the tissue recovery at later timepoints. Overall, it appears that the impact of neutrophilia and eosinophilia in T‐bet−/− lungs is not permanently detrimental following virus infection, and tissue homeostasis is not adversely affected with the loss of T‐bet functions.
Influenza–bacterial superinfection is a common complication of flu pandemic and a major cause of severe pneumonia and morbidity in the human population (Metersky et al, 2012). One of the causes of lethal synergism between influenza viruses and respiratory bacterial pathogens involves suppression of immune responses to bacteria by prior influenza virus infection (McNamee & Harmsen, 2006; Sun & Metzger, 2008; Shahangian et al, 2009; Ghoneim et al, 2013; Cao et al, 2014). Therefore, reactivating pulmonary immune defences in influenza‐infected lungs appears to be an outstanding opportunity against bacterial superinfection. The marked increase in neutrophil infiltration as well as production of Th17 cytokines IL‐17 and IL‐22 in influenza‐infected T‐bet−/− lungs at a timepoint where hosts are most susceptible to bacterial infection (Sun & Metzger, 2008; Chertow & Memoli, 2013) prompted us to examine antibacterial defences of T‐bet‐deficient hosts during secondary infection. Furthermore, previous studies have attributed the importance of neutrophils and Th17 immune responses in this infection model (McNamee & Harmsen, 2006; Shahangian et al, 2009; Kudva et al, 2011; Ivanov et al, 2013; Cao et al, 2014). We found that decreased bacterial load in T‐bet−/− superinfected lungs and increased survival were attributable to IL‐17‐mediated neutrophil recruitment. While IL‐17 has also been shown to induce antibacterial defence through upregulating antimicrobial peptides (Liang et al, 2006; Archer et al, 2016), our observation that there is no considerable increased expression in T‐bet−/− lungs at day 3 post‐bacterial infection suggests that this mechanism may only be applicable to early bacterial load control. In addition, IL‐22 neutralization did not abrogate enhanced bacterial clearance in T‐bet−/− mice. Hence, we propose IL‐17‐mediated neutrophil recruitment as the major mechanism conferring survival advantage to secondary infection in T‐bet−/− mice. As it is known that T‐bet inhibits development of Th17 cells (Villarino et al, 2010; Lazarevic et al, 2011), our results also suggest suppressed Th17‐mediated antibacterial immunity as an underlying mechanism to lethal synergism between influenza and bacterial superinfection in T‐bet sufficient hosts. Inhibition of T‐bet thus confers protection against secondary bacterial infection through reactivating Th17‐mediated antibacterial defences.
Enhanced type 17 responses, due to the lack of T‐bet, has been described in a number of cell types, including CD8+ T cells (Burrell et al, 2008; Intlekofer et al, 2008), CD4+ T cells (Rangachari et al, 2006; Yuan et al, 2008; Guo et al, 2009; Lazarevic et al, 2011) and γδ T cells (Barros‐Martins et al, 2016). Of these, Lazarevic et al described a mechanism wherein T‐bet deficiency allowed for the expression of Th17 transcription factor Rorγt in Th1 cells, driving naïve CD4+ T cells into Th17 differentiation instead of Th1 lineage. We observed increased number of IL‐17‐producing immune cells with a corresponding reduction in IFNγ‐producing cells in T‐bet‐deficient infected lungs, suggesting that a similar mechanism of T‐bet‐mediated Th17 suppression might be involved. While it has been shown that cell intrinsic loss of IL‐10 promotes CD4+ T cell differentiation into the Th17 lineage with a corresponding reduction in T‐bet expression (McKinstry et al, 2009), we did not identify a reciprocal reduction of IL‐10 in BAL of infected T‐bet‐deficient lungs in our cytokine screen eluding to elevated type 17 immunity. Considering that a significant proportion of the collective type 17 immune response involves CD8+ and γδ T cells and are protective against pathogen infection, future studies might consider a systematic delineation of the molecular and cellular events underlying T‐bet‐mediated type 17 immunity in these other cell types.
B cells were observed to be increased in T‐bet−/− lungs at day 15 post‐influenza infection and day 3 post‐secondary bacterial infection. Current knowledge of T‐bet function in B cells are limited to its role in promoting IgG2a class switching leading to the control of viral infections (Peng et al, 2002; Rubtsova et al, 2013; Barnett et al, 2016) and development of memory B cells (Wang et al, 2012; Knox et al, 2017). Gene expression analysis also shows reduced transcripts that are linked to proliferation and regulation of tissue migration in T‐bet‐deficient B cells (Barnett et al, 2016). Hence, the increase in B cells in T‐bet‐deficient lungs shown in this study may be due to dysregulation in B cell trafficking or aberrant cell proliferation. However, such mechanisms need to be further tested, and the physiological significance of increased T‐bet‐deficient B cells post‐infection remains to be determined.
