

Abstract
Cancer is a disease of the genome. Whereas efforts to understand the molecular basis of cancer have in the past largely focused on the role of mutations, recent evidence points to a strong epigenetic component in tumorigenesis, and epigenetic defects have been linked to loss of cell cycle control and cell survival. Here, we discuss the possibility that epigenetic alterations may promote tumor formation by an alternative mechanism. We speculate that epigenetic changes in stem cells and somatic cells contribute significantly to carcinogenesis by disruption of cellular differentiation programs. Epigenetic interference and loss of cellular identity may be particularly relevant for the emergence of cancer stem cells.
Cancer evolves from a combination of epigenetic and genetic abnormalities resulting in misregulated gene expression and disruption of cell homeostasis. Although cancer initiation appears to be predominantly driven by acquired genetic alterations such as point mutations or more complex genomic rearrangements, during the last decades it has become clear that aberrant epigenetic changes also have a major role at various stages of disease development. In particular, epigenetic mechanisms such as DNA methylation, histone modifications, and higher-order chromatin structure control expression of genes involved in proliferation, survival, and cell differentiation and are often targets of misregulation in cancer.
Recent observations suggest that subversion of proper control of cell proliferation may only be one aspect of epigenetic misregulation and that epigenetic effects may have more far-reaching consequences in the etiology of cancer than anticipated. We explore here the possibility that epigenetic alterations are key in the reprogramming of stem and somatic cells into cancer cells, including cancer stem cells.
EPIGENETIC COMPONENTS IN THE ETIOLOGY OF CANCER
One of the major epigenetic processes linked to cancer is DNA methylation. Its implication in cancer is based on the observation that normal patterns of DNA methylation are altered in virtually all cancer types. Global loss of DNA methylation in intergenic and heterochromatic regions is a general feature of neoplasia and has been suggested to lead to alterations in proper packaging of DNA and to increased genomic instability (Feinberg et al. 2006; Brock et al. 2007). In addition, gene-specific hypermethylation and hypomethylation of promoter regions mimics genetic mutations by silencing expression of tumor suppressors and activating transcription of oncogenes, respectively (Herman and Baylin 2003; Oshimo et al. 2003; Sato et al. 2003; Nishigaki et al. 2005). In many cases, DNA methylation changes occur during the early stages of carcinogenesis, as suggested by the presence of altered methylation patterns in preneoplastic lesions (Goelz et al. 1985; Feinberg et al. 1988; Brock et al. 2007) as well as in normal tissue adjacent to the cancer (Guo et al. 2004). These observations have suggested that altered DNA methylation might occur in the very early stages of carcinogenesis, acting as a primary transforming event (Fein-berg et al. 2006) or predisposing cells to malignant transformation (Baylin and Ohm 2006).
Despite the early onset of methylation changes, it has become clear that hypermethylation of promoters per se cannot induce silencing of the corresponding genes (Veigl et al. 1998; Bird and Wolffe 1999; Clark and Melki 2002) and that chromatinrelated events, such as altered post-translational histone modifications, chromatin remodeling, and more global changes in genome organization, cooper-ate and likely precede the DNA methylation process (Jones et al. 1998; Wade et al. 1999; Brock et al. 2007). In light of these observations, it has been proposed that DNA hypermethylation during tumorigenesis acts as a means to ensure permanent silencing of tumor suppressors, initiated by repressive chromatin configurations, thereby locking cells in an irreversible neoplastic state (Ting et al. 2006).
Chromatin structure and histone modifications have also been implicated in carcinogenesis. Several trans-acting proteins that modulate chromatin structure are misregulated or mutated in several cancers. Loss-of-function mutations in histone acetyltransferases (HATs) are found in both solid tumors and lymphomas (Gayther et al. 2000; Davis and Brackmann 2003; Gorrini et al. 2007), whereas augmented activity of histone deacetylases (HDACs), such as HDAC1, HDAC2, HDAC5, and SIRT proteins, characterizes a variety of cancers including colon cancer, prostate cancer, and leukemia (Botrugno et al. 2009). Experimental evidence for a causal role of these defects has provided a molecular rationale for targeting HDAC activity in cancers, and promising results have been obtained in clinical trials for several cancers including lymphoma, sarcoma, and ovarian cancer (Botrugno et al. 2009; Ellis et al. 2009). Similarly, aberrant expression or activity of enzymes re-sponsible for histone methylation in tumor cells has been documented (Chi et al. 2010). MLL and EZH2 are histone methyltransferases that catalyze the addition of methyl groups to histone H3 lysine 4 (H3K4) and lysine 27 (H3K27), respectively, and are involved in the maintenance of cell memory (Breiling et al. 2007). Mixed lineage leukemia (MLL) rearrangements altering enzyme activity are the most common mutations in leukemia, whereas overexpression of EZH2 is frequently observed in a variety of solid tumors (Chi et al. 2010). Furthermore, aberrant function of proteins involved in reading, rather than establishing, histone methylation patterns has been documented in various cancers. Mutations in several plant homeodomain (PHD) finger-containing proteins that bind to methylated histone H3K4 have a causal role in the development of myeloid leukemia and various types of carcinomas (Chi et al. 2010), and the heterochromatin protein HP1, which promotes and maintains heterochromatin do-mains by binding to methylated histone H3K9, acts as a metastasis suppressor in breast cancer (Dialynas et al. 2008). These observations have strengthened the notion that disrupted regulation and interpretation of histone modifications and consequent effects on higher-order chromatin organization actively contribute to human cancer.
