

Abstract
Breast cancer metastasis to gynaecological organs is an understudied pattern of tumour spread. We explored clinico‐pathological and molecular features of these metastases to better understand whether this pattern of dissemination is organotropic or a consequence of wider metastatic dissemination. Primary and metastatic tumours from 54 breast cancer patients with gynaecological metastases were analysed using immunohistochemistry, DNA copy‐number profiling, and targeted sequencing of 386 cancer‐related genes. The median age of primary tumour diagnosis amongst patients with gynaecological metastases was significantly younger compared to a general breast cancer population (46.5 versus 60 years; p < 0.0001). Median age at metastatic diagnosis was 54.4, time to progression was 4.8 years (range 0–20 years), and survival following a diagnosis of metastasis was 1.95 years (range 0–18 years). Patients had an average of five involved sites (most frequently ovary, fallopian tube, omentum/peritoneum), with fewer instances of spread to the lungs, liver, or brain. Invasive lobular histology and luminal A‐like phenotype were over‐represented in this group (42.8 and 87.5%, respectively) and most patients had involved axillary lymph nodes (p < 0.001). Primary tumours frequently co‐expressed oestrogen receptor cofactors (GATA3, FOXA1) and harboured amplifications at 8p12, 8q24, and 11q13. In terms of phenotype conversion, oestrogen receptor status was generally maintained in metastases, FOXA1 increased, and expression of progesterone receptor, androgen receptor, and GATA3 decreased. ESR1 and novel AR mutations were identified. Metastasis to gynaecological organs is a complication frequently affecting young women with invasive lobular carcinoma and luminal A‐like breast cancer, and hence may be driven by sustained hormonal signalling. Molecular analyses reveal a spectrum of factors that could contribute to de novo or acquired resistance to therapy and disease progression.
Keywords: breast cancer, metastasis, immunophenotyping, luminal subtype, genomics, ovary
Introduction
Metastatic spread is the single most significant predictor of poor survival in breast cancer. In order to metastasise, tumour cells must develop the necessary biological capabilities to overcome extrinsic selection pressure, and thus clonal evolution is driven by the selective acquisition of somatic mutations, as well as dynamic interactions with the microenvironment 1. The extent and overall clinical significance of diversity in metastatic progression is still being elucidated, partly because metastatic deposits are not routinely biopsied and the availability of samples for molecular analysis is limited.
The most common sites of breast cancer metastasis are the bone, lung, liver, and brain 2, 3. Analyses of autopsy and surgical series have compared organ‐specific spread of the two main histological types, invasive carcinoma of no special type (IC‐NST) and invasive lobular carcinoma (ILC). IC‐NST spreads more frequently to the lung/pleura, liver, and brain, and whilst ILC can spread to these organs too, it has a propensity to spread to unusual sites, including peritoneum, gynaecological organs, gastrointestinal (GI) tract, adrenal glands, and skin 4, 5, 6, 7. In addition to histological type, tumour grade, expression of receptors for oestrogen and progesterone and human epidermal growth factor (ER/PR/HER2) and molecular subtype are also associated with different organ tropism and latency 6, 8, 9, 10, 11, 12.
Gynaecological metastases (GMs) are relatively rare and thus this pattern of spread is poorly understood. Breast cancer is the second most common primary tumour type to spread to gynaecological organs following colorectal cancer, raising the possibility that dissemination from the breast may be targeted rather than simply due to proximity 13, 14, 15. Available data suggest that patients present with a primary breast cancer at a young age (range 46–54 years 13, 16, 17, 18, 19), and usually have an ER‐positive, HER2‐negative, and ILC phenotype 20, 21. Hormone signalling and/or an infiltrative pattern of tumour growth may be important contributors to this pattern of dissemination.
Here we present a comprehensive analysis of a unique cohort of breast cancer patients with GMs, providing insight into the natural history and heterogeneity of breast cancer progression.
Materials and methods
Patient cohort and statistical analyses
Analysis of the Queensland Centre for Gynaecological Cancer and Pathology Queensland databases identified 54 breast cancer patients who were diagnosed with metastasis to gynaecological tissues in Queensland between 1982 and 2015. This cohort is referred to as the GM cohort. Clinical and pathology data were obtained from pathology reports, clinical charts, and the Queensland Cancer Registry. Archival formalin‐fixed, paraffin‐embedded (FFPE) tissue blocks were available for 39 cases: 15 cases with matched primary and metastatic tumours; 4 with only the primary available; 20 with only metastases available. The Queensland Follow‐Up (QFU) cohort 22, 23, 24 was used to compare various clinico‐pathological parameters. This cohort contains 449 consecutive, unselected primary breast cancer cases diagnosed between 1986 and 1993 at the Royal Brisbane and Women's Hospital, with detailed clinical data and a minimum of 25 years clinical follow‐up data. Due to the historic nature of the cohort, distant metastasis data are unreliable for the QFU cohort. The Harrell data 25 were used to compare the frequency of metastasis and expression of ER/HER2 between the GM primary tumours and those that spread specifically to the brain, lung, bone, and liver. Human research ethics committees of The University of Queensland (ref. 2005000785) and The Royal Brisbane and Women's Hospital (2005/022) approved the study. All statistical analyses were carried out using GraphPad Software, La Jolla, CA, USA. Fisher's exact and Chi‐square tests were used and a P value of <0.05 was considered significant. Supplementary material, Figure S1, illustrates the number of GM cases that underwent immunophenotyping and molecular profiling.
Tissue microarray construction and immunohistochemical analysis
All FFPE blocks were recut, stained with haematoxylin and eosin (H&E) and reviewed by a pathologist SS, GM, SP, MC to identify tumour‐rich areas for sampling in tissue microarrays (TMAs) (duplicate 1 mm biopsy cores). TMAs were stained for ER, PR, HER2, Ki67, EGFR, CK8/18, CK5/6, CK14, p53, androgen receptor (AR), FOXA1, and GATA3 (see supplementary material, Table S1 for technical details and scoring systems used for each antibody). The antibodies were detected using the MACH1 Universal HRP Detection Kit (Biocare Medical, LLC, Concord, CA 94520, USA). For cases where blocks were unavailable, ER, PR, and HER2 data were extracted from diagnostic pathology reports. Ki67 staining was also undertaken on whole sections of tumour (for comparison to TMAs), with both manual scoring and digital image analysis using the Leica Aperio Scanscope (Leica Biosystems, Melbourne, Australia) and the algorithm Nuclear v.9. Supplementary material, Figure S2 illustrates the immunophenotyping classification systems utilised for the GM and QFU cohorts.
Array‐based comparative genomic hybridisation and targeted sequencing
TMA cores (1 mm, 4–6 cores per tumour) were taken and DNA was extracted using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Pty Ltd, VIC, Australia), following overnight sodium thiocyanate (1 M) incubation and 3 days proteinase K (ThermoFisher Scientific, Australia) digestion at 55 °C. Qiagen AW2 buffer was replaced with two 70% ethanol washes as recommended by Agilent Technologies to remove impurities that interfere with the labelling protocol for array‐based comparative genomic hybridisation (aCGH). DNA yield and quality were assessed using the Nanodrop spectrophotometer, Qubit BR DNA kit (both ThermoFisher Scientific), and the Illumina Infinium HD FFPE QC Assay (Illumina, Scoresby, VIC, Australia) following the manufacturer's protocols. DNA samples with delta Cq values <5 were considered suitable for downstream microarray applications. Whole genome DNA copy‐number changes for 84 samples from 27 cases were measured on the Agilent SurePrint G3 Human CGH Microarray 4 × 180 K format, following the ULS labelling protocol (Agilent Technologies, Mulgrave, VIC, Australia) 16. Penetrance was calculated using Agilent Cytogenomics software version 3.0 and presented alongside centromere positions calculated from the Genome Reference Consortium Human Build 37 (GRCh37‐HG19) chromosome assembly using base and graphics packages within the R programming environment (version 3.4.1); R Core Team (2017). Thirty‐five samples (41.6%) from 14 cases yielded satisfactory hybridisation data for analysis (supplementary material, Table S2 ), consistent with our previous experience of aCGH profiled archival samples 16. Array CGH data are available at the Gene Expression Omnibus (GEO) website, accession number GSE115080.
