

Abstract
The abundance of any protein is determined by the balance of protein synthesis and protein degradation. Regulated protein degradation has emerged as a powerful means of precisely controlling individual protein abundance within cells and is largely mediated by the ubiquitin-proteasome system (UPS). By controlling the levels of key regulatory proteins, the UPS contributes to nearly every aspect of cellular function. The UPS also functions in protein quality control, rapidly identifying and destroying misfolded or otherwise aberrant proteins that may be toxic to cells. Increasingly, we understand that dysregulation of protein degradation pathways is critical for many human diseases. Conversely, the versatility and scope of the UPS provides opportunities for therapeutic intervention. In this review, we will discuss the basic mechanisms of protein degradation by the UPS. We will then consider some paradigms of human disease related to protein degradation using selected examples. Finally, we will highlight several established and emerging therapeutic strategies based on altering pathways of protein degradation.
The precise control of protein abundance represents a major mechanism of cellular regulation and is determined by the parallel action of pathways of protein synthesis and protein degradation. Protein degradation was initially thought to be a relatively nonspecific process. The discovery of the ubiquitin-proteasome system (UPS) dramatically changed this view. In this pathway, proteins destined for degradation are first labeled with the small protein ubiquitin. This ubiquitin tag targets the substrate protein to the proteasome, a large multisubunit protease that recognizes, unfolds, and ultimately destroys the substrate protein (Figure 1). Work over several decades has established the UPS as a complex, potent, and versatile means for regulating protein abundance. Many cellular pathways are controlled by the abundance of key or rate-determining factors. Targeted destruction of these critical proteins thus serves as a powerful means of regulating cellular function (Figure 1). Misfolded proteins represent a second major class of substrates for the UPS. Given their potential toxicity to cells, their timely recognition and destruction by the UPS are critical to cellular well-being (Figure 1).
Figure 1.

Overview of the ubiquitin-proteasome system for protein degradation. Many proteins misfold either during or immediately after protein synthesis (upper pathway). These proteins (red) are recognized by a series of ubiquitinating enzymes, known as E1, E2, and E3, that covalently attach the small protein ubiquitin (green) to the misfolded protein. This ubiquitin signal allows the protein to be recognized by the proteasome. The proteasome binds the ubiquitinated substrate, unfolds it, removes its ubiquitin molecules, and injects the protein into the central core of the proteasome, where the protein is degraded into small polypeptides. Properly folded proteins are also subject to degradation (lower pathway). In these cases, by controlling the abundance of key regulatory proteins, the ubiquitin-proteasome system contributes to nearly every aspect of cell biology. Some well-characterized examples are listed. Moreover, any properly folded protein can be converted into a misfolded protein at any time because of exposure to any of the numerous stressors that cause protein misfolding, only a few of which are listed.
As the basic mechanisms of the UPS have been unraveled, there has been an ever-increasing understanding of the relevance of protein degradation for human disease. In this review, we will briefly summarize the basic mechanisms of the UPS. We will then discuss how defects of protein degradation directly cause human disease, using several paradigmatic examples. Finally, we will discuss some established and emerging therapeutic strategies that are based on altering endogenous pathways of protein degradation.
The Ubiquitin System
Ubiquitin is a small 76–amino acid protein and is among the most highly conserved proteins ever described. Ubiquitin is also remarkably stable, maintaining its folded structure at temperatures up to 85°C and across a pH spectrum of 1 to 13.1 Ubiquitin invariably ends with a diglycine (GG) motif, which is critical for its ability to bind to target proteins. The C-terminal glycine of ubiquitin is joined to the ε-amino group of a lysine residue within the acceptor protein through an isopeptide bond. This reaction, termed ubiquitination, is mediated by a family of proteins known as ubiquitinating enzymes. The first, known as E1, activates ubiquitin for transfer by using ATP to generate a thioester linkage between itself and ubiquitin. This activated ubiquitin can then be transferred to a second enzyme, E2. In many cases, the E2 enzyme directly transfers ubiquitin to the target protein, typically with the assistance of a third protein, E3. In other cases, the ubiquitin is transferred from E2 to E3, and the E3 is responsible for conjugation to the target protein. The discovery of the pathway of ubiquitination formed the basis for the 2004 Nobel Prize in Chemistry.2
Ubiquitin can be attached to a protein as a single molecule (monoubiquitination) at one or more lysines within the target protein. However, because ubiquitin itself contains seven lysines, ubiquitin can also be conjugated to itself, resulting in a chain of covalently linked ubiquitin molecules (polyubiquitination). These different ubiquitin chains take on unique structures and thus may signal different events within the cell. Some chain types, particularly lysine-48, are potent signals for destruction by the proteasome, whereas other chains signal nondegradative extraproteasomal regulatory functions. Although the complexity of these pathways is beyond the scope of the current review, the capacity to generate such a diversity of modifications contributes to ubiquitin's remarkable versatility as a cellular signaling molecule.3
The UPS controls the degradation of thousands of substrates, and as such, it must be not only comprehensive but exquisitely specific. The hierarchical organization of ubiquitination contributes to these aspects of the UPS. Most cells contain one or two E1 enzymes that perform all ubiquitin activation. By contrast, there are approximately 40 E2 enzymes and >600 E3 enzymes.4 The cooperation between distinct E2-E3 enzyme pairs allows for the specific degradation of thousands of individual substrates. The existence of a large family of deubiquitinating enzymes (approximately 80 to 90) adds yet another layer of regulation to ubiquitination pathways. Indeed, the rate of degradation of some substrates reflects a dynamic balance between pathways of ubiquitination and deubiquitination, each of which, in turn, may be regulated by distinct stimuli or changing conditions within the cell.5 Finally, there is a large and ever-growing family of proteins that binds to ubiquitinated substrates. There are >150 of these ubiquitin receptors, and they typically play key roles in determining the fate of their respective target substrates.6 All told, the UPS comprises approximately a thousand or more individual components, highlighting its remarkable complexity.