Novel players in carcinogenesis are microRNAs (miRNAs), which are epigenetically regulated but also control epigenetic events. miRNAs comprise a class of small noncoding RNA that is involved in posttranscriptional regulation of gene expression. miRNAs act by inhibiting translation of target mRNAs, and it is estimated that one-third of protein-coding mRNAs are subjected to regulation by miRNA (Visone and Croce 2009). Misexpression of miRNAs has been implicated in cancer development, and both oncogenic and tumor-suppressor miRNAs have been identified, many of which act through inhibition of translation of proteins controlling cell proliferation, survival, and development (Visone and Croce 2009). Misexpression of miRNAs in cancer can be due to genetic alterations (Calin and Croce 2006), but epigenetic changes may also have an important role in aberrantly activating or inhibiting miRNA expression. DNA methylation at miRNA genes is detectable at high frequencies, both in normal and malignant cells (Weber et al. 2007). Studies of various types of cancer, including ovarian cancer, breast cancer, and leukemia, have uncovered how perturbation of the methylation status of miRNA genes leads to pathological up- or down-regulation of the corresponding miRNAs and the consequent misregulation of their protein-coding mRNA targets (Guil and Esteller 2009). Similar to what happens at protein-coding genes, DNA methylation at miRNA genes is usually mediated by repressive chromatin conformations and histone modifications. For example, inhibition of miR-223 in leukemia through aberrant recruitment of the corepressor SMRT, N-CoR repressors, HDACs, or DNA methyltransferase by the fusion protein AML1/ETO illustrates this paradigm (Fabbri et al. 2007). Interestingly, miRNAs may also act as upstream regulatory factors of key chromatin-modifying en-zymes (Lewis et al. 2005), thereby creating an epigenetic regulatory network that controls gene expression at multi-ple levels. Several recent reports have highlighted the central roles that particular miRNAs have as direct or indirect regulators of DNA methyltransferase activity (Fabbri et al. 2007; Benetti et al. 2008; Ting et al. 2008). Furthermore, histone methyltransferases, methyl CpG-binding proteins, chromodomain-containing proteins, and histone deacety-lases have been predicted to be potential targets of miRNA regulation by computational methods (Lewis et al. 2005). Thus, misregulation of miRNAs in cancer might have major and pleiotropic effects by inducing broad alterations of epigenetic control of gene expression.
EPIGENETIC DISRUPTION OF DIFFERENTIATION PROGRAMS IN CANCER
Epigenetic changes are a hallmark of cancer and likely contribute to disease development. Like cancer-causing genetic defects, epigenetic cancer mechanisms may cause defects in cell cycle control and survival (Brock et al. 2007). However, in addition to these traditional modes of action, epigenetic alterations may also act by an alternative, less explored mechanism, namely, the interference with cellular differentiation programs.
Indirect evidence that epigenetic changes might corrupt the differentiation state of cancer cells comes from the observation that many oncogenes, c-myc being the most no-table example, enhance cellular plasticity and interfere with cellular differentiation (Andres et al. 1988; Rapp et al. 2008). In addition to promoting cell proliferation and regulating apoptosis, c-myc is also a potent inhibitor of cell differentiation, and its action is partially mediated by histone deacetylation complexes and chromatin remodeling (Cartwright et al. 2005; Liu et al. 2007). Furthermore, genomic-wide studies have revealed that the influence of c-myc on gene expression is not limited to target promot-ers but extends to both genic and intragenic regions, suggesting that the oncoprotein may have a more complex regulatory role than a classical transcription factor, likely involving epigenetic mechanisms (Varlakhanova and Knoepfler 2009).