Targeted sequence analysis was performed on DNA extracted from matched tumour and normal DNA from non‐involved lymph nodes from seven cases (a total of 37 samples, see supplementary material, Table S3). The Comprehensive Capture Panel v2 (Peter MacCallum Cancer Centre, Melbourne, Australia) screens for mutations in the coding regions and splice sites of 386 cancer‐related genes. DNA libraries were prepared using the KAPA Hyper Library Preparation Kit (KAPA Biosystems, Illumina) and enriched using a custom designed SureSelect XT Target Enrichment assay (Agilent Technologies). Samples were uniquely indexed, pooled, and sequenced on an Illumina NextSeq500 (Illumina) to generate 2 × 75 bp reads at a target germline coverage of 100 reads/base and target tumour coverage of 500 reads/base. Sequence performance was assessed using FastQC, GATK DepthOfCoverage, and custom scripts. A bioanalysis pipeline consisting of read trimming with cutadapt, alignment to hg19 with BWA‐MEM, base quality score recalibration and indel realignment with GATK, and removal of duplicate reads with Picard was performed. Variants were called with GATK HaplotypeCaller for normal DNA, or MuTect, GATK Indellocator, and VarScanSomatic for tumour. Annotation was performed with Variant Effect Predictor from Ensembl Release 73. Variant filtering was performed by identifying rare (population allele frequency < 0.01), non‐synonymous variants, detected in tumour tissue but deficient in matching normal DNA, and were further refined by manual variant curation (see supplementary material, Table S4 for sequencing statistics). A custom filter was applied to capture variants meeting the following criteria: present in tumour and not matched normal; present in <10% of samples tested with this panel; present in <0.4% of individuals in the Exome Server Project; present in <0.4% of individuals in the 1000 Genomes study; not intronic (excluding splicing regions); not synonymous; not in an untranslated region; and not in an intergenic region.
Validation of CNAs with nanoString cancer CNV panel
Copy‐number alterations were validated in the primary tumour and multiple metastases from two cases (GM63 and GM78) using the nanoString nCounter v2 Cancer CN Assay, following the manufacturer's instructions (MAN‐C0024‐02, nanoString, Bio‐strategy, VIC, Australia). Briefly, 600 ng of DNA was fragmented using the Alu1 restriction enzyme prior to hybridisation. The probes were processed on the nCounter FLEX prep station and probes counted on the nCounter Digital Analyzer (GEN2). The data were analysed following the nCounter Data Analysis Guidelines for CNV (MAN‐C0014‐02), using matched normal DNA for data normalisation.
Results
Clinical characteristics of breast cancer patients with GMs
The clinical and pathology features of the cohort of 54 GM patients were compared to those of an unselected breast cancer cohort (QFU; Table 1). The median age at diagnosis of the primary tumour was significantly lower in the GM cohort (46.5 versus 60 years; p < 0.0001). Using a cut‐off of 51 years as the average age of menopause in Australia 26, GM patients were more likely to be pre‐menopausal at the time of diagnosis compared to QFU patients (57.4% versus 32.9%; p = 0.0004). There was no difference in tumour grade or size, however the GM cohort was enriched for lobular histology (42.6% versus 14.4% ILC; p < 0.0001) and ILC primary tumours were larger than IC‐NST (p = 0.0075; supplementary material, Table S5). The patients were also more likely to have regional lymph node metastases at the time of primary diagnosis (83.3% in GM cohort versus 46.2% in the QFU cohort; p < 0.0001).
Table 1.
Clinico‐pathological characteristics of breast cancer metastatic to gynaecological organs
| GM (n = 54) | QFU (n = 445) | P value | |
|---|---|---|---|
| Histological type | n (%) | n (%) | |
| IC‐NST | 26 (48.1) | 256 (57.5) | <0.0001 |
| ILC | 23 (42.6) | 64 (14.4) | |
| Other | 5 (9.3) | 124 (27.9) | |
| Age of breast cancer diagnosis | Years (95% CI) | Years (95% CI) | |
| Median | 46.5 (43–54) | 60 (57–62) | <0.0001* |
| Median ILC cases | 44.8 (41.6–53.9) | 62.5 (57–66) | <0.0001* |
| Median IC‐NST cases | 46 (41.8–56.8) | 58 (55–63) | 0.0022* |
| Range | 30–79 | 27–88 | |
| Menopausal status | n (%) | n (%) | |
| Premenopausal | 31 (57.4) | 141 (32.9) | 0.0004 |
| Postmenopausal | 23 (42.6) | 287 (67.1) | |
| Tumour size | n (%) | n (%) | |
| <2 cm | 14 (43.8) | 177 (46.6) | 0.1922 |
| 2–5 cm | 12 (37.5) | 169 (44.5) | |
| >5 cm | 6 (18.8) | 34 (8.9) | |
| Unknown | 22 | ||
| Tumour grade | n (%) | n (%) | |
| Grade 1 | 2 (6.6) | 63 (14.2) | 0.3622 |
| Grade 2 | 14 (46.7) | 220 (49.5) | |
| Grade 3 | 14 (46.7) | 161 (36.3) | |
| Unknown | 24 | ||
| Lymph node status | n (%) | n (%) | |
| Positive | 30 (83.3) | 116 (46.2) | <0.0001 |
| Negative | 6 (16.7) | 135 (53.8) | |
| Unknown | 18 | ||
| Age of first metastasis diagnosis | Years (95% CI) | ||
| Median | 54.4 (49.8–59.3) | ||
| Range | 35–82 | ||
| Number of metastatic sites | n (%) | ||
| 1–3 | 22 (40.7) | ||
| 4–11 | 32 (59.3) | ||
| Median | 5 | ||
| Range | 1–11 |
t‐test; all other P values were derived using Chi‐square analysis. Significant P values are underlined.
The median age at diagnosis of metastatic disease was 54.4 years (range 35–82 years). There were 258 metastatic deposits recorded for the 54 patients (median five metastases/patient; range 1–11, Table 1). The most common gynaecological organs involved were the ovaries (46/54; 85.1%), fallopian tubes (29/54; 53.7%), and uterus (20/54; 37%; Figure 1A; supplementary material, Table S6). Uterine metastases were more frequent in ILC cases than IC‐NST (p = 0.0214; Figure 1A). Most cases (48/54; 88.9%) also had other organs involved, particularly the peritoneum/omentum (26/48; 54.2%), bone (22/48; 45.8%), and regions of the GI tract (appendix, stomach, colon, but excluding liver; 14/48; 29.5%). Bone metastases were more frequent in IC‐NST cases, and GI metastases were more frequent in ILC cases, but these frequencies did not reach statistical significance (Figure 1B). The more usual sites of breast cancer metastasis, such as lung, liver, and brain were less frequently involved in this cohort (16.7, 14.6, and 14.6%, respectively; Figure 1B; supplementary material, Table S6), however they were more frequently involved in IC‐NST compared to ILC cases. Furthermore, in 70.4% of cases, the GMs were the first metastatic site detected (supplementary material, Table S6).
Figure 1.
