

Abstract
Sensitive glycomics analysis by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) is of great importance but significantly hampered by their low ionization efficiency and labile sialic acid moieties. Chemical derivatization offers a viable way to improve both the ionization efficiency and analytical sensitivity of the glycans in MS analysis by altering their hydrophobicity or charge property. Here we employed Girard's reagent T (GT) for on-target derivatization (GTOD) of reducing glycan under mild acid condition to form stable hydrazones at room temperature, allowing rapid and sensitive identification of neutral and sialylated glycans in positive-ion mode as only permanently positive charged molecular ions without multiple ion adducts by MALDI-TOF-MS. The MS signal intensities of lactose, sialylated N-glycans derived from bovine fetuin and neutral N-glycans derived from RNaseB and ovalbumin were boosted by 7.44, 9.13, 12.96 and 13.47 folds on average (n = 3), respectively. More importantly, after GTOD strategy, unwanted desialylation of sialylated glycans during MS was suppressed. The detection limit of the assay is desirable since the nanogram of N-glycans derived from 0.16 μg ovalbumin could be detected. The assay demonstrated good stability (RSD≤2.95%, within 10 days), reliable reproducibility (RSD = 2.96%, n = 7) and a desirable linear dynamic range from 78 nmol/mL to 10 μmol/mL. The strategy has been successfully applied to MS analysis of reducing glycans from human milks, neutral and sialylated O-, N-glycans from glycoproteins, and reducing glycans derived from glycosphingolipids, presenting neater [M]+ signals which allow detection of more low-abundance glycans and assignation of Neu5Ac vs. Neu5Gc or fucose vs. hexose in glycans due to the absence of the ambiguous interpretation from multiple peaks (ion adducts [M+Na]+ and [M+K]+). Moreover, the GTOD assay prevents desialylation during MALDI-TOF-MS profiling and enables distinct linkage-specific characterization of terminal sialic acids of N-glycans derived from human serum protein when combines with an esterification.
Keywords: Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS), Reducing glycan, Derivatization, Girard's reagent T (GT), Sialic acids, Hydrazide
GRAPHICAL ABSTRACT
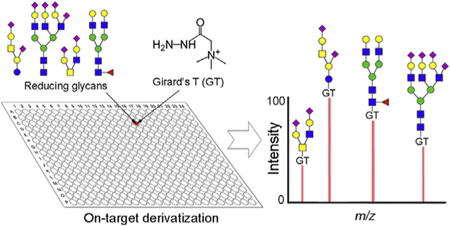
1. Introduction
Glycans including free reducing glycans or constituents of glycoproteins, glycolipids and proteoglycans, play fundamental roles in many biological processes including extracellular or intracellular interaction [1], communication [2], signaling [3], receptor interaction [4–6] and disease development [7–10]. Glycomics is the systematic study of glycan structures, including monosaccharide compositions, glycan sequence, branching pattern and linkage specifications, which are essential to the functions of glycoconjugates [11–14]. Nevertheless, glycomics analysis is inherently challenging due to their structure complexity and heterogeneity, low natural abundance and the lack of amplification methods [5,14,15].
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) has become widely used for high throughput profiling of glycans with high heterogeneity and low abundance [14,16–21]. MALDI-TOF-MS generates dominantly singly charged ions and detects a large mass range. Sample preparation is simple and high throughput analysis can be carried out easily and quickly [14,22,23]. However, MALDI-MS analysis of underivatized glycans is limited by their low ionization efficiency.
For purpose of improving MS detection sensitivity, many chemical derivatizations of free glycans have been developed to improve the detection sensitivity during MS analysis [24–26], including widely used permethylation [27], reductive amination with various type of hydrophobic aromatic amines [26], Michael's addiction with 1-phenyl-3-methyl-5-pyrazolone (PMP) [28] and acid catalyzed condensation with hydrazides or hydroxylamines to form hydrazones or oximes [29–32]. These derivatizations are usually applied to significant amounts of samples (μg to mg), often under relatively strong reaction conditions involving heating with acid or base. Lengthy and tedious clean-up procedures are often required to remove the excess labeling reagents.