Genome-wide analysis of DNA methylation in colon cancer samples has also more directly linked global epigenetic changes in cancer cells and possible subversion of entire differentiation programs. An interesting correlation has been observed between hypermethylated and hypomethylated regions in colon cancer and genome regions whose methylation status varies during normal tissue differentiation (Irizarry et al. 2009). Approximately 50% of genome locations where methylation changes occur in colon cancer overlap with regions showing tissue-specific methylation patterns in normal cells. These domains are mostly found in regions adjacent to CpG islands, do not coincide with promoters, and have been named CpG island shores (Irizarry et al. 2009). Based on these observations, a model, referred to as the epigenetic progenitor model of cancer, has been proposed. This model suggests that epigenetic alterations affecting tissue-specific differentiation are the primary and predominant mechanism by which epigenetic changes contribute to cancer (Feinberg et al. 2006). Although these findings do not negate a large body of evidence supporting the importance of promoter-associated hypermethylation and hypomethylation in genes critically involved in regulation of cell proliferation and cell cycle control, they add an additional layer of com-plexity to the effect of epigenetic alterations in cancer and suggest an intimate link between altered differentiation programs and cancer. Whether the altered methylome in colon cancer is a primary cause or secondary effect of the disease is currently unclear.
In line with the observation that altered epigenetic con-trol of gene expression in cancer cells might translate into abnormal control of differentiation, gene-expression sig-natures reminiscent of embryonic stem cells (ESCs) have been found in various types of cancers (Ben-Porath et al. 2008; Wong et al. 2008), with poorly differentiated tumors showing higher similarity with ESCs than well-differentiated tumors (Ben-Porath et al. 2008). Interestingly, differentiated epithelial cells acquire such ESC-like transcriptional programs following experimental transformation, suggesting that molecular imprints of stemness may be acquired through an epigenetically based reprogramming event associated with loss of cellular identity during transformation (Wong et al. 2008).
LESSONS FROM INDUCED PLURIPOTENT STEM CELLS (IPSCS): COOPTING DIFFERENTIATION PROGRAMS TO EPIGENETICALLY REPROGRAM CELLS
Unexpected insights from reprogramming of somatic cells into ESC-like cells support the notion that loss of cell identity might be an important mechanism in tumorigenesis. Ectopic expression of four transcription factors, c-MYC, OCT4, SOX2, and KLF4, is sufficient to erase the epigenetic memory of somatic cells and to revert the differentiation process in both mice and humans (Takahashi and Yamanaka 2006; Takahashi et al. 2007). These and similar studies demonstrate that differentiated somatic cells possess great plasticity and that a few factors are sufficient to induce a global reprogramming event that resets the epigenetic constraints imposed during development. Interestingly, all reprogramming factors used to date have been previously implicated in cancer. Ectopic expression of OCT4 and SOX2 induce dysplasia and tumorigenesis (Hochedlinger et al. 2005; Chen et al. 2008) and their aberrant expression is observed in various cancers (Liu et al. 2010; Peng et al. 2010; Ugolkov et al. 2010). KLF4 is overexpressed in colorectal tumors and early-stage laryngeal squamous cell carcinoma (Foster et al. 1999) and is a marker of poor prognosis in earlystage ductal carcinoma of the breast (Pandya et al. 2004). NANOG and Lin-28, which have been used as substitutes of c-MYC and KLF4 to reprogram cells (Yu et al. 2007), have also been associated with oncogenesis (Liu et al. 2010; Viswanathan and Daley 2010). These observations suggest that experimental induction of pluripotency and tumorigenesis might entail similar pathways and that a pathological reprogramming event induced by oncogenic determinants might turn a fully differentiated cell into a cancer cell. In further support of this possibility, recent reports have indicated that inhibition of the p53 network enhances the efficiency of generation of iPSCs, directly linking disruption of tumor-suppressor mechanisms and reprogramming of somatic cells (Hong et al. 2009; Kawamura et al. 2009; Li et al. 2009; Marion et al. 2009; Utikal et al. 2009). p53-defective iPSCs are genetically unstable and either fail to generate mice or give rise to tumor-prone mice (Hong et al. 2009; Marion et al. 2009). These observations indicate that preservation of genomic integrity limits cellular reprogramming and suggest that, in the context of cancer, loss of tumor suppressors might synergize with activation of oncogenes to enhance cellular plasticity. Interestingly, disruption of the p53 net-work not only releases cellular constraints imposing the differentiated state and makes cells more competent to reprogramming, but also simplifies the process by enabling production of iPSCs with only two factors: OCT4 and SOX2 (Kawamura et al. 2009). It is therefore possible that loss of p53 actively promotes cellular reprogramming, mimicking the effect of reprogramming factors, such as NANOG and KLF4. Regardless of the mechanistic details of how p53 inhibits iPSC generation, it can be speculated that dedifferentiation and loss of cellular identity might be among the pleiotropic effects induced by the loss of this key tumor suppressor during oncogenic transformation.