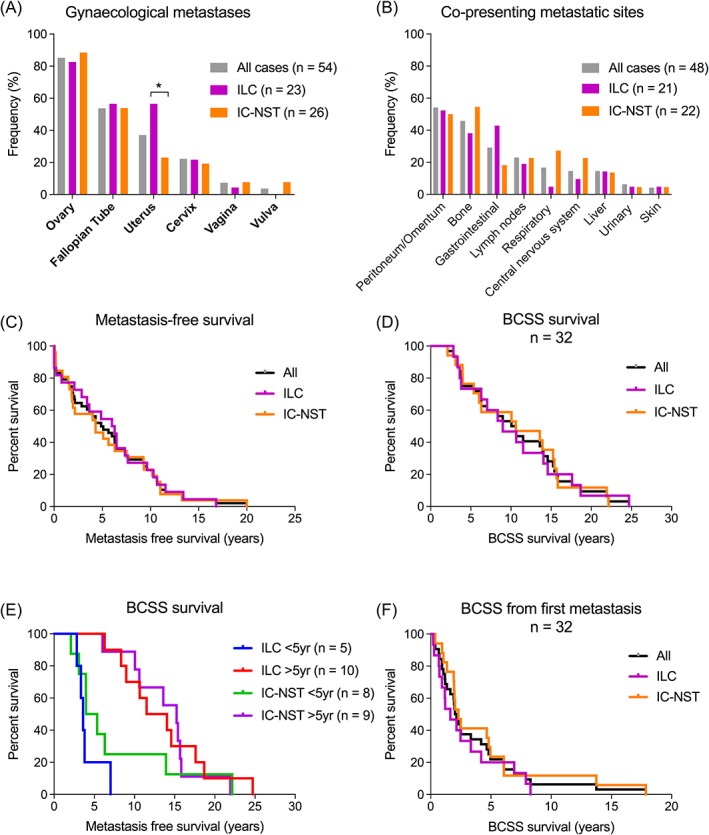
(A) Distribution of gynaecological organs involved by metastatic disease, stratified by primary tumour type. The ovary was the most common site involved (85.1%, 46/54 patients). Metastasis to the uterus was more frequent in ILC than IC‐NST (p = 0.0214). Forty‐eight cases (88%) presented with multiple metastases. (B) Frequencies of co‐presenting metastatic sites. The most common sites involved were peritoneum/omentum (54.2%, 27/48 patients), followed by bone (45.8%, 22/48), and GI tract (29.2%, 12/48). Common sites of breast cancer metastasis (lungs, liver, brain) rarely coincided with GMs. Bone, lung, and brain metastases were more frequent in IC‐NST and GI metastases were more frequent in ILC, but the differences were not statistically significant. (C) Metastasis‐free survival in the GM cohort. The median time to metastasis was 5 years. Approximately 25% of patients developed metastases after 10 years from primary tumour diagnosis. (D, E) BCSS in the GM cohort (n = 36): 50% of patients were alive at 10 years post‐primary diagnosis, and BCSS was not affected by primary tumour type or time to metastasis (<5 years versus >5 years). (F) Median survival after first metastasis diagnosis was 1.9 years (ILC: 1.6 years, IC‐NST: 2.3 years). 75% of patients died within 5 years of metastatic diagnosis.
The median metastasis‐free survival was 5 years (6 years for ILC cases, 4.3 years for IC‐NST cases, p = ns; Figure 1C). Interestingly, we found that ILC patients who relapsed after 5 years were significantly younger than those who relapsed within 5 years (median age at primary tumour diagnosis 41.8 years versus 50.9 years, p = 0.0364, supplementary material, Table S5).
Breast cancer‐specific survival (BCSS) data were available for 32 cases. At 10 years, 50% of patients were still alive (Figure 1D) and survival was not affected by primary tumour type (albeit small sample sizes. Figure 1E). When considering survival after metastasis diagnosis, the median survival time was only 1.95 years (ILC: 1.6 years, IC‐NST: 2.3 years, Figure 1F).
Biomarker profile of primary tumours that spread to gynaecological organs
Compared to the QFU cohort of unselected primary breast cancers, primary tumours in the GM cohort more frequently expressed ER (93.5%; p < 0.0001); were less frequently positive for HER2 (0%, p < 0.0001), basal markers (CK14: 0%, p = 0.0564; CK5/6: 0%, p = 0.0016; EGFR: 0%, p = 0.096), and p53 [immunohistochemical (IHC) score < 180 in 89.5% of cases; p = 0.0147]; and had a lower Ki67 proliferative index (all primary tumours were <10% positive; p = 0.0259, Table 2 , supplementary material, Figure S3 , Tables S7 and S8). In comparison to the primary tumours profiled in the Harrel et al dataset 25, GM primary tumours more frequently expressed ER, and lacked HER2 expression compared to primary tumours that spread to the brain (p < 0.0001 and ns, respectively), lung (p < 0.0001 and p = 0.0085, respectively), bone (p = 0.0055 and p = 0.001, respectively), and liver (p < 0.0001 and p < 0.0001, respectively) (supplementary material, Figure S4).
Table 2.
Biomarker expression of primary tumours that spread to gynaecological sites
| Primary tumours GM cohort | Primary tumours QFU cohort | ||
|---|---|---|---|
| Positive (%) | Positive (%) | P value | |
| ER | 43/46 (93.5) | 333/433 (76.9) | 0.0075 |
| PR | 23/35 (65.7) | 267/427 (62.5) | ns |
| HER2 | 0/33 (0) | 43/441 (9.8) | 0.06 |
| CK14 | 0/18 (0) | 27 394 (6.9) | ns |
| CK5/6 | 0/19 (0) | 43/407 (10.6) | ns |
| EGFR | 0/17 (0) | 31/340 (9.2) | ns |
| AR | 17/19 (89.5) | 358/407 (88.0) | ns |
| GATA3 | 19/19 (100) | 356/421 (84.6) | 0.091 |
| FOXA1 | 19/19 (100) | 317/408 (77.7) | 0.018 |
| p53 low (<180*) | 17 (89.5) | 205 (62.3) | 0.015 |
| p53 high (>180*) | 2 (10.5) | 124 (37.7) | nd |
| Ki67 low (0–10%) | 19/19 (100) | 255 (71.9) | 0.026 |
| Ki67 moderate (10–30%) | 0 (0) | 87 (17.2) | nd |
| Ki67 high (>30%) | 0 (0) | 55 (10.9) | nd |
| Luminal A‐like† | 14/16 (87.5) | 261/398 (65.5) | 0.0001 |
The QFU cohort of unselected breast cancer cases were used for comparison. P values were calculated using Fisher's exact tests.
IHC score. A cut‐off of 180 was used to define high/low expression.
Luminal A‐like: ER positive, HER2 negative, Ki67 < 20%. Significant P values are underlined. ns, not significant; nd, not determined.
Given that luminal‐like phenotype is usually associated with favourable clinical outcomes including long life expectancy, we investigated whether aberrant expression of AR and ER pathway regulators could account for the paradoxically poor outcome of GM cases. AR was expressed at similar frequencies in the GM and QFU cohorts (89.5% versus 88%, p = ns). FOXA1 and GATA3, key regulators of ER transcriptional activity 27, 28, were both highly expressed in primary tumours in the GM cohort (FOXA1: 100% versus 77.7%, p = 0.0181; GATA3: 100% versus 84.6%, p = ns). The biomarker profile was not significantly different between primary ILC and IC‐NST (supplementary material, Figure S5 ).
