

Abstract
Kinetochores are supramolecular assemblies that link centromeres to microtubules for sister chromatid segregation in mitosis. For this, the inner kinetochore CCAN/Ctf19 complex binds to centromeric chromatin containing the histone variant CENP‐A, but whether the interaction of kinetochore components to centromeric nucleosomes is regulated by posttranslational modifications is unknown. Here, we investigated how methylation of arginine 37 (R37Me) and acetylation of lysine 49 (K49Ac) on the CENP‐A homolog Cse4 from Saccharomyces cerevisiae regulate molecular interactions at the inner kinetochore. Importantly, we found that the Cse4 N‐terminus binds with high affinity to the Ctf19 complex subassembly Okp1/Ame1 (CENP‐Q/CENP‐U in higher eukaryotes), and that this interaction is inhibited by R37Me and K49Ac modification on Cse4. In vivo defects in cse4‐R37A were suppressed by mutations in OKP1 and AME1, and biochemical analysis of a mutant version of Okp1 showed increased affinity for Cse4. Altogether, our results demonstrate that the Okp1/Ame1 heterodimer is a reader module for posttranslational modifications on Cse4, thereby targeting the yeast CCAN complex to centromeric chromatin.
Keywords: Ame1, centromere, Gcn5, Okp1, posttranslational modification
Subject Categories: Cell Cycle; Chromatin, Epigenetics, Genomics & Functional Genomics; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The proper segregation of chromosomes during mitosis is essential for the faithful transmission of genetic information from the mother to the daughter cell, and errors in this process contribute to carcinogenesis and sterility in humans. For segregation, kinetochores are assembled at the centromeres of the chromosomes and provide the contact to the microtubules that pull the sister chromatids apart during mitosis (Musacchio & Desai, 2017). Centromeric chromatin is defined by the presence of the histone H3 variant CENP‐A (centromere protein A; Earnshaw & Migeon, 1985), which replaces canonical H3 at centromeric nucleosomes that are interspersed with H3‐containing nucleosomes in higher eukaryotes (Bodor et al, 2014). The yeast Saccharomyces cerevisiae possesses a point centromere with a single nucleosome containing the CENP‐A homolog Cse4, around which ~125 bp of centromeric DNA (CEN) are wrapped (Stoler et al, 1995; Meluh et al, 1998). Cse4 contains a histone fold domain that is homologous to CENP‐A in other organisms. In addition, it has an extended N‐terminal domain of 135 amino acids that is specific to closely related fungi and is essential for centromere function (Keith et al, 1999). The yeast kinetochore serves as a model for kinetochores from higher eukaryotes, which are thought to consist of modular repeats of this kinetochore unit (Westermann & Schleiffer, 2013).
The core kinetochore is a large multiprotein complex consisting of ~30 unique proteins in S. cerevisiae (Musacchio & Desai, 2017) that are assembled in modules, several of which are present in multiple copies (Joglekar et al, 2009). At the outer kinetochore, the KMN network provides the contact to the microtubule (Fig 1; Varma & Salmon, 2012). Here, several copies of the Ndc80 complex (Wigge & Kilmartin, 2001), which is a dumbbell‐shaped structure, interact with the Dam1 ring (Nogales & Ramey, 2009). This ring encircles the microtubule and links the kinetochore to the mitotic spindle. At its centromere‐proximal side, the Ndc80 complex contacts the MIND complex (Kudalkar et al, 2015).
Figure 1. Overview of proteins of the yeast kinetochore.
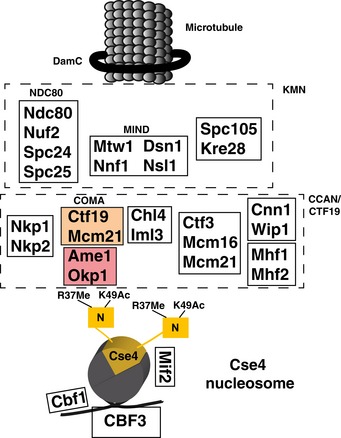
The Cse4‐containing nucleosome is depicted with the extended N‐terminus, which carries R37Me and K49Ac. COMA subunits are shown in colour.
On the chromatin‐proximal side of the kinetochore, the Ctf19 complex is an important component of the inner kinetochore that interacts with centromeric chromatin and serves as an assembly platform for the outer kinetochore (Foltz et al, 2006; Izuta et al, 2006; Okada et al, 2006). The Ctf19 complex is the yeast equivalent of the constitutive centromere‐associated network (CCAN) in higher eukaryotes and contains 11 other proteins (Hornung et al, 2014). The COMA complex represents a subassembly within the Ctf19 complex, and it links the inner kinetochore to the MIND complex (De Wulf et al, 2003). COMA was named for the S. cerevisiae components Ctf19CENP‐P (Hyland et al, 1999), Okp1CENP‐Q (Ortiz et al, 1999), Mcm21CENP‐O (Poddar et al, 1999) and Ame1CENP‐U (Burns et al, 1994; Cheeseman et al, 2002; Gavin et al, 2002; superscript indicates the mammalian orthologs). The mammalian equivalent of yeast COMA is the CENP‐O/P/Q/U complex (Musacchio & Desai, 2017).
Ctf19 and Mcm21 both contain RWD domains and together form a Y‐shaped heterodimer with flexible N‐terminal extensions (Schmitzberger & Harrison, 2012). This heterodimer interacts with the Okp1/Ame1 heterodimer via Okp1 residues 317–343 (Schmitzberger et al, 2017). Loss of this contact results in the disruption of binding of two Ctf19 components, Chl4CENP‐N and Iml3CENP‐L, at the centromere. Next to its binding to Ctf19/Mcm21, Okp1/Ame1 furthermore forms a platform for interactions with other components of the Ctf19 complex, for instance Nkp1/Nkp2, as well as to Mif2CENP‐C, which binds the Cse4‐containing nucleosome (Kato et al, 2013; Falk et al, 2016; Xiao et al, 2017). Okp1/Ame1 also connects COMA to the outer kinetochore by interacting with the MIND complex (Hornung et al, 2014).
Apart from the interaction with Mif2, little is known about how COMA is recruited to the centromeric nucleosome and how this may be regulated during the cell cycle. Ctf19 was shown to interact by two‐hybrid analysis with Cse4 (Ortiz et al, 1999), but this has not been confirmed using biochemical methods. Furthermore, Okp1/Ame1 interacts with DNA, but the interaction is not specific to centromeric sequences (Hornung et al, 2014). Of note, the genes encoding Okp1, Ame1 and Mif2, but not other components of the Ctf19 complex, are essential for viability, implying that they perform non‐redundant functions at the centromere.
One possibility to regulate protein–protein interactions is by posttranslational modification (PTM). PTMs on histones are widely used to regulate chromatin function by altering interactions with chromatin‐binding proteins (“reader modules”; Patel & Wang, 2013). Such readers include bromo‐ or YEATS domains for histone acetylation and chromo‐, tudor‐ and PWWP domains for arginine or lysine methylation. However, this type of regulation has not been described for centromeric chromatin, and no reader modules are known within kinetochore proteins.
Next to canonical histone modifications, PTMs have also been discovered in CENP‐A homologs. In human cells, serine 7 (S7) in the N‐terminus of CENP‐A is phosphorylated by Aurora kinase during mitosis (Zeitlin et al, 2001; Kunitoku et al, 2003), and CENP‐A is furthermore phosphorylated at S16 and S18 (Bailey et al, 2013). Also, the initiating methionine of CENP‐A is cleaved, and the α‐amino group of glycine 1 is trimethylated by NRMT1 (Sathyan et al, 2017). The absence of this modification reduces the recruitment of CCAN components to the centromere.
Furthermore, several PTMs are known on Cse4 from S. cerevisiae. It is ubiquitylated by the ubiquitin ligase Psh1, which controls the levels of Cse4 in the cell and thus restricts its localization to centromeres (Hewawasam et al, 2010; Ranjitkar et al, 2010). In our earlier work, we identified the methylation of arginine 37 (R37Me), which lies in the essential N‐terminal domain (END) of Cse4 (Keith et al, 1999), as a modification whose absence causes severe synthetic genetic interactions in combination with deletions or mutations in genes encoding proteins of the COMA subcomplex of the Ctf19 complex, arguing that mutation of this site disturbs the interaction of Cse4 with a kinetochore component (Samel et al, 2012). Cse4 is furthermore acetylated on lysine 49 and phosphorylated on S22, S33, S40 and S105 (Boeckmann et al, 2013). While Cse4‐S33 phosphorylation plays a role in histone deposition at the centromere (Hoffmann et al, 2018), the functional role of the other PTMs has not been resolved.
Here, we have investigated the molecular mechanism by which Cse4‐R37 methylation and Cse4‐K49 acetylation regulate the recruitment of the kinetochore to the centromere. Through genetic screens, we identified mutations in OKP1 (among others, okp1‐R164C) and AME1 (among others, ame1‐273*) that suppress synthetic growth defects of cse4‐R37A. This suggested a) that Okp1/Ame1 binds the Cse4 N‐terminus, and b) that Okp1/Ame1 binding to mutant Cse4 is restored by the suppressor mutations. In agreement with this, we found Okp1/Ame1 to form a stable complex in vitro with the Cse4 N‐terminus, whereas no binding to Ctf19/Mcm21 was observed. Importantly, in vitro studies using microscale thermophoresis showed that the binding affinity of Cse4 to Okp1/Ame1 was reduced by R37 methylation and K49 acetylation, and this defect in binding to modified Cse4 was suppressed by Okp1‐R164C. Altogether, our results demonstrate that Okp1/Ame1 is a reader module for the N‐terminus of Cse4. It tethers the kinetochore to centromeric chromatin by binding to Cse4, and this interaction is inhibited by Cse4‐R37 methylation and Cse4‐K49 acetylation.
Results
Suppression of Cse4‐R37 mutation by okp1‐R164C
Cse4 is methylated on R37 (Fig 2A), and mutation of this site (cse4‐R37A) causes a synthetic growth defect in cells lacking Cbf1 (cbf1∆; Samel et al, 2012). Cbf1 binds the CDEI element of centromeric sequences (Cai & Davis, 1990). Posttranslational modifications on proteins can exert their function by regulating the interaction with another protein. We hypothesized that the growth defect of cbf1∆ cse4‐R37A is caused by the abrogation of an interaction between Cse4 and an interaction partner, possibly a kinetochore protein. To identify this factor, we isolated mutations that suppress the temperature‐sensitive growth defect of cbf1∆ cse4‐R37A. We surmised that some of these mutations might improve the binding of the hypothesized factor to the mutant N‐terminus of Cse4 and therefore restore centromere function. Several mutants were isolated, and the site of the extragenic suppressor was determined by whole‐genome sequencing. Interestingly, we obtained one isolate that carried a mutation in OKP1, which encodes the COMA component Okp1 (Ortiz et al, 1999). The mutation causes the change of arginine 164 of Okp1 to cysteine (okp1‐R164C), a residue that lies within the “core” region (amino acids 166–211) that is essential for viability (Schmitzberger et al, 2017). We constructed the okp1‐R164C mutation de novo and tested its ability to suppress the temperature sensitivity of cbf1∆ cse4‐R37A. Significantly, while cbf1∆ cse4‐R37A cells showed a pronounced growth defect at elevated temperatures, this growth defect was suppressed by okp1‐R164C (Fig 2B). A tentative biochemical interpretation of this genetic suppression is that Okp1 interacts with the N‐terminus of Cse4, and that this interaction is regulated by methylation of R37.