In summary, our study demonstrates that T‐bet is dispensable for influenza virus clearance and loss of T‐bet functions is not adverse to disease outcomes. However, the increased Th17 immune responses and pulmonary neutrophil recruitment in influenza‐infected T‐bet−/− host proved to be beneficial through increased antibacterial immunity. These results support the suppression of T‐bet as a potential treatment option against lethal bacterial superinfections, without affecting protection against primary influenza virus.
Materials and Methods
Mice and pathogens
Age‐ (8–13 weeks) and gender‐matched C57BL/6 wild‐type and T‐bet−/− mice (004648) from Jackson's laboratory were used in all experiments. Influenza virus used for infection is the mouse‐adapted H1N1 PR/8/34 (PR8) strain (ATCC, VR‐95). Propagation of viruses was performed in embryonated chicken eggs at 37°C for 72 h. Allantoic fluid was harvested and viral titres were determined by standard plaque assay in Madin–Darby canine kidney (MDCK) cells. Streptococcus pneumoniae serotypes 19F and S3 used for infection were originally clinical isolates from National University Hospital, Singapore, provided by A/Prof Vincent Chow (NUS Department of Microbiology and Immunology). Bacterial stocks were cultured to mid‐log phase in brain–heart infusion broth (Sigma) supplemented with 5% foetal bovine serum (Hyclone), at 37°C under anaerobic conditions. Bacteria counts were determined by plating on trypticase soy agar with 5% sheep blood (TSA II, Becton Dickinson) following serial dilution and cultured for 18 h at 37°C under anaerobic conditions. All mice experiments were performed under guidelines and protocols (R15‐1141, BR15‐1142) approved by the NUS Institutional Animal Care and Use Committee.
Infection
Administration of pathogens to mice was performed through the intratracheal route. Specifically, mice anaesthetized with ketamine and medetomidine cocktail were kept upright with the tongue held out tightly to prevent swallowing of the inoculum. Influenza virus diluted in 75 μl phosphate‐buffered saline (PBS) or S. pneumoniae in 25 μl of PBS was introduced into mouth to be breathed into lungs directly through the trachea. Reversal of anaesthesia was achieved by intraperitoneal injection of atipamezole hydrochloride. Pathogen doses were empirically determined to cause almost no mortality when given alone (Figs 1A and EV3A) and more than 50% mortality in wild‐type mice following post‐influenza secondary bacterial infection (Figs 3B and EV3B). Weight and survival were monitored daily, and mice with significant morbidity or more than 30% weight loss were euthanized and considered having succumbed to infection. Recovery in mice was assessed as having gained more than 0.3 g in weight for two consecutive days and displaying improved clinical characteristics.
In vivo cytokine neutralization
Cytokine neutralization was performed by administering a total of 100 μg of anti‐IL‐17A monoclonal antibody (BioXCell, Clone 17F3), 250 μg of anti‐IL‐22 monoclonal antibody (Genentech, Clone 8E11) or equal amounts of mouse IgG1 isotype control antibody (Genentech) per mouse on each dose. For depletion of IL‐17 and IL‐22 in post‐secondary bacterial superinfection, antibody treatments were given every other day from 2 days before bacterial infection (or day 5 post‐influenza virus infection). Treatments were administered through both intranasal (20 μl) and intraperitoneal (200 μl) routes. However, after day 7 post‐influenza virus infection where lungs are severely inflamed, the full dose of antibody was administered intraperitoneally in 200 μl volume.
Neutrophil depletion
For in vivo neutrophil depletion, 150 μg anti‐Ly6G antibody (BioXCell, Clone 1A8) or rat IgG2a isotype control antibody (BioXCell) was administered in 100 μl volume intravenously by tail vein injection. Treatment was given from day 5 post‐influenza infection and every 24 h thereafter. Up to 70% depletion was confirmed by staining and enumeration of CD45+CD11b+Ly6Cmid cells, while binding of blocking antibody to remaining neutrophils was observed by negative staining to Ly6G antibody (Fig EV5A), as assessed by flow cytometry.