Immunophenotype of GMs
Overall, the expression of all markers investigated was similar in primary and metastatic tumours (supplementary material, Figure S3 and Table S7). A case‐by‐case assessment of 13 individual cases with complete sample sets showed that while expression of ER and FOXA1 was maintained or increased during progression to gynaecological organs, expression of accessory proteins involved in hormonal regulation decreased during progression, with the percentage of tumour cells expressing PR, AR, and GATA3 in distant metastases lower in 69.2% (9/13), 38.5% (5/13), and 46% (6/13) of cases, respectively (Figure 2).
Figure 2.
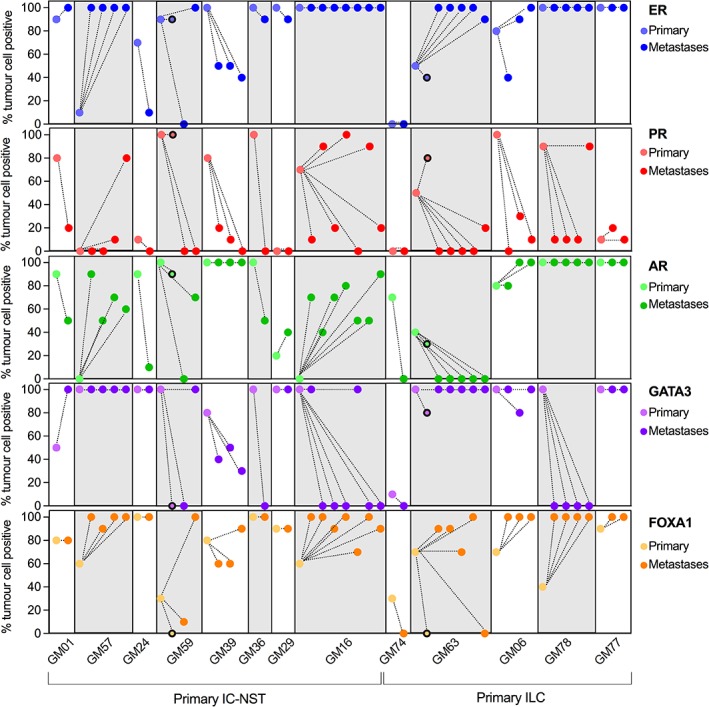
Immunophenotyping of endocrine‐related biomarkers between the primary tumour and matched metastases in 13 cases. Each case displayed a variety of changes in expression of these biomarkers within metastatic deposits in a case; the changes in expression also varied between cases. Data points with a black border indicate lymph node metastases that were removed at primary tumour diagnosis in cases GM59 and GM63.
DNA copy‐number alterations in metastasis to gynaecological organs
The primary tumours (n = 9) displayed copy‐number alterations (CNAs) consistent with ER positive primary tumours 29, 30, with gains (1q, 7q, 8q, 11q, 16p, and 17q) and losses (8p, 16q, 22q, and Xq) identified in over 50% of the samples (supplementary material, Figure S6). The most common alterations in ovarian metastases (n = 17, CNAs identified in over 50% of samples) included gain at 1p/q, 3p, 6p, 7p/q, 8q, 12q, 15q, and 17q, 19p/q; and loss at 8p, 13p/q, 16q, 22q and Xq (supplementary material, Figure S6). Frequent gains were identified in loci encoding MDM4, CDK6, FGFR1, MYC, CCND1, CDK4, and MDM2 (supplementary material, Table S9). We observed instances of both concordance and discordance in CNA profiles of matched primary and metastatic tumours, reflecting clonal progression and divergence (Figures 3 and 4, supplementary material, Figures S7–S11).
Figure 3.
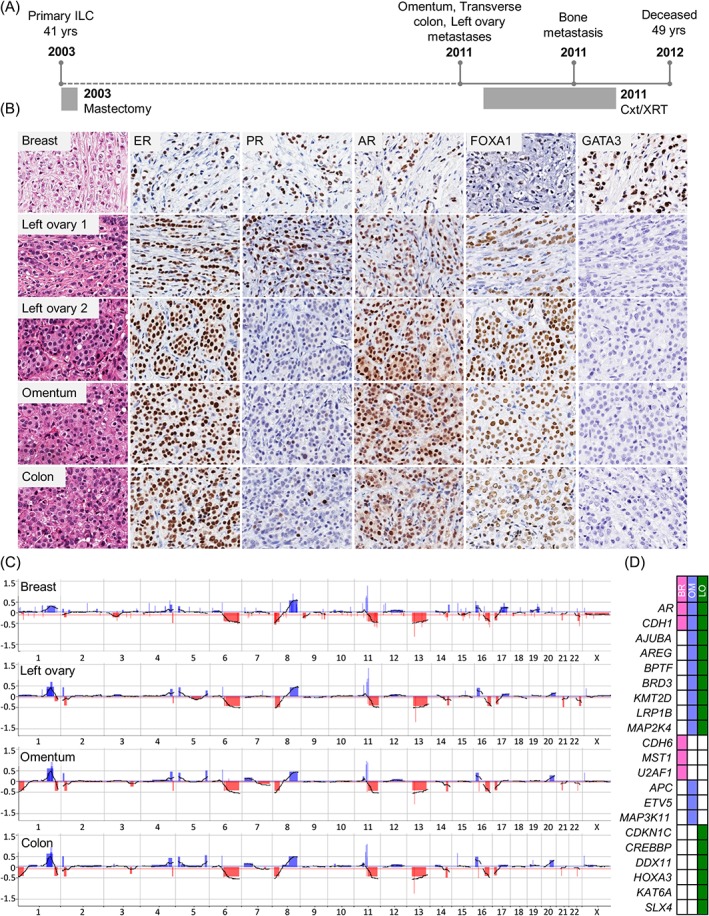
Case GM78. (A) The patient presented with a lymph node positive, grade 2 ILC at age 41; metastases to the left ovary, transverse colon, and omentum were diagnosed 8 years later. (B) Morphology and immunophenotype of tumours: the ovarian metastasis displayed two distinct morphological patterns; PR and GATA3 were lost during metastasis, while ER, AR, and FOXA1 remained positive. (C) Copy‐number profiling highlights the clonal nature of all tumours, together with evidence of intra‐tumour heterogeneity (e.g. 1p−, 1q−, and 20q+ in the metastases). (D) Targeted gene sequencing demonstrated shared mutations included CDH1 and AR (white boxes indicate that no mutation was detected). Of note, AR protein was still expressed at 3+ intensity in 100% of the tumour cells. The colon sample failed sequencing. BR, breast; Cxt, chemotherapy; LO, left ovary; OM, Omentum; XRT, radiation therapy.
Figure 4.