Figure 2. Mutations in OKP1 suppress growth defects of mutation of Cse4‐R37.
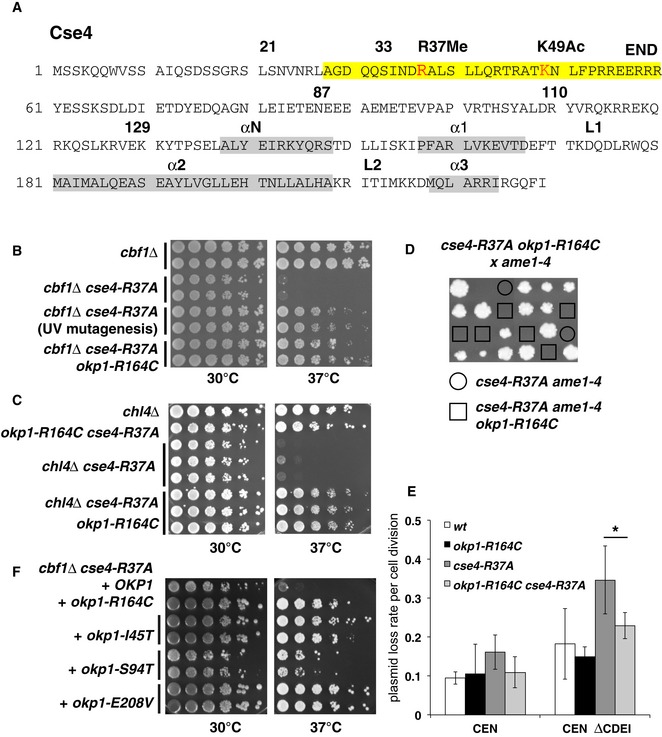
- Overview of the amino acid sequence of Cse4. R37Me and K49Ac sites are part of the essential N‐terminal domain (END, aa 28–60, yellow) and are indicated in red. The localization of α‐helices in the histone fold domain is shown in grey. Amino acid residues that are relevant for this study are indicated with numbers.
- okp1‐R164C suppressed the temperature‐sensitive growth defect of cbf1∆ cse4‐R37A. Serial dilutions of strains with the indicated genotypes were spotted on full medium and grown for 3 days at 30 or 37°C. The original okp1‐R164C isolate from the suppressor screen is indicated as “UV mutagenesis”.
- okp1‐R164C suppressed the growth defect of cse4‐R37A with chl4∆. Representation as in (B).
- okp1‐R164C was unable to suppress the lethality of cse4‐R37A with ame1‐4. Tetrad dissection of a genetic cross between a cse4‐R37A okp1‐R164C and an ame1‐4 strain is shown. The four spores from individual asci are aligned in vertical rows.
- okp1‐R164C suppressed the maintenance defect of cse4‐R37A for plasmids lacking the CDEI sequence of CEN6 (CEN ∆CDEI, at 37°C). Error bars give SD of at least three independent transformants. *P = 0.03.
- The okp1 mutations I45T, S94T and E208V suppressed the temperature‐sensitive growth defect of cbf1∆ cse4‐R37A. Strains with the indicated okp1 alleles on a plasmid (derivatives of AEY5584) are shown as in (B).
We further determined whether other defects of cse4‐R37A (Samel et al, 2012) were also suppressed by okp1‐R164C. Indeed, okp1‐R164C also suppressed the growth defect of cse4‐R37A with mutations/deletions of other Ctf19 complex components (Fig 2C and D, Table 1). There was one notable exception to this, which is that the lethality of cse4‐R37A with ame1‐4 was not suppressed by okp1‐R164C (Fig 2D). This observation is interesting in light of the fact that Okp1 and Ame1 are Ctf19 complex components that are essential for viability, and they form a heterodimer in vitro (Hornung et al, 2014).
Table 1.
Overview of the suppression of cse4‐R37A phenotypes by okp1‐R164C and ame1‐273*
| Kinetochore component/complex | Allele | Synthetic phenotype with cse4‐R37A (Samel et al, 2012) | Suppression by okp1‐R164C | Suppression by by ame1‐273* |
|---|---|---|---|---|
| Cbf1 | cbf1∆ | Growth defect | Suppression | Suppression |
| COMA | ctf19∆ | Lethality | Partial suppression | Partial suppression |
| COMA | mcm21∆ | Lethality | Partial suppression | Partial suppression |
| COMA | ame1‐4 | Lethality | No suppression | n. d. |
| COMA | okp1‐5 | Growth defect | n.d. | No suppression |
| Ctf19 complex | iml3∆ | Growth defect | Suppression | Suppression |
| Ctf19 complex | chl4∆ | Growth defect | Suppression | Suppression |
| Ctf19 complex | ctf3∆ | Growth defect | Suppression | Suppression |
n.d., not determined.
cse4‐R37A causes defects in chromosome and mini‐chromosome segregation at centromeres that are compromised for CDEI function, which can be measured as an increased loss of plasmids that lack the CDEI sequence (Samel et al, 2012). Since okp1‐R164C suppressed the temperature sensitivity of cbf1∆ cse4‐R37A, we tested whether it also suppressed the plasmid maintenance defect of cse4‐R37A. Indeed, while cse4‐R37A caused an increased loss rate of a plasmid lacking CDEI (CEN ∆CDEI), this defect was decreased by additional okp1‐R164C mutation (Fig 2E), showing that okp1‐R164C suppressed the segregation defect of cse4‐R37A at centromeres lacking CDEI function.
cse4‐R37A furthermore causes a cell‐cycle arrest at the G2/M phase transition in cbf1∆ at the restrictive temperature (Samel et al, 2012), which reflects its defect in chromosome segregation. We therefore tested the effect of okp1‐R164C on cell‐cycle progression in cbf1∆ cse4‐R37A by measuring the DNA content of cells by FACS analysis. Importantly, while cbf1∆ cse4‐R37A cells showed an accumulation of cells with a 2n DNA content at the restrictive temperature, this arrest was partially suppressed in cbf1∆ cse4‐R37A okp1‐R164C (Fig EV1A), indicating that okp1‐R164C partially restored centromere function and chromosome segregation to cse4‐R37A.
Figure EV1. okp1‐R164C partially suppressed the G2/M arrest of cbf1Δ cse4‐R37A at the restrictive temperature (related to Fig 2).
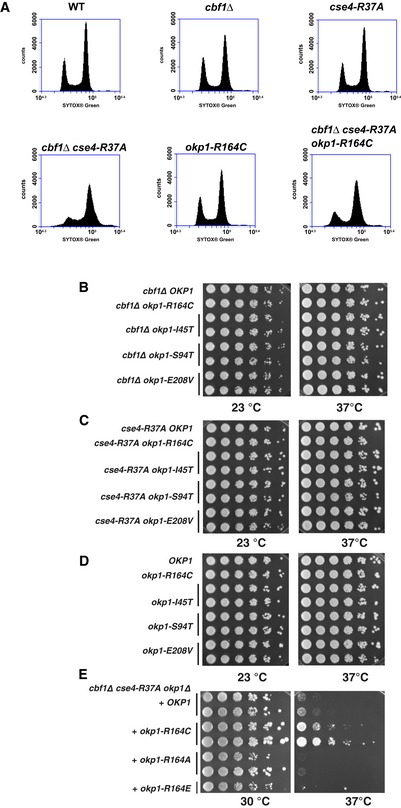
-
AWT (AEY4), cbf1Δ (AEY4816), cse4‐R37A (AEY4965), cbf1Δ cse4‐R37A (AEY4985), okp1‐R164C (AEY5594) and cbf1Δ cse4‐R37A okp1‐R164C (AEY5974) cells were grown to early logarithmic phase at 23°C and shifted to 37°C for 5 h. DNA content of cells stained with SYTOX Green was measured by FACS analysis.
-
B–DCharacterization of okp1 alleles. Alleles of okp1 that suppress cbf1∆ cse4‐R37A defects had no defect with cbf1∆ (B), cse4‐R37A (C) or in a wild‐type background (D). Strains carrying the indicated alleles were serially diluted and grown on full medium at the indicated temperatures for 3 days.
-
EMutation Okp1‐R164 to alanine or glutamate did not suppress the temperature‐sensitive defect of cbf1∆ cse4‐R37A.
We next asked whether other mutations in OKP1 could be identified that suppressed the defect of cbf1∆ cse4‐R37A. Through a random mutagenesis of OKP1, two strong suppressor alleles, okp1‐E208V and okp1‐I45T, and one weaker suppressor, okp1‐S94T, were isolated (Fig 2F). None of the okp1 alleles caused a growth defect on their own, nor in the presence of cbf1∆ or cse4‐R37A alone (Fig EV1B–D). Okp1‐E208, like R164, lies in the Okp1 core (166–211), the deletion of which is lethal, but is not required for the interaction with Ame1 (Schmitzberger et al, 2017), suggesting that it may be an interaction site with Cse4. Okp1‐I45 and Okp1‐S94 are in the flexible N‐terminal region that is dispensable for binding to Ame1 and therefore may also represent a Cse4 binding region. We also tested substitutions of Okp1‐R164 to alanine or glutamate, but both were unable to suppress the growth defect of cbf1∆ cse4‐R37A (Fig EV1E), indicating that the mutation to cysteine bestowed special binding properties upon Okp1. Taken together, this showed that multiple alleles of OKP1 exist that suppress the defects of cse4‐R37A and suggested that these alleles restore the hypothesized interaction of Okp1 with the Cse4 N‐terminus.
cse4‐R37A reduces the recruitment of the MIND complex component Mtw1 to centromeric sequences under restrictive conditions (Samel et al, 2012). Since okp1‐R164C suppressed the growth defect of cse4‐R37A, we asked whether it also restored binding of Okp1 (Fig 3A) and Ame1 (Fig 3B) to the centromere as measured by chromatin immunoprecipitation (ChIP). Importantly, while the amount of Okp1 and Ame1 associated with CEN4 was reduced in cbf1∆ cse4‐R37A cells grown at 37°C, their association was partially restored in okp1‐R164C cells (Fig 3A and B). Furthermore, okp1‐R164C restored the association of the MIND component Mtw1 to the centromere (Fig 3C). This indicated that the improved growth of the cells by the okp1 mutation was achieved by restoring the recruitment of other kinetochore components to the centromeric sequence.
Figure 3. okp1‐R164C restored binding of kinetochore components to centromeric sequences.