Histology
Mouse left lung lobes were fixed in 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS) at 4°C for 24–48 h. Following dehydration and embedding in paraffin, 5 μm of lung sections was excised onto glass slides. For assessing overall lung pathology, haematoxylin and eosin (H&E) staining and pathological evaluation were performed at the Advanced Molecular Pathology Laboratory, IMCB, A*STAR, Singapore. Scoring was performed in a double‐blind manner from the assessment of 3–5 infected lungs per experimental group. Pathology was based on assessment for inflammation, pulmonary injury and tissue response, with specific criteria detailed in Fig EV1C. Immunohistochemistry was performed by antigen retrieval at 95°C in 10 mM citrate buffer, pH 6 for 20 min and subsequently stained with anti‐podoplanin antibody (Rnd #AF3244), anti‐goat HRP secondary antibody (Sigma) and liquid DAB+ substrate kit (GBI labs). Images were captured with Axio Observer Z1 microscope (Carl Zeiss) and Axiocam 105 colour (Carl Zeiss) camera under 20× magnification. Tile images were then stitched using the Zen 2 core imaging software (Carl Zeiss).
Processing of lungs for single‐cell suspensions
Lung lobes were cut into 0.5‐ to 1‐mm pieces in Roswell Park Memorial Institute media (RPMI 1640, Thermofisher Scientific) supplemented with 0.5 mg/ml collagenase D (Roche) and 20 U/ml Dnase I (Roche) and further dissociated using GentleMACS C tubes (Miltenyi Biotec) through running of program Lung_01, followed by incubation for 15 min at 37°C, and then the program Lung_02. Single cells were filtered through 70‐μm strainers, re‐suspended in 30% Percoll (GE Healthcare) and centrifuged at 500 g (with brakes off) for 10 min at 4°C to remove cell debris. Single‐cell suspensions were subjected to red blood cell lysis using ACK lysis buffer (Thermofisher Scientific). Resultant cells were stained with trypan blue (Gibco) and counted in a haemocytometer before flow cytometry analysis.
Flow cytometry
Antibodies used for flow cytometry analysis were as follows: CD45.2 (104), NK1.1 (PK136), CD11b (M1/70), CD11c (N418), CD16/32 (93), Ly6C (HK1.4), F4/80 (BM8), MHCII (M5/114.15.2), Ly6G (1A8), CD8a (53‐6.7), B220 (RA3‐6B2), TCRβ (H57‐597), CD4 (GK1.5), CD3e (145‐2C11), IL‐17A (eBio17B7) were from eBioscience. Antibodies targeting CD24 (M1/69), γδ TCR (GL3) and IFNγ (XMG1.2) were from Biolegend. Antibody targeting SiglecF (E50‐2440) was from Becton Dickinson, while anti‐CD64 (REA286) was from Miltenyi Biotec. All surface antigen staining was performed in 1× PBS supplemented with 2% FBS on ice after staining with fixable viability dye (eBioscience) and Fc receptor blocking. IC fixation buffer (eBioscience) was used to fix cells after staining. For detection of intracellular cytokines, single‐cell suspensions were incubated in RPMI‐1640 media (Gibco) supplemented with 10% FBS (Hyclone) and GolgiPlug protein transport inhibitor (Becton Dickinson) in the absence (for unstimulated controls) or presence of 30 ng/ml Phorbol 12‐Myristate 13‐Acetate (Sigma) and 500 ng/ml ionomycin (Thermofisher Scientific) for 5 h at 37°C. Following cell surface staining, samples were fixed and permeabilized with FoxP3 transcription factor staining set (eBioscience) and stained with antibodies against intracellular cytokines in permeabilization buffer. Flow cytometry data were acquired on BD LSRFortessa with compensation applied using stained compensation beads (eBioscience). Analysis was performed on FlowJo V10 software (TreeStar), and cells were gated using parallelly stained fluorescence minus one (FMO) controls or unstimulated cells for intracellular cytokine stains. Gating strategies are provided in Fig EV2C.
Bronchoalveolar lavage
Mice were euthanized with overdose of anaesthetic (ketamine and medetomidine cocktail), and peripheral blood was drained by cutting the posterior vena cava. Mouse lungs were perfused with three separate washes of 1, 0.5 and 0.5 ml ice‐cold PBS, respectively, through a 23‐G cannula inserted into the trachea. Cells in BAL were pelleted by centrifugation at 800 g for 10 min at 4°C and supernatant was collected for subsequent analysis.
