

Abstract
Checkpoint blockade immunotherapies have revolutionised cancer treatment in the last decade. Nevertheless, these are only beneficial for a small proportion of cancer patients. Important prognosticators for response to immunotherapy are the mutation burden of tumours as well as the quality and quantity of tumour‐infiltrating immune cells. High‐throughput multiplex immunophenotyping technologies have a central role in deciphering the complexity of anti‐tumour immune responses. Current techniques for the immunophenotyping of solid tumours are held back by the lack of spatial context, limitations in the number of targets that can be visualised simultaneously, and/or cumbersome protocols. We developed a tyramide signal amplification‐free method for the simultaneous detection of seven cellular targets by immunofluorescence. This method overcomes limitations posed by most widespread techniques and provides a unique tool for extensive phenotyping by multispectral fluorescence microscopy. Furthermore, it can be easily implemented as a high‐throughput technology for validation of discovery sets generated by RNA sequencing or mass cytometry and may serve in the future as a complementary diagnostic tool.
Keywords: multispectral immunofluorescence, cancer microenvironment, T‐cells, myeloid cells, biomarkers, immune infiltration, immunotherapy
Introduction
Immunotherapy has emerged as one of the most promising treatments for cancer 1, 2, 3, 4. However, a substantial proportion of patients are not responsive to current immunotherapies and, therefore, the discovery of (immune) biomarkers with predictive clinical value has become a major research avenue in cancer immunology 5, 6, 7, 8. It has long been recognised that the type and density of tumour‐infiltrating immune cells is an important prognostic indicator in several cancer types 9, 10, 11, 12. High‐throughput and multiplex immunophenotyping technologies are, thus, fundamental for research purposes but also carry great potential for implementation in a diagnostic setting. However, few tools are currently available for extensive immunophenotyping of tumour tissues in readily available clinical samples (often formalin‐fixed, paraffin‐embedded [FFPE] tissues).
Singleplex or multiplex immunohistochemistry (IHC) and immunofluorescence (IF) are the gold standard for the analysis of archival tumour tissues, but a major drawback of these techniques is the limited number of targets that can be detected simultaneously. Flow cytometry, on the other hand, has been successfully implemented for diagnostic purposes as well as for monitoring treatment responses 13, 14. However, the lack of tissue‐context data generated by flow cytometry remains a limitation for the analysis of solid tumours 15. Mass cytometry and transcriptomic technologies have demonstrated great value for characterisation of cancer immune landscapes but are better applicable in a discovery setting and less suitable for high‐throughput investigation of biomarkers 16, 17, 18. An emerging tool that can overcome these limitations is sequential IHC of tissue sections with alternating immune detection and bleaching steps. Later, images are digitally overlaid to construct a ‘multiparametric’ image 19, 20. Furthermore, the Opal™ 6‐plex quantitative IF assay, in combination with multispectral imaging systems, allows for multiplex IF detection on a single tissue section through sequential steps of tyramide signal amplification (TSA), followed by microwave‐mediated antibody stripping 21, 22, 23. Nevertheless, sequential immune detection methods are time‐consuming and antibody stripping can compromise tissue integrity and morphology, which limits its application in certain tissue types (e.g. bone or adipose tissue).
We developed a seven‐colour IF method for the analysis of FFPE tissues, which can be used for extensive phenotyping by multispectral fluorescence microscopy. This protocol overcomes limitations posed by the previous techniques by using a combination of both indirect and direct fluorescence detection of antibody binding. Furthermore, we demonstrate the flexibility of the system as it is easily combined with the TSA method for the improved detection of low‐abundance antigens.
Materials and methods
Tissue material
FFPE blocks from tonsil and colorectal cancer tissues were obtained from the Department of Pathology of the Leiden University Medical Centre (Leiden, The Netherlands). Mismatch repair (MMR) status was determined as either MMR proficient or deficient by immunohistochemical detection of PMS2 and MSH6 proteins 24. Patient samples were anonymised and handled according to the medical ethical guidelines described in the Code of Conduct for Proper Secondary Use of Human Tissue of the Dutch Federation of Biomedical Scientific Societies.