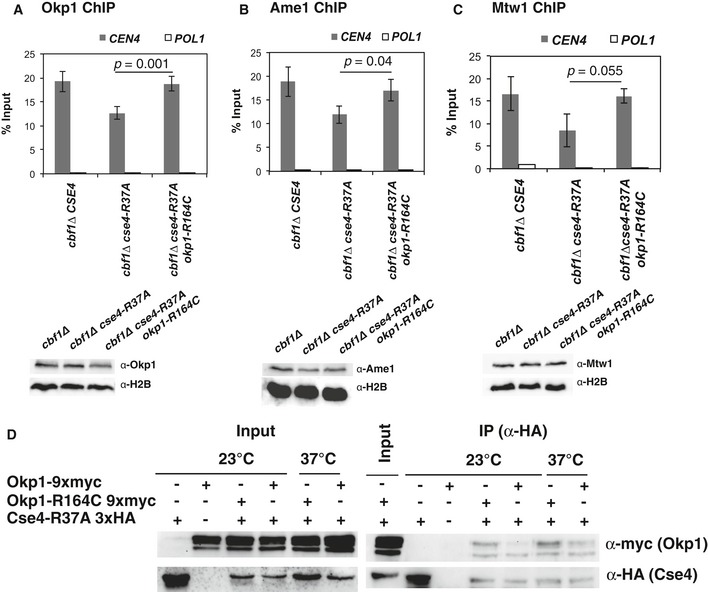
- okp1‐R164C restored binding of Okp1 to centromeric sequences. ChIP of 9xmyc‐tagged Okp1 was performed in strains with the indicated genotypes that were grown at 37°C for 4 h before ChIP. Enrichment of CEN4 and POL1 (as a control) relative to input is given. Below, Western blot analysis of the amounts of 9xmyc‐Okp1 and histone H2B (loading control) in whole cell extracts.
- okp1‐R164C restored binding of Ame1 to centromeric sequences. ChIP of 9xmyc‐tagged Ame1 was performed as in (A). Below, Western blot analysis of the amounts of 9xmyc‐Ame1 and H2B as in (A).
- okp1‐R164C restored binding of Mtw1 to centromeric sequences. ChIP of 9xmyc‐Mtw1 and Western blots (right) are presented as in (A) and (B).
- okp1‐R164C improved in vivo association of Okp1 with Cse4‐R37A. Co‐immunoprecipitation of Cse4‐R37A and Okp1 was carried out in cells carrying cse4‐R37A with (+) or without (−) 3xHA‐tag and OKP1 or okp1‐R164C with (+) or without (−) 9xmyc‐tag (AEY5040, AEY5972, AEY5973, AEY6589, AEY6592). Cells were grown at 23°C or were shifted to 37°C for 5 h prior to harvesting. Cse4‐R37A was immunoprecipitated using an α‐HA antibody, and the presence of Cse4‐R37A and Okp1 or Okp1‐R164C in the immunoprecipitate was tested by Western blotting with α‐HA and α‐myc antibody, respectively. Left, inputs (the two bands indicate Okp1 and a shorter degradation product); right, α‐HA immunoprecipitates.
We next asked whether okp1‐R164C affected the in vivo association of Okp1 and Cse4. To test this, co‐immunoprecipitation (co‐IP) was conducted between Cse4 and Okp1. Wild‐type Okp1 was readily co‐IPed with wt Cse4 (Fig EV2A). Upon IP of HA‐tagged mutant Cse4‐R37A, more Okp1‐R164C than Okp1 was recovered in the immunoprecipitates (Fig 3D). We observed several bands for Okp1 (the lower bands most likely are proteolytic degradation products of Okp1). Altogether, we conclude that the in vivo association between Okp1 and Cse4‐R37A (which could be direct or indirect) is improved by okp1‐R164C.
Figure EV2. Interaction between Cse4 and Okp1 (related to Figs 3D and 4).
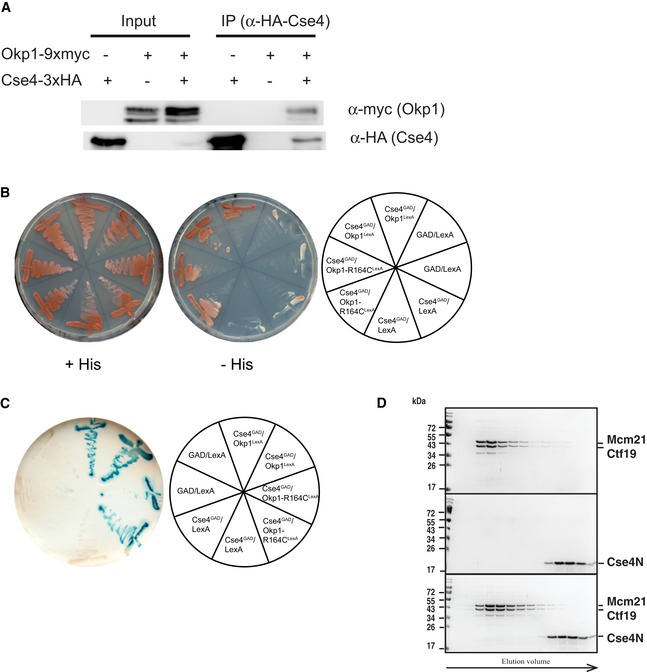
- Co‐immunoprecipitation of Okp1 and Cse4. Co‐IP was carried out in cells carrying CSE4 with (+) or without (−) 3xHA‐tag and OKP1 with (+) or without (−) 9xmyc‐tag. Cells were grown at 30°C, and Cse4 was immunoprecipitated using an α‐HA antibody (AEY2781, AEY5972, AEY6578). The presence of Cse4 and Okp1 in the immunoprecipitate was tested by Western blotting with α‐HA and α‐myc antibody, respectively. Left: inputs of the indicated strains; right: α‐HA immunoprecipitates of the indicated strains.
- Two‐hybrid analysis of the interaction between Cse4 (aa 11–139) and Okp1. Cells carrying the indicated constructs were streaked on minimal medium (+His) or on medium lacking histidine (−His) to test for activation of the two‐hybrid reporter.
- Activity of the lacZ two‐hybrid reporter of strains carrying the indicated two‐hybrid constructs.
- Absence of in vitro interaction between Ctf19/Mcm21 and Cse4N. Recombinant Ctf19/Mcm21 and Cse4N were co‐incubated prior to size‐exclusion chromatography (SEC) as for Okp1/Ame1 in Fig 4A. Coomassie Blue‐stained SDS–PAGE gels of SEC runs with Mcm21/Ctf19 (top), Cse4N (middle) and co‐incubation of Ctf19/Mcm21 with Cse4N (bottom) are shown.
Source data are available online for this figure.
The Cse4 N‐terminus binds Okp1/Ame1
The suppression of cse4‐R37A defects by mutations in OKP1 suggests that cse4‐R37A causes a loss of interaction of the Cse4 N‐terminus with Okp1, and that the mutations in Okp1 restore binding of Okp1 to Cse4. We therefore sought to test whether there is a direct interaction between Okp1 and Cse4. To do so, we asked whether a recombinant N‐terminal fragment of Cse4 (amino acids 21–129, Cse4N, Fig 2A) interacted in vitro with Okp1. Okp1 as obtained by recombinant expression in bacteria was only soluble upon co‐expression with Ame1 (Hornung et al, 2014), and the two proteins formed a dimer in solution (Okp1/Ame1) as determined by size‐exclusion chromatography (SEC, Fig 4A). Importantly, incubation of purified Okp1/Ame1 with recombinant Cse4N yielded a complex that eluted at higher molecular weight by SEC and contained all three proteins (Fig 4A). This demonstrated that Cse4N interacted in vitro with Okp1/Ame1, which was in line with our observation of a two‐hybrid interaction between Cse4 and Okp1 (Fig EV2B and C).
Figure 4. The N‐terminus of Cse4 interacts in vitro with Okp1/Ame1.
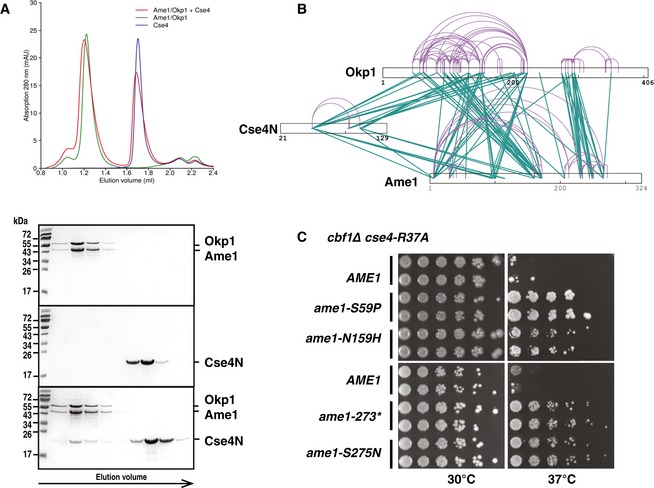
- Cse4 (21–129, Cse4N) and Okp1/Ame1 were expressed and purified separately from bacteria and co‐incubated before separation by analytical gel filtration. Top, representative size‐exclusion chromatography (SEC) chromatogram with absorbance measurements at 280 nanometres (nm) is shown. Below, image of Coomassie Blue‐stained SDS–PAGE gels with fractions from principal SEC peaks of runs with Okp1/Ame1 alone (top), Cse4N alone (middle) and Okp1/Ame1 preincubated with Cse4N before SEC (bottom). Molecular weight standards are shown in kilodalton (kDa).
- Intersubunit and self‐link map for the Cse4N/Okp1/Ame1 complex formed in vitro show direct contacts of Cse4N with both Okp1 and Ame1. Self‐links and intersubunit cross‐links are coloured in purple and green, respectively.
- The ame1 mutations S59P, N159H, S275N and a C‐terminal truncation after aa 273 (ame1‐273*) suppressed the growth defect of cbf1∆ cse4‐R37A. cbf1∆ cse4‐R37A ame1∆ strains carrying the indicated plasmid‐borne ame1 alleles are shown as in Fig 2B.
Source data are available online for this figure.
Since direct contacts between Cse4 and Ctf19 have previously been suggested by two‐hybrid analysis (Ortiz et al, 1999), we tested whether Cse4N was capable of forming a complex in vitro with Ctf19. Recombinant Ctf19 expressed in bacteria was insoluble, but could be obtained in a soluble complex by co‐expression with Mcm21 (Schmitzberger & Harrison, 2012; Schmitzberger et al, 2017). However, no complex between Cse4N and Ctf19/Mcm21 could be obtained using the same conditions as for Okp1/Ame1 interaction with Cse4N (Fig EV2D). This suggested either that there is no direct interaction, or that the interaction is instable under the conditions used here for SEC. It furthermore indicated that the interaction between the COMA complex and Cse4 is primarily mediated by the Okp1 and Ame1 subunits.