Protein assays
Lactate dehydrogenase levels in BAL were assessed using LDH cytotoxicity assay kit (Thermofisher Scientific #88953). Total protein in BAL was assessed using Bradford assay (Bio‐Rad #5000006). Concentration of albumin in BAL was determined by ELISA (Bethyl Laboratories # E90‐134_32). Concentrations of IL‐17 and IL‐22 were determined by ELISA (Thermofisher Scientific IL‐17 #88‐7371, IL‐22 #88‐7422). Optical density readings were acquired through a microplate reader (Biotek). Concentrations of IFNα and IFNβ were determined by ProcartaPlex assay (Thermofisher Scientific IFNα #EPX01A‐26027‐901, IFNβ #EPX01A‐26044‐901). Cytokine and chemokine screening in BAL was performed using the mouse cytokine 20‐plex for Luminex platform (Thermofisher Scientific #LMC0006M). Concentrations of IL‐9, IL‐18, IL‐22 and IL‐28 were determined using ProcartaPlex Mix&match mouse panel (Thermofisher Scientific #MXCE3XC). ProcartaPlex and protein bead array data were acquired through the Magpix system and xPONTENT v3.1 software. All assays were carried out according to respective manufacturer's instructions.
Immunoblotting
Right superior lobe of mouse lung was homogenized in ice‐cold 500 μl RIPA buffer (50 mM Tris pH 7.5, 0.5% sodium deoxycholate, 150 mM NaCl, 0.1% SDS, 1 mM EDTA, 1% NP‐40) supplemented with 1× protease inhibitor (Roche #05056489001) in a 2‐ml Eppendorf tube. The homogenate was clarified by centrifugation at 16,000 g for 10 min at 4°C, and supernatant was collected and quantitated using Bradford assay (Bio‐Rad). To detect target proteins, 10 μg of total protein was loaded onto 12% SDS–PAGE gels, followed by transfer to PVDF membrane using the Trans‐Blot Turbo RTA transfer kit (Bio‐Rad, #170‐4273) on the Trans‐Blot Turbo Transfer System (Bio‐Rad) using the “Standard SD” program. Membranes were blocked in 5% skimmed milk (Difco #232100) in TBST (TBS + 0.05% Tween‐20) for 1 h at room temperature and incubated with anti‐podoplanin antibody (Rnd systems, #AF3244) at 1:3,000 dilution in 5% milk in TBST at 4°C. Blots were washed three times for 10 min each in TBST and incubated with rabbit anti‐goat HRP secondary antibody for 1 h at 1:3,000 dilution in 5% milk in TBST for 1 h at room temperature. Subsequently, blots were washed three times for 5 min each in TBST and once for 10 min in TBST. Detection of secondary antibody was performed by incubation in WesternBright ECL HRP substrate (Advansta #K‐12045‐D20) and imaged using ImageQuant LAS 4000 Mini (GE Healthcare). For detection of loading controls, membranes were stripped in Restore Western Blot stripping buffer (Thermofisher Scientific #21059) and re‐probed with anti‐β tubulin antibody (Cell Signaling Technologies, #2146).
RNA extraction and quantitative PCR
Right inferior lobe of mouse lungs was homogenized in 500 μl TRIzol reagent (Thermofisher Scientific #15596026) in a 2‐ml Eppendorf tube and 100 μl of chloroform added with 15 s of vigorous shaking. Sample was then incubated for 2 min at room temperature and centrifuged at 12,000 g, 10 min at 4°C. The aqueous phase was then transferred to a fresh tube, and 250 μl of isopropanol was added to each sample, vortexed briefly and incubated at −80°C for 1 h or kept overnight. Thereafter, RNA was precipitated by centrifugation at 18,000 g, 10 min, 4°C and supernatant was aspirated. To reduce DNA contamination and further purify RNA, RNA pellet was re‐dissolved in 500 μl of TRIzol and the extracted using the procedures as described above. RNA pellet from the second round of extraction was washed in 500 μl of ice‐cold 70% ethanol and pelleted at 8,000 g, 5 min, 4°C. RNA pellet was vacuum‐dried and dissolved in 20 ml nuclease‐free water. 2.5 μg of RNA was used for reverse transcription using SuperScript™ III First‐Strand Synthesis System (Thermofisher Scientific #18080051) according to manufacturer's instructions. The resulting cDNA was diluted 10 times in nuclease‐free water. Two microliter of cDNA was used for quantitation of the following genes using the given primers listed in Appendix Table S1. Quantitative PCR (qPCR) was performed using GoTaq qPCR Master Mix (Promega #A6002) under the following parameters: Denaturation at 95°C for 2 min, 45 cycles of amplification at 95°C for 10 s, annealing at 60°C for 1 min. Primers used were verified to produce single peaks under melting curve analyses. mRNA expression of target genes was expressed relative to the expression of Rpl27.
Viral and bacterial load determination
For assessing viral loads, total RNA from homogenized mouse inferior right lung lobes were extracted and reverse‐transcribed to cDNA (Thermofisher Scientific) as described above. H1N1 PR8 viral NP mRNA was then quantified by qPCR. For assessing bacterial loads, BAL fluid or lung homogenates (2 ml) were serially diluted with 1× PBS and plated onto trypticase soy agar with 5% sheep blood (TSA II, Becton Dickinson). The number of cfu was determined after culturing for 18 h at 37°C under anaerobic conditions.