The Proteasome
The best-known function of ubiquitination is to direct proteins to the proteasome for destruction. The proteasome is the most complex protease ever described and bears little resemblance to typical proteases, like trypsin or chymotrypsin. Rather, the proteasome is a large (2.5 million Da) multisubunit complex that consists of approximately 35 different components.7 Unlike a typical protease, the proteasome harbors three different active sites. Its tryptic sites cleave after basic residues; its chymotryptic sites cleave after hydrophobic residues; and its caspase-like sites cleave after acidic residues. This multiplicity of active sites ensures that the proteasome will be able to degrade its thousands of client substrates. However, it is not enough for the proteasome to be potent. It must also be highly specific given that promiscuous degradation of other proteins could wreak havoc within the cell. In fact, the proteasome's active sites are sequestered within a closed cylindrical chamber known as the core particle (CP8). At either end of the CP is a small pore through which proteins can enter, but only if they have been unfolded first. Such access to the interior of the CP is controlled by a subcomplex of the proteasome known as the regulatory particle. The regulatory particle first recognizes and binds to the ubiquitinated substrate using its own ubiquitin receptors. The regulatory particle then unfolds the substrate using the energy of ATP and injects it into the CP for degradation. The CP degrades the protein into small peptides. These can then be further broken down by other proteases into single amino acids that can be reused for new protein synthesis.
Unlike its target substrates, ubiquitin is generally spared from degradation by the proteasome. This is achieved through the presence of multiple deubiquitinating enzymes that reside in the proteasome itself.9 This recycling of ubiquitin is advantageous for many reasons. As mentioned above, ubiquitin is tightly folded, and so its presence would likely prevent the substrate from translocating through the narrow pore into the CP. In principle, ubiquitin could be unfolded along with the substrate, but given ubiquitin's extreme stability, this would likely require significant energy expenditures. Furthermore, given that ubiquitin chains can be long, it would be wasteful to destroy multiple ubiquitin molecules every time a substrate was processed.
The complexity of the proteasome is further underlined by the existence of at least 10 other proteins whose sole function is to aid in the process of assembling new proteasomes.10 These proteasome chaperones help to orchestrate a highly complex stepwise pathway by which proteasome precursors are assembled into increasingly more complex structures until the proteasome reaches its final mature state.
Paradigms of Disease Related to Protein Degradation
Given the versatility and power of the UPS in shaping the cellular protein repertoire, it is not surprising that alterations in pathways of protein degradation lie at the heart of many human diseases. Indeed, so many examples have now been discovered that an exhaustive cataloging is no longer practical. Instead, some major paradigmatic themes in this area have been highlighted below using selected examples.
Excess Degradation of Tumor Suppressor Proteins
Tumor suppressor proteins typically exert their antioncogenic effects by inhibiting growth. Evasion of this inhibitory effect is critical for the initiation and maintenance of many cancer types.11 It is not surprising, then, that cancers have developed diverse mechanisms to neutralize key tumor suppressor proteins. Among the simplest and most effective strategies is to destroy the tumor suppressor protein. A prime example is well-differentiated liposarcoma, the most common form of liposarcoma. This tumor has a high propensity for local recurrence but little or no metastatic risk. The term atypical lipomatous tumor is used at surgically amenable locations to reflect this behavior. By contrast, dedifferentiated liposarcoma shows a significant rate of metastasis and death. Well-differentiated and dedifferentiated liposarcoma share a common molecular pathogenesis characterized by giant marker and ring chromosomes, the effect of which is to amplify sequences on the long arm of chromosome 12.12, 13 Among the genes amplified is Mdm2. The accumulation of Mdm2 is routinely used as a diagnostic adjunct: increased protein abundance can be monitored using immunohistochemistry, whereas gene amplification can be detected using fluorescence in situ hybridization.14, 15 Mdm2 functions as an E3 ubiquitin ligase, and its best-known substrate is the tumor suppressor p53.16, 17 Thus, the amplification of Mdm2 results in increased ubiquitination and proteasomal degradation of p53. Loss of the growth inhibitory function of p53 plays a crucial role in the development of this sarcoma.
Targeted elimination of p53 is apparently such an effective oncogenic strategy that it appears to have evolved separately in multiple tumor types. Human papillomavirus is responsible for several benign and malignant epithelial proliferations, including warts, condylomas, cervical carcinoma, and various head and neck carcinomas. In general, malignancies tend to be associated with high-risk serotypes, including human papillomaviruses 16 and 18. The oncogenic potential of these serotypes is attributable, in large part, to the presence of their E6 and E7 proteins.18 The viral E6 protein interacts with and stimulates the function of a human protein known as E6-associated protein. Like Mdm2, E6-associated protein is also an E3 ubiquitin ligase and its primary target is also p53.19, 20 By destroying this cellular guardian, the virus can proliferate unchecked within cells and, with the help of other genetic changes, can promote their transformation. Thus, liposarcoma and human papillomavirus–associated carcinomas use molecularly distinct approaches to achieve the same outcome (namely, the induced degradation of p53 protein).
Failure to Properly Destroy Oncoproteins
In addition to evading tumor suppressors, another of Hanahan and Weinberg's11 original hallmarks of cancer is sustaining proliferative signaling. Not surprisingly, cells have developed complex mechanisms to restrain the activity of these powerful growth-promoting factors, sometimes referred to as oncoproteins. Spatiotemporal control of gene expression is a common and powerful approach. However, many of these proteins are also subject to tight control of their abundance through regulated protein degradation. β-Catenin is an important example and a major driver of numerous benign and malignant neoplasms. Its cellular levels are normally kept low through continuous protein degradation in the cytoplasm.21 Progrowth signaling attenuates this degradation, allowing β-catenin to translocate to the nucleus, where it functions as a transcription factor. Defects in this degradation pathway result in the accumulation of nuclear β-catenin protein and constitutive progrowth signaling.