To further query the contacts between the three proteins, we performed chemical cross‐linking combined with mass spectrometry (CLMS, Fig 4B, Table EV1; Chen et al, 2010). A total of 182 cross‐links between the three proteins in the complex formed in vitro were mapped, which means that 182 unique residue pairs were identified as being chemically linked, with one residue being in one protein and the other in a different protein. In addition to extensive cross‐links between Okp1 and Ame1, we identified several cross‐links between Okp1 and Cse4, thus suggesting a close spatial proximity of the two proteins in the complex formed in vitro and accessibility to the cross‐linker. Notably, Cse4 lysine 49 (K49) was involved in the formation of seven cross‐links to Okp1. Interestingly, the same K49 also showed seven cross‐links to Ame1, thus revealing proximity of Cse4 to Ame1 in the in vitro complex and suggesting that Cse4N binds in a cleft between Okp1 and Ame1.
Mutations in AME1 suppress defects of cse4‐R37A
Next to direct contacts between Cse4N and Okp1, the CLMS analysis above also revealed interactions to Ame1, suggesting that mutations in AME1 may also be able to suppress the defects of cse4‐R37A, as is the case for the mutations described above in OKP1. To test this, a random mutagenesis of AME1 was performed in order to isolate suppressor mutations of the temperature‐sensitive growth defect of cbf1∆ cse4‐R37A. Significantly, several alleles were retrieved that caused suppression, namely ame1‐S59P, ‐N159H, ‐S275N and a C‐terminal truncation after aa 273 (codon 274 mutated to a stop codon, total length of Ame1 is 324 aa, ame1‐273*, Fig 4C). Residues I273 and S275 are located within segment 1 of Ame1, which interacts with Nkp1/Nkp2, suggesting that abrogation of Nkp1/Nkp2 binding suppresses cse4‐R37A defects (Schmitzberger et al, 2017). Ame1‐N159 lies within the Ame1 core, the region that interacts with Okp1, and the mutation therefore may therefore strengthen the interaction to Okp1, or it increases the affinity for Cse4N. Ame1‐S59 is in the flexible N‐terminal region, but outside of the MIND interaction site at the extreme N‐terminus of Ame1.
ame1‐273* was furthermore tested for suppression of other cse4‐R37A phenotypes. Interestingly, suppression was found for all cse4‐R37A phenotypes, with the exception of the lethality of cse4‐R37A with okp1‐5 (Table 1), thus revealing a reciprocal relationship between the suppressor alleles in AME1 and OKP1. Furthermore, our ability to isolate AME1 suppressor alleles was consistent with the existence of a physical interaction between Ame1 and Cse4N as shown by CLMS.
Antagonistic relationship between Cse4‐R37 mutation and acetylation on lysine 49
Direct contacts between Cse4N and Okp1/Ame1 involve Cse4 lysine 49. Interestingly, this site is subject to acetylation (K49Ac; Figs 2A and EV3A; Boeckmann et al, 2013). Our semi‐quantitative analysis showed that 58% of Cse4 from asynchronous cells carried K49Ac (Fig EV3B). To further characterize the modification, we generated an antibody specific for Cse4‐K49Ac (Fig EV3C). This antibody recognized wild‐type (wt) Cse4 purified from yeast, but not Cse4 in which K49 was mutated to arginine (K49R; Fig EV3D), and was able to immunoprecipitate Cse4, but not a Cse4 version in which K49 is mutated to arginine (Cse4‐K49R; Fig EV4A), further confirming that Cse4 is acetylated on Cse4 in vivo. Interestingly, Cse4‐K49Ac was enriched in cells arrested in S‐phase and G2/M phase as compared to G1‐arrested cells (Fig 5A, left), suggesting that Cse4‐K49Ac has a regulatory function during the S‐phase of the cell cycle. Cse4‐R37 methylation was also mildly increased in S‐phase (Fig 5A, right), arguing for a potential coordination of the appearance of K49 acetylation and R37 methylation during S‐phase.
Figure EV3. Cse4 is acetylated on lysine 49 (related to Fig 5).
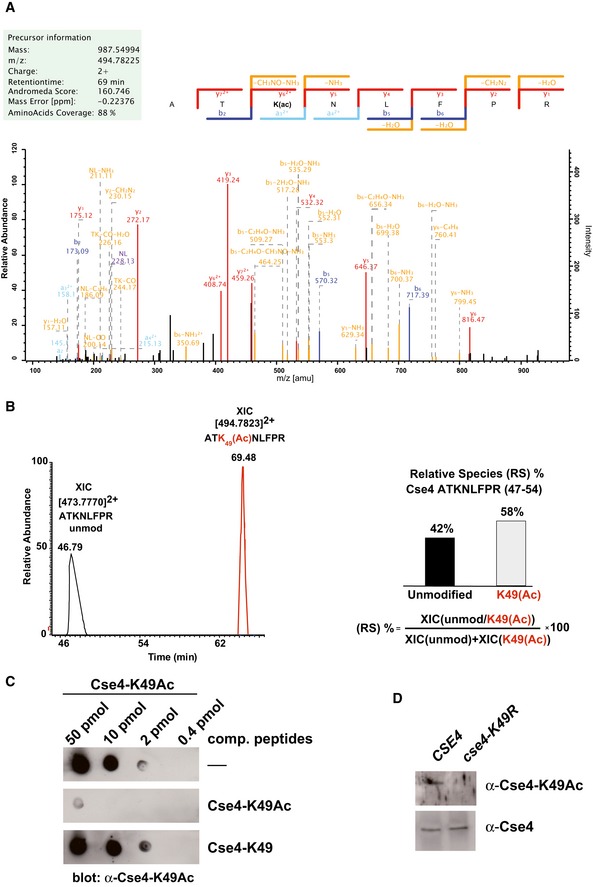
- Collision‐induced dissociation analysis and full annotation of the parent ion (Cse4 47‐ATK(Ac)NLFPR‐54) with m/z = 494.78. Score and expected values as calculated by Mascot are reported, and fragments detected in the experiment are indicated in colour (blue for b ions and red for y ions).
- Extract ion chromatograms (XIC) relative to each form of the peptide (left) were used to estimate relative abundance of the two species (right).
- Specificity of the α‐Cse4‐K49Ac antibody. Immuno‐dot blot showing the specific reactivity of the antibody. The indicated amounts of Cse4‐K49Ac peptide were spotted on a nitrocellulose membrane. Antibodies were preincubated with the competing peptides indicated to the right of the blots.
Figure EV4. Cse4‐K49 is acetylated by the Gcn5/SAGA complex (related to Fig 5).
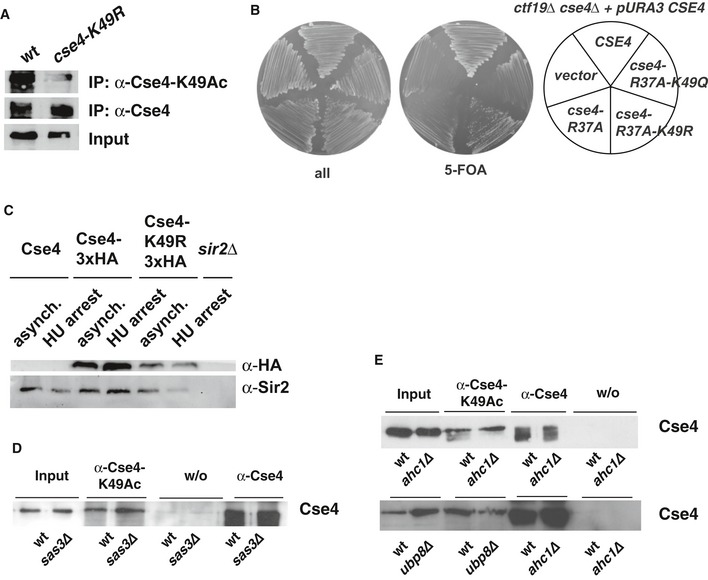
- Cse4 is not precipitated with the α‐Cse4‐K49Ac in cells carrying cse4‐K49R. Cells carrying HA‐tagged Cse4 were used to immunoprecipitate Cse4 with an α‐Cse4‐K49Ac (top) and an α‐HA antibody (bottom).
- The growth defect of cbf1∆ cse4‐R37A was not suppressed by additional cse4‐K49Q mutation.
- Mutation of Cse4‐K49R did not affect Cse4 degradation. Western blot analysis of the amounts of Cse4‐3xHA and Sir2 (loading control) in whole cell extracts of asynchronous cultures or cells arrested with hydroxyurea (HU, 3 h arrest). Strains used were as follows: Cse4, AEY1; Cse4‐3xHA, AEY2781; Cse4‐K49R‐3xHA, AEY2960; sir2∆, AEY13. Extract of a sir2∆ strain was included to show specificity of the Sir2 antibody.
- Acetylation of Cse4‐K49 is independent of the HAT Sas3. Immunoprecipitations (IP) were performed as in (A) using cells with sas3∆.
- Ahc1 and Ubp8 are not required for Cse4‐K49Ac in vivo. IPs were performed as in (A) using ahc1∆ and ubp8∆ strains.
Figure 5. Acetylation of Cse4 K49 counteracts mutation of Cse4‐R37.
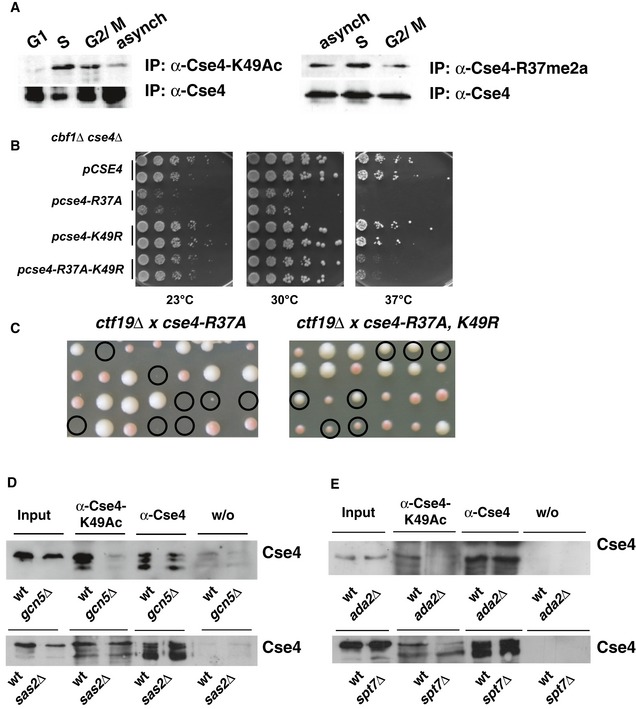
- Cse4‐K49Ac and –R37Me are increased in S‐phase of the cell cycle. Cells carrying 3xHA‐tagged Cse4 were used to immunoprecipitate Cse4 with an α‐Cse4‐K49Ac (left, top) and an α‐HA antibody (left, bottom). To detect Cse4‐R37 methylation, 3xHA‐tagged Cse4 was immunoprecipitated with an α‐Cse4‐R37Me2a (right, top; Samel et al, 2012) and an α‐HA antibody (right, bottom). Western blots were probed for the presence of Cse4 with the α‐HA antibody. G1, treatment with α‐factor; S, arrest with hydroxyurea; G2/M, arrest in nocodazole; asynch, asynchronous culture.
- Mimicking the unacetylated state of Cse4‐K49 (cse4‐K49R) partially suppressed the temperature sensitivity of cbf1∆ cse4‐R37A. cbf1∆ cse4∆ strains carrying the indicated cse4 alleles on a plasmid were spotted on full medium and grown as in Fig 2B.