To overcome these limitations, derivatizations of reducing sugar with suitable tags directly on target plate has been exploited to simplify and increase the sensitivity of MALDI-MS analysis of glycans. 3-Aminoquinoline/p-coumaric acid [33] and phenylhydrazine [34–36] have been employed to on-target derivatization of the reducing glycans through acid catalyzed condensation reaction. However, the consistency of these derivatizations of various samples for MALDI-MS analysis has not been well-established, possibly due to the instability of Schiff-base formation. Furthermore, sialic acids are functionally important terminal components of many glycans but a significant portion of sialic acids falls off during MALDI-MS analysis [37]. Many derivatizations including methylation [38] and amidation [39] have been developed to stabilize sialic acids but none of these methods are compatible with on-target derivatization. Therefore, developing a robust on-target derivatization of reducing glycans including neutral and particularly sialylated glycans for highly-sensitive MALDI-TOF-MS analysis is greatly needed.
Positively charged reagents including Girard's reagent T/P (GT, GP) and other cationic tags [40,41] have been employed as MS detection enhancers to analyze glycan after acid catalyzed non-reductive condensation [42–44]. The yields are high and only [M]+ ions were observed in MS analysis, avoiding multiple ion adducts such as the [M+Na]+, [M+NH4]+ and [M+K]+, which simplifies the MS data interpretation and increases the detection sensitivity [40]. Moreover, the positively charged tags can promote the ionization efficiency of the tagged glycans [42,44], and thus further boost the detection sensitivity of glycans in MS analysis. However, these derivatizations were carried out independently from MS analysis using μg even mg amount of glycan samples, often with undesired loss of labile sialic acid due to heating [43]. Nevertheless, many reports suggest that hydrazides react with glycan reducing end quickly under room temperature or even at 4 °C [45–47]. We therefore explored Girard's reagent T (GT) for on-target derivatization (GTOD) under neutral or mild acid conditions at room temperature to facilitate MALDI-TOF-MS analysis neutral and sialylated glycans in positive-ion mode, as shown in Scheme 1.
Scheme 1.

The strategy for GT on-target derivatization (GTOD) of reducing glycans and subsequent positive-mode MALDI-TOF-MS analysis. (A) The chemical reaction of free reducing glycans with GT form stable hydrazones under room temperature. (B) The schematic presentation of GTOD.
In our GTOD strategy, glycans, GT and matrix (DHB) were sequentially spotted on the MALDI target and gently mixed. The spotted glycans were efficiently derivatized on-target with GT in the presence of DHB matrix under room temperature over normal drying time. After drying, MALDI-MS analysis of the derivatized glycans are directly carried out. Only [M]+ type molecular ions were observed, greatly improving spectral quality and subsequent MS data analysis. This strategy has been applied to a variety of neutral and sialylated glycans including those released from glycoproteins and glycosphingolipids (GSLs) and proved to be an effective and reproducible procedure for highly sensitive glycomics analysis by MALDI-TOF-MS at micro-scale level. Additionally, when combined with an esterification [48], the assay can be used to characterize linkage-specificity of glycan terminal sialic acids by MALDI-TOF-MS.
2. Experimental section
2.1. Chemicals
Girard's reagent T (GT), phenylhydrazine, 2,5-dihydroxybenzoic acid (DHB), lactose (Lac), ribonuclease B (RNase B), ovalbumin (chicken egg white albumin), bovine fetuin, 3’-sialyllactose (3’-SL, α2,3-linked sialic acid), 6’-sialyllactose (6’-SL, α2,6-linked sialic acid), 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC) hydrochloride 1-hydroxybenzotriazole (HOBt) hydrate, DL-dithiothreitol (DTT), sodium dodecyl sulfate (SDS), β-cyclodextrin and peptide-N-glycosidase F (PNGase F; supplied glycerol free) were obtained from Sigma (St. Louis, MO, USA). HPLC-grade solvents (acetonitrile, ethanol) were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Water was obtained through a Milli-Q water purification system (Millipore, Bedford, MA, USA). Nonporous graphitized carbon (150 mg/4 mL) solid phase extraction (SPE) columns were obtained from Alltech Associates (Deerfield, IL, USA). Sep-Pak C18 (3 cc) solid phase extraction (SPE) cartridges were obtained from Water Corporation (Milford, Massachusetts, USA). Human serum was obtained from a healthy volunteer in Xi'an Jiaotong Hospital. The research investigations were performed after obtained the informed consent and in accordance with an institutional review board protocol approved by the Human Research Ethic Committee at the Jiaotong Hospital. All other chemicals were purchased from Aladdin Inc. (Shanghai, China).