Familial adenomatous polyposis is a cancer predisposition syndrome associated with a variety of benign and malignant tumors, including colorectal cancer, desmoid fibromatosis, Gardner fibroma, and others. It is inherited in an autosomal dominant manner, with subsequent loss of heterozygosity driving tumor formation. The term familial adenomatous polyposis reflects that these individuals have numerous colonic adenomas, and affected individuals have a lifetime risk for colorectal cancer that approaches 100%. These patients are frequently treated by prophylactic colectomy. The genetic defect in familial adenomatous polyposis was mapped to chromosome 5, where the culprit gene was named APC for adenomatous polyposis coli.22 This protein functions as a positive regulator of β-catenin degradation. Thus, loss of APC function leads to accumulation of β-catenin and constitutive progrowth signaling.21 This accumulation of nuclear β-catenin is routinely used as a diagnostic adjunct in desmoid fibromatosis and other related tumor types. Interestingly, some familial adenomatous polyposis patients do not have APC mutations. Rather, these patients have mutations within β-catenin itself that render it refractory to degradation,23 resulting in the same final outcome (namely, an excess of β-catenin signaling).
Failure to Destroy Toxic Misfolded Proteins
The proper function of most proteins requires them to attain their precise three-dimensional folded shape. Even under ideal conditions, this process can be inherently difficult, particularly for larger or more complex proteins. In reality, there exists a seemingly endless array of cellular and environmental factors that complicate protein folding. In addition to inherited and acquired mutations, numerous environmental exposures contribute to protein misfolding. These include heat, heavy metals, metalloids (eg, arsenic), oxidizing agents, translational inhibitors, noncanonical amino acids, and others. These factors, coupled with the reality that the intracellular space is extremely crowded, make protein misfolding an ever-present threat to all cells.
Protein misfolding is toxic for two reasons. The more obvious reason is that the normal function of any protein may be lost if the protein cannot assume its proper shape. However, there is also gain-of-function toxicity that is unrelated to the protein's normal cellular function and that has become increasingly relevant to human disease. Hydrophobic regions are most often buried deep within the core of proteins to prevent energetically unfavorable interactions with the polar aqueous environment. Misfolding tends to expose these hydrophobic regions, which then tend to make promiscuous interactions with exposed hydrophobic regions in other proteins. This process leads to aberrant protein-protein interactions and may ultimately culminate in protein aggregation.
Many human diseases are now thought to be directly caused by protein misfolding and are sometimes termed proteinopathies.24, 25 Perhaps the most prominent among these are the neurodegenerative diseases, including Parkinson, Huntington, and Alzheimer diseases, amyotrophic lateral sclerosis, and many others. Many of these diseases show both sporadic and inherited forms. The inherited forms are illuminating because transmission tends to be autosomal dominant.26 This underscores the gain-of-function nature of this toxicity. These diseases are not due to loss of the normal function of the affected protein. Indeed, knocking out the relevant gene in animal models tends not to recapitulate the disease.
A common feature of most neurodegenerative diseases is the presence of conspicuous intracellular inclusions, either nuclear or cytoplasmic. Pathologists have visualized these cellular inclusions since the 19th century. Parkinson disease is an important example. This common disease affects approximately 1% of all Americans aged >60 years. Motor defects, including bradykinesia, rigidity, and tremor, are well recognized, but this disease may also result in devastating dementia.27
The pathologic hallmark of Parkinson disease is the Lewy body, a cytoplasmic aggregate seen in the substantia nigra and other affected cells (Figure 2). There has been controversy over whether these inclusions are themselves toxic to cells or rather a protective sequestration of toxic misfolded proteins. In either case, their major constituent is the small protein α-synuclein. Immunohistochemistry is not typically necessary for diagnosis but may be used to highlight these structures. Interestingly, Lewy bodies (like most cellular inclusions associated with neurodegenerative disease) tend to stain intensely with antiubiquitin antibodies as well. In rare cases, inherited mutations in α-synuclein lead to early-onset Parkinson disease.28 These tend to be point mutations. A rare but particularly interesting genetic form of Parkinson disease is characterized not by mutations in α-synuclein itself, but by duplications or triplications of a part of chromosome 4 that includes the α-synuclein gene.29 This remarkable gene dosage effect suggests that the simple abundance of α-synuclein protein (even in the absence of a mutation) is a critical determinant of Parkinson disease. This, in turn, implies that α-synuclein levels must be regulated within a relatively narrow range, and failure to do so may be at the heart of this disease. A similar gene dosage effect is also known in Alzheimer disease. Patients with trisomy 21 (Down syndrome) show a markedly increased risk of developing early-onset Alzheimer disease. Herein, the gene dosage effect relates to the amyloid precursor protein, which resides on chromosome 21 and is the major constituent of the amyloid plaques that are thought to be major contributors to Alzheimer disease.30
Figure 2.
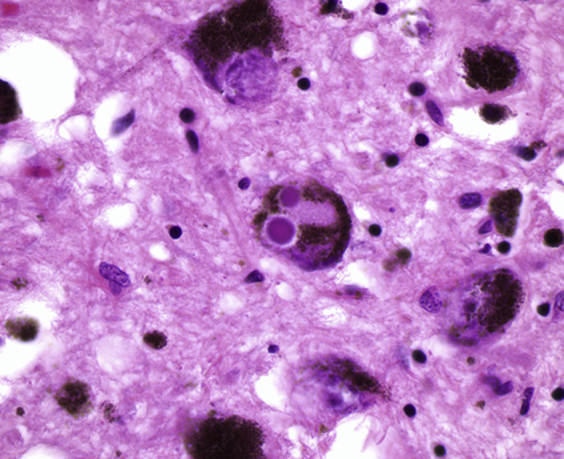
Lewy bodies associated with Parkinson disease. A hematoxylin and eosin–stained section from the substantia nigra is shown. Two Lewy bodies are present within the cytoplasm of a dopaminergic neuron present at the center of the image. Original magnification, ×400.
Excess Degradation of Misfolded but Partially Functional Proteins
Given the toxicity of misfolded proteins, cells have developed complex stress responses to identify and destroy misfolded proteins. These pathways are sometimes referred to as protein quality control. Despite their obvious utility, they are responsible for a somewhat counterintuitive group of diseases related to protein misfolding. In these cases, a protein may misfold but does not necessarily lose all its functional activity. Nevertheless, the protein is recognized by the cell as misfolded (ie, potentially dangerous) and is rapidly destroyed. These diseases, then, are caused by the preemptive degradation of what would otherwise have been a partially functional protein.