The ability to generate iPSCs has generated much excitement, mainly because of its potential therapeutic applications. It may also turn out to be a valuable tool to obtain novel insights into basic biological processes. Although there is no evidence that reversion of cellular ontogeny to an embryonic stage ever occurs in an organism, it may be possible that the experimental reprogramming into ESC-like cells mimics, and possibly exaggerates, what happens in certain physiological and pathological conditions that take advantage of the intrinsic plasticity of cells (Stoll and Muller 1999; Lang et al. 2004; Owens et al. 2004). In this view, differentiation pathways that are active during development may be coopted in cancer as a consequence of oncogenic insults and, in the absence of functional tumor suppressors, may lead to loss of epigenetic memory and reversion to a dedifferentiated cell: a “pseudo” iPSC endowed with unrestricted proliferation ability but lacking full pluripotent potential. Interestingly, the process of iPSC generation is highly inefficient and most cells become trapped into partially reprogrammed states characterized by immortality but not full pluripotency (Mikkelsen et al. 2008). These cells might be similar to cancer cells. Notably, a key functional hallmark of iPSCs is the ability to form germ cell tumors when injected into immunocompromised mice (Takahashi et al. 2007). In line with the possibility that pathways and epigenetic events associated with reprogramming into iPSCs might be active in cancer, CpG is-land shores with altered DNA methylation in cancer are enriched among the regions that revert their methylation status during reprogramming into iPSCs (Doi et al. 2009). The use of controlled in vitro systems that recapitulate neo-plastic transformation might allow a more direct comparison between epigenetic mechanisms associated with tumorigenesis and those responsible for somatic-cell reprogramming.
CANCER EPIGENETICS IN THE CONTEXT OF THE CANCER STEM CELL MODEL
Most studies aimed at uncovering possible roles of epigenetic mechanisms in cancer development, particularly in solid cancers, have analyzed tumors in their entirety, disregarding intratumor heterogeneity. Tumors contain heterogeneous populations of cancer cells with distinct phenotypes and biological properties, and these differences likely have both a genetic and epigenetic basis (Heppner 1984). Cancer-related epigenetic alterations documented to date are therefore an average of multiple cellular states within a tumor (Fig. 1). Although it is obviously impossible to dissect a tumor into all of its different cellular components, a distinction between a least two subpopulations might be important: cancer stem cells and the rest of the tumor. Cancers have been traditionally regarded as populations of cells with equal proliferating activity. However, a growing body of evidence in both hematological and solid cancers suggests that only a subset of cancer cells, the cancer stem cells (CSCs), are truly immortal and sup-port cancer progression, whereas the rest of the cells have only limited proliferative capacity and do not significantly contribute to tumor growth (Reya et al. 2001; Clarke et al. 2006). In support of the CSC model, in various types of cancers only a subpopulation of cells, identifiable by markers, are able to repopulate the tumor in of all its complexity when injected into immunocompromised mice, whereas the vast majority of tumor cells lack this tumormaintaining and regeneration capability (Zhou et al. 2009). These observations have been interpreted as evidence that CSCs have the ability to self-renew and differentiate similarly to somatic stem cells, whereas their progeny lose tumorigenic potential (Reya et al. 2001; Clarke et al. 2006). According to the CSC model, tumors are therefore hierarchically organized in a similar way as normal tissues, and differences between tumorigenic CSCs and their nontumorigenic heterogeneous progeny result from a differentiation process (Fig. 1) (Reya et al. 2001). A key corollary of the CSC model, although at present unproven, is that epigenetic mechanisms that drive the differentiation process ultimately determine the tumorigenic potential of a cell (Fig. 1) (Reya et al. 2001; Shackleton et al. 2009).
Figure 1.

Epigenetic heterogeneity of cancer cells. Tumors comprise phenotypically and functionally different cell types. In some cancers, tumor growth is maintained by tumorigenic CSCs, endowed with self- renewal and differentiation abilities. Histone modifications (cylinders), DNA methylation (green circles), and higher-order chromatin structure define the epigenetic state of CSCs and contribute to the maintenance of CSC properties. The bulk of the tumor arises through differentiation of CSCs into heterogeneous cell types, characterized by various epigenetic states that activate or silence distinct sets of genes (arrows). Changes in the epigenetic landscape occur during differentiation and are associated with loss of self-renewal and tumorigenicity.
In the context of the CSC model, epigenetic alterations seen in tumors may in part be a reflection of the aberrant differentiation of CSCs. In tumors where CSCs represent only a small fraction of cancer cells, analysis of the bulk of the tumor predominantly reveals the epigenetic state of nontumorigenic cells, but not of CSCs. Epigenetic changes may have arisen as a consequence of the differentiation of CSCs and might be associated with loss of tumorigenicity. Therefore, care should be taken in assigning casual roles to abnormal patterns of DNA methylation or histone modifications observed in cancers. Direct comparison between the epigenetic states of CSCs and their derivatives will be crucial to identify the epigenetic basis of tumorigenicity and to assess what alterations are cause, primarily or secondarily, of cancer and what are simply biomarkers of the disease arising in non-CSCs. On the other hand, such studies will directly test whether tumors indeed form through a differentiation process in a hierarchical fashion and will quantify the contribution of epigenetic changes to tumor complexity. Although the phenotypic heterogeneity of the non-CSC population might complicate the analysis, if basic epigenetic differences defining tumorigenic and nontumorigenic cells exist, they will likely emerge.