2.2. Preparation of free oligosaccharides from human milks
This procedure was conducted according to a previously reported paper [49]. Human milk was from a healthy volunteer within 20 days after giving birth. 0.5 mL of milk was centrifuged at 4 °C and 13000 rpm for 30 min. The clear solution in the middle layer containing oligosaccharides was collected and loaded onto a graphitized carbon SPE column pre-washed with 5 mL of acetonitrile (ACN) and equilibrated with 5 mL of water. After a wash of the graphitized carbon column with 10 mL of water to remove salts, glycans were eluted with 2 mL of 25% ACN containing 0.1% tri-fluoroacetic acid (TFA) [50]. The elutes were dried under a stream of nitrogen gas and dissolved in 50 μL water for further derivatization or analysis by MS.
2.3. Preparation of reducing N-glycans from glycoprotein by PNGase F digestion
This procedure was based on previously reported papers [42]. 100 μL human serum or human milk was dialyzed in an 8000-12,000 KDa dialysis membrane against fresh double distilled water for 3 days at 4 °C. The dialysate was dried to afford protein using a vacuum evaporator. 100-500 μg of protein sample was dissolved in 200 μL of protein denaturation solution and denatured. After the sample was cooled down, the denatured sample was subjected to the PNGase F (10 μL, 30-60 U/mL) digestion at 37 °C for 24 h. Then sample was subjected to a glycan purification process using C18 SPE cartridge combined with graphitized carbon SPE column [50]. Briefly, the sample solution was loaded onto a C18 cartridge pre-washed with 5 mL of ACN and equilibrated with 5 mL of water. The target glycans were eluted with 4 mL of water, and the eluted residue was collected. Then the elutes were loaded onto a graphitized carbon SPE column and purified as described above. The purified glycans were dried under a stream of nitrogen gas and dissolved in 50 μL of water for further use.
2.4. Preparation of reducing O-glycans from glycoproteins
This procedure was operated according to reference [51]. Briefly, 500 μg porcine stomach mucin (PSM) was incubated in 500 μL of 26–28% ammonium hydroxide at 45 °C for 24 h. After reaction was completed, the mixture was purified by C18 SPE cartridge combined with graphitized carbon SPE column as described in section 2.2 and 2.3. The purified glycans were dried and dissolved in 50 μL of water for further use.
2.5. Preparation of GSLs-glycan from glycosphingolipids (GSLs)
The GSLs-glycan were released from glycosphingolipids according to the procedure reported in Ref. [52]. Briefly, 100 μg of glycosphingolipids were treated with ozone in 2 mL chloroform/methanol (1:1) about 3 min in a white tube until blue color occurs. To this solution, 1/3 volume of 100 mM ammonium bicarbonate buffer (pH = 8.0) was added and the mixture is heated at 65 °C for 16 h without a cap. The resulting mixture was lyophilized and purified by C18 and graphitized carbon SPE columns as described in section 2.2 and 2.3. The purified glycans were dried and dissolved in 50 μL of water for further use.
2.6. GT on-target derivatization (GTOD) of reducing glycans
Glycans were dissolved in double distilled water. DHB matrix solution was prepared by dissolving 20 mg of DHB in 1 mL of 1:1 (v/v) acetonitrile/0.1% TFA mixture. GT was dissolved in a solvent of water/methanol/glacial acetic acid mixture (6:1:3, v/v/v) at 0.05 M as the glycan-labeling solution. Briefly, 0.5 μL DHB matrix solution, 0.5 μL glycan solution and 0.5 μLGT solution were deposited sequentially and gently mixed on MALDI target. After the deposited sample was naturally dried at room temperature, the on-target labeling reaction is completed. Then the target is directly inserted for MALDI-TOF-MS analysis. Linkage specific derivatization of sialylated glycans was achieved by one-pot on-target derivatization with GT combined with a linkage specific derivatization via the ethyl esterification [48]. EDC and HOBt, respectively, were dissolved in ethanol at 0.25 M as the esterification buffer. Then 0.5 μL DHB mixture, 0.5 μL glycans liquor, 0.5 μL esterification buffer and 0.5 μL GT glycans-labeling solution were sequentially deposited and gently mixed on MALDI target. After the spotted mixture was dried at room temperature, the labeled glycans could be submitted to linkage-specific characterization of glycans terminal sialic acids by MALDI-TOF-MS.