Cystic fibrosis is an instructive example. This disease is the most common autosomal recessive disease in Caucasians, with approximately 30,000 cases in the United States.31 The disease is typically fatal, although recent advances have pushed the average life expectancy into the mid fourth decade. The disease is characterized by thick viscous secretions throughout the body, but most prominent in the lungs and pancreas. In the lungs, this leads to obstruction, inflammation, and infection, ultimately resulting in loss of respiratory capacity. As the disease progresses, lung transplant may become necessary for survival. Pancreatic exocrine dysfunction can be partially treated with digestive enzyme supplements, although pancreatitis and diabetes remain major complications.
The cystic fibrosis gene was identified in 1989 and encodes a chloride ion channel known as cystic fibrosis transmembrane conductance regulator (CFTR).32 This is a large and difficult to fold protein with 10 transmembrane domains. It is synthesized at the endoplasmic reticulum (ER) and must traverse the secretory pathway from the ER to the Golgi to secretory vesicles, which then fuse with plasma membrane where CFTR functions. The ER is a particularly important protein quality control checkpoint. Proteins that are not properly folded do not advance through the secretory pathway. Instead, they are transported out of the ER and into the cytoplasm, where they are ubiquitinated and destroyed by the proteasome. This complex process is known as ER-associated degradation.33
The most common mutation in cystic fibrosis is a deletion of a single residue, phenylalanine, at position 508.34 This mutation accounts for approximately 90% of cystic fibrosis cases in the United States and is associated with a marked decrease in delivery of CFTR protein to the cell surface. This is because the ΔF508 mutant is recognized in the ER as misfolded and is rapidly destroyed by the proteasome.35, 36 The irony is that the mutant protein is not functionally dead. In fact, it retains significant chloride ion channel function: simply lowering the temperature, for example, reduces misfolding and allows the mutant protein to traffic through the ER to the plasma membrane, where the protein performs its normal function, albeit at a reduced level compared with wild type.37, 38 This surprising discovery served as the basis for the development of a new class of drugs that have been termed correctors (eg, lumacaftor) and that promote delivery of CFTR to the cell surface, where they can be further activated by small-molecule potentiators of CFTR's chloride transport function (eg, ivacaftor39, 40). These new US Food and Drug Administration (FDA)–approved therapies represent the first molecularly targeted approaches for cystic fibrosis and are revolutionizing the treatment of this disease.
Therapeutic Approaches Based On Protein Degradation
Although defects in regulated protein degradation may contribute to disease, the versatility and comprehensive scope of the UPS also provide opportunities for therapeutic development. As an increasingly detailed understanding of specific pathways of protein degradation is developed, opportunities arise to engineer precise changes in protein abundance that may result in clinical benefit. A few highly successful examples already exist, with a number of potentially new approaches under investigation. Four instructive examples are discussed below.
Proteasome Inhibitors as a Therapeutic Modality
Proteasome inhibitors were originally developed as research tools. Given the essential role the proteasome plays in so many cellular processes, there was some initial skepticism that these inhibitors could be safely used as clinical drugs. However, the FDA approval of bortezomib (Velcade; Takeda, Tokyo, Japan) in 2003 proved that proteasome inhibitors could be used in a safe and effective manner in patients.41
Bortezomib is a modified dipeptide that primarily targets the chymotryptic proteolytic active site within the proteasome's core particle. Bortezomib's initial approval was for multiple myeloma, and, along with a second drug, thalidomide (see below), it has helped revolutionize treatment of this aggressive disease. The basis for bortezomib's specific efficacy in multiple myeloma remains somewhat unclear. Plasma cells produce immense amounts of immunoglobulin protein and, therefore, may be particularly dependent on pathways of protein quality control. Bortezomib has also been approved for mantle cell lymphoma, and many other potential clinical indications for proteasome inhibition are currently under study. Since the development of bortezomib, other proteasome inhibitors have been developed and approved, including carfilzomib in 2012 and ixazomib, which is the first orally available proteasome inhibitor, in 2015.
Induced Degradation of Oncoproteins
Acute promyelocytic leukemia (APML) is characterized by a cytogenetic translocation between chromosomes 15 and 17. This translocation results in a novel fusion protein, promyelocytic leukemia–retinoic acid receptor-α (PML-RARα), which plays a critical role in the initiation and maintenance of this cancer. The introduction of arsenic, in combination with a second drug, all-trans-retinoic acid (ATRA), has transformed this disease from one that was typically fatal to one in which cure rates now exceed 90%.42 In fact, APML is one of the most remarkable success stories of cancer medicines.
Arsenic is a ubiquitous environmental toxin, and its use as an anticancer modality is surprising because arsenic itself is a well-established cause of various cancers. Its role in cutaneous squamous cell carcinoma is perhaps best known, but it has also been linked to various other cancer types, including bladder, lung, and kidney. The precise mechanism of arsenic's role in these cancer types is not well understood. Arsenic is a potent oxidizing agent and may contribute to DNA damage. Recent evidence suggests that arsenic is also a potent inducer of protein misfolding.43, 44 Arsenic is known to covalently bind to free cysteine residues within proteins, and this may distort their shape, inducing misfolding and subsequent recognition and degradation by the UPS.
The clinical utility of arsenic in APML was first suggested by Chinese medicine.45 This effect was validated in several studies, ultimately leading to FDA approval in 2000. However, its mechanism of action remained unclear until recently. Arsenic covalently binds to thiol groups within cysteine residues of the PML-RARα fusion protein. This arsenic binding triggers the ubiquitination of the fusion protein, leading to its destruction by the proteasome.46 When PML-RARα levels decrease below a certain threshold, the leukemic cells are released from the inhibitory effect of this protein and are able to mature along their intended hematopoietic lineage.47 This lost capacity for self-renewal irreversibly eliminates the oncogenic potential of these cells, resulting in the dramatic and durable observed responses rates.
Arsenic is typically given in conjunction with ATRA. The RARα functions as a transcriptional regulator. It was initially thought that ATRA functioned by altering RARα's transcriptional activity. Recent work, however, suggests that ATRA's mechanism of action is similar to that of arsenic. de Thé and colleagues48 studied several retinoic acid variants and were able to identify molecules that retained the capacity to alter RARα's transcriptional activity, but lost the capacity to promote protein degradation. Surprisingly, these variants showed markedly reduced efficacy against APML. Thus, the therapeutic effect for both arsenic and ATRA in APML is based on the induced degradation of the PML-RARα oncoprotein.