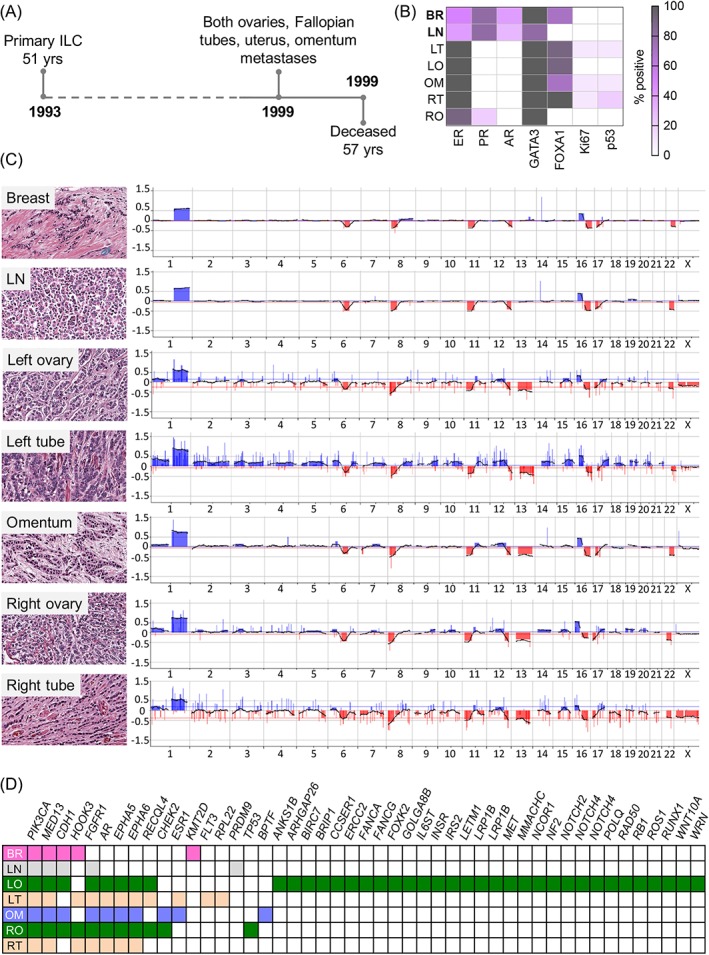
Case GM63. (A) The patient presented with a lymph node positive, grade 2 ILC at age 51; metastases to the left and right ovaries and Fallopian tubes, uterus, and omentum were detected 6 years later. (B) Immunophenotype of the tumours: white = negative, grey = 100% positive. (C) Copy‐number analysis highlights clonal relatedness of all tumours (shared alterations include 1+, 6q−, 8p−, 16p+, 16q−, 17q−). The axillary lymph node metastasis (detected at the time of primary tumour diagnosis) is more similar to the primary tumour than to the distant metastases; note 13q loss in distant metastases but not the primary tumour or lymph node. (D) Targeted gene sequencing reveals shared mutations between tumours, including PIK3CA, MED13, and CDH1 (not detected in the Fallopian tube metastases; may be due to technical limitations of the DNA in this tumour). Note: (1) an AR mutation in the metastases but not the primary tumour mirrors loss of protein expression during progression; (2) an ESR1 mutation (S463P) detected in the left Fallopian tube and omental tumours. The metastasis in the uterus was too small for analysis. BR, breast; BSO, bilateral salpingo‐oophorectomy; LN, lymph node; LO, left ovary; LT, left Fallopian tube; OM, omentum; RO, right ovary; RT, right Fallopian tube; TAH, total abdominal hysterectomy.
Amplification of the region containing CCND1 (11q13) was identified in the metastases in two cases but not in the primary tumour (GM59 and GM63) and, similarly, FGFR1 (8p11.23) was amplified only in the metastases in one case (GM16). FGFR1 was amplified in the primary tumour alone in cases GM78 and GM74. Co‐amplification of 8p12 and 11q13 has been associated with resistance to endocrine therapy 31 and was present in 2 of 9 (22%) of the primary tumours and 3 of 17 (17.6%) of the ovarian metastases (supplementary material, Figure S12). Among the matched cases, FGFR1 and CCND1 were co‐amplified in the primary tumour only in GM78, and the metastases only in GM59. The nanoString Cancer CNV panel of 88 cancer‐related loci, performed on GM63 and GM78, validated the presence of gains and losses in these cases (supplementary material, Table S10).
Targeted sequencing of matched primary tumours and GMs
Seven cases with DNA available from normal tissue, primary tumour, and metastases were analysed by targeted next‐generation sequencing of 386 genes (supplementary material, Table S3). The most frequently mutated genes were CDH1 and PIK3CA (supplementary material, Figure S13). CDH1 was mutated in five of seven (71.4%) primary tumours, all five of which were of lobular histological type. Metastases from these cases harboured the same CDH1 mutation as the primary tumours. PIK3CA was mutated in four of seven primary tumours; GM74 had two PIK3CA mutations in the primary tumour, yet only one (c.3140A>G) was shared with the ovarian metastasis (supplementary material, Figure S10).
An ESR1 mutation (c.1387T>C, S463P) was identified in two of six metastases sampled from case GM63 (Figure 4, supplementary material, Table S11), and AR was mutated in two cases. In case GM63 a novel nonsense mutation (c.2011C>T, Gln671*; Figure 4D) was identified in all the distant metastases, but not the primary tumour or lymph node metastasis. This was corroborated by IHC, where AR protein expression was observed in the primary and lymph node metastasis, but not the distant metastases (Figure 4B). Missense mutations at this codon (Q671L and Q671R) have been reported for oesophageal and prostate cancer respectively (COSMIC 32). All tumours in GM78 harboured a novel AR mutation (c.869G>T; C290F), and all were positive for AR by IHC (Figure 3B). Again, this point mutation has not been previously reported (COSMIC 32, however a c.867G>A mutation resulting in the same amino acid change has been reported in one primary breast cancer 33.
All cases shared at least one mutation between the primary tumour and metastases (supplementary material, Figures S7–S11, individual case reports), along with unique mutations present in either the primary tumour alone (e.g. TBX3 in GM06BR), or metastases alone (e.g. RB1, TP53 in GM74LO). Despite being a relatively homogeneous cohort in terms of metastatic spread, we observed substantial inter‐case diversity (Figures 3 and 4, supplementary material, Figures S7–S11), with no two cases showing similarity in genomic or phenotypic characteristics during progression to gynaecological organs.
Discussion
Substantial evidence now supports the theory of organotropism, where breast cancers with different biological properties have a predilection to spread to different organs. To further understand this concept, we studied a unique cohort of 54 breast cancer cases with metastases to gynaecological organs. We demonstrated that relative to a general, unselected breast cancer cohort from the same region of Australia, patients with tumours metastatic to gynaecological organs presented at a significantly younger age (likely pre‐menopausal), with a higher frequency of lymph node metastases and were more likely to exhibit invasive lobular histology. From a biological point of view, the primary tumours were all of luminal‐like phenotype with high expression of ER, PR, AR, FOXA1, and GATA3, suggesting that patients would be likely to respond to endocrine therapy 34. Interestingly, they rarely expressed biomarkers typically associated with aggressive clinical behaviour (e.g. HER2, basal markers, p53), and expressed Ki67 at relatively low frequency. Thus, histopathologically, the primary tumours exhibit features consistent with a favourable prognosis, and yet paradoxically these patients had very poor overall survival, in agreement with previous reports demonstrating that young age is an independent prognostic factor 35. It is also important to highlight that population‐based studies of young breast cancer patients describe higher proportions of ER−, HER2+, and triple negative disease 36, 37, which we did not observe in this cohort.
The enrichment for ILC is consistent with previous studies investigating features of tumours spreading to gynaecological sites or comparing patterns of metastasis between ILC and IC‐NST 4, 5, 6, 17, 20. Collectively, these and our data support the idea that gynaecological organs are metastasis targets for ILC. Given that ILC and IC‐NST are biologically and clinically different diseases with divergent long‐term outcomes 38, 39, 40, 41, we investigated the clinical features of breast cancer metastatic to gynaecological organs stratified by histological type. While ILC generally affects older women 7, 41, patients in the GM cohort were relatively young (median 44 years), suggesting that young age of ILC diagnosis may be a risk factor for GM, and raising the possibility that these women could benefit from closer clinical surveillance. Moreover, ILC patients who relapsed after 5 years were significantly younger than those who relapsed within 5 years, suggesting that these women may be suitable candidates for extended endocrine therapy 42, 43, addition of palbociclilb 44, or inclusion in emerging clinical trials specifically for E‐cadherin defective ILC 45. We also observed that ILC was more likely to colonise the uterus than IC‐NST and, amongst non‐GMs, ILC more frequently colonised the GI tract and less frequently the lungs and CNS compared to IC‐NST.