- cse4‐K49R suppressed the lethality of cse4‐R37A with ctf19∆. Tetrad dissection of a ctf19∆ strain crossed with cse4‐R37A (left) and cse4‐R37A K49R (right). Representation as in Fig 2D.
- Acetylation of Cse4‐K49 was reduced in the absence of Gcn5 and the SAGA complex, but not in the absence of the HAT Sas2. Cse4 was precipitated as in (A) from strains wild‐type (wt) or gcn5∆ (top) or sas2∆ strains (bottom) carrying HA‐tagged Cse4. w/o, control immunoprecipitation without antibody.
- Cse4‐K49Ac depended on the SAGA components Ada2 and Spt7. Cse4‐K49Ac was determined as in (A, left) with cells carrying ada2∆ or spt7∆.
Source data are available online for this figure.
We next asked how the absence of K49Ac affected Cse4 function. Mutation of K49 to arginine (cse4‐K49R), which imitates the deacetylated state, did not affect cell viability (Fig 5B), nor did we observe synthetic genetic interactions with deletions or mutations in kinetochore components. Importantly, however, we observed a genetic interaction with cse4‐R37A. While cse4‐K49R on its own in cbf1∆ did not cause a growth defect, simultaneous mutation of K49R in cse4‐R37A (cse4‐R37A‐K49R) partially suppressed the growth defect of cbf1∆ cse4‐R37A (Fig 5B). Furthermore, cse4‐R37A‐K49R suppressed the lethality of cse4‐R37A with the deletion of CTF19, which encodes a component of the COMA complex (Fig 5C). This suggested that when the interaction of Cse4 with Okp1 is disrupted by cse4‐R37A, acetylation of K49 has a detrimental effect on Cse4 function. To further test this notion, we tested the effect of mutation of K49 to glutamine (K49Q), which mimics the acetylated state of K49. Interestingly, cse4‐K49Q was unable to suppress the lethality of ctf19∆ cse4‐R37A (cse4‐R37A‐K49Q, Fig EV4B), suggesting that the absence of acetylation suppressed the defects of cse4‐R37A.
One potential explanation for the rescue of cse4‐R37A phenotypes by cse4‐K49R is that this is because Cse4‐K49R is not degraded during S‐phase (Wisniewski et al, 2014). However, we observed similar levels of both Cse4 and Cse4‐K49R in asynchronous and in S‐phase‐arrested cells (Fig EV4C), which argued against a role for K49Ac in Cse4 degradation.
Cse4‐K49Ac depends on the Gcn5/SAGA histone acetyltransferase complex
The acetylation of histones and non‐histone proteins is performed by (histone) acetyltransferase (HAT) complexes (Roth et al, 2001), and we next sought to address which of the known HATs/HAT complexes in yeast were responsible for Cse4‐K49 acetylation. Importantly, we observed that the absence of the HAT Gcn5 (Brownell et al, 1996) abrogated the ability of Cse4 to be immunoprecipitated with the α‐Cse4‐K49Ac antibody (Fig 5D), thus showing that Gcn5 was responsible for a large part of Cse4‐K49Ac. In contrast, no effect was observed in the absence of the HATs Sas2 (Fig 5D; Reifsnyder et al, 1996; Ehrenhofer‐Murray et al, 1997), Sas3 (John et al, 2000), Hpa2 (Angus‐Hill et al, 1999), Hat1 and Elp3 (Wittschieben et al, 1999; Fig EV4D and E), indicating that they are not major HATs for Cse4‐K49.
Gcn5 is part of three multiprotein complexes, SAGA, ADA and SLIK/SALSA. SAGA is composed of several smaller modules (Daniel & Grant, 2007). Interestingly, the absence of Ada2 and Spt7, components of the SAGA HAT and architecture unit, respectively (Grant et al, 1997), abrogated Cse4‐K49Ac (Fig 5E). In contrast, the absence of Ubp8, a component of the DUB module (Henry et al, 2003), and Ahc1, a component unique to the ADA complex (Eberharter et al, 1999), did not affect Cse4‐K49Ac (Fig EV4E). Taken together, these results showed that the HAT activity of SAGA/SLIK, but not the deubiquitination activity or the ADA complex, was required for Cse4‐K49 acetylation in vivo.
Inhibition of Cse4 interaction with Okp1/Ame1 by Cse4‐R37 methylation and K49 acetylation
The biochemical analysis above showed that the N‐terminal region of Cse4 interacts with Okp1/Ame1. Furthermore, genetic interactions between mutations of Cse4‐R37, Cse4‐K49 and Okp1 or Ame1 suggested that this interaction is modulated by posttranslational modifications on the Cse4 residues. To investigate this, we sought to determine the binding of Cse4 to Okp1/Ame1 in dependence of R37 and K49 PTMs. Since recombinant Cse4 from E. coli is unmodified, we obtained a synthetic Cse4 (aa 33–110) peptide that includes presumed binding regions based on CLMS (Figs 4B and 2A). This peptide bound to Okp1/Ame1 in a pull‐down assay as well as by microscale thermophoresis (MST; Figs 6A and EV5, K D 129 ± 35 nM). Unfortunately, due to the complexity of the amino acid sequence and the length, synthesis of modified versions of Cse4 (33–110) failed. However, a shorter Cse4 (33–87) peptide that includes one possible binding site could be synthesized with R37Me and K49Ac (or both; Fig 2A). Therefore, to test the effect of these modifications, we performed competition assays to measure the replacement of labelled Cse4 (33–110) by unlabelled Cse4 (33–87).
Figure 6. Methylation of Cse4‐R37 and acetylation of Cse4‐K49 inhibit binding of Cse4 to Okp1/Ame1.
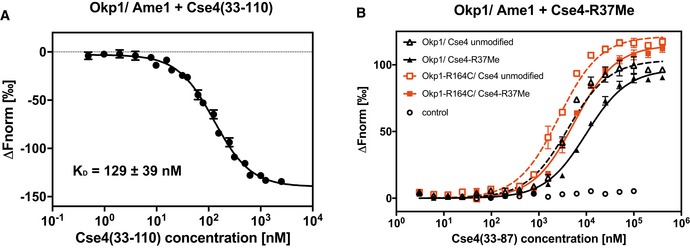
- A synthetic 78‐aa Cse4 peptide (aa 33–110) binds to Okp1/Ame1 in vitro. Microscale thermophoresis (MST) measurement of binding of fluorescently labelled Cse4 (33–110) to Okp1/Ame1 is shown. Normalized fluorescence, from which the fluorescence of unbound Okp1/Ame1 was subtracted (∆F norm), is plotted against Cse4 (33–110) concentration (logarithmic scale). Mean values ± SD (n = 3) are shown.
- Plot of MST measuring the competition of fluorescently labelled Cse4 (33–110) bound to Okp1/Ame1 (black; or Okp1‐R164C/Ame1, red) with unlabelled Cse4 (33–87) that is unmodified or monomethylated on R37 (R37Me). Normalized fluorescence from which the fluorescence of unbound Cse4 (33–110) was subtracted (∆F norm) is plotted against peptide concentration. Mean values ± standard deviation (n = 3) are given. Control, normalized fluorescence of Cse4 (33–110) alone at increasing concentrations.
Figure EV5. Binding of Okp1/Ame1 to Cse4N is inhibited by Cse4‐R37Me and Cse4‐K49Ac (related to Fig 6).
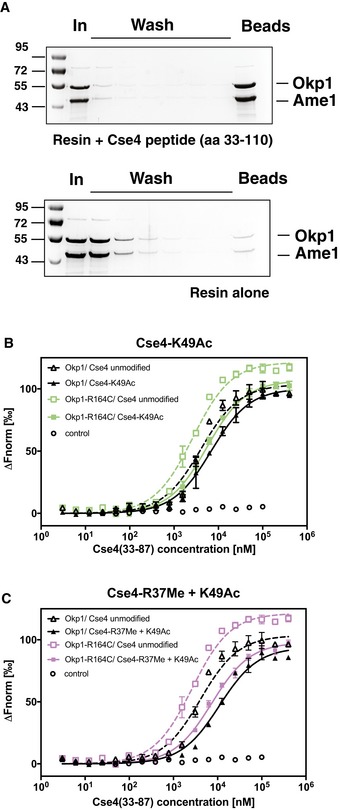
- A synthetic 78‐aa Cse4 peptide (aa 33–110) binds to Okp1/Ame1 in vitro. Pull‐down of Okp1/Ame1 by Cse4 (33–110). The Cse4 peptide was coupled to sepharose beads, and recombinant Okp1/Ame1 was incubated with beads with (top) or without (bottom) coupled Cse4 peptide. Beads were washed, and remaining bound Okp1/Ame1 amounts were determined. Samples of input (In), washes and beads were separated on SDS–PAGE gels, which were stained with Coomassie Blue.
- The binding of Okp1/Ame1 to Cse4N was inhibited by Cse4‐K49Ac, and Okp1‐R164C improved binding to Cse4N. Plot of MST measuring the competition of fluorescently labelled Cse4 (33–110) bound to Okp1/Ame1 (black; or Okp1‐R164C/Ame1, green) with unlabelled Cse4 (33–87) that is unmodified or acetylated on K49 (K49Ac). Data are represented as in Fig 6B.
- Binding of Okp1/Ame1 to Cse4N was inhibited by simultaneous Cse4‐R37Me and ‐K49Ac. Representation as in (B), but using purple for Okp1‐R164C/Ame1.
The inhibition constant (K I) of unmodified Cse4 (33–87) to Okp1/Ame1 was 18.3 ± 1.49 nM. Importantly, R37 methylation decreased the binding by ~13‐fold to a K I of 238.9 ± 24.51 nM (Fig 6B, Table 2). Furthermore, K49Ac reduced binding to Okp1/Ame1 by ~6.7‐fold, as shown by a K I of the K49‐acetylated Cse4 (33–87) peptide of 122.4 ± 19.07 nM. A peptide carrying both R37Me and K49Ac showed lower binding than R37Me or K49Ac alone (299.0 ± 50.84 nM, Fig EV5B and C, Table 2). This demonstrated that both R37Me and K49Ac inhibited the binding of Cse4 to Okp1/Ame1. The reduction of binding by K49Ac was consistent with the genetic observation that mutation of Cse4‐K49 to imitate the unacetylated state (K49R) suppressed the defect of cse4‐R37A.
Table 2.