Immunohistochemistry
Antibody specificity and optimal conditions for antigen retrieval were assessed by singleplex IHC. FFPE 4‐μm tissue sections were deparaffinised with xylene and washed in ethanol, after which endogenous peroxidase was blocked by incubation in a 0.3% hydrogen peroxide/methanol (Merck Millipore, Burlington, MA, USA) solution for 20 min. Heat‐induced antigen retrieval was done with either citrate buffer (10 mm, pH 6.0) or Tris–EDTA buffer (10 mm/1 mm, pH 9.0). After cooling, non‐specific antibody binding sites were blocked for 30 min with Superblock solution (Thermo Fisher Scientific, Waltham, MA, USA) and incubated overnight with a primary antibody (Table 1). After washing in PBS, 1 h incubation with poly‐horseradish peroxidase solution (Immunologic, Duiven, The Netherlands) was performed at room temperature (RT). The slides were developed with the DAB+ chromogen (DAKO, Agilent Technologies, Santa Clara, CA, USA) solution and counterstained with haematoxylin (Thermo Fisher Scientific). Optimal IHC conditions were evaluated by light microscopy.
Table 1.
Antibodies included in the T‐cell and myeloid panels
| Target | Clone | Species | Isotype | Detection | Fluorochrome | Supplier |
|---|---|---|---|---|---|---|
| T‐cell panel | ||||||
| Pan‐Cytokeratin | AE1/AE3 and C11 | Mouse | IgG1 | Direct | Alexa 488 | Thermo Fisher Scientific |
| Cell Signaling Technology | ||||||
| CD8 | 4B11 | Mouse | IgG2b | Indirect | CF555 | DAKO |
| CD3 | D7A6E | Rabbit | IgG | Direct | Alexa 594 | Cell Signaling Technology |
| FOXP3 | 236A/E7 | Mouse | IgG1 | Indirect | CF633 | Thermo Fisher Scientific |
| CD45RO | UCHL1 | Mouse | IgG2a | Indirect | CF647 | Cell Signaling Technology |
| GZMB | EPR8260 | Rabbit | IgG | Indirect | Alexa 680 | Abcam |
| DAPI | ||||||
| Myeloid panel | ||||||
| PDL1 | SP‐142 | Rabbit | IgG | Opal | Opal 520 | Spring Bioscience |
| CD11c | EP1347Y | Rabbit | IgG | Direct | Alexa 546 | Abcam |
| HLA‐DR | TAL 1B5 | Mouse | IgG1 | Direct | Alexa 594 | Thermo Fisher Scientific |
| CD163 | 10D6 | Mouse | IgG1 | Indirect | CF633 | Thermo Fisher Scientific |
| Pan‐Cytokeratin | AE1/AE3 and C11 | Mouse | IgG1 | Direct | Alexa 647 | Novus Biologicals/Biolegend |
| CD68 | D4B9C | Rabbit | IgG | Indirect | Alexa 680 | Cell Signaling Technology |
| DAPI | ||||||
Direct and indirect immunofluorescent detection
Seven‐target IF detection was performed on 4‐μm FFPE tissue sections which were deparaffinised with xylene and washed in ethanol. Heat‐induced antigen retrieval in citrate buffer (10 mm, pH 6) was performed and the slides were allowed to cool down to RT. Subsequently, the tissues were blocked with Superblock buffer (Thermo Fisher Scientific) and incubated overnight with all primary antibodies to be detected indirectly (Table 1). After incubation, detection with corresponding fluorescent secondary antibodies (CF dye conjugated secondary antibodies, Sigma–Aldrich, Saint Louis, MO, USA; Alexa Fluor dye conjugated secondary antibodies, Thermo Fisher Scientific) was performed followed by incubation of the tissue with directly conjugated primary antibodies. Directly conjugated antibodies were obtained with the corresponding Alexa Fluor (546, 594) antibody labelling kits (Thermo Fisher Scientific). Furthermore, two pre‐conjugated (Alexa Fluor 488 for the T‐cell panel and Alexa Fluor 647 for the myeloid panel) anti‐keratin monoclonal antibodies that detect different keratins were used simultaneously to enhance the signal detection of epithelial tumour cells. Finally, the tissues were incubated with DAPI (1 μm) as nuclear counterstain and mounted with Prolong® Gold Antifade Reagent (Cell Signaling Technology, Danvers, MA, USA).