The interval between diagnosis of primary and metastatic disease can be between 5 and 20 years 17, 20, and so there may be opportunities for prevention therapy for patients deemed to be at high risk, and/or earlier detection and surgical/medical intervention for metastatic disease. Survival after metastatic diagnosis in this group of patients is poor (median of 1.9 years). This is likely to be due to the late presentation of disease owing to both the lack of apparent symptoms associated with metastases to gynaecological organs, GI tract, peritoneum, and so on 14, 17, 18, 20, and the widespread nature of dissemination (we recorded a median of five distinct sites of metastasis; range 1–11).
The luminal phenotype of all primary tumours in the cohort suggests that progression to gynaecological sites may be hormonally driven, and indeed we found high expression of hormone receptors and co‐factors. Gynae‐tropic metastatic behaviour might be explained by the tumour cells seeking out the oestrogen‐rich environment of the ovaries 20, particularly in pre‐menopausal women, and colonising other sites en route. There are significant data to suggest that the molecular phenotype of the primary tumour impacts the pattern of metastasis 6, 8, 9, 10, 11, 12. For instance, in contrast to the current study, we have previously showed that primary breast cancers that spread to the brain are ER and/or PR negative in up to 76% of cases 46. We and others have also previously shown that ER and PR are frequently down‐regulated during spread to distant organs, with a specific association with metastasis to lungs, liver, and bone 16, 47, suggesting hormonal signalling is less important in the progression of disease. However, in line with the idea that ER plays an important role in spread to the ovaries, the level of expression of ER seen in the primary tumour remained high in most cases, and in most metastases within individual cases.
The interplay between ER, PR, and AR and pioneer factors GATA3 and FOXA1 is complex, with growing evidence to suggest that the various members of this axis can alter tumour cell transcription programmes with unpredictable outcomes – expression has been associated with both good prognosis and endocrine therapy resistance 28, 48. Expression of PR, AR, FOXA1, and GATA3 in our GM cohort was extremely variable, both between and within cases. For instance, down‐regulation of one or more of these five key factors was observed in every case with matching primary and metastatic tissue available. To illustrate, case GM78 had high expression of ER and AR in all samples, yet PR expression decreased from 80% in the primary tumour to 10% in three of four metastases; expression of GATA3 went from 100% positive to negative in all metastases; and FOXA1 expression went from ~40% positive to 100% positive in all metastases. This suggests that the hormonally driven transcriptional programme of the primary tumour is significantly altered during metastasis to gynaecological sites and appears heterogeneous between metastases from the same patient (Figure 2). Such evolution in the dynamic interactions between these receptors and co‐factors may contribute to treatment‐resistant tumour cell clones. These intriguing findings require independent validation and comparison to other metastatic sites, when such cohorts become available.
Genomic alterations may also contribute to the aggressive behaviour of these tumours. The recurrent amplification of genes found at 8p11‐12 and 11q13 (e.g. FGFR1 and CCND1, respectively) are associated with resistance to therapy in ER‐positive breast cancer 31, 49, 50. In a group of mostly older women treated with letrozole in the neoadjuvant setting, Giltnane et al 31, 50 identified the pro‐survival effects of amplified FGFR1 and/or CCND1 in oestrogen‐deprived ER+/HER2− breast cancer, and that the combination of ER antagonists with FGFR1 or CDK4/6 inhibitors could increase cancer cell death. Several cases harboured amplification of these regions, and in some cases the amplifications were identified in metastases and not the primary tumour, suggesting they were present in a minor sub‐clone not sampled from the primary tumour 16 or were acquired to enhance survival during progression/treatment.
Mutations in ESR1 are particularly important in endocrine therapy resistance, producing protein products that function independently of ligand binding 51, 52, 53. Detecting mutant ESR1 in circulating DNA may therefore prove to be an important biomarker of relapse 54, 55, though heterogeneity between metastases in the same patient may be confounding. Indeed, we identified a previously reported ESR1 mutation (S463P 56) in just two of six metastases from one case. Mutation of AR is a mechanism of treatment resistance in castrate‐resistant prostate cancer 57, with most variants occurring in the ligand binding domain (analogous to ESR1 mutations). The gene is rarely mutated in primary breast cancer (7/977, 0.7% 29) and has not yet been reported in metastatic breast disease. We identified two cases with AR mutations. In one case, all tumours harboured the variant, and in the other only the distant metastases harboured the variant, correlating with loss of protein expression. These novel mutations reside in exon 1, which encodes the N‐terminal Activator Function‐1 (AF‐1) domain (C290F), and in exon 4 at the beginning of the ligand binding domain (LBD, G671*) 57. The functional roles of these alterations in breast cancer are yet to be elucidated, though we hypothesise they will impair functional AR owing to: (1) the importance of exon 1 for wild‐type AR activity and (2) loss of the LBD 58, 59. These changes are worthy of further investigation given the analogous mechanisms of ESR1 mutations in endocrine resistance, and the lack of understanding of the role of AR in ER‐positive metastatic breast cancer.
We also cannot exclude the contributions of other mechanisms of metastasis driving this pattern of dissemination, such as tumour dormancy 60, 61, or microenvironmental cues from the bone marrow or ‘pre‐metastatic niche’ 62, 63, 64; it is unlikely that the mechanisms are mutually exclusive. Epithelial to mesenchymal transition is not a defining feature of ILC 65 and may play a less overt role in this context.
In summary, this study adds to our understanding of the clinical and phenotypic characteristics of breast cancer metastases to gynaecological organs. We acknowledge the challenges of collating such cohorts for analysis and that there may be bias towards certain patient subsets in which tissue is available for analysis. Nevertheless, combined with available clinical data and comparison to other available cohort data, we identify some findings that may be of clinical relevance. The patients were frequently young and suffered from widespread metastatic disease after variable latency, suggesting they may benefit from extended clinical surveillance. Progression of disease was characterised by the maintenance of ER signalling, but with apparent changes in hormone signalling through associated co‐factors, and acquisition of genomic alterations characteristic of aggressive clinical behaviour. Where possible, sampling and analysis of metastatic deposits may be beneficial for treatment planning, for example changing endocrine therapy if metastatic deposits harbour unique ESR1/AR mutations or other hormone signalling‐associated changes, or consideration of CDK4/6 or FGFR inhibitors for cases harbouring amplification of CCND1 and/or FGFR1.
Author contributions statement
JRK performed experiments, analysed data, and wrote the paper. AMR designed experiments, analysed the data, and wrote the paper. RM, LD, CRM, and AF performed experiments and analysed the data. SS performed experiments and pathology review. AD and SL analysed the data. JMS designed experiments and analysed the data. GM and SP performed pathology review. LR performed experiments. CN and KF provided clinical data. FAE designed experiments and provided resources. SRL provided funding, tissue resources, supervised pathology review, and conceived the study. MC provided resources, pathology review, and conceived the study. PTS analysed the data, wrote the paper, provided resources, and conceived the study.
Supporting information
Figure S1. Venn diagram illustrating the number of cases that underwent immunophenotyping and molecular profiling
Figure S2. Flow chart illustrating the classification system of the GM and QFU cohorts
Figure S3. Immunophenotyping of primary breast tumours and metastatic deposits.
Figure S4. Comparison of GM primary tumours with primary tumours that have spread to the brain, lung, bone, and liver.
Figure S5. Immunophenotype of the GM cohort comparing primary tumour type.
Figure S6. Whole genome frequency plots showing copy‐number alterations derived from array comparative genomic hybridisation (aCGH) in primary breast tumours and ovarian metastases.
Figure S7. Case GM06.
Figure S8. Case GM16.
Figure S9. Case GM59.
Figure S10. Case GM74.
Figure S11. Case GM77.