2.7. MALDI-TOF-MS conditions
MS signal was obtained on an AXIMA instrument (Shimadzu, Tokyo, Japan) according to the conditions described previously [23,45]. The matrix was DHB (20 mg/mL, in 50% ACN containing 0.01% TFA). The sample in water was loaded onto target with GT labeling buffer and DHB matrix solution. The target was directly subjected into MALDI-TOF-MS profiling after drying in ambient air. The MS data acquisition was carried out in positive-ion polarities, using reflectron time-of-flight (TOF) mode. The instrument was equipped with a 337 nm nitrogen laser capable of operating at repetition rates of 5 Hz and the 90-110 mV power level. The acceleration potential was set to be 20 kV. All data was acquired in the low-mass range (m/z 200-4000). For each sample, 200 laser shots per point were irradiated and acquired. For the post-source decay (PSD) experiments, the PSD fragmentation conditions were adjusted in reflectron positive-ion mode with ion accelerating voltage of 20 kV to get the enhanced intensity and (signal/noise) S/N ratios of ionized peaks in spectra.
3. Results and discussions
3.1. GTOD improves detection sensitivity of reducing glycans in MALDI-TOF-MS analysis
We first tested GTOD on reducing glycans under room temperature using maltodextrin (glucose polymers), followed by direct MALDI-TOF-MS analysis. As shown in Fig. 1, after GTOD, reducing glycan such as G12 was labeled with GT through oximation to form corresponding hydrazone product G12-GT, leading to mass increments of m/z 114 addition per sugar observed in MS. All maltodextrin oligosaccharides with different DPs (degree of polymerizations) were efficiently derivatized and only [M]+ type molecular ion peaks with mass increments of m/z 114 addition were observed in MS profiles (Fig. 1B) comparatively to the underivatized group (Fig. 1A, mainly [M+Na]+ peaks), correspondingly. Remarkable increase in S/N (signal/noise) ratios of [M]+ peaks was observed compared to [M+Na]+ and [M+K]+ peaks of underivatized sample (Fig. 1A). Significantly higher DP oligosaccharides can be detected after GTOD than those without derivatization.
Fig. 1.

MALDI-TOF-MS spectra of maltodextrins without (A) or with (B) GTOD treatment. Both spectra were acquired under the same concentration and MS conditions. (DP: degree of polymerization; G1: saccharide with DP of 1; G1-GT: G1 labeled with GT).
3.2. Effect of molar ratios of GT to glycans on reaction efficiency of GTOD by MALDI-TOF-MS
To determine the amount of labeling reagent required for efficient derivatization, we applied GTOD on lactose (Lac) and 3’-sialyllactose (3’-SL) using different molar ratios of GT/glycan (1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1 9:1 and 10:1) and measured the increase of MS peak intensity after derivation (Fig. S1). β-Cyclodextrin, which is a non-reducing sugar and can not be derivatized, was used as an internal standard for relative quantification. It is shown that a molar ratio of GT to glycans larger than 7:1 is sufficient to achieve significant amplification of MS peak intensities. While it is impossible to determine concentrations of each glycan sample, the universal GTOD protocol described in experimental section provides sufficient amount of GT for derivatizations.
3.3. GTOD suppresses desialylation of sialylated glycans in MALDI-TOF-MS analysis
Fig. 2 shows MALDI-MS profiles of Lac (Fig. 2A) and sialylated N-glycans released from bovine fetuin (Fig. 2B) before and after GTOD at the same concentration. MS profiles annotated with [M]+ ion peaks after GTOD show obvious increase in the S/N ratios and detection sensitivity, especially when compared to the β-cyclodextrin internal standard. After GTOD labeling, S/N ratios of lactose-GT conjugates ionized at m/z 456 (S/N = 69, Fig. 2A, lower layer) as well as one random H5N4S2-GT conjugates ionized at m/z 2336 (S/N = 49, Fig. 2B, lower layer) were enhanced about 11 and 10 folds over those of native glycans (Fig. 2A, upper layer, lactose, m/z 365, S/N = 6; Fig. 2B, upper layer, H5N4S2, m/z 2245, S/N = 5), respectively (H, hexose; N, N-acetylhexosamine; S, Neu5Ac). The formation of [M]+ ions and the suppression of other metal ion adduct helps preventing possible misinterpretation from multiple metal molecular ion peaks and improving the detection sensitivity and S/N ratios, consistent with our previous work [42]. Moreover, before GTOD, peak at m/z 1662.93 was assigned to desialylated product of the sialylated glycans (m/z 1954.31) during ionization. However, after GTOD, no corresponding desialylation peak was observed, suggesting that GTOD stabilize sialylated glycans and prevent desialylation during MALDI-MS analysis (Fig. 2B). Although detailed mechanism is not fully understood, the stabilization of sialylated glycans after GTOD is likely due to the neutralization of negative charge, the enhancement of ionization efficiency and the increase of ion abundance by cationic GT tag [53–58].