TSA in combination with direct and indirect immunofluorescent detection
For the detection of low abundance proteins, TSA signal amplification was performed with the Opal 7‐colour manual IHC kit (Perkin Elmer, Waltham, MA, USA). Deparafinisation, blocking of endogenous peroxidase, and antigen retrieval in citrate buffer were performed as described previously. Slides were incubated for 1 h with anti‐PD‐L1 (Clone SP142, Spring Bioscience, Pleasanton, CA, USA) followed by a 30 min incubation with poly‐horseradish peroxidase solution (Immunologic). Fluorescence detection was achieved by using the Opal 520 reagent (Perkin Elmer). Finally, after microwave‐mediated antibody stripping, the slides were sequentially incubated with primary antibodies, secondary fluorescent antibodies, and directly conjugated antibodies (Table 1), using the previously described method.
Image acquisition and analysis
Tissue slides were imaged at ×40 magnification with the Vectra 3.0 Automated Quantitative Pathology Imaging System (Perkin Elmer). Image analysis and spectral separation of dyes was performed with the InForm Cell Analysis software (Perkin Elmer) by using spectral libraries defined with single‐marker IF detection. To ensure the signals of the single markers were specific, the immunofluorescent detection was compared to chromogenic IHC. Tissue segmentation was trained manually with DAPI and keratin to segment images into tumour, stroma, and ‘no tissue’ areas. Next, cellular segmentation was performed using a counterstain‐based approach with DAPI to segment nuclei and membrane markers (CD8, CD3, and CD45RO for the T‐cell panel and HLA‐DR, CD163, and CD68 for the myeloid panel) to detect cell contours. Phenotyping was trained manually per tumour where 20 cells per phenotype were defined (Supplementary material, Figure S1). The following phenotypes were identified for the T‐cell panel: total T‐cells (CD3+), CD8− T‐cells (CD3+CD8−, mostly T‐helper cells), cytotoxic T‐cells (CD3+CD8+), regulatory T‐cells (CD3+FOXP3+), memory T‐cells (CD3+CD45RO+), and activated, cytotoxic T‐cells (CD3+GZMB+). All images were visually inspected to confirm the correct attribution and quantification of phenotypes. For each case, cell counts were normalised by tissue area (number of cells/mm2).
Results
The combination of indirect and direct IF detection allows for the simultaneous assessment of seven markers
To assess seven markers in a single tissue slide, a method was developed in which four targets were detected with primary antibodies and isotype/species‐specific secondary fluorescent antibodies (Figure 1A–C) followed by the detection of two additional cellular markers with directly conjugated antibodies in order to overcome limitations posed by species/isotype overlap (Figure 1D). Finally, nuclear counterstain was obtained with DAPI (Figure 1E). As a proof of principle, tumour tissues were evaluated for the expression of CD3 and pan‐keratin with directly conjugated antibodies while the expression of CD8, FOXP3, CD45RO, and Granzyme B was assessed by indirect IF (Figure 2). After the development of a spectral library with single IF assays, multiplexed images were obtained with the following filter‐dye combinations: DAPI, FITC/AF488 (keratin), Cy3/TRITC (CD8‐CF555), Texas Red (CD3‐AF594), and Cy5 (FOXP3‐CF633, CD45RO‐CF647, GZMB‐AF680). Importantly, antibodies were titrated so that spectral overlap was limited by using comparable exposure times (400–500 ms) in all non‐DAPI filters. This was particularly relevant for the Cy5 filter where three different spectra were being deconvoluted. Specificity and correct separation of spectra was assessed by comparison to single marker patterns (Supplementary material, Figure S2). Autofluorescence of erythrocytes in FFPE material is a notorious issue for the evaluation of IF signals. Erythrocytes, however, were morphologically distinct from cells of interest and lacked expression of membrane markers. We addressed this issue through extensive training and segmentation with the InForm software, enabling the system to differentiate between cells of interest and erythrocytes.