Figure S12. Co‐amplification of CCND1 and FGFR1.
Figure S13. Commonly mutated genes in primary breast tumours (left panel) and ovarian metastases
Table S1. Antibody details and scoring methods 66
Table S2. Cases that underwent aCGH
Table S3. Characteristics of matched cases
Table S4. Sequencing statistics
Table S5. Clinical and pathology characteristics – comparison of ILC versus IC‐NST
Table S6. Distribution of GMs
Table S7. All immunohistochemistry data
Table S8. Ki67 scores of primary breast tumours
Table S9. Frequent copy‐number gains
Table S10. Nanostring cancer copy‐number variation (CNV) panel integer copy‐number results
Table S11. Sequencing data
Acknowledgements
We thank the patients and their families for the tissues donated for research through the Brisbane Breast Bank and to clinical staff involved in the collection and retrieval of tissue resources. The project was supported by funding from the Pathology Queensland Study Education and Research fund (Grant no. 4757_SimS) awarded to SS, JK, PTS, and MC and from the Australian National Health and Medical Research Council awarded to SRL (APP1113867). PTS was supported in part by a fellowship from National Breast Cancer Foundation, Australia; FAE is supported by a fellowship from the Australian Research Council. We thank Andrew Fellowes and team at the Peter MacCallum Cancer Centre and staff at the Ramaciotti Centre for Genomics at the University of NSW for their technical expertise in performing genomic analyses. We thank the many staff at the Queensland Centre for Gynaecological Cancer for providing access to their clinical database. We also acknowledge the many researchers whose contributions are not cited here due to space limitations.
No conflicts of interest were declared.
References
- 1. Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med 2008; 359: 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee YT. Breast carcinoma: pattern of metastasis at autopsy. J Surg Oncol 1983; 23: 175–180. [DOI] [PubMed] [Google Scholar]
- 3. Hess KR, Varadhachary GR, Taylor SH, et al Metastatic patterns in adenocarcinoma. Cancer 2006; 106: 1624–1633. [DOI] [PubMed] [Google Scholar]
- 4. Borst MJ, Ingold JA. Metastatic patterns of invasive lobular versus invasive ductal carcinoma of the breast. Surgery 1993; 114: 637–641 discussion 41‐2. [PubMed] [Google Scholar]
- 5. Lamovec J, Bracko M. Metastatic pattern of infiltrating lobular carcinoma of the breast: an autopsy study. J Surg Oncol 1991; 48: 28–33. [DOI] [PubMed] [Google Scholar]
- 6. Harris M, Howell A, Chrissohou M, et al A comparison of the metastatic pattern of infiltrating lobular carcinoma and infiltrating duct carcinoma of the breast. Br J Cancer 1984; 50: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arpino G, Bardou VJ, Clark GM, et al Infiltrating lobular carcinoma of the breast: tumor characteristics and clinical outcome. Breast Cancer Res 2004; 6: R149–R156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porter GJ, Evans AJ, Pinder SE, et al Patterns of metastatic breast carcinoma: influence of tumour histological grade. Clin Radiol 2004; 59: 1094–1098. [DOI] [PubMed] [Google Scholar]
- 9. Fulford LG, Reis‐Filho JS, Ryder K, et al Basal‐like grade III invasive ductal carcinoma of the breast: patterns of metastasis and long‐term survival. Breast Cancer Res 2007; 9: R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maki DD, Grossman RI. Patterns of disease spread in metastatic breast carcinoma: influence of estrogen and progesterone receptor status. AJNR Am J Neuroradiol 2000; 21: 1064–1066. [PMC free article] [PubMed] [Google Scholar]
- 11. Sihto H, Lundin J, Lundin M, et al Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: a nationwide cohort study. Breast Cancer Res 2011; 13: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smid M, Wang Y, Zhang Y, et al Subtypes of breast cancer show preferential site of relapse. Cancer Res 2008; 68: 3108–3114. [DOI] [PubMed] [Google Scholar]
- 13. Kondi‐Pafiti A, Kairi‐Vasilatou E, Iavazzo C, et al Metastatic neoplasms of the ovaries: a clinicopathological study of 97 cases. Arch Gynecol Obstet 2011; 284: 1283–1288. [DOI] [PubMed] [Google Scholar]
- 14. Moore RG, Chung M, Granai CO, et al Incidence of metastasis to the ovaries from nongenital tract primary tumors. Gynecol Oncol 2004; 93: 87–91. [DOI] [PubMed] [Google Scholar]
- 15. Yada‐Hashimoto N, Yamamoto T, Kamiura S, et al Metastatic ovarian tumors: a review of 64 cases. Gynecol Oncol 2003; 89: 314–317. [DOI] [PubMed] [Google Scholar]
- 16. Cummings MC, Simpson PT, Reid LE, et al Metastatic progression of breast cancer: insights from 50 years of autopsies. J Pathol 2014; 232: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Demopoulos RI, Touger L, Dubin N. Secondary ovarian carcinoma: a clinical and pathological evaluation. Int J Gynecol Pathol 1987; 6: 166–175. [DOI] [PubMed] [Google Scholar]
- 18. de Waal YR, Thomas CM, Oei AL, et al Secondary ovarian malignancies: frequency, origin, and characteristics. Int J Gynecol Cancer 2009; 19: 1160–1165. [DOI] [PubMed] [Google Scholar]
- 19. Salamalekis E, Bakas P, Sykiotis K, et al Outcome of patients with ovarian metastatic tumors. Report of 83 cases and review. Eur J Gynaecol Oncol 2004; 25: 713–715. [PubMed] [Google Scholar]
- 20. Bigorie V, Morice P, Duvillard P, et al Ovarian metastases from breast cancer: report of 29 cases. Cancer 2010; 116: 799–804. [DOI] [PubMed] [Google Scholar]
- 21. St Romain P, Madan R, Tawfik OW, et al Organotropism and prognostic marker discordance in distant metastases of breast carcinoma: fact or fiction? A clinicopathologic analysis. Hum Pathol 2012; 43: 398–404. [DOI] [PubMed] [Google Scholar]
- 22. Raghavendra A, Kalita‐de Croft P, Vargas AC, et al Expression of MAGE‐A and NY‐ESO‐1 cancer/testis antigens is enriched in triple‐negative invasive breast cancers. Histopathology 2018; 73: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Al‐Ejeh F, Simpson PT, Saunus JM, et al Meta‐analysis of the global gene expression profile of triple‐negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogene 2014; 3: e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Junankar S, Baker LA, Roden DL, et al ID4 controls mammary stem cells and marks breast cancers with a stem cell‐like phenotype. Nat Commun 2015; 6: 6548. [DOI] [PubMed] [Google Scholar]
- 25. Harrell JC, Prat A, Parker JS, et al Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat 2012; 132: 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Do KA, Treloar SA, Pandeya N, et al Predictive factors of age at menopause in a large Australian twin study. Hum Biol 1998; 70: 1073–1091. [PubMed] [Google Scholar]
- 27. Asselin‐Labat ML, Sutherland KD, Barker H, et al Gata‐3 is an essential regulator of mammary‐gland morphogenesis and luminal‐cell differentiation. Nat Cell Biol 2007; 9: 201–209. [DOI] [PubMed] [Google Scholar]
- 28. Hurtado A, Holmes KA, Ross‐Innes CS, et al FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet 2011; 43: 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. The Cancer Genome Atlas Network . Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Curtis C, Shah SP, Chin SF, et al The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giltnane JM, Hutchinson KE, Stricker TP, et al Genomic profiling of ER(+) breast cancers after short‐term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med 2017; 9: eaai7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Forbes SA, Beare D, Boutselakis H, et al COSMIC: somatic cancer genetics at high‐resolution. Nucleic Acids Res 2017; 45: D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stephens PJ, Tarpey PS, Davies H, et al The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rakha EA, Reis‐Filho JS, Ellis IO. Combinatorial biomarker expression in breast cancer. Breast Cancer Res Treat 2010; 120: 293–308. [DOI] [PubMed] [Google Scholar]