Statistics
All statistical analysis was performed using the Prism 7.04 software (GraphPad). A P‐value of < 0.05 was considered significant.
Author contributions
JZE conceived, designed and performed experiments with intellectual input from JLD; RAGK provided assistance to the performing of experiments and analysis of data. JLD supervised and coordinated the study. JZE and JLD wrote the manuscript with input from RAGK.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Acknowledgements
This work was supported by the National Medical Research Council (NMRC/CBRG/0055/2013), Ministry of Education (MOE2013‐T2‐2‐007 and R‐154‐000‐A76‐114), Singapore. J.Z.E. is a graduate scholar of the NUS Graduate School of Integrative Sciences and Engineering Scholarship. We fully acknowledge A/Prof Vincent Chow's lab (NUS Department of Microbiology and Immunology) and Dr Li Liang (NTU School of Biological Sciences) for preparation of stock aliquots of influenza and S. pneumoniae, as well as providing help and advice in setting up of the infection model. We thank the NUHS flow cytometry laboratory unit for their service and technical help.
The EMBO Journal (2019) 38: e99176
References
- Archer NK, Adappa ND, Palmer JN, Cohen NA, Harro JM, Lee SK, Miller LS, Shirtliff ME (2016) Interleukin‐17A (IL‐17A) and IL‐17F are critical for antimicrobial peptide production and clearance of Staphylococcus aureus nasal colonization. Infect Immun 84: 3575–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, Gunn B, Chen D, Paley MA, Alter G, Reiner SL, Lauer GM, Teijaro JR, Wherry EJ (2016) Cutting edge: B cell‐intrinsic T‐bet expression is required to control chronic viral infection. J Immunol 197: 1017–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros‐Martins J, Schmolka N, Fontinha D, Pires de Miranda M, Simas JP, Brok I, Ferreira C, Veldhoen M, Silva‐Santos B, Serre K (2016) Effector γδ T cell differentiation relies on master but not auxiliary Th cell transcription factors. J Immunol 196: 3642–3652 [DOI] [PubMed] [Google Scholar]
- Bedoya F, Cheng G‐S, Leibow A, Zakhary N, Weissler K, Garcia V, Aitken M, Kropf E, Garlick DS, Wherry EJ, Erikson J, Caton AJ (2013) Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus‐infected mice. J Immunol 190: 6115–6125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braciale TJ, Sun J, Kim TS (2012) Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol 12: 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK (2008) CD8+ Th17 mediate costimulation blockade‐resistant allograft rejection in T‐bet‐deficient mice. J Immunol 181: 3906–3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Wang D, Xu F, Gong Y, Wang H, Song Z, Li D, Zhang H, Li D, Zhang L, Xia Y, Xu H, Lai X, Lin S, Zhang X, Ren G, Dai Y, Yin Y (2014) Activation of IL‐27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO Mol Med 6: 120–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) (2009) Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1) ‐ United States, May–August 2009. MMWR Morb Mortal Wkly Rep 58: 1071–1074 [PubMed] [Google Scholar]
- Chen L, He W, Kim ST, Tao J, Gao Y, Chi H, Intlekofer AM, Harvey B, Reiner SL, Yin Z, Flavell RA, Craft J (2007) Epigenetic and transcriptional programs lead to default IFN‐γ production by γ∆ T cells. J Immunol 178: 2730–2736 [DOI] [PubMed] [Google Scholar]
- Chertow DS, Memoli MJ (2013) Bacterial coinfection in influenza: a grand rounds review. JAMA 309: 275–282 [DOI] [PubMed] [Google Scholar]
- Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM (2007) Transcription factors T‐bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol 8: 145–153 [DOI] [PubMed] [Google Scholar]
- Dolfi DV, Mansfield KD, Polley AM, Doyle SA, Freeman GJ, Pircher H, Schmader KE, Wherry EJ (2013) Increased T‐bet is associated with senescence of influenza virus‐specific CD8 T cells in aged humans. J Leukoc Biol 93: 825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Miaw S‐C, Yu J‐S, Chen T‐C, Lin C‐Y, Lin Y‐C, Chang C‐S, He Y‐C, Chuang S‐H, Yen M‐I, Huang C‐T (2013) Altered T‐bet dominance in IFN‐γ‐decoupled CD4+ T cells with attenuated cytokine storm and preserved memory in influenza. J Immunol 190: 4205–4214 [DOI] [PubMed] [Google Scholar]
- Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A (2015) TRAIL+ monocytes and monocyte‐related cells cause lung damage and thereby increase susceptibility to influenza‐Streptococcus pneumoniae coinfection. EMBO Rep 16: 1203–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa H (2008) Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol 82: 2772–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim HE, Thomas PG, McCullers JA (2013) Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol 191: 1250–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville N, Peel TJ, Schröder A, Aniscenko J, Walton RP, Finotto S, Johnston SL (2016) Tbet deficiency causes T helper cell dependent airways eosinophilia and mucus hypersecretion in response to rhinovirus infection. PLoS Pathog 12: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MB, Dalton DK, Giltinan D, Braciale VL, Stewart TA, Braciale TJ (1993) Response to influenza infection in mice with a targeted disruption in the interferon gamma gene. J Exp Med 178: 1725–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Cobb D, Smeltz RB (2009) T‐bet inhibits the in vivo differentiation of parasite‐specific CD4+ Th17 cells in a T cell‐intrinsic manner. J Immunol 182: 6179–6186 [DOI] [PubMed] [Google Scholar]
- Hua L, Yao S, Pham D, Jiang L, Wright J, Sawant D, Dent AL, Braciale TJ, Kaplan MH, Sun J (2013) Cytokine‐dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J Virol 87: 11884–11893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford MM, Richardson G, Zhou H, Manicassamy B, García‐Sastre A, Enelow RI, Braciale TJ (2012) Influenza‐infected neutrophils within the infected lungs act as antigen presenting cells for anti‐viral CD8+ T cells. PLoS One 7: e46581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultgren OH, Verdrengh M, Tarkowski A (2004) T‐box transcription‐factor‐deficient mice display increased joint pathology and failure of infection control during staphylococcal arthritis. Microbes Infect 6: 529–535 [DOI] [PubMed] [Google Scholar]
- Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH (2005) T helper cell fate specified by kinase‐mediated interaction of T‐bet with GATA‐3. Science 307: 430–433 [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL (2005) Effector and memory CD8+ T cell fate coupled by T‐bet and eomesodermin. Nat Immunol 6: 1236–1244 [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, Reiner SL (2007) Requirement for T‐bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med 204: 2015–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, DeJong CS, Shin H, Hunter CA, Wherry EJ, Lindsten T, Reiner SL (2008) Anomalous Type 17 response to viral infection by CD8+ T cells lacking T‐bet and eomesodermin. Science 321: 408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, Blanc F, De Trez C, Van Maele L, Dumoutier L, Huerre M‐R, Eberl G, Si‐Tahar M, Gosset P, Renauld JC, Sirard JC, Faveeuw C, Trottein F (2013) Interleukin‐22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol 87: 6911–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Pillai PS (2014) Innate immunity to influenza virus infection. Nat Rev Immunol 14: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata S, Mikami Y, Sun H‐W, Brooks SR, Jankovic D, Hirahara K, Onodera A, Shih H‐Y, Kawabe T, Jiang K, Nakayama T, Sher A, O'Shea JJ, Davis FP, Kanno Y (2017) The transcription factor T‐bet limits amplification of type I IFN transcriptome and circuitry in T helper 1 cells. Immunity 46: 983–991.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon EJ, Kim KH, Min KH (2010) Acute eosinophilic pneumonia associated with 2009 influenza A (H1N1). Thorax 65: 268–270 [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM (2007) Inflammation directs memory precursor and short‐lived effector CD8+ T cell fates via the graded expression of T‐bet transcription factor. Immunity 27: 281–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, D'Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A (2013) A T‐bet gradient controls the fate and function of CCR6‐RORγt+ innate lymphoid cells. Nature 494: 261–265 [DOI] [PubMed] [Google Scholar]
- Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, Robb ML, Ostrowski MA, Deeks SG, Slifka MK, Tomaras GD, Moir S, Moody MA, Betts MR (2017) T‐bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight 2: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, Alcorn JF (2011) Influenza A inhibits Th17‐mediated host defense against bacterial pneumonia in mice. J Immunol 186: 1666–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Thakar MS, Ouyang W, Malarkannan S (2013) IL‐22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol 6: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrañaga JM, Marcos PJ, Pombo F, Otero‐González I (2016) Acute eosinophilic pneumonia as a complication of influenza A (H1N1) pulmonary infection. Sarcoidosis Vasc Diffuse Lung Dis 33: 95–97 [PubMed] [Google Scholar]
- Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH (2011) T‐bet represses TH 17 differentiation by preventing Runx1‐mediated activation of the gene encoding RORγt. Nat Immunol 12: 96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH (2011) T‐bet in disease. Nat Immunol 12: 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH, Lord GM (2013) T‐bet: a bridge between innate and adaptive immunity. Nat Rev Immunol 13: 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KN, Ada GL (1981) Induction of natural killer cells during murine influenza virus infection. Immunobiology 160: 352–366 [DOI] [PubMed] [Google Scholar]