Figure 1.
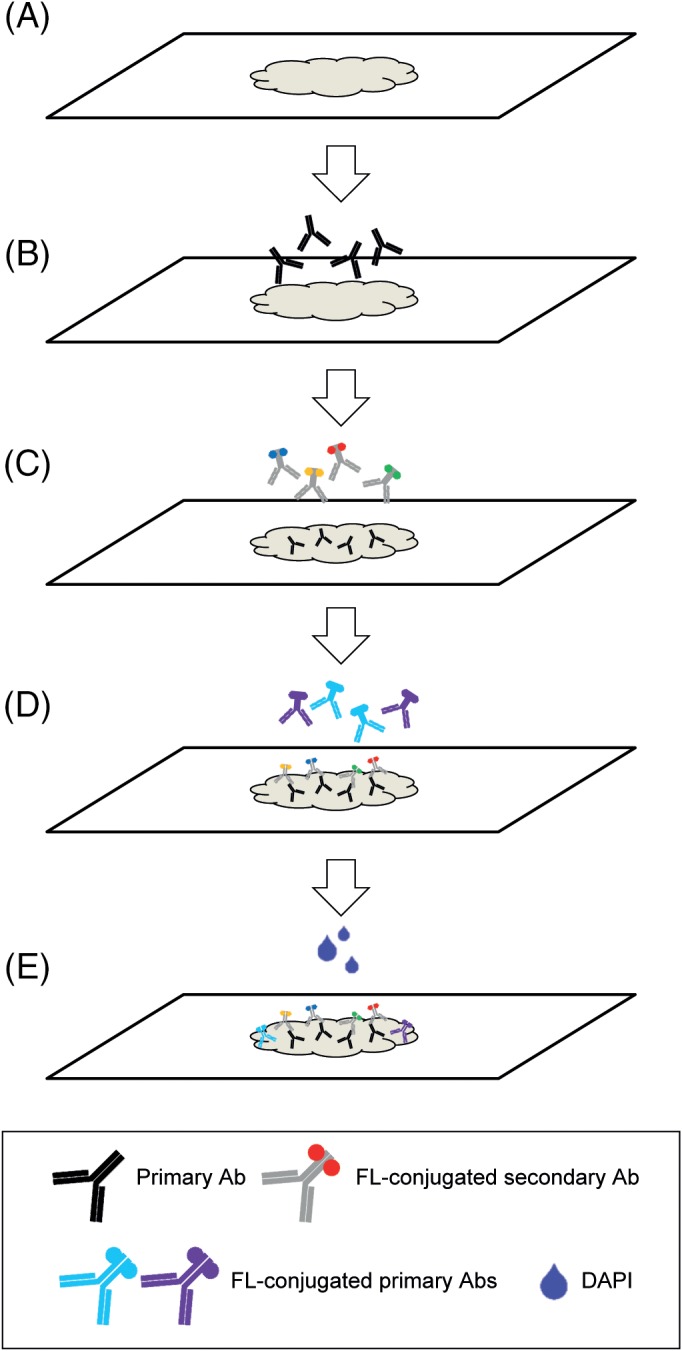
Workflow of seven‐target IF detection starting with tissue preparation (A), followed by detection with primary antibodies (B), which are subsequently detected with secondary antibodies (C). Afterwards, the tissue is incubated with directly conjugated antibodies (D), and incubated with DAPI as nuclear counterstain (E).
Figure 2.

Immunofluorescent detection of T‐cell markers in MMR‐proficient (A) and MMR‐deficient (B) colorectal cancer tissues, namely CD3 (red), CD8 (blue), FOXP3 (cyan), CD45RO (orange), GZMB (green), keratin (white) to distinguish tumour tissue, and DAPI (grey) as nuclear counterstain. Panels C–I show DAPI, keratin, CD8, CD3, FOXP3, CD45RO, and GZMB, respectively, detected in the MMR‐deficient tumour presented in panel B.