Cse4‐R37 methylation and K49 acetylation inhibit the in vitro binding of Cse4 to Okp1/Ame1, and binding is restored by Okp1‐R164C. Mean ± SD values (n = 3) of measurements by microscale thermophoresis as performed in Fig 6 are given
| Peptide | K I [nM] | |
|---|---|---|
| Cse4 (33–87) | Okp1/Ame1 | Okp1‐R164C/Ame1 |
| Unmodified | 18.3 ± 1.49 | < 0.1 |
| R37Me | 238.9 ± 24.51 | 68.5 ± 10.67 |
| K49Ac | 122.4 ± 19.07 | 66.3 ± 6.77 |
| R37Me + K49Ac | 299.0 ± 50.84 | 130.1 ± 20.27 |
The genetic suppression of the cse4‐R37A mutation by okp1‐R164C suggested that the mutant Okp1‐R164C version restores centromere function by showing increased binding to Cse4. To test this hypothesis, we determined the affinity of Okp1‐R164C/Ame1 to the unmodified and R37Me/K49Ac‐modified Cse4 peptides. Significantly, while wild‐type Okp1/Ame1 interacted with R37Me‐Cse4 peptide with a K I of 238.9 ± 24.51 nM, Okp1‐R164C/Ame1 showed a higher affinity of 68.5 ± 10.67 nM (Fig 6B, Table 2), thus showing that the R164C mutation in Okp1 indeed increased binding of Okp1/Ame1 to the R37‐methylated Cse4 N‐terminus. R164C also resulted in a higher binding affinity of Okp1/Ame1 to the R37Me + K49Ac as well as the K49Ac Cse4 peptide (Fig EV5B and C, Table 2). Taken together, these results demonstrated that the binding of Cse4 to Okp1/Ame1 is reduced by both R37 methylation and K49 acetylation. Furthermore, the mutation Okp1‐R164C improves binding to Cse4, which results in a more efficient recruitment of the Ctf19 complex to the centromeric chromatin in vivo and thus in restoration of kinetochore function.
Discussion
Posttranslational modifications on histones regulate gene expression states by modulating the interaction of specific protein domains of chromatin modifiers with the nucleosome (Patel & Wang, 2013). Here, we have identified the Okp1/Ame1 complex from S. cerevisiae as a novel reader module for the unmodified N‐terminus of the centromeric H3 variant Cse4CENP‐A. Specifically, Okp1/Ame1 preferentially binds to Cse4, and the binding is inhibited by two modifications, methylation on R37 and acetylation on K49. Hence, Okp1/Ame1 is sensitive to the modification state of Cse4 in the nucleosome and thus regulates the assembly of the kinetochore on centromeric chromatin. We suggest that this regulates the dynamic association of kinetochores to chromatin during the cell cycle.
The discovery of a reader module in the inner kinetochore is unexpected in several ways. Firstly, Okp1/Ame1 contains no sequence motifs that are related to known chromatin readers, such that its identity as a reader of PTMs was unanticipated. Okp1/Ame1 was previously shown to bind DNA in vitro, but the in vivo relevance of this observation was unknown (Hornung et al, 2014). Our results provide clear evidence for a dominant role of Okp1/Ame1 in recognizing centromeric chromatin via the Cse4 N‐terminus, thereby linking this to the recruitment of the Ctf19 complex and the hierarchical assembly of the kinetochore.
Secondly, the effect of Cse4 PTMs on Okp1/Ame1 binding is an ~7–13‐fold reduction in affinity, which may seem like a relatively subtle change, but this change clearly is sufficient in vivo to disrupt binding. Accordingly, the restoration of in vitro binding by Okp1‐R164C is ~2–4‐fold, and again, this seemingly moderate change of in vitro binding is sufficient to strongly suppress growth defects in vivo. We note that the affinity constants measured here were determined with a short (55 aa) Cse4 peptide and lie in the micromolar range, which is a binding strength comparable to that of other PTM readers like the bromodomain of the transcriptional co‐activator P/CAF (Dhalluin et al, 1999), or tudor domains to methyl‐lysine (Lu & Wang, 2013). As expected, the binding of a longer (78 aa) Cse4 peptide to Okp1/Ame1 is stronger (~130 nM), and the full Cse4N (aa 21–121) interacts with low nanomolar affinity. This robust binding of Okp1/Ame1 to Cse4 as a critical contact point at the base of the kinetochore ensures that the contact can resist the strong pulling forces that are exerted on the single Cse4 nucleosome during chromosome segregation.
Thirdly, based on earlier observations, the Cse4 N‐terminus was expected to interact with Ctf19/Mcm21 (Ortiz et al, 1999), but we were unable to detect such an interaction with purified components. Rather, we identified a tight association to Okp1/Ame1. Our results thus demonstrate that the predominant contact of COMA to chromatin occurs via Okp1/Ame1. This is consistent with the fact that Cse4N, Okp1 and Ame1, but not Ctf19, Mcm21 or other Ctf19 components, are essential for viability.
What is the function of the two PTMs in the Cse4 N‐terminus? Yeast kinetochores are associated with centromeric sequences throughout most of the cell cycle, except for a brief period within S‐phase where they are released as the centromeric DNA is replicated (Kitamura et al, 2007). Also, Cse4 is replaced in early S‐phase at the time of DNA synthesis (Wisniewski et al, 2014). We speculate that the R37 methylation and K49 acetylation in Cse4N mediate this process by impairing the interaction of COMA to the Cse4‐containing nucleosome. In agreement with this notion, K49Ac and R37Me are increased in cells arrested in S‐phase. How the respective modifying enzymes (Gcn5/SAGA for K49Ac and an unknown arginine methyltransferase for R37) access their target sites in this context, and whether this is coupled to the deposition of new Cse4, remains to be determined.
With respect to cell‐cycle dynamics, it is interesting to note that there has been some debate regarding the exact stoichiometry of Cse4 within the centromeric nucleosome across the cell cycle, or whether it constitutes a hemisome (Henikoff & Furuyama, 2012). The Cse4 content has been proposed to oscillate during mitosis from one to two molecules per centromeric nucleosome (Shivaraju et al, 2012), whereas another study observed replacement of Cse4 during S‐phase, and it remained stably present at the centromere for the rest of the cell cycle until the next S‐phase (Wisniewski et al, 2014). If two Cse4 molecules are in the nucleosome, presumably two Okp1/Ame1 entities are bound, raising the question of the stoichiometry of other Ctf19 components relative to Okp1/Ame1. Of note, Okp1 and Ame1 are more abundant in the nucleus than Ctf19 and Mcm21 (Dhatchinamoorthy et al, 2017), arguing that not all cellular Okp1/Ame1 is part of COMA. Thus, further work is required to elucidate the precise architecture of the kinetochore at its interface with chromatin.
Our discovery of an Okp1/Ame1 axis for kinetochore recruitment by interaction with Cse4 raises the question how it translates to higher eukaryotic systems, since the Cse4 N‐terminus is specific to yeasts (Boeckmann et al, 2013) and is not homologous to the N‐terminus of CENP‐A, e.g. from humans. It will therefore be interesting to determine how CENP‐Q/CENP‐U makes contacts at its centromere‐proximal side, and whether this likewise is regulated by modifications on that protein. On the other hand, a yeast‐specific Okp1/Ame1 recruitment mechanism may extend to pathological yeasts like Candida albicans, which shares with S. cerevisiae an extended N‐terminus on Cse4. Our observations therefore afford a possibility to design molecules that specifically disrupt this interaction and inhibit chromosome segregation and cell growth selectively in yeasts, but not in the human host. Such molecules provide a therapeutic opportunity for the treatment of yeast pathogens.
Materials and Methods
Saccharomyces cerevisiae strains and yeast methods
Yeast strains and plasmids used in this study are described in Tables EV2 and EV3. Yeasts were grown and manipulated using standard genetic techniques (Sherman, 1991). Plasmid‐borne alleles of cse4, okp1 and ame1 were constructed using the gap repair method and verified by sequence analysis. For chromosomal integration of cse4 and okp1 alleles, the respective allele was transferred into a URA3‐marked integrating vector and introduced into the corresponding strain by integrative transformation followed by loop‐out on 5‐fluoroorotic acid (5‐FOA) medium (Boeke et al, 1984). For genetic crosses, cse4 and okp1 were marked with HISMX or URAMX by integration of the respective resistance cassettes downstream of the open reading frame. Deletions of chromosomal genes were performed using the integration of knockout cassettes (Longtine et al, 1998).
UV mutagenesis of cbf1∆ cse4‐R37A cells (AEY4984), isolation of suppressor mutants of the temperature sensitivity and identification of the causative mutation were carried out as described (Samel et al, 2017). Briefly, DNA of a temperature‐resistant derivative (AEY5438) was subjected to whole‐genome sequencing, and single‐nucleotide polymorphisms were identified by comparing the sequence to that of AEY4984. AEY5438 carried the mutation okp1‐R164C as well as non‐synonymous mutations in two other genes. To determine whether okp1‐R164C caused the suppression phenotype, the mutation was created by site‐directed mutagenesis in an integrative URA3‐marked plasmid and introduced into AEY4984 by integrative transformation and loop‐out as described above. cbf1∆ cse4‐R37A okp1‐R164C strains were identified by sequence analysis of the okp1 gene and tested for temperature resistance by growth assays on full medium at elevated temperatures.
Plasmid loss was measured in a wt (AEY1), cse4‐R37A (AEY5596), okp1‐R164C (AEY5594) and okp1‐R164C cse4‐R37A (AEY5586) strain carrying a CEN6‐TRP1 plasmid containing either a functional centromere sequence (pAE264) or a centromere sequence without CDEI element (CEN∆CDEI, pAE1771) as previously described (McNally & Rine, 1991).
For FACS analysis, strains were grown in YPD at 23°C and shifted for 5 h to 37°C. Samples of 0.2 OD were harvested in mid‐exponential phase at both temperatures. After washing the samples with ddH2O, cells were resuspended in 400 μl ddH2O, sonicated briefly and fixed with 950 μl 100% EtOH at −20°C. For FACS analysis, the cells were washed with 800 μl of 50 mM sodium citrate (pH 7.2) and incubated with 500 μl RNase A solution [0.25 mg/ml in 50 mM sodium citrate (pH 7.2)] overnight at 37°C. The cells were subsequently incubated with 20 μl of 20 mg/ml proteinase K for 2 h at 49°C, sonicated briefly and stained with 500 μl Sytox Green solution [4 μM Sytox Green in 50 mM sodium citrate (pH 5.2)]. FACS analysis was performed with a BD Accuri flow cytometer.
Screen for suppressor alleles of OKP1 and AME1
Plasmid‐borne OKP1 (pAE2134) was mutagenized by error‐prone PCR and introduced by gap repair into a cbf1∆ cse4‐R37A okp1∆ strain that carried OKP1 on a URA3‐marked plasmid (AEY5584). Transformants were plated at the restrictive temperature, and temperature‐resistant candidates were isolated. The URA3‐marked plasmid with wild‐type OKP1 was subsequently eliminated on 5‐FOA medium and the temperature resistance verified. For mutagenesis of AME1, plasmid‐borne AME1 (pAE2456) was mutagenized by error‐prone PCR and introduced into the cbf1∆ cse4‐R37A strain AEY4984 as above. Mutant okp1 or ame1 plasmids, respectively, were isolated, amplified in E. coli and subjected to sequence analysis. Plasmids from temperature‐resistant ame1 isolates were further phenotypically tested in a cbf1∆ cse4‐R37A ame1∆ strain (AEY6091) after counter‐selection of the pURA3‐AME1 plasmid on 5‐FOA medium. For plasmid isolates that carried several mutations, the causative mutation was identified by constructing plasmids carrying a single mutation by site‐directed mutagenesis and testing its ability to suppress the temperature sensitivity of the cbf1∆ cse4‐R37A okp1∆ or ame1∆ strain by plasmid shuffle as described above.