APML is just one of many cancers driven by an oncogenic fusion protein. In addition to other hematolymphoid malignancies, many sarcomas and even carcinomas are now associated with cytogenetic translocations, many of which result in abnormal fusion proteins. An important question is to what extent the arsenic-induced degradation of PML-RARα represents a unique situation, or whether it might represent a proof of principle for a more generalizable therapeutic approach. This question will have to be answered empirically. In principle, the capacity of arsenic to modify proteins is relatively nonspecific, its main basis being the availability of reactive cysteine residues within a target protein. Indeed, arsenic induces widespread proteomic changes within cells.44 Thus, it is certainly possible that other oncoproteins, particularly translocation-derived fusion proteins, may be subject to therapeutically beneficial induced degradation.
Targeted Ubiquitination of Key Disease Mediators
In 1957, the drug thalidomide was released and marketed to pregnant women as a sedative and an antiemetic. Within just a few years, reports began to appear suggesting an increased rate of birth defects in the children of women who had taken thalidomide.49, 50 These defects typically involved shortening of the proximal limbs, or phocomelia, along with other congenital abnormalities. Thalidomide was pulled from the market in 1961, but not before some 10,000 children had been affected by its teratogenic effects. The drug was never approved in the United States, largely sparing US families from this tragedy.
In recent years, thalidomide has had a remarkable second act as an anticancer therapy. Some evidence had emerged that thalidomide might have antiangiogenic properties,51 and in the late 1990s, a small number of patients were treated with the drug on a compassionate use basis. Early dramatic responses led to larger trials, ultimately culminating in FDA approval of thalidomide for multiple myeloma in 2006.52 A more potent version of the drug, known as lenalidomide (REVLIMID; Celgene, Summit, NJ), was subsequently developed and, along with the proteasome inhibitors described above, remains a mainstay of myeloma treatment to this day.
Much like arsenic, the mechanism of action for thalidomide was unknown at the time of its FDA approval and it was used empirically for many years. A major advance came in 2010 when thalidomide's cellular target was identified as the protein cereblon.53 Cereblon is part of a large E3 ubiquitin ligase and specifically functions to recruit substrates to the complex for ubiquitination. Remarkably, thalidomide did not function to inhibit this E3 enzyme. Rather, it specifically stimulated the ubiquitination of two proteins, IKZF1 and IKZF3, which are oncogenic transcription factors critical for multiple myeloma.54, 55 Increased ubiquitination of these proteins, in turn, drives their destruction by the proteasome. Structural analysis showed that thalidomide and its analogs achieve this by directly binding cereblon and enhancing its ability to bind IKZF1 or IKZF3.56
Remarkably, lenalidomide was later found to be highly effective in a subset of myelodysplastic syndrome (namely, those associated with chromosome 5q deletion). Herein, lenalidomide did not operate via IKZF1 or IKZF3, but through a third novel substrate, casein kinase 1A1, which is itself located on chromosome 5q.57 Nevertheless, the mechanism of action in myelodysplastic syndrome is essentially the same as in multiple myeloma: lenalidomide increases recruitment of casein kinase 1A1 to cereblon, resulting in its ubiquitination and subsequent degradation by the proteasome.58
Thalidomide and its analogs demonstrate how altering the activity of a ubiquitinating enzyme can result in new therapeutic opportunities. However, the example of thalidomide was fortuitous. The real challenge will be to do this in a prospective manner, selecting disease-related proteins and targeting them to specific enzymes for ubiquitination. In principle, any disease highly dependent on a single protein or a small number of proteins may be amenable to such intervention, raising the possibility that this could be a broadly applicable therapeutic strategy.
Enhancement of Proteasome Activity
As described earlier, ubiquitin targets proteins for destruction at the proteasome, but is itself largely spared from degradation. This is achieved through the activity of three deubiquitinating enzymes that are part of the proteasome itself. In the absence of one of these enzymes, Ubp6, the proteasome erroneously destroys ubiquitin along with its substrates.59, 60, 61, 62, 63 Surprisingly, under these conditions, the proteasome showed an enhanced protein-degrading capacity.62, 63, 64, 65 This suggested that Ubp6 is an endogenous inhibitor of the proteasome, which was unexpected because proteasomes were previously thought to operate at maximal speed under normal conditions. These results implied an elegant balance at the proteasome between speed and accuracy: Ubp6 enhances the recycling of ubiquitin at the proteasome, but only at the cost of decreased speed in degrading proteins.
As described earlier, many diseases are thought to be caused by the accumulation of misfolded or otherwise aberrant proteins, and elimination of these proteins might be therapeutic. The identification of an endogenous proteasome inhibitor suggested a potential strategy to enhance proteasome function in cells. Indeed, a small-molecule inhibitor of Usp14 (the human version of Ubp6) stimulated proteasome function in vitro and in vivo.66 This small molecule was able to enhance the degradation of at least some disease-related proteins, including tau (Alzheimer disease) and TDP-43 (amyotrophic lateral sclerosis).66, 67, 68 Subsequent work from other groups showed that chemical or genetic inhibition of Usp14 can also accelerate degradation of prion protein as well as many other proteins.69, 70, 71, 72, 73, 74, 75 The full extent of Usp14's substrate specificity remains to be determined. Nevertheless, in some cell types, overall protein degradation rates were increased by as much as 45% in the absence of Usp14.76 Although still at a relatively early stage, this work suggests a potentially novel therapeutic approach based on proteasome enhancement. Given the complexity of the proteasome, there may be other routes besides Usp14 to enhancing proteasome function and protein degradation more generally.77, 78, 79
Conclusion
The ubiquitin-proteasome system is the major pathway for regulated protein degradation and allows for precise control of the abundance of individual proteins within cells. Dysregulation of protein degradation pathways plays a critical role in many common and rare human diseases. Conversely, the versatility and scope of this pathway provide opportunities for therapeutic intervention. Many diseases are highly dependent on one or more proteins that may be either key regulators or toxic aberrant species. For such diseases, targeted protein degradation may result in clinical benefit (Figure 3). In other cases, a deficiency of functional protein may be the result of protein misfolding and/or excessive protein degradation. For these diseases, interventions that increase the abundance of functional protein may be therapeutic (Figure 3). A comprehensive understanding of protein degradation pathways, coupled with detailed knowledge of molecular disease pathogenesis, may allow us to design and engineer cellular outcomes that have clinical benefit.