After optimisation, all markers could be discriminated in the tumour tissues. Immune cell populations defined as CD3+CD8− T‐cells (mostly T‐helper cells), cytotoxic T‐cells (CD3+CD8+), regulatory T‐cells (CD3+FOXP3+), memory T‐cells (CD3+CD45RO+), and activated, cytotoxic T‐cells (CD3+GZMB+) could be successfully identified and counted using the InForm software (Supplementary material, Figure S3). Furthermore, phenotype counts were compared between seven‐marker and single‐marker detection in consecutive tissue sections of a MMR‐deficient (MMR‐D) and MMR‐proficient MMR‐P CRC, to exclude biases introduced by the combination of antibodies and separation of spectra (Supplementary material, Table S1). After this, the T‐cell panel was applied for the characterisation of 19 colorectal cancer tissues, of which four were MMR‐D. Using the approach described here it was possible to phenotype and count the different T‐cell phenotypes at various cellular densities across the tissues (Figure 3). Furthermore, the T‐cell phenotypes were compared between MMR‐D and MMR‐P tumours which showed distinctly higher cytotoxic and memory T‐cell densities in MMR‐D tumours (Figure 3).
Figure 3.
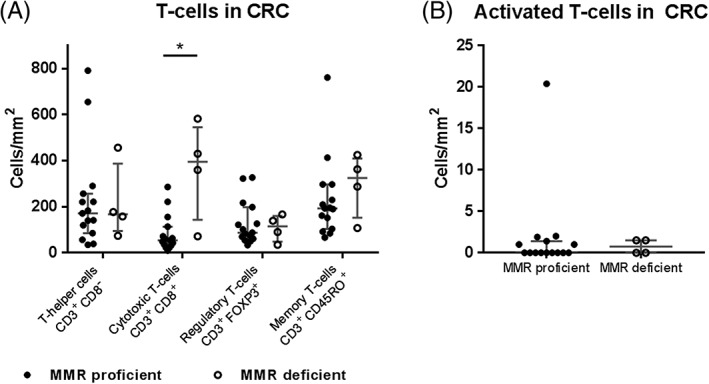
Prevalence of T‐cell immunophenotypes in colorectal cancer (CRC) tissue. Phenotypes are defined as T‐helper cells (CD3+CD8−), cytotoxic T‐cells (CD3+CD8+), regulatory T‐cells (CD3+FOXP3+), memory T‐cells (CD3+CD45RO+), and activated T‐cells (CD3+GZMB+).
TSA can be specifically employed for the detection of low abundance antigens in combination with direct and indirect IF
For the detection of antigens with low expression level, fluorescence signal amplification might be desirable. Therefore, we assessed if our methodology was compatible with applying TSA for selected markers. Here, we assessed the expression of PD‐L1, CD11c, HLA‐DR, CD163, keratin, and CD68 (myeloid panel) in a colorectal cancer tissue. PD‐L1 detection was challenging with both direct and indirect IF and therefore TSA with Opal 520 was applied to assess the expression of this molecule. Figure 4 demonstrates the successful detection of PD‐L1 together with six other markers detected by indirect and direct IF. Thus, we show that TSA detection can be easily combined with the current method for the detection of low abundance proteins without requiring subsequent steps of TSA‐based detection.
Figure 4.