- Li L, Chong HC, Ng SY, Kwok KW, Teo Z, Tan EHP, Choo CC, Seet JE, Choi HW, Buist ML, Chow VTK, Tan NS (2015) Angiopoietin‐like 4 increases pulmonary tissue leakiness and damage during influenza pneumonia. Cell Rep 10: 654–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Tan X‐Y, Luxenberg DP, Karim R, Dunussi‐Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203: 2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Ohnishi N, Ni L, Akira S, Bacon KB (2003) CpG directly induces T‐bet expression and inhibits IgG1 and IgE switching in B cells. Nat Immunol 4: 687–693 [DOI] [PubMed] [Google Scholar]
- Lugo‐Villarino G, Maldonado‐Lopez R, Possemato R, Penaranda C, Glimcher LH (2003) T‐bet is required for optimal production of IFN‐γ and antigen‐specific T cell activation by dendritic cells. Proc Natl Acad Sci USA 100: 7749–7754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM (2011) Differential expression of Ly6C and T‐bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity 35: 633–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Moriya O, Yoshimoto T, Akatsuka T (2005) T‐bet is required for protection against vaccinia virus infection. J Virol 79: 12798–12806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauad T, Hajjar LA, Callegari GD, da Silva LFF, Schout D, Galas FRBG, Alves VAF, Malheiros DMAC, Auler JOC, Ferreira AF, Borsato MRL, Bezerra SM, Gutierrez PS, Caldini ETEG, Pasqualucci CA, Dolhnikoff M, Saldiva PHN (2010) Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med 181: 72–79 [DOI] [PubMed] [Google Scholar]
- Mayer KD, Mohrs K, Reiley W, Wittmer S, Kohlmeier JE, Pearl JE, Cooper AM, Johnson LL, Woodland DL, Mohrs M (2008) Cutting edge: T‐bet and IL‐27R are critical for in vivo IFN‐γ production by CD8 T cells during infection. J Immunol 180: 693–697 [DOI] [PubMed] [Google Scholar]
- McCullers JA (2014) The co‐pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol 12: 252–262 [DOI] [PubMed] [Google Scholar]
- McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, Tighe M, Hamada H, Sell S, Dutton RW, Swain SL (2009) IL‐10 deficiency unleashes an influenza‐specific Th17 response and enhances survival against high‐dose challenge. J Immunol 182: 7353–7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee LA, Harmsen AG (2006) Both influenza‐induced neutrophil dysfunction and neutrophil‐independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun 74: 6707–6721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo AA, Foreman O, Bosio CM, Elkins KL (2014) T‐bet regulates immunity to Francisella tularensis live vaccine strain infection, particularly in lungs. Infect Immun 82: 1477–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metersky ML, Masterton RG, Lode H, File TM, Babinchak T (2012) Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int J Infect Dis 16: e321–e331 [DOI] [PubMed] [Google Scholar]
- Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A (2003) Endogenous IL‐17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol 170: 4665–4672 [DOI] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, Fauci AS (2008) Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 198: 962–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, Bengsch B, Linderman SL, Stelekati E, Spolski R, Wherry EJ, Hunter C, Hensley SE, Leonard WJ, Cancro MP (2016) Cutting edge: IL‐4, IL‐21, and IFN‐γ interact to govern T‐bet and CD11c expression in TLR‐activated B cells. J Immunol 197: 1023–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, Van Rooijen N, Chow VT (2011) Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179: 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AH, Cardani A, Braciale TJ (2016) The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol 38: 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HH, van Ginkel FW, Vu HL, Novak MJ, McGhee JR, Mestecky J (2000) Gamma interferon is not required for mucosal cytotoxic T‐lymphocyte responses or heterosubtypic immunity to influenza A virus infection in mice. J Virol 74: 5495–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Szabo SJ, Glimcher LH (2002) T‐bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci USA 99: 5545–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, Alcorn JF (2013) IL‐22 is essential for lung epithelial repair following influenza infection. Am J Pathol 182: 1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope SM, Brandt EB, Mishra A, Hogan SP, Zimmermann N, Rothenberg ME, Pope SM, Hogan SP, Matthaei KI, Foster PS (2001) IL‐13 induces eosinophil recruitment into the lung by an IL‐5‐ and eotaxin‐dependent mechanism. J Allergy Clin Immunol 108: 594–601 [DOI] [PubMed] [Google Scholar]