Figure 3.

Simple schematic for therapeutic intervention based on protein degradation. Left panel: Many diseases are driven by the accumulation of one or more proteins. These may be key regulatory proteins or toxic misfolded proteins. Targeted degradation of these proteins may lead to disease amelioration (red arrow). Right panel: Other diseases may be caused by a relative lack of functional protein. This may be due to partial misfolding and degradation, or otherwise excessive degradation of key mediators. In these cases, interventions to increase the yield of properly folded functional protein may be effective (red arrow).
Acknowledgments
J.H. thanks Jeffrey Golden, Mel Feany, Daniel Finley, and Christopher Fletcher for helpful discussions and support; we thank Monica Boselli and Matthew Stachler for comments on the manuscript.
Footnotes
ASIP 2019 Cotran Early Career Investigator Award
Supported by the NIH Early Independence Award DP5-OD019800 (J.H.).
Disclosures: J.H. receives royalties from Harvard Medical School related to US patent 9,201,073.
The American Society for Investigative Pathology (ASIP) Cotran Early Career Investigator Award recognizes early career investigators with demonstrated excellence as an investigator with recently established or emerging independence and with a research focus leading to an improved understanding of the conceptual basis of disease. John Hanna, recipient of the ASIP 2019 Cotran Early Career Investigator Award, delivered a lecture entitled “Protein Degradation and the Pathologic Basis of Disease” on October 22, 2018, at the Pathobiology for Investigators, Students, and Academicians (PISA) meeting in Ann Arbor, MI.
References
- 1.Lenkinski R.E., Chen D.M., Glickson J.D., Goldstein G. Nuclear magnetic resonance studies of the denaturation of ubiquitin. Biochim Biophys Acta. 1977;494:126–130. doi: 10.1016/0005-2795(77)90140-4. [DOI] [PubMed] [Google Scholar]
- 2.Ciechanover A. Intracellular protein degradation: from a vague idea, through the lysosome and the ubiquitin-proteasome system, and onto human diseases and drug targeting (Nobel lecture) Angew Chem Int Ed Engl. 2005;44:5944–5967. doi: 10.1002/anie.200501428. [DOI] [PubMed] [Google Scholar]
- 3.Yau R., Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18:579–586. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 4.Clague M.J., Heride C., Urbé S. The demographics of the ubiquitin system. Trends Cell Biol. 2015;25:417–426. doi: 10.1016/j.tcb.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Crosas B., Hanna J., Kirkpatrick D.S., Zhang D.P., Tone Y., Hathaway N.A., Buecker C., Leggett D.S., Schmidt M., King R.W., Gygi S.P., Finley D. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell. 2006;127:1401–1413. doi: 10.1016/j.cell.2006.09.051. [DOI] [PubMed] [Google Scholar]
- 6.Randles L., Walters K.J. Ubiquitin and its binding domains. Front Biosci. 2012;17:2140–2157. doi: 10.2741/4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lander G.C., Estrin E., Matyskiela M.E., Bashore C., Nogales E., Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groll M., Ditzel L., Löwe J., Stock D., Bochtler M., Bartunik H.D., Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 9.de Poot S.A.H., Tian G., Finley D. Meddling with fate: the proteasomal deubiquitinating enzymes. J Mol Biol. 2017;429:3525–3545. doi: 10.1016/j.jmb.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Budenholzer L., Cheng C.L., Li Y., Hochstrasser M. Proteasome structure and assembly. J Mol Biol. 2017;429:3500–3524. doi: 10.1016/j.jmb.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Pedeutour F., Suijkerbuijk R.F., Van Gaal J., Van de Klundert W., Coindre J.M., Van Haelst A., Collin F., Huffermann K., Turc-Carel C. Chromosome 12 origin in rings and giant markers in well-differentiated liposarcoma. Cancer Genet Cytogenet. 1993;66:133–134. doi: 10.1016/0165-4608(93)90245-h. [DOI] [PubMed] [Google Scholar]
- 13.Sreekantaiah C., Karakousis C.P., Leong S.P., Sandberg A.A. Cytogenetic findings in liposarcoma correlate with histopathologic subtypes. Cancer. 1992;69:2484–2495. doi: 10.1002/1097-0142(19920515)69:10<2484::aid-cncr2820691017>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Leach F.S., Tokino T., Meltzer P., Burrell M., Oliner J.D., Smith S., Hill D.E., Sidransky D., Kinzler K.W., Vogelstein B. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res. 1993;53:2231–2234. [PubMed] [Google Scholar]
- 15.Binh M.B., Sastre-Garau X., Guillou L., de Pinieux G., Terrier P., Lagacé R., Aurias A., Hostein I., Coindre J.M. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol. 2005;29:1340–1347. doi: 10.1097/01.pas.0000170343.09562.39. [DOI] [PubMed] [Google Scholar]
- 16.Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 17.Kubbutat M.H., Jones S.N., Vousden K.H. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 18.Stanley M.A. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev. 2012;25:215–222. doi: 10.1128/CMR.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheffner M., Huibregtse J.M., Vierstra R.D., Howley P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Zapien D., Ruiz F.X., Poirson J., Mitschler A., Ramirez J., Forster A., Cousido-Siah A., Masson M., Vande Pol S., Podjarny A., Travé G., Zanier K. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature. 2016;529:541–545. doi: 10.1038/nature16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munemitsu S., Albert I., Souza B., Rubinfeld B., Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinzler K.W., Nilbert M.C., Su L.K., Vogelstein B., Bryan T.M., Levy D.B., Smith K.J., Preisinger A.C., Hedge P., McKechnie D., Finniear R., Markham A., Groffen J., Boguski M.S., Alstchul S.F., Horii A., Ando H., Miyoshi Y., Miki Y., Nishisho I., Nakamura Y. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 23.Sparks A.B., Morin P.J., Vogelstein B., Kinzler K.W. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 24.Hipp M.S., Park S.H., Hartl F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Labbadia J., Morimoto R.I. The biology of proteostasis in aging and disease. Annu Rev Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin J.B. Molecular basis of the neurodegenerative disorders. N Engl J Med. 1999;340:1970–1980. doi: 10.1056/NEJM199906243402507. [DOI] [PubMed] [Google Scholar]