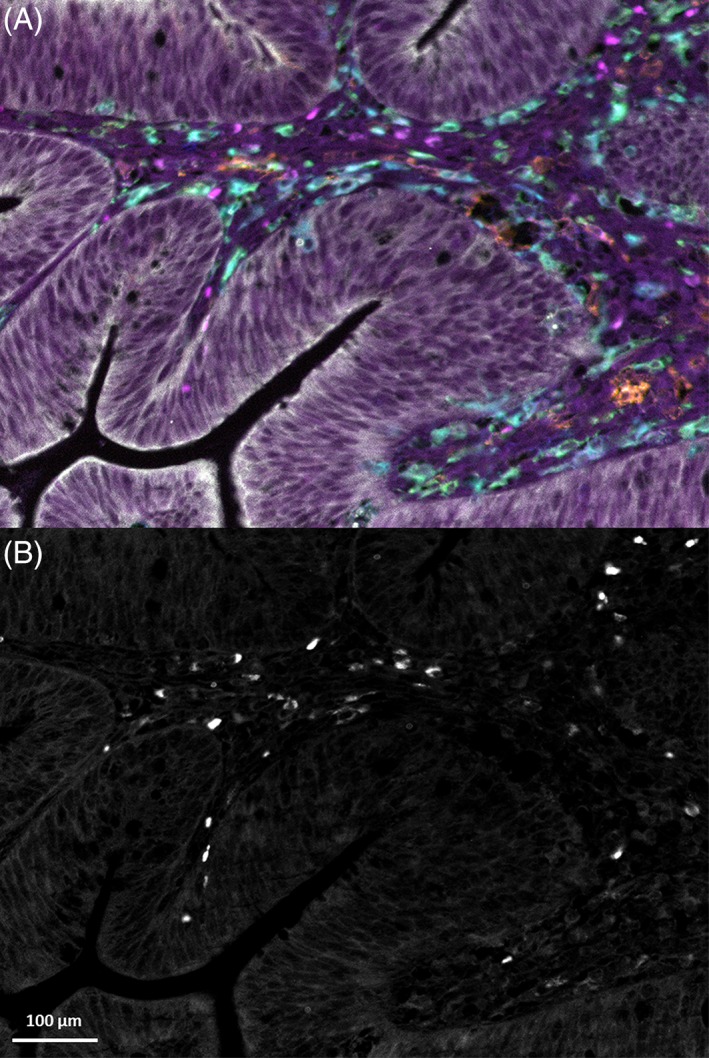
(A) Immunofluorescent detection of myeloid‐related markers in colorectal cancer tissue in which PD‐L1 was enhanced by TSA. Markers are PD‐L1 (magenta), HLA‐DR (orange), CD11c (blue), CD163 (green), CD68 (cyan), and keratin (white). (B) TSA‐enhanced PD‐L1 only, in the same tissue.
Discussion
The increasing amount of data highlighting the importance of cancer immunophenotypes in predicting patient prognosis and response to immunotherapies underlines the need for high‐throughput immunophenotyping tools that can be easily applied in a clinical context. Here, we describe an efficient and fast seven‐colour IF method for the analysis of tumour tissues by multispectral microscopy. The use of primary antibodies that are detected with fluorescent secondary antibodies, followed by the detection of antigens by directly conjugated antibodies, allows the use of two or more antibodies of the same isotype in one sample. The limitations of standardising exposure times and spectral overlap in different or the same optical filters can be easily overcome. Moreover, this method is time‐efficient when compared to the consecutive detection of single antigens, which is currently applied in other multiplex immunodetection techniques. Furthermore, the challenges in evaluating the expression of low expressed antigens can be addressed by combining the current method with TSA‐based antigen detection.
Efficient multiplex IF detection methods are becoming essential in the assessment of immune cell infiltration, which can have large implications for patient prognosis. For instance, targeting the PD‐L1/PD1 axis has shown great applicability for the treatment of tumours with high mutation burden 5, 6, 8. Nevertheless, not all patients respond to checkpoint blockade and this differential sensitivity to treatment might be explained by the immune cell composition of the tumour microenvironment 25, 26. Besides aiding patient selection for current (immuno‐) therapies, multiplex immunophenotyping can lead to the identification of distinct immunophenotypes that pose potential targets for immunotherapy 27. In this work we describe two panels that can be readily applied for the detection of lymphoid and myeloid immune cells.
High cytotoxic T‐cell densities have been associated with a better prognosis in solid cancers 9, 10, 11, 12 while the presence of regulatory T‐cells has been associated with improved clinical outcome when cells reside inside the tumour tissue but with a worse prognosis when present in the tumour surroundings 28, 29. Importantly, the ratio between cytotoxic T‐cells and regulatory T‐cells appears to be particularly relevant for patient prognosis 30, 31. Furthermore, increased infiltration of memory T‐cells (CD45RO+) in tumours has also been associated with a favourable clinical outcome 11, 32. All these parameters can be readily investigated in retrospective cohorts with the T‐cell panel described here.