- Price GE, Gaszewska‐Mastarlarz A, Moskophidis D (2000) The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol 74: 3996–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U (2006) T‐bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med 203: 2009–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran R, Foley J, Stoklasek T, Glimcher LH, McSorley SJ (2005) Expression of T‐bet by CD4 T cells is essential for resistance to Salmonella infection. J Immunol 175: 4603–4610 [DOI] [PubMed] [Google Scholar]
- Rice TW, Rubinson L, Uyeki TM, Vaughn FL, John BB, Miller RR, Higgs E, Randolph AG, Smoot BE, Thompson BT, NHLBI ARDS Network (2012) Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit Care Med 40: 1487–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P (2013) T‐box transcription factor T‐bet, a key player in a unique type of B‐cell activation essential for effective viral clearance. Proc Natl Acad Sci USA 110: E3216–E3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasinghe AE, Melo RCN, Duan S, LeMessurier KS, Liedmann S, Surman SL, Lee JJ, Hurwitz JL, Thomas PG, McCullers JA (2017) Eosinophils promote antiviral immunity in mice infected with influenza A virus. J Immunol 198: 3214–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC (2009) Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 119: 1910–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein‐Streilein J, Guffee J (1986) In vivo treatment of mice and hamsters with antibodies to asialo GM1 increases morbidity and mortality to pulmonary influenza infection. J Immunol 136: 1435–1441 [PubMed] [Google Scholar]
- Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH (2003) Antigen‐driven effector CD8 T cell function regulated by T‐bet. Proc Natl Acad Sci USA 100: 15818–15823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BM, Jobe O, Lazarevic V, Vasquez K, Bronson R, Glimcher LH, Kramnik I (2005) Increased susceptibility of mice lacking T‐bet to infection with Mycobacterium tuberculosis correlates with increased IL‐10 and decreased IFN‐γ production. J Immunol 175: 4593–4602 [DOI] [PubMed] [Google Scholar]
- Sun K, Metzger DW (2008) Inhibition of pulmonary antibacterial defense by interferon‐γ during recovery from influenza infection. Nat Med 14: 558–564 [DOI] [PubMed] [Google Scholar]
- Svensson A, Nordstrom I, Sun J‐B, Eriksson K (2005) Protective immunity to genital herpes simpex virus type 2 infection is mediated by T‐bet. J Immunol 174: 6266–6273 [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH (2002) Distinct effects of T‐bet in TH1 lineage commitment and IFN‐γ production in CD4 and CD8 T cells. Science 295: 338–343 [DOI] [PubMed] [Google Scholar]
- Tate MD, Ioannidis LJ, Croker B, Brown LE, Brooks AG, Reading PC (2011) The role of neutrophils during mild and severe influenza virus infections of mice. PLoS One 6: 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM (2008) The pathology of influenza virus infections. Annu Rev Pathol Mech Dis 3: 499–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Kash JC (2010) Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 7: 440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH (2004) T‐bet regulates the terminal maturation and homeostasis of NK and Vα14i NKT cells. Immunity 20: 477–494 [DOI] [PubMed] [Google Scholar]
- Tumpey TM, Garcia‐Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin‐Jackwood MJ, Schultz‐Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF (2005) Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79: 14933–14944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino AV, Gallo E, Abbas AK (2010) STAT1‐activating cytokines limit Th17 responses through both T‐bet‐dependent and ‐independent mechanisms. J Immunol 185: 6461–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang NS, McHeyzer‐Williams LJ, Okitsu SL, Burris TP, Reiner SL, McHeyzer‐Williams MG (2012) Divergent transcriptional programming of class‐specific B cell memory by T‐bet and RORα. Nat Immunol 13: 604–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M‐L, Wang C‐T, Yang S‐J, Leu C‐H, Chen S‐H, Wu C‐L, Shiau A‐L (2017) IL‐6 ameliorates acute lung injury in influenza virus infection. Sci Rep 7: 43829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Garvey P, Zhang P, Nelson S, Bagby G, Summer W (2001) Interleukin‐17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol 25: 335–340 [DOI] [PubMed] [Google Scholar]
- Yuan X, Paez‐Cortez J, Schmitt‐Knosalla I, D'Addio F, Mfarrej B, Donnarumma M, Habicht A, Clarkson MR, Iacomini J, Glimcher LH, Sayegh MH, Ansari MJ (2008) A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med 205: 3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Zhang T, Wu DZA, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F (2015) P63+ Krt5+ distal airway stem cells are essential for lung regeneration. Nature 517: 616–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1