Molecular cloning
The open reading frames corresponding to the Cse4 (21–129), Ctf19 and Mcm21 proteins were amplified from genomic DNA or cDNA (in the case of Mcm21). CSE4 was cloned into expression vector pETM11 using NcoI/SalI, CTF19 and MCM21 were subcloned from the monocistronic pET3aTr vector into the polycistronic expression vector pST39 (cassettes 3 (SacI/KpnI) and 4 (BspEI/MluI), respectively; Tan et al, 2005). The pST39/Okp1‐R164C‐Ame1 construct was obtained based on pST39/Okp1‐Ame1 by site‐directed mutagenesis using primers GATCCATCTTTGTTTATTAGAGACTAACACTGTAAGC and AGTCTCTAATAAACAAAGTAGGATCCTATCATTATTTT. Constructs were verified by DNA sequencing. The vectors pST39 and pET3aTr were kindly provided by Song Tan (The Pennsylvania State University, USA).
For construction of pAE2123 (= pRS426‐OKP1), an OKP1 fragment with 372 basepairs (bp) upstream and 485 bp downstream of the open reading frame (ORF) was PCR‐amplified from genomic DNA and cloned with SalI/SacI in pRS426 (Sikorski & Hieter, 1989). To generate pAE2455 (= pRS426‐AME1), a PCR‐amplified AME1 fragment including 324 bp upstream and 321 bp downstream of ORF was cloned with NotI/SalI into pRS426.
Recombinant protein expression and purification
Plasmids containing the genes of interest were transformed into the E. coli strain Rosetta. Cells containing plasmids with Cse4N were grown in auto‐induction media to an optical density at 600 nm (OD600) of 0.3 at 37°C and incubated for 24 h at 18°C (Studier, 2005). Ctf19‐Mcm21 complex was expressed at 18°C overnight after induction at an OD600 of 0.6 with 0.5 mM IPTG.
For Cse4 (21–129) purification, cells pellet was resuspended in 50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 10 mM imidazole, 0.01% Tween‐20, 1 mM β‐mercaptoethanol, 1 mM PMSF, protease inhibitors. Bacteria were lysed by sonication, and protein was isolated using Ni‐NTA agarose beads (Qiagen). Washing of beads was performed in 50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole, 1 mM β‐mercaptoethanol. Cse4 (21–129) was eluted from Ni‐NTA beads with 50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 250 mM imidazole, 1 mM β‐mercaptoethanol. Fractions containing Cse4 (21–129) were pooled, supplemented with TEV protease and dialysed against 50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM β‐mercaptoethanol at 4°C overnight. Cse4 (21–129) protein without 6× HisTag and TEV protease were separated using Ni‐NTA beads. Fractions containing Cse4 (21–129) were concentrated and further separated via Superdex 75 size‐exclusion column (GE Healthcare) in 30 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM DTT. Peak fractions were pooled, concentrated and stored at −80°C.
Ame1/Okp1 complex purification was performed as described (Hornung et al, 2014) using plasmid pST39 carrying OKP1 and AME1 (kindly provided by S. Westermann, University of Duisburg‐Essen, Germany).
For Ctf19/Mcm21 complex purification, cells were lysed by sonication in 50 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol, 10 mM imidazole, 1 mM β‐mercaptoethanol, protease inhibitors. The lysate was incubated with Ni‐NTA beads, and bound protein complex was washed and eluted with 50 mM HEPES, pH 7.5, 300 mM NaCl, 10% glycerol, 250 mM imidazole, 1 mM β‐mercaptoethanol. The protein solution was diluted to 100 mM NaCl and applied to a MonoQ anion exchange column (GE Healthcare) in 50 mM HEPES, pH 7.5, 100 mM NaCl, 10% glycerol and 1 mM β‐mercaptoethanol and eluted with a gradient of 100 mM–1 M NaCl. Pooled fractions were concentrated and further purified on a Superdex 200 size‐exclusion column (GE Healthcare) in 30 mM HEPES, pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM DTT. Peak fractions were pooled, concentrated and stored at −80°C.
Interaction studies using SEC
Analytical gel filtration was performed on a Superdex 200 Increase 3.2/300 column under isocratic conditions at 4°C. Proteins were mixed in equimolar ratio in binding buffer (50 mM HEPES, pH 7.5, 200 mM NaCl, 1 mM DTT) and incubated for 20 min on ice. Elution of proteins was monitored by absorbance at a wavelength of 280 nm. The eluates were fractionated, loaded onto SDS–PAGE, and the gels were stained with Coomassie Blue.
Microscale thermophoresis
Measurements were performed using a Monolith NT.115 instrument in Premium coated glass capillaries (Nanotemper). Okp1/Ame1 or Okp1‐R164C/Ame1 (starting at 2 μM) was titrated against of 150 nM of labelled Cse4 peptide (33–110) in MST buffer (50 mM HEPES, pH 7.5, 200 mM NaCl, 1 mg/ml BSA and 0.05% Tween‐20). The Cse4 (33–110) peptide (Table EV4, 50 μM, CASLO, Kongens Lyngby, Denmark) was labelled with Alexa 647‐NHS (600 μM) in the dark overnight at 4°C. Unbound Alexa 647 was removed with a G25 column (GE Healthcare). The labelling of peptide yielded a dye to peptide ratio of 3:1. Okp1/Ame1 or Okp1‐R164C/Ame1 complex was incubated with Cse4 (33–110) peptide at RT for 10 min in dark. Then, the mixture was loaded into glass capillaries and measured at 25°C for initial fluorescence and change in fluorescence upon a thermophoresis measurement (setup: MST power: 40%, red LED power: 40%). For analysis, the T‐jump with thermophoresis (for 21 s) was used. In order to determine the noise level, triplicates of the same dilution of labelled peptide and protein complex were measured leading to ΔF≈ FU. The change in fluorescence was expressed as fraction bound of the maximum fluorescence in the bound state. Dissociation constants were deduced from a binding saturation fit of mean values of independent experiments (n = 3). The function for fitting Equation (1) is derived from the law of mass action,
| (1) |
with L being the concentration of labelled peptide, B being the concentration of protein with constant concentration and Kdapp being the apparent dissociation constant. Data were analysed using Affinity Analysis Software (Nanotemper, v2.0) and plotted with Graphpad Prism 5.
For competition assays, a complex between Ame1/Okp1 dimer (4 μM) and labelled Cse4 peptide (600 nM) was assembled in MST buffer at RT for 10 min in dark. Then, shorter Cse4 (33–87) peptide versions (initial concentration is 800 μM) were titrated against of Okp1‐Ame1/Cse4 (33–110) peptide complex in order to replace labelled peptide (Table EV4). Mixtures were loaded on glass capillaries and measured at 25°C for initial fluorescence and change in fluorescence with the thermophoresis measurement (MST power 40% and LED power 40%). Results from competition experiments were analysed according to the same formula (Equation (1)); however, the half maximum saturation point was described by a half maximum inhibition value (IC50). In order to convert IC50 values into inhibitor constants (K I), Equation (1) was implemented, which is based on (Nikolovska‐Coleska et al, 2004) and can be computed with an online tool (http://sw16.im.med.umich.edu/software/calc_ki/).
| (2) |
With [I]50 being the concentration of free inhibitor at 50% inhibition, [L]50 being the concentration of free labelled peptide and [P]0 being the concentration of the free protein at 0% inhibition.
Data availability
The mass spectrometry data from CLMS of Okp1/Ame1/Cse4N have been deposited to the ProteomeXchange Consortium database (dataset identifier PXD009648).
For further experimental details and Materials and Methods, please consult the Appendix.
Author contributions
EAA, AS‐P, TMTN, SS‐B and JP performed experiments. AC, TB and JR performed experiments and data analysis. DL and AH performed data analysis. AEE‐M conceived and planned experiments, supervised the study and wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank S. Tan, J. Vogel and S. Westermann for generously providing reagents, J. Hamann, S. Steinborn and A.‐K. Schulze for technical assistance and M. Boltengagen for help with graphs. This work was supported by Deutsche Forschungsgemeinschaft to A. E.‐M. (GRK1431 and EH237/12‐1) and by the Wellcome Trust to J. R. (103139). The Wellcome Trust Centre for Cell Biology is supported by core funding from the Wellcome Trust (203149).