Although epigenetic alterations in cancers might be the result of aberrant differentiation of CSCs, epigenetic defects are also likely to contribute to the genesis of CSCs and their evolution during disease progression. The extent and types of epigenetic alterations involved in the generation of CSCs likely depend on the nature of the cells undergoing transformation (Fig. 2). CSCs share many functional and molecular similarities with normal stem cells (Cicalese et al. 2009; Pece et al. 2010), and it is generally assumed that adult stem cells are the primary source of CSCs (Pardal et al. 2003; Wang 2010). Evidence from hematological human cancers and transgenic mice models has demonstrated that in some cancers, this is indeed the case (Fialkow et al. 1978; Passegué et al. 2004; Barker et al. 2009; Perez-Caro et al. 2009; Zhu et al. 2009). In this scenario, epigenetic mechanisms may contribute to the transformation of normal stem cells by acting locally at specific genes and mimicking the effect of genetic mutations. In particular, aberrant activation or silencing of genes controlling self-renewal and differentiation is a likely mechanism driving transformation of adult stem cells (Fig. 2). There is evidence, however, that normal stem cells are not the only source of CSCs and that progenitors can ac-quire stem-cell-like properties following mutation and act as CSCs (Jamieson et al. 2004; Krivtsov et al. 2006). A no-table example comes from MLL-driven leukemia, in which disruption of MLL expression in myeloid progenitors leads to aberrant acquisition of self-renewal ability that correlates with a reversion to an embryonic transcriptional program (Somervaille et al. 2009). The molecular mechanism responsible for this reversion has been characterized and, not surprisingly, considering the histone methyltransferase activity of MLL and its role in maintaining cell memory, involves aberrant activation of transcription and chromatin regulatory factors responsible for self-renewal in normal hematopoietic stem cells, which are epigenetically silenced in normal progenitors (Somervaille et al. 2009). Thus, following mutation, a reprogramming event can lead to the loss of epigenetic memory of committed cells and confer stem-cell-like properties (Fig. 2). Although not experimentally tested, it is conceivable that fully differentiated cells may also undergo a similar reprogramming, presumably through multiple mutagenic events and changes to their epigenetic landscape, and then act as CSCs. In this scenario, acquisition of self-renewal and differentiation abilities by somatic cells would result from dedifferentiation to a more primitive cell state induced by transformation (Fig. 2). Studies on somatic reprogramming into iPSCs support this scenario. If ectopic expression of as few as two proteins is sufficient to erase the epigenetic memory of fully differentiated cells and revert them to an embryonic stem-cell-like state, it does not appear unlikely that combinations of multiple mutations might turn a somatic cell into a CSC. In all cases in which CSCs derive from committed or differentiated cells, epigenetic alterations observed in cancer are therefore a superposition between early events mediating cellular reprogramming and later events mediating evolution of CSCs and differentiation into nontumorigenic cells.
Figure 2.
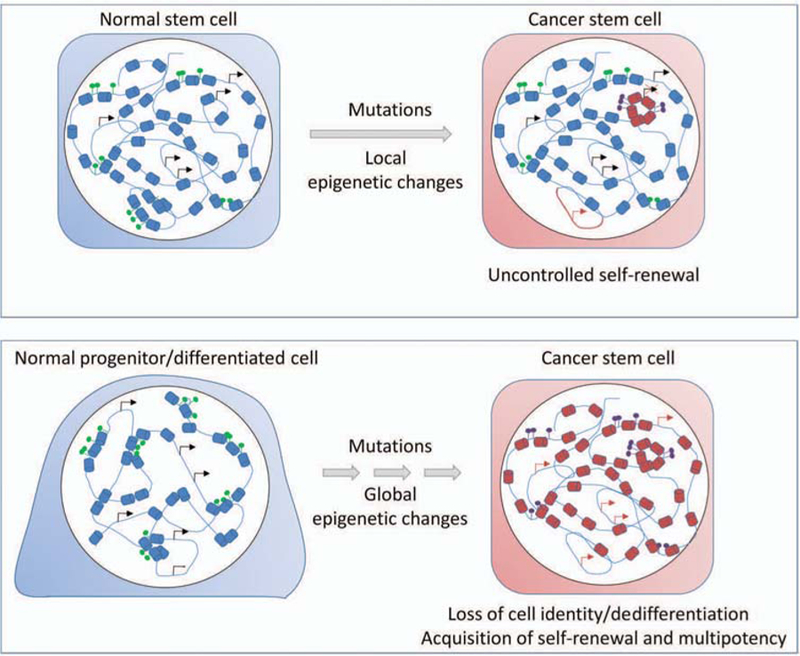
Epigenetic mechanisms of CSC formation. CSCs may arise from transformed adult stem cells or by reprogramming of com-mitted progenitors or fully differentiated cells. Epigenetic changes may contribute to the transformation of normal stem cells (upper panel) by inducing local changes in histone modifications (red cylinders), DNA methylation (purple circles), and activation or silencing of genes involved in proper control of self-renewal (arrows). Global epigenetic changes induced by oncogenic determinants may induce reprogramming of progenitors and differentiated cells (lower panel), leading to loss of epigenetic memory and cell identity. A global reorganization of the genome is associated with acquisition of self-renewal and multipotency.