Fig. 2.

Positive-ion mode spectra of lactose (A) and sialylated N-glycans released from bovine fetuin (B) before and after GTOD by MALDI-TOF-MS. Lac, lactose; Structure formulas: green circle, mannose; blue square, N-acetylglucosamine; yellow circle, galactose; purple diamond, N-acetylneuraminic acid (Neu5Ac). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Evaluation of GTOD assay for reducing glycans MALDI-TOF-MS analysis
Compared with the underivatized reducing glycans, the MS signal intensities after GTOD of four groups of glycan samples (lactose; sialylated N-glycans derived from bovine fetuin; neutral N-glycans derived from RNaseB and ovalbumin, respectively) in Fig. 2 and S2-3 were boosted by 7.44, 9.13, 12.96 and 13.47 folds on average (n = 3, Fig. S4), based on quantification using β-cyclodextrin peaks as an internal standard. Importantly, desialylation of sialylated glycans during MALDI-TOF-MS was suppressed and greater MS signals were observed (Fig. 2B and S2-3), presumably due to the permanent positive charge promoting more efficient ionization and ion abundance [53–58].
To assess the minimal detection limitation using GTOD strategy, equal amounts of N-glycans released from chicken ovalbumin was treated without or with GTOD and analyzed by MALDI-TOF-MS in parallel. As shown in Fig. S5, the S/N ratios of native glycans as well as glycan-GT [M]+ peaks decreased with decreasing amount of original glycoprotein used for glycan release. Equal amount of N-glycans after GTOD derivatization demonstrated higher and neater MS signals over that of the underivatized ones, correspondingly. Major N-glycans derived from as little as 0.16 μg ovalbumin can be successfully detected after GTOD, whereas major peaks in same amount of N-glycan before GTOD could hardly be monitored. Additionally, a total of 17 N-glycans were detected before GTOD (Fig. S5A, upper layer). In contrast, 20 N-glycans identified with evidently improved signal intensity were achieved after GTOD (Fig. S5B, upper layer) when equal amount of sample used under same experimental conditions. This result suggests that GTOD greatly enhance glycan detection sensitivity, which is of great importance for highly sensitive identification of glycans derived from microscale biological specimen.
When different concentrations of lactose (0.078, 0.156, 0.312, 0.625, 1.25, 2.5, 5.0, 8.0 and 10.0 μM/mL) were treated with GTOD and analyzed by MALDI-TOF-MS, a linear concentration dependence of MS ion relative abundance normalized based on β-cyclodextrin internal standard was observed (Fig. S6, y = 1.4687x+0.2215, R2 = 0.9992), suggesting the potential of GTOD for quantitative or semi-quantitative analysis of glycans which don't require high quantification accuracy. When the analysis is repeated multiple times, the relative abundance is highly reproducible (Table S1, RSD = 2.96%, n = 7), suggesting a robust derivatization by GTOD. Furthermore, when GTOD treated lactose was stored at room temperature for a period of time (1, 2, 3, 4, 5, 7, 10 and 14 d) and then submitted to MALDI-MS analysis, no significant deterioration of MS peaks was observed, suggesting the glycan-GT derivatives are rather stable (RSD≥2.95% within 10 days, n = 3) after GTOD (Table S2).
We also compared the GTOD approach with a previous method described by Erika et al. [34], which employed a hydrazide reagent phenylhydrazine (PHN) for on-target derivatization of reducing glycans to improve MS analysis of reducing sugars and simplify the MALDI-MS procedure (Fig. 3). While both GT and PHN on-target derivatization improved MS spectra quality over un-derivatized glycans, in our hands, the GTOD approach provides stronger signals and simpler MS profile than PHN on-target derivatization, presumably due to the permanent positive charge installed.
Fig. 3.

Positive-ion spectra of N-glycans released from ribonuclease B (RNase B) before (A) or after on-target derivatization with phenylhydrazine (PHN) (B) or after GTOD (C) by MALDI-TOF-MS at the same concentration and MS conditions.