The EMBO Journal (2019) 38: e98991
References
- Angus‐Hill ML, Dutnall RN, Tafrov ST, Sternglanz R, Ramakrishnan V (1999) Crystal structure of the histone acetyltransferase Hpa2: a tetrameric member of the Gcn5‐related N‐acetyltransferase superfamily. J Mol Biol 294: 1311–1325 [DOI] [PubMed] [Google Scholar]
- Bailey AO, Panchenko T, Sathyan KM, Petkowski JJ, Pai PJ, Bai DL, Russell DH, Macara IG, Shabanowitz J, Hunt DF, Black BE, Foltz DR (2013) Posttranslational modification of CENP‐A influences the conformation of centromeric chromatin. Proc Natl Acad Sci USA 110: 11827–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Mata JF, Sergeev M, David AF, Salimian KJ, Panchenko T, Cleveland DW, Black BE, Shah JV, Jansen LE (2014) The quantitative architecture of centromeric chromatin. Elife 3: e02137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann L, Takahashi Y, Au WC, Mishra PK, Choy JS, Dawson AR, Szeto MY, Waybright TJ, Heger C, McAndrew C, Goldsmith PK, Veenstra TD, Baker RE, Basrai MA (2013) Phosphorylation of centromeric histone H3 variant regulates chromosome segregation in Saccharomyces cerevisiae . Mol Biol Cell 24: 2034–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine‐5′‐phosphate decarboxylase activity in yeast: 5‐fluoro‐orotic acid resistance. Mol Gen Genet 197: 345–346 [DOI] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD (1996) Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84: 843–851 [DOI] [PubMed] [Google Scholar]
- Burns N, Grimwade B, Ross‐Macdonald PB, Choi EY, Finberg K, Roeder GS, Snyder M (1994) Large‐scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae . Genes Dev 8: 1087–1105 [DOI] [PubMed] [Google Scholar]
- Cai M, Davis RW (1990) Yeast centromere binding protein CBF1, of the helix‐loop‐helix protein family, is required for chromosome stability and methionine prototrophy. Cell 61: 437–446 [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR III, Chan CS, Drubin DG, Barnes G (2002) Phospho‐regulation of kinetochore‐microtubule attachments by the Aurora kinase Ipl1p. Cell 111: 163–172 [DOI] [PubMed] [Google Scholar]
- Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski‐Wills JC, Nilges M, Cramer P, Rappsilber J (2010) Architecture of the RNA polymerase II‐TFIIF complex revealed by cross‐linking and mass spectrometry. EMBO J 29: 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Grant PA (2007) Multi‐tasking on chromatin with the SAGA coactivator complexes. Mutat Res 618: 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wulf P, McAinsh AD, Sorger PK (2003) Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev 17: 2902–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399: 491–496 [DOI] [PubMed] [Google Scholar]
- Dhatchinamoorthy K, Shivaraju M, Lange JJ, Rubinstein B, Unruh JR, Slaughter BD, Gerton JL (2017) Structural plasticity of the living kinetochore. J Cell Biol 216: 3551–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Migeon BR (1985) Three related centromere proteins are absent from the inactive centromere of a stable isodicentric chromosome. Chromosoma 92: 290–296 [DOI] [PubMed] [Google Scholar]
- Eberharter A, Sterner DE, Schieltz D, Hassan A, Yates JR III, Berger SL, Workman JL (1999) The ADA complex is a distinct histone acetyltransferase complex in Saccharomyces cerevisiae . Mol Cell Biol 19: 6621–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenhofer‐Murray AE, Rivier D, Rine J (1997) The role of Sas2, an acetyltransferase homolog of Saccharomyces cerevisiae, in silencing and ORC function. Genetics 145: 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Lee J, Sekulic N, Sennett MA, Lee TH, Black BE (2016) CENP‐C directs a structural transition of CENP‐A nucleosomes mainly through sliding of DNA gyres. Nat Struct Mol Biol 23: 204–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR III, Cleveland DW (2006) The human CENP‐A centromeric nucleosome‐associated complex. Nat Cell Biol 8: 458–469 [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T et al (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147 [DOI] [PubMed] [Google Scholar]
- Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen‐Hughes T, Allis CD, Winston F, Berger SL, Workman JL (1997) Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev 11: 1640–1650 [DOI] [PubMed] [Google Scholar]
- Henikoff S, Furuyama T (2012) The unconventional structure of centromeric nucleosomes. Chromosoma 121: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA‐associated Ubp8. Genes Dev 17: 2648–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewawasam G, Shivaraju M, Mattingly M, Venkatesh S, Martin‐Brown S, Florens L, Workman JL, Gerton JL (2010) Psh1 is an E3 ubiquitin ligase that targets the centromeric histone variant Cse4. Mol Cell 40: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann G, Samel‐Pommerencke A, Weber J, Cuomo A, Bonaldi T, Ehrenhofer‐Murray AE (2018) A role for CENP‐A/Cse4 phosphorylation on serine 33 in deposition at the centromere. FEMS Yeast Res 18: fox094 [DOI] [PubMed] [Google Scholar]
- Hornung P, Troc P, Malvezzi F, Maier M, Demianova Z, Zimniak T, Litos G, Lampert F, Schleiffer A, Brunner M, Mechtler K, Herzog F, Marlovits TC, Westermann S (2014) A cooperative mechanism drives budding yeast kinetochore assembly downstream of CENP‐A. J Cell Biol 206: 509–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland KM, Kingsbury J, Koshland D, Hieter P (1999) Ctf19p: a novel kinetochore protein in Saccharomyces cerevisiae and a potential link between the kinetochore and mitotic spindle. J Cell Biol 145: 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuta H, Ikeno M, Suzuki N, Tomonaga T, Nozaki N, Obuse C, Kisu Y, Goshima N, Nomura F, Nomura N, Yoda K (2006) Comprehensive analysis of the ICEN (Interphase Centromere Complex) components enriched in the CENP‐A chromatin of human cells. Genes Cells 11: 673–684 [DOI] [PubMed] [Google Scholar]
- Joglekar AP, Bloom K, Salmon ED (2009) In vivo protein architecture of the eukaryotic kinetochore with nanometer scale accuracy. Curr Biol 19: 694–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Howe L, Tafrov ST, Grant PA, Sternglanz R, Workman JL (2000) The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30‐containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)‐FACT complex. Genes Dev 14: 1196–1208 [PMC free article] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y (2013) A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP‐C. Science 340: 1110–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith KC, Baker RE, Chen Y, Harris K, Stoler S, Fitzgerald‐Hayes M (1999) Analysis of primary structural determinants that distinguish the centromere‐specific function of histone variant Cse4p from histone H3. Mol Cell Biol 19: 6130–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura E, Tanaka K, Kitamura Y, Tanaka TU (2007) Kinetochore microtubule interaction during S phase in Saccharomyces cerevisiae . Genes Dev 21: 3319–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar EM, Scarborough EA, Umbreit NT, Zelter A, Gestaut DR, Riffle M, Johnson RS, MacCoss MJ, Asbury CL, Davis TN (2015) Regulation of outer kinetochore Ndc80 complex‐based microtubule attachments by the central kinetochore Mis12/MIND complex. Proc Natl Acad Sci USA 112: E5583–E5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitoku N, Sasayama T, Marumoto T, Zhang D, Honda S, Kobayashi O, Hatakeyama K, Ushio Y, Saya H, Hirota T (2003) CENP‐A phosphorylation by Aurora‐A in prophase is required for enrichment of Aurora‐B at inner centromeres and for kinetochore function. Dev Cell 5: 853–864 [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lu R, Wang GG (2013) Tudor: a versatile family of histone methylation “readers”. Trends Biochem Sci 38: 546–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally FJ, Rine J (1991) A synthetic silencer mediates SIR‐dependent functions in Saccharomyces cerevisiae . Mol Cell Biol 11: 5648–5659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meluh PB, Yang P, Glowczewski L, Koshland D, Smith MM (1998) Cse4p is a component of the core centromere of Saccharomyces cerevisiae . Cell 94: 607–613 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Desai A (2017) A molecular view of kinetochore assembly and function. Biology 6: E5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolovska‐Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S (2004) Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem 332: 261–273 [DOI] [PubMed] [Google Scholar]
- Nogales E, Ramey VH (2009) Structure‐function insights into the yeast Dam1 kinetochore complex. J Cell Sci 122: 3831–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Cheeseman IM, Hori T, Okawa K, McLeod IX, Yates JR III, Desai A, Fukagawa T (2006) The CENP‐H‐I complex is required for the efficient incorporation of newly synthesized CENP‐A into centromeres. Nat Cell Biol 8: 446–457 [DOI] [PubMed] [Google Scholar]
- Ortiz J, Stemmann O, Rank S, Lechner J (1999) A putative protein complex consisting of Ctf19, Mcm21, and Okp1 represents a missing link in the budding yeast kinetochore. Genes Dev 13: 1140–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DJ, Wang Z (2013) Readout of epigenetic modifications. Annu Rev Biochem 82: 81–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar A, Roy N, Sinha P (1999) MCM21 and MCM22, two novel genes of the yeast Saccharomyces cerevisiae are required for chromosome transmission. Mol Microbiol 31: 349–360 [DOI] [PubMed] [Google Scholar]
- Ranjitkar P, Press MO, Yi X, Baker R, MacCoss MJ, Biggins S (2010) An E3 ubiquitin ligase prevents ectopic localization of the centromeric histone H3 variant via the centromere targeting domain. Mol Cell 40: 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifsnyder C, Lowell J, Clarke A, Pillus L (1996) Yeast silencing genes and human genes associated with AML and HIV‐1 Tat interactions are homologous with acetyltransferases. Nat Genet 14: 42–49 [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases. Annu Rev Biochem 70: 81–120 [DOI] [PubMed] [Google Scholar]
- Samel A, Cuomo A, Bonaldi T, Ehrenhofer‐Murray AE (2012) Methylation of CenH3 arginine 37 regulates kinetochore integrity and chromosome segregation. Proc Natl Acad Sci USA 109: 9029–9034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samel A, Nguyen TKL, Ehrenhofer‐Murray AE (2017) Defects in methylation of arginine 37 on CENP‐A/Cse4 are compensated by the ubiquitin ligase complex Ubr2/Mub1. FEMS Yeast Res 17: fox009 [DOI] [PubMed] [Google Scholar]
- Sathyan KM, Fachinetti D, Foltz DR (2017) alpha‐amino trimethylation of CENP‐A by NRMT is required for full recruitment of the centromere. Nat Commun 8: 14678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitzberger F, Harrison SC (2012) RWD domain: a recurring module in kinetochore architecture shown by a Ctf19‐Mcm21 complex structure. EMBO Rep 13: 216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitzberger F, Richter MM, Gordiyenko Y, Robinson CV, Dadlez M, Westermann S (2017) Molecular basis for inner kinetochore configuration through RWD domain‐peptide interactions. EMBO J 36: 3458–3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F (1991) Getting started with yeast. Methods Enzymol 194: 3–21 [DOI] [PubMed] [Google Scholar]
- Shivaraju M, Unruh JR, Slaughter BD, Mattingly M, Berman J, Gerton JL (2012) Cell‐cycle‐coupled structural oscillation of centromeric nucleosomes in yeast. Cell 150: 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler S, Keith KC, Curnick KE, Fitzgerald‐Hayes M (1995) A mutation in CSE4, an essential gene encoding a novel chromatin‐associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev 9: 573–586 [DOI] [PubMed] [Google Scholar]
- Studier FW (2005) Protein production by auto‐induction in high density shaking cultures. Protein Expr Purif 41: 207–234 [DOI] [PubMed] [Google Scholar]
- Tan S, Kern RC, Selleck W (2005) The pST44 polycistronic expression system for producing protein complexes in Escherichia coli . Protein Expr Purif 40: 385–395 [DOI] [PubMed] [Google Scholar]
- Varma D, Salmon ED (2012) The KMN protein network–chief conductors of the kinetochore orchestra. J Cell Sci 125: 5927–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterborg JH (2000) Steady‐state levels of histone acetylation in Saccharomyces cerevisiae . J Biol Chem 275: 13007–13011 [DOI] [PubMed] [Google Scholar]
- Westermann S, Schleiffer A (2013) Family matters: structural and functional conservation of centromere‐associated proteins from yeast to humans. Trends Cell Biol 23: 260–269 [DOI] [PubMed] [Google Scholar]
- Wigge PA, Kilmartin JV (2001) The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J Cell Biol 152: 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J, Hajj B, Chen J, Mizuguchi G, Xiao H, Wei D, Dahan M, Wu C (2014) Imaging the fate of histone Cse4 reveals de novo replacement in S phase and subsequent stable residence at centromeres. Elife 3: e02203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittschieben BO, Otero G, de Bizemont T, Fellows J, Erdjument‐Bromage H, Ohba R, Li Y, Allis CD, Tempst P, Svejstrup JQ (1999) A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol Cell 4: 123–128 [DOI] [PubMed] [Google Scholar]
- Xiao H, Wang F, Wisniewski J, Shaytan AK, Ghirlando R, FitzGerald PC, Huang Y, Wei D, Li S, Landsman D, Panchenko AR, Wu C (2017) Molecular basis of CENP‐C association with the CENP‐A nucleosome at yeast centromeres. Genes Dev 31: 1958–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin SG, Shelby RD, Sullivan KF (2001) CENP‐A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J Cell Biol 155: 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The mass spectrometry data from CLMS of Okp1/Ame1/Cse4N have been deposited to the ProteomeXchange Consortium database (dataset identifier PXD009648).
For further experimental details and Materials and Methods, please consult the Appendix.