CONCLUSIONS
In addition to genetic mechanisms, epigenetic defects are now recognized as key contributors to cancer. Whereas epigenetic mechanisms may trigger similar disease pathways as genetic cancer mutations, particularly affecting cellular proliferation and survival mechanisms, epigenetic alterations may also act via an entirely distinct route by com-promising cellular differentiation pathways and facilitating the emergence of tumorigenic cells. This view is in line with the notion that cancer development entails not only the loss of proliferation control, but also the loss of cellular identity and abnormal cellular differentiation during growth of the tumor mass. Loss of cellular identity by alteration of its epigenetic landscape may be induced by oncogenic mutations and facilitate the growth and spread of tumor cells by activating self-renewal pathways. Alternatively, epigenetic changes induced by extracellular cues may precede genetic abnormalities and be a primary cause of transformation. Interference with differentiation programs by epigenetic events may be particularly relevant for the emergence of CSCs because epigenetic mechanisms are largely responsible for determining stem cell status and potential.
The role of epigenetics in altering cellular identity in cancer is poorly understood at present. Key questions now are What is the interplay between genetic and epigenetic events in determining the heterogeneous cellular pheno-types of cancer cells? What is the epigenetic basis for the difference in tumorigenicity that distinguishes CSCs and the bulk of the tumor? What is the contribution of reprogramming events in cancer development? Can aberrant epigenetic mechanisms be sufficient to induce normal somatic or tissue stem cells to take on CSC properties? These are challenging topics. Major obstacles in their successful investigation are the complex nature of primary tumors and the uncertainty with regard to their origin and history of the cells that initiate and sustain tumor growth. It stands to reason that a major focus of the field should be the development of methods to manipulate tumor-causing cells, including CSCs, in controlled conditions. This will provide much-needed tools to allow the experimental exploration of epigenetic mechanisms in cancer formation and as such will facilitate the discovery of the epigenetic dimension to cancer.
REFERENCES
- Andres AC, van der Valk MA, Schonenberger CA, Fluckiger F, LeMeur M, Gerlinger P, Groner B. 1988. Ha-ras and c-myc oncogene expression interferes with morphological and func-tional differentiation of mammary epithelial cells in single and double transgenic mice. Genes Dev 2: 1486–1495. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of in-testinal cancer. Nature 457: 608–611. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Ohm JE. 2006. Epigenetic gene silencing in cancer: A mechanism for early oncogenic pathway addiction? Nat Rev Cancer 6: 107–116. [DOI] [PubMed] [Google Scholar]
- Benetti R, Gonzalo S, Jaco I, Munoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, et al. 2008. A mammalian microRNA cluster controls DNA methylation and telomere re-combination via Rbl2-dependent regulation of DNA methyl-transferases. Nat Struct Mol Biol 15: 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. 2008. An embryonic stem cell-like gene expres-sion signature in poorly differentiated aggressive human tumors. Nat Genet 40: 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. 1999. Methylation-induced repression: Belts, braces, and chromatin. Cell 99: 451–454. [DOI] [PubMed] [Google Scholar]
- Botrugno OA, Santoro F, Minucci S. 2009. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation ther-apies of cancer. Cancer Lett 280: 134–144. [DOI] [PubMed] [Google Scholar]
- Breiling A, Sessa L, Orlando V. 2007. Biology of Polycomb and Trithorax group proteins. Int Rev Cytol 258: 83–136. [DOI] [PubMed] [Google Scholar]
- Brock MV, Herman JG, Baylin SB. 2007. Cancer as a manifesta-tion of aberrant chromatin structure. Cancer J 13: 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Croce CM. 2006. MicroRNAs and chromosomal ab-normalities in cancer cells. Oncogene 25: 6202–6210. [DOI] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. 2005. LIF/STAT3 controls ES cell self-renewal and pluripo-tency by a Myc-dependent mechanism. Development 132: 885–896. [DOI] [PubMed] [Google Scholar]
- Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, et al. 2008. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem 17969–17978. [DOI] [PubMed]
- Chi P, Allis CD, Wang GG. 2010. Covalent histone modifications: Miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10: 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. 2009. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138: 1083–1095. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Melki J. 2002. DNA methylation and gene silencing in cancer: Which is the guilty party? Oncogene 21: 5380–5387. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. 2006. Cancer stem cells: Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 66: 9339–9344. [DOI] [PubMed] [Google Scholar]