- 35. Lian W, Fu F, Lin Y, et al The impact of young age for prognosis by subtype in women with. Early Breast Cancer Sci Rep 2017; 7: 11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Azim HA Jr, Michiels S, Bedard PL, et al Elucidating prognosis and biology of breast cancer arising in young women using gene expression profiling. Clin Cancer Res 2012; 18: 1341–1351. [DOI] [PubMed] [Google Scholar]
- 37. Keegan TH, DeRouen MC, Press DJ, et al Occurrence of breast cancer subtypes in adolescent and young adult women. Breast Cancer Res 2012; 14: R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Metzger Filho O, Giobbie‐Hurder A, Mallon E, et al Relative effectiveness of Letrozole compared with tamoxifen for patients with lobular carcinoma in the BIG 1‐98 trial. J Clin Oncol 2015; 33: 2772–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ciriello G, Sinha R, Hoadley KA, et al The molecular diversity of luminal a breast tumors. Breast Cancer Res Treat 2013; 141: 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rakha EA, El‐Sayed ME, Powe DG, et al Invasive lobular carcinoma of the breast: response to hormonal therapy and outcomes. Eur J Cancer 2008; 44: 73–83. [DOI] [PubMed] [Google Scholar]
- 41. Pestalozzi BC, Zahrieh D, Mallon E, et al Distinct clinical and prognostic features of infiltrating lobular carcinoma of the breast: combined results of 15 international breast cancer study group clinical trials. J Clin Oncol 2008; 26: 3006–3014. [DOI] [PubMed] [Google Scholar]
- 42. Goss PE, Ingle JN, Pritchard KI, et al Extending aromatase‐inhibitor adjuvant therapy to 10 years. N Engl J Med 2016; 375: 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davies C, Pan H, Godwin J, et al Long‐term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor‐positive breast cancer: ATLAS, a randomised trial. Lancet 2013; 381: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finn RS, Crown JP, Ettl J, et al Efficacy and safety of palbociclib in combination with letrozole as first‐line treatment of ER‐positive, HER2‐negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA‐1/TRIO‐18. Breast Cancer Res 2016; 18: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bajrami I, Marlow R, van de Ven M, et al E‐cadherin/ROS1 inhibitor synthetic lethality in breast cancer. Cancer Discov 2018; 8: 498–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Da Silva L, Simpson PT, Smart CE, et al HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res 2010; 12: R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Idirisinghe PK, Thike AA, Cheok PY, et al Hormone receptor and c‐ERBB2 status in distant metastatic and locally recurrent breast cancer. Pathologic correlations and clinical significance. Am J Clin Pathol 2010; 133: 416–429. [DOI] [PubMed] [Google Scholar]
- 48. Ross‐Innes CS, Stark R, Teschendorff AE, et al Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012; 481: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Turner N, Pearson A, Sharpe R, et al FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010; 70: 2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Formisano L, Stauffer KM, Young CD, et al Association of FGFR1 with ERalpha maintains ligand‐independent ER transcription and mediates resistance to estrogen deprivation in ER(+) breast cancer. Clin Cancer Res 2017; 23: 6138–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toy W, Shen Y, Won H, et al ESR1 ligand‐binding domain mutations in hormone‐resistant breast cancer. Nat Genet 2013; 45: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robinson DR, Wu YM, Vats P, et al Activating ESR1 mutations in hormone‐resistant metastatic breast cancer. Nat Genet 2013; 45: 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jeselsohn R, Yelensky R, Buchwalter G, et al Emergence of constitutively active estrogen receptor‐alpha mutations in pretreated advanced estrogen receptor‐positive breast cancer. Clin Cancer Res 2014; 20: 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schiavon G, Hrebien S, Garcia‐Murillas I, et al Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 2015; 7: 313ra182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spoerke JM, Gendreau S, Walter K, et al Heterogeneity and clinical significance of ESR1 mutations in ER‐positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 2016; 7: 11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toy W, Weir H, Razavi P, et al Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov 2017; 7: 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brooke GN, Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Curr Genomics 2009; 10: 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jenster G, van der Korput HA, Trapman J, et al Identification of two transcription activation units in the N‐terminal domain of the human androgen receptor. J Biol Chem 1995; 270: 7341–7346. [DOI] [PubMed] [Google Scholar]
- 59. Tilley WD, Buchanan G, Hickey TE, et al Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res 1996; 2: 277–285. [PubMed] [Google Scholar]
- 60. Riethmuller G, Klein CA. Early cancer cell dissemination and late metastatic relapse: clinical reflections and biological approaches to the dormancy problem in patients. Semin Cancer Biol 2001; 11: 307–311. [DOI] [PubMed] [Google Scholar]
- 61. Suzuki M, Mose ES, Montel V, et al Dormant cancer cells retrieved from metastasis‐free organs regain tumorigenic and metastatic potency. Am J Pathol 2006; 169: 673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Janni W, Vogl FD, Wiedswang G, et al Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse‐‐a European pooled analysis. Clin Cancer Res 2011; 17: 2967–2976. [DOI] [PubMed] [Google Scholar]
- 63. Kaplan RN, Riba RD, Zacharoulis S, et al VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 2005; 438: 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peinado H, Zhang H, Matei IR, et al Pre‐metastatic niches: organ‐specific homes for metastases. Nat Rev Cancer 2017; 17: 302–317. [DOI] [PubMed] [Google Scholar]
- 65. McCart Reed AE, Kutasovic JR, Vargas AC, et al An epithelial to mesenchymal transition programme does not usually drive the phenotype of invasive lobular carcinomas. J Pathol 2016; 238: 489–494. [DOI] [PubMed] [Google Scholar]
- 66. Gudlaugsson E, Ivar S, Janssen EAM, et al Comparison of the effect of different techniques for measurement of Ki67 proliferation on reproducibility and prognosis prediction accuracy in breast cancer. Histopathology 2012; 61: 1134–1144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Venn diagram illustrating the number of cases that underwent immunophenotyping and molecular profiling
Figure S2. Flow chart illustrating the classification system of the GM and QFU cohorts
Figure S3. Immunophenotyping of primary breast tumours and metastatic deposits.
Figure S4. Comparison of GM primary tumours with primary tumours that have spread to the brain, lung, bone, and liver.
Figure S5. Immunophenotype of the GM cohort comparing primary tumour type.
Figure S6. Whole genome frequency plots showing copy‐number alterations derived from array comparative genomic hybridisation (aCGH) in primary breast tumours and ovarian metastases.
Figure S7. Case GM06.
Figure S8. Case GM16.
Figure S9. Case GM59.
Figure S10. Case GM74.
Figure S11. Case GM77.
Figure S12. Co‐amplification of CCND1 and FGFR1.
Figure S13. Commonly mutated genes in primary breast tumours (left panel) and ovarian metastases
Table S1. Antibody details and scoring methods 66
Table S2. Cases that underwent aCGH
Table S3. Characteristics of matched cases
Table S4. Sequencing statistics
Table S5. Clinical and pathology characteristics – comparison of ILC versus IC‐NST
Table S6. Distribution of GMs
Table S7. All immunohistochemistry data
Table S8. Ki67 scores of primary breast tumours
Table S9. Frequent copy‐number gains
Table S10. Nanostring cancer copy‐number variation (CNV) panel integer copy‐number results
Table S11. Sequencing data