- 27.Nutt J.G., Wooten G.F. Clinical practice: diagnosis and initial management of Parkinson's disease. N Engl J Med. 2005;353:1021–1027. doi: 10.1056/NEJMcp043908. [DOI] [PubMed] [Google Scholar]
- 28.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E.S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W.G., Lazzarini A.M., Duvoisin R.C., Di Iorio G., Golbe L.I., Nussbaum R.L. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 29.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M.R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 30.Wiseman F.K., Al-Janabi T., Hardy J., Karmiloff-Smith A., Nizetic D., Tybulewicz V.L., Fisher E.M., Strydom A. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16:564–574. doi: 10.1038/nrn3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cutting G.R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16:45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rommens J.M., Iannuzzi M.C., Kerem B., Drumm M.L., Melmer G., Dean M., Rozmahel R., Cole J.L., Kennedy D., Hidaka N., Zsiga M., Buchwald M., Riordan J.R., Tsui L.C., Collins F.S. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 33.Berner N., Reutter K.R., Wolf D.H. Protein quality control of the endoplasmic reticulum and ubiquitin-proteasome-triggered degradation of aberrant proteins: yeast pioneers the path. Annu Rev Biochem. 2018;87:751–778. doi: 10.1146/annurev-biochem-062917-012749. [DOI] [PubMed] [Google Scholar]
- 34.Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L., Drumm M.L., Iannuzzi M.C., Collins F.S., Tsui L.C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 35.Jensen T.J., Loo M.A., Pind S., Williams D.B., Goldberg A.L., Riordan J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 36.Ward C.L., Omura S., Kopito R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 37.Denning G.M., Anderson M.P., Amara J.F., Marshall J., Smith A.E., Welsh M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y., Devor D.C., Engelhardt J.F., Ernst S.A., Strong T.V., Collins F.S., Cohn J.A., Frizzell R.A., Wilson J.M. Molecular basis of defective anion transport in L cells expressing recombinant forms of CFTR. Hum Mol Genet. 1993;2:1253–1261. doi: 10.1093/hmg/2.8.1253. [DOI] [PubMed] [Google Scholar]
- 39.Accurso F.J., Rowe S.M., Clancy J.P., Boyle M.P., Dunitz J.M., Durie P.R., Sagel S.D., Hornick D.B., Konstan M.W., Donaldson S.H., Moss R.B., Pilewski J.M., Rubenstein R.C., Uluer A.Z., Aitken M.L., Freedman S.D., Rose L.M., Mayer-Hamblett N., Dong Q., Zha J., Stone A.J., Olson E.R., Ordoñez C.L., Campbell P.W., Ashlock M.A., Ramsey B.W. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wainwright C.E., Elborn J.S., Ramsey B.W. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:1783–1784. doi: 10.1056/NEJMc1510466. [DOI] [PubMed] [Google Scholar]
- 41.Richardson P.G., Barlogie B., Berenson J., Singhal S., Jagannath S., Irwin D., Rajkumar S.V., Srkalovic G., Alsina M., Alexanian R., Siegel D., Orlowski R.Z., Kuter D., Limentani S.A., Lee S., Hideshima T., Esseltine D.L., Kauffman M., Adams J., Schenkein D.P., Anderson K.C. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 42.Rice K.L., de Thé H. The acute promyelocytic leukaemia success story: curing leukaemia through targeted therapies. J Intern Med. 2014;276:61–70. doi: 10.1111/joim.12208. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson T., Navarrete C., Sharma S.K., Sideri T.C., Ibstedt S., Priya S., Grant C.M., Christen P., Goloubinoff P., Tamás M.J. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J Cell Sci. 2012;125:5073–5083. doi: 10.1242/jcs.107029. [DOI] [PubMed] [Google Scholar]
- 44.Guerra-Moreno A., Isasa M., Bhanu M.K., Waterman D.P., Eapen V.V., Gygi S.P., Hanna J. Proteomic analysis identifies ribosome reduction as an effective proteotoxic stress response. J Biol Chem. 2015;290:29695–29706. doi: 10.1074/jbc.M115.684969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kogan S.C. Medicine: poisonous contacts. Science. 2010;328:184–185. doi: 10.1126/science.1189198. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X.W., Yan X.J., Zhou Z.R., Yang F.F., Wu Z.Y., Sun H.B., Liang W.X., Song A.X., Lallemand-Breitenbach V., Jeanne M., Zhang Q.Y., Yang H.Y., Huang Q.H., Zhou G.B., Tong J.H., Zhang Y., Wu J.H., Hu H.Y., de Thé H., Chen S.J., Chen Z. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328:240–243. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 47.de Thé H., Pandolfi P.P., Chen Z. Acute promyelocytic leukemia: a paradigm for oncoprotein-targeted cure. Cancer Cell. 2017;32:552–560. doi: 10.1016/j.ccell.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 48.Ablain J., Leiva M., Peres L., Fonsart J., Anthony E., de Thé H. Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J Exp Med. 2013;210:647–653. doi: 10.1084/jem.20122337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McBride W.G. Thalidomide and congenital abnormalities. Lancet. 1961;278:1358. [Google Scholar]
- 50.Lenz W., Pfeiffer R.A., Kosenow W., Hayman D.J. Thalidomide and congenital abnormalities. Lancet. 1962;279:45–46. [Google Scholar]
- 51.D'Amato R.J., Loughnan M.S., Flynn E., Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajkumar S.V., Blood E., Vesole D., Fonseca R., Greipp P.R., Eastern Cooperative Oncology Group Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24:431–436. doi: 10.1200/JCO.2005.03.0221. [DOI] [PubMed] [Google Scholar]