- Davis PK, Brackmann RK. 2003. Chromatin remodeling and can-cer. Cancer Biol Ther 2: 22–29. [DOI] [PubMed] [Google Scholar]
- Dialynas GK, Vitalini MW, Wallrath LL. 2008. Linking Hete-rochromatin Protein 1 (HP1) to cancer progression. Mutat Res 13–20. [DOI] [PMC free article] [PubMed]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. 2009. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 41: 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis L, Atadja PW, Johnstone RW. 2009. Epigenetics in cancer: Targeting chromatin modifications. Mol Cancer Ther 8: 1409–1420. [DOI] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. 2007. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci 104: 15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. 1988. Reduced ge-nomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 48: 1159–1161. [PubMed] [Google Scholar]
- Feinberg AP, Ohlsson R, Henikoff S. 2006. The epigenetic progen-itor origin of human cancer. Nat Rev Genet 7: 21–33. [DOI] [PubMed] [Google Scholar]
- Fialkow PJ, Denman AM, Jacobson RJ, Lowenthal MN. 1978. Chronic myelocytic leukemia. Origin of some lymphocytes from leukemic stem cells. J Clin Invest 62: 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, Hayes MR, Broker TR, Chow LT, Ruppert JM. 1999. Oncogene expression cloning by retroviral transduction of ade-novirus E1A-immortalized rat kidney RK3E cells: Transforma-tion of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ 10: 423–434. [PubMed] [Google Scholar]
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, et al. 2000. Muta-tions truncating the EP300 acetylase in human cancers. Nat Genet 24: 300–303. [DOI] [PubMed] [Google Scholar]
- Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. 1985. Hy-pomethylation of DNA from benign and malignant human colon neoplasms. Science 228: 187–190. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Mar-tinato F, Sardella D, Verrecchia A, Bennett S, et al. 2007. Tip60 is a haplo insufficient tumour suppressor required for an onco-gene-induced DNA damage response. Nature 448: 1063–1067. [DOI] [PubMed] [Google Scholar]
- Guil S, Esteller M. 2009. DNA methylomes, histone codes and miR-NAs: Tying it all together. Int J Biochem Cell Biol 41: 87–95. [DOI] [PubMed] [Google Scholar]
- Guo M, House MG, Hooker C, Han Y, Heath E, Gabrielson E,Yang SC, Baylin SB, Herman JG, Brock MV. 2004. Promoter hypermethylation of resected bronchial margins: A field defect of changes? Clin Cancer Res 10: 5131–5136. [DOI] [PubMed] [Google Scholar]
- Heppner GH. 1984. Tumor heterogeneity. Cancer Res 44: 2259–2265. [PubMed] [Google Scholar]
- Herman JG, Baylin SB. 2003. Gene silencing in cancer in associ-ation with promoter hypermethylation. N Engl J Med 349: 2042–2054. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. 2005. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121: 465–477. [DOI] [PubMed] [Google Scholar]
- Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. 2009. Suppression of induced pluripo-tent stem cell generation by the p53-p21 pathway. Nature 460: 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. 2004. Granulo-cyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351: 657–667. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Lands-berger N, Strouboulis J, Wolffe AP. 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19: 187–191. [DOI] [PubMed] [Google Scholar]
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Belmonte JC. 2009. Linking the p53 tumour sup-pressor pathway to somatic cell reprogramming. Nature 460: 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. 2006. Trans-formation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442: 818–822. [DOI] [PubMed] [Google Scholar]
- Lang B, Liu HL, Liu R, Feng GD, Jiao XY, Ju G. 2004. Astrocytes in injured adult rat spinal cord may acquire the potential of neu-ral stem cells. Neuroscience 128: 775–783. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20. [DOI] [PubMed] [Google Scholar]
- Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, Blasco MA, Serrano M. 2009. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460: 1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Tee AE, Porro A, Smith SA, Dwarte T, Liu PY, Iraci N, Sekyere E, Haber M, Norris MD, et al. 2007. Activation of tis-sue transglutaminase transcription by histone deacetylase inhi-bition as a therapeutic approach for Myc oncogenesis. Proc Natl Acad Sci 104: 18682–18687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Cheng L, Du J, Peng Y, Allan RW, Wei L, Li J, Cao D. 2010. Diagnostic utility of novel stem cell markers SALL4, OCT4, NANOG, SOX2, UTF1, and TCL1 in primary mediastinal germ cell tumors. Am J Surg Pathol 34: 697–706. [DOI] [PubMed] [Google Scholar]
- Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fer-nandez-Capetillo O, Serrano M, Blasco MA. 2009. A p53-me-diated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460: 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. 2008. Dis-secting direct reprogramming through integrative genomic analysis. Nature 454: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigaki M, Aoyagi K, Danjoh I, Fukaya M, Yanagihara K, Sakamoto H, Yoshida T, Sasaki H. 2005. Discovery of aberrant expression of R-RAS by cancer-linked DNA hypomethylation in gastric cancer using microarrays. Cancer Res 65: 2115–2124. [DOI] [PubMed] [Google Scholar]
- Oshimo Y, Nakayama H, Ito R, Kitadai Y, Yoshida K, Chayama K, Yasui W. 2003. Promoter methylation of cyclin D2 gene in gas-tric carcinoma. Int J Oncol 23: 1663–1670. [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. 2004. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801. [DOI] [PubMed] [Google Scholar]
- Pandya AY, Talley LI, Frost AR, Fitzgerald TJ, Trivedi V, Chakravarthy M, Chhieng DC, Grizzle WE, Engler JA, Kronti-ras H, et al. 2004. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res 10: 2709–2719. [DOI] [PubMed] [Google Scholar]
- Pardal R, Clarke MF, Morrison SJ. 2003. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 3: 895–902. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagner EF, Weissman IL. 2004. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 119: 431–443. [DOI] [PubMed] [Google Scholar]
- Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP. 2010. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 140: 62–73. [DOI] [PubMed] [Google Scholar]
- Peng S, Maihle NJ, Huang Y. 2010. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 29: 2153–2159. [DOI] [PubMed] [Google Scholar]
- Perez-Caro M, Cobaleda C, Gonzalez-Herrero I, Vicente-Duenas C, Bermejo-Rodriguez C, Sanchez-Beato M, Orfao A, Pintado B, Flores T, Sanchez-Martin M, et al. 2009. Cancer induction by restriction of oncogene expression to the stem cell compart-ment. EMBO J 28: 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp UR, Ceteci F, Schreck R. 2008. Oncogene-induced plasticity and cancer stem cells. Cell Cycle 7: 45–51. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature 414: 105–111. [DOI] [PubMed] [Google Scholar]
- Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA, Rosty C, Goggins M. 2003. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res 63: 4158–4166. [PubMed] [Google Scholar]
- Shackleton M, Quintana E, Fearon ER, Morrison SJ. 2009. Het-erogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 138: 822–829. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM, Chang HY, Shurtleff SA, Downing JR, Cleary ML. 2009. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 4: 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Muller HW. 1999. Nerve injury, axonal degeneration and neural regeneration: Basic insights. Brain Pathol 9: 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by de-fined factors. Cell 126: 663–676. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872. [DOI] [PubMed] [Google Scholar]
- Ting AH, McGarvey KM, Baylin SB. 2006. The cancer epigenome: Components and functional correlates. Genes Dev 20: 3215–3231. [DOI] [PubMed] [Google Scholar]
- Ting AH, Suzuki H, Cope L, Schuebel KE, Lee BH, Toyota M, Imai K, Shinomura Y, Tokino T, Baylin SB. 2008. A requirement for DICER to maintain full promoter CpG island hypermethy-lation in human cancer cells. Cancer Res 68: 2570–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolkov AV, Eisengart LJ, Luan C, Yang XJ. 2010. Expression analysis of putative stem cell markers in human benign and ma-lignant prostate. Prostate (in press). [DOI] [PubMed]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K. 2009. Immor-talization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460: 1145–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlakhanova NV, Knoepfler PS. 2009. Acting locally and globally: Myc’s ever-expanding roles on chromatin. Cancer Res 69: 7487–7490. [DOI] [PubMed] [Google Scholar]
- Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, et al. 1998. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci 95: 8698–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visone R, Croce CM. 2009. MiRNAs and cancer. Am J Pathol 1131–1138. [DOI] [PMC free article] [PubMed]
- Viswanathan SR, Daley GQ. 2010. Lin28: A microRNA regulator with a macro role. Cell 140: 445–449. [DOI] [PubMed] [Google Scholar]
- Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. 1999. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet 23: 62–66. [DOI] [PubMed] [Google Scholar]
- Wang JC. 2010. Good cells gone bad: The cellular origins of cancer. Trends Mol Med 16: 145–151. [DOI] [PubMed] [Google Scholar]
- Weber B, Stresemann C, Brueckner B, Lyko F. 2007. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 6: 1001–1005. [DOI] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. 2008. Module map of stem cell genes guides creation of epithe-lial cancer stem cells. Cell Stem Cell 2: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. 2009. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov 8: 806–823. [DOI] [PubMed] [Google Scholar]
- Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. 2009. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 457: 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]