The current myeloid panel was conceptualised to test the combination of the TSA method with indirect and direct IF. Tumour‐associated macrophages (TAMs) play an important role in cancer pathogenesis 33. In several cancer types, the polarisation between classically (M1) and alternatively (M2) activated macrophages is thought to be crucial for disease progression 34, 35. Our myeloid panel is able to discern M1 (CD68+HLA‐DR+) and M2 (CD68+CD163+PD‐L1+) related phenotypes in cancer tissues as well as to assess tumour infiltration by dendritic cells (CD11c+).
In conclusion, we have developed a method for IF detection of seven markers in FFPE tissue that can be directly applied for extensive immunophenotyping. The method is time‐efficient because it does not involve the use of TSA‐based detection protocols which also prevents tissue damage as a consequence of repeated boiling or bleaching for antibody stripping. This technique may serve as a diagnostic tool and is also highly complementary to techniques such as (single cell) RNA sequencing or mass cytometry that require validation in extensive cohorts of archival tissue.
Author contributions statement
MEIJ, TPB, and NFM planned experiments, analysed data, and wrote the manuscript. MEIJ, TPB, ZA, ER, AR, and AMH conducted experiments; AV, ESJ, and NFM supervised all immunofluorescent experiments. NFM designed and directed the project. All authors contributed to and had final approval of the submitted manuscript.
Supporting information
Figure S1. Workflow for image analysis by the InForm software
Figure S2. Single marker immunofluorescent detection of T‐cell markers in tonsil tissue
Figure S3. Detection of immunophenotypes in tumour tissue by the InForm software
Table S1. Comparison of phenotype counts between seven‐marker labelled and single‐marker labelled consecutive tissue sections
Acknowledgements
This work was supported by the Fight Colorectal Cancer‐Michael's Mission‐AACR Fellowship (2015), Alpe d'HuZes/KWF Bas Mulder Award (UL2015‐7664), Veni ZonMw grant (91617144), and the European Commission H2020 MSCA‐ETN grant under proposal number 675743 (project acronym: ISPIC).
No conflicts of interest were declared.
References
- 1. Brahmer JR, Tykodi SS, Chow LQ, et al Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hodi FS, O'Day SJ, McDermott DF, et al Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Le DT, Uram JN, Wang H, et al PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015; 372: 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Topalian SL, Hodi FS, Brahmer JR, et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor‐based immunotherapy. Lancet Oncol 2016; 17: e542–e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le DT, Durham JN, Smith KN, et al Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017; 357: 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riaz N, Havel JJ, Makarov V, et al Tumor and microenvironment evolution during immunotherapy with Nivolumab. Cell 2017; 171: 934–949 e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizvi NA, Hellmann MD, Snyder A, et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015; 348: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Miranda NF, Goudkade D, Jordanova ES, et al Infiltration of lynch colorectal cancers by activated immune cells associates with early staging of the primary tumor and absence of lymph node metastases. Clin Cancer Res 2012; 18: 1237–1245. [DOI] [PubMed] [Google Scholar]
- 10. Fridman WH, Pages F, Sautes‐Fridman C, et al The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012; 12: 298–306. [DOI] [PubMed] [Google Scholar]
- 11. Galon J, Costes A, Sanchez‐Cabo F, et al Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964. [DOI] [PubMed] [Google Scholar]
- 12. Punt S, Dronkers EA, Welters MJ, et al A beneficial tumor microenvironment in oropharyngeal squamous cell carcinoma is characterized by a high T cell and low IL‐17(+) cell frequency. Cancer Immunol Immunother 2016; 65: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martens A, Wistuba‐Hamprecht K, Geukes Foppen M, et al Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res 2016; 22: 2908–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Dongen JJ, Lhermitte L, Bottcher S, et al EuroFlow antibody panels for standardized n‐dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012; 26: 1908–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan J, Hegde PS, Clynes R, et al Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J Immunother Cancer 2016; 4: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Unen V, Li N, Molendijk I, et al Mass cytometry of the human mucosal immune system identifies tissue‐ and disease‐associated immune subsets. Immunity 2016; 44: 1227–1239. [DOI] [PubMed] [Google Scholar]