- 53.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 54.Krönke J., Udeshi N.D., Narla A., Grauman P., Hurst S.N., McConkey M., Svinkina T., Heckl D., Comer E., Li X., Ciarlo C., Hartman E., Munshi N., Schenone M., Schreiber S.L., Carr S.A., Ebert B.L. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu G., Middleton R.E., Sun H., Naniong M., Ott C.J., Mitsiades C.S., Wong K.K., Bradner J.E., Kaelin W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fischer E.S., Böhm K., Lydeard J.R., Yang H., Stadler M.B., Cavadini S., Nagel J., Serluca F., Acker V., Lingaraju G.M., Tichkule R.B., Schebesta M., Forrester W.C., Schirle M., Hassiepen U., Ottl J., Hild M., Beckwith R.E., Harper J.W., Jenkins J.L., Thomä N.H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krönke J., Fink E.C., Hollenbach P.W., MacBeth K.J., Hurst S.N., Udeshi N.D., Chamberlain P.P., Mani D.R., Man H.W., Gandhi A.K., Svinkina T., Schneider R.K., McConkey M., Järås M., Griffiths E., Wetzler M., Bullinger L., Cathers B.E., Carr S.A., Chopra R., Ebert B.L. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petzold G., Fischer E.S., Thomä N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 59.Hanna J., Leggett D.S., Finley D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. 2003;23:9251–9261. doi: 10.1128/MCB.23.24.9251-9261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leggett D.S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R.T., Walz T., Ploegh H., Finley D. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- 61.Shabek N., Iwai K., Ciechanover A. Ubiquitin is degraded by the ubiquitin system as a monomer and as part of its conjugated target. Biochem Biophys Res Commun. 2007;363:425–431. doi: 10.1016/j.bbrc.2007.08.185. [DOI] [PubMed] [Google Scholar]
- 62.Hanna J., Hathaway N.A., Tone Y., Crosas B., Elsasser S., Kirkpatrick D.S., Leggett D.S., Gygi S.P., King R.W., Finley D. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 63.Hanna J., Meides A., Zhang D.P., Finley D. A ubiquitin stress response induces altered proteasome composition. Cell. 2007;129:747–759. doi: 10.1016/j.cell.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 64.Koulich E., Li X., DeMartino G.N. Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell. 2008;19:1072–1082. doi: 10.1091/mbc.E07-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bashore C., Dambacher C.M., Goodall E.A., Matyskiela M.E., Lander G.C., Martin A. Ubp6 deubiquitinase controls conformational dynamics and substrate degradation of the 26S proteasome. Nat Struct Mol Biol. 2015;22:712–719. doi: 10.1038/nsmb.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee B.H., Lee M.J., Park S., Oh D.C., Elsasser S., Chen P.C., Gartner C., Dimova N., Hanna J., Gygi S.P., Wilson S.M., King R.W., Finley D. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee J.H., Shin S.K., Jiang Y., Choi W.H., Hong C., Kim D.E., Lee M.J. Facilitated tau degradation by USP14 aptamers via enhanced proteasome activity. Sci Rep. 2015;5:10757. doi: 10.1038/srep10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boselli M., Lee B.H., Robert J., Prado M.A., Min S.W., Cheng C., Silva M.C., Seong C., Elsasser S., Hatle K.M., Gahman T.C., Gygi S.P., Haggarty S.J., Gan L., King R.W., Finley D. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem. 2017;292:19209–19225. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Homma T., Ishibashi D., Nakagaki T., Fuse T., Mori T., Satoh K., Atarashi R., Nishida N. Ubiquitin-specific protease 14 modulates degradation of cellular prion protein. Sci Rep. 2015;5:11028. doi: 10.1038/srep11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKinnon C., Goold R., Andre R., Devoy A., Ortega Z., Moonga J., Linehan J.M., Brandner S., Lucas J.J., Collinge J., Tabrizi S.J. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol. 2016;131:411–425. doi: 10.1007/s00401-015-1508-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakashima A., Ohnuma S., Kodani Y., Kaneko Y.S., Nagasaki H., Nagatsu T., Ota A. Inhibition of deubiquitinating activity of USP14 decreases tyrosine hydroxylase phosphorylated at Ser19 in PC12D cells. Biochem Biophys Res Commun. 2016;472:598–602. doi: 10.1016/j.bbrc.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 72.Sareen-Khanna K., Papillon J., Wing S.S., Cybulsky A.V. Role of the deubiquitinating enzyme ubiquitin-specific protease-14 in proteostasis in renal cells. Am J Physiol Renal Physiol. 2016;311:F1035–F1046. doi: 10.1152/ajprenal.00252.2016. [DOI] [PubMed] [Google Scholar]
- 73.Xu D., Shan B., Lee B.H., Zhu K., Zhang T., Sun H., Liu M., Shi L., Liang W., Qian L., Xiao J., Wang L., Pan L., Finley D., Yuan J. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife. 2015;4:e10510. doi: 10.7554/eLife.10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liao Y., Liu N., Hua X., Cai J., Xia X., Wang X., Huang H., Liu J. Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor. Cell Death Dis. 2017;8:e2585. doi: 10.1038/cddis.2016.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen L., Zhu G., Johns E.M., Yang X. TRIM11 activates the proteasome and promotes overall protein degradation by regulating USP14. Nat Commun. 2018;9:1223. doi: 10.1038/s41467-018-03499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim H.T., Goldberg A.L. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J Biol Chem. 2017;292:9830–9839. doi: 10.1074/jbc.M116.763128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo X., Engel J.L., Xiao J., Tagliabracci V.S., Wang X., Huang L., Dixon J.E. UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc Natl Acad Sci U S A. 2011;108:18649–18654. doi: 10.1073/pnas.1113170108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lokireddy S., Kukushkin N.V., Goldberg A.L. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A. 2015;112:E7176–E7185. doi: 10.1073/pnas.1522332112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raina K., Crews C.M. Targeted protein knockdown using small molecule degraders. Curr Opin Chem Biol. 2017;39:46–53. doi: 10.1016/j.cbpa.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]