- 17. Angelova M, Charoentong P, Hackl H, et al Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol 2015; 16: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bendall SC, Nolan GP, Roederer M, et al A deep profiler's guide to cytometry. Trends Immunol 2012; 33: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsujikawa T, Kumar S, Borkar RN, et al Quantitative multiplex immunohistochemistry reveals myeloid‐inflamed tumor‐immune complexity associated with poor prognosis. Cell Rep 2017; 19: 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glass G, Papin JA, Mandell JW. SIMPLE: a sequential immunoperoxidase labeling and erasing method. J Histochem Cytochem 2009; 57: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parra ER, Uraoka N, Jiang M, et al Validation of multiplex immunofluorescence panels using multispectral microscopy for immune‐profiling of formalin‐fixed and paraffin‐embedded human tumor tissues. Sci Rep 2017; 7: 13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toth ZE, Mezey E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. J Histochem Cytochem 2007; 55: 545–554. [DOI] [PubMed] [Google Scholar]
- 23. Huang W, Hennrick K, Drew S. A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum Pathol 2013; 44: 29–38. [DOI] [PubMed] [Google Scholar]
- 24. Hall G, Clarkson A, Shi A, et al Immunohistochemistry for PMS2 and MSH6 alone can replace a four antibody panel for mismatch repair deficiency screening in colorectal adenocarcinoma. Pathology 2010; 42: 409–413. [DOI] [PubMed] [Google Scholar]
- 25. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res 2016; 22: 1865–1874. [DOI] [PubMed] [Google Scholar]
- 26. Wei SC, Levine JH, Cogdill AP, et al Distinct cellular mechanisms underlie anti‐CTLA‐4 and anti‐PD‐1 checkpoint blockade. Cell 2017; 170: 1120–1133, e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whiteside TL, Demaria S, Rodriguez‐Ruiz ME, et al Emerging opportunities and challenges in cancer immunotherapy. Clin Cancer Res 2016; 22: 1845–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saito T, Nishikawa H, Wada H, et al Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016; 22: 679–684. [DOI] [PubMed] [Google Scholar]
- 29. Salama P, Phillips M, Grieu F, et al Tumor‐infiltrating FOXP3(+) T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 2009; 27: 186–192. [DOI] [PubMed] [Google Scholar]
- 30. Jordanova ES, Gorter A, Ayachi O, et al Human leukocyte antigen class I, MHC class I chain‐related molecule A, and CD8+/regulatory T‐cell ratio: which variable determines survival of cervical cancer patients? Clin Cancer Res 2008; 14: 2028–2035. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki H, Chikazawa N, Tasaka T, et al Intratumoral CD8(+) T/FOXP3(+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol Immun 2010; 59: 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu GM, Wang SM. Tumor‐infiltrating CD45RO(+) memory T lymphocytes predict favorable clinical outcome in solid tumors. Sci Rep 2017; 7: 10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer 2016; 16: 447–462. [DOI] [PubMed] [Google Scholar]
- 34. Kurahara H, Shinchi H, Mataki Y, et al Significance of M2‐polarized tumor‐associated macrophage in pancreatic cancer. J Surg Res 2011; 167: E211–E219. [DOI] [PubMed] [Google Scholar]
- 35. Yuan A, Hsiao YJ, Chen HY, et al Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci Rep 2015; 5: 14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Workflow for image analysis by the InForm software
Figure S2. Single marker immunofluorescent detection of T‐cell markers in tonsil tissue
Figure S3. Detection of immunophenotypes in tumour tissue by the InForm software
Table S1. Comparison of phenotype counts between seven‐marker labelled and single‐marker labelled consecutive tissue sections