3.5. Application of GTOD to a variety of reducing glycans derived from biological specimens
We then applied the GTOD approach to a variety of glycans from biological specimens, including free oligosaccharides, N-, O-glycans released from glycoproteins and glycans derived from GSLs. Released and purified reducing glycans were processed with or without GTOD and analyzed by MALDI-TOF-MS (Fig. 4 and S7). All MS profiles generated with GTOD show greatly enhanced mass signals and neater pattern than MS profiles without GTOD, resulting more glycan compositions detected. In GTOD-MALDI-MS profiles of human milk N-glycans (Fig. 4A) and free oligosaccharides (Fig. 4B), porcine brain (Fig. 4C) and liver (Fig. 4D) GSLs-glycans, 37, 19, 13 and 10 different compositions were identified, whereas only 29, 11, 7 and 9 compositions were identified in the MS profiles without GTOD treatment. The compositions assigned based on MS data are listed in Tables S4–8, which are consistent with previous reports [14,52,59,60]. The compositions identified only with GTOD (labeled with blue characters) were all assigned to low-abundance glycans. This brings significant advantage in the analysis of minor components of a glycome, which are often overlooked due to less sensitive MS detection.
Fig. 4.

Positive-ion MS profiles of a variety of reducing glycans derived from biological specimens with or without GTOD at the same concentration. (A) MS profiles of N-glycans released from glycoproteins in human milk before and after GTOD; (B) MS profiles of free oligosaccharides from human milk before and after GTOD; (C) MS profiles of GSLs-glycan released from porcine brain glycosphingolipids before and after GTOD; (D) MS profiles of GSLs-glycan released from porcine liver glycosphingolipids before and after GTOD. Abbreviations used are hexose (H), N-acetylhexosamine (N), fucose (F), Neu5Ac (S) and Neu5Gc (G) with unspecified linkage. * indicates the presence of [M+K]+ ion peak. Blue characters numbered peaks were low abundance glycans observed after GTOD only. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Interestingly and importantly, many of these compositions identified only with GTOD are sialylated glycans, suggesting that sialylation is significantly stabilized by the positive charge introduced by GTOD. It is well known that sialylated glycans are prone to desialylation during MALDI-TOF-MS analysis and often need to be chemically stabilized before MS analysis [23,44,48,61]. The stabilization of sialic acid moieties by GTOD provides a one-pot procedure for MALDI-TOF-MS analysis of sialylated glycans. Furthermore, the 16 Da mass difference between Neu5Ac and Neu5Gc cannot be discriminated from the 16 Da mass difference between [M+Na]+ and [M+K]+ adducts under normal resolution, which often leads to misinterpretation of Neu5Gc. Since GTOD-MALDI-MS only exhibits the [M]+ ions without the interference by [M+Na]+ and [M+K]+ ions, it provides a simple and unique solution for the discrimination of Neu5Ac (S) and Neu5Gc (G), as shown in Fig. 4D. Peaks signals (Fig. 4D) at m/z 1112.12 and 1128.23 of in GTOD-MALDI-MS profile were unambiguously assigned to H3N1S1 and H3N1G1 (H, hexose; N, N-acetylhexosamine; F, fucose). Similarly, peaks signals at m/z 1403.23, 1419.32 and 1435.12 (Fig. 4D) were assigned to H3N1S2, H3N1S1G1 and H3N1G2. These compositions were confirmed by MS/MS analysis in Figs. S8–9.
Besides discriminating Neu5Ac and Neu5Gc, the exhibition of only [M]+ ions by GTOD also helps discrimination of other compositions that can potentially be confused by [M+Na]+ and [M+K]+ in MS profiles of underivatized glycans, such as fucose vs. hexose. For instance, as shown in Fig. 4A, three groups of peaks of m/z 1576.74 and 1593.04, 1738.29 and 1754.88, 1779.87 and 1795.19 with m/z difference of 16 were assigned to six compositions as N4H3F1 and N4H4, N4H4F1 and N4H5, N5H3F1 and N5H4 definitively, as presented in Table S4, whereas in the profile of underivatized glycans, three compositions (N4H3F1, N4H4F1 and N5H3F1) as [M+Na]+ and [M+K]+ adducts could be wrongly assigned. The same phenomena was also observed in Fig. 4D and Table S7, where m/z 1258.23 and 1274.47 with difference (ΔDa = 16) were undoubtedly assigned to be two compositions H3N1F1S1 and H4N1S1. Whereas, in the MS profile of underivatized glycans, corresponding m/z 1167.08 and 1183.09 (ΔDa = 16) could be mis-interpreted as a single composition H3N1F1S1 as [M+Na]+ and [M+K]+ adducts.
3.6. Linkage-specific analysis of glycan terminal sialic acids using GTODE
Due to the pathological importance of the glycan terminal sialic acids, it is of great necessity to develop high-throughput method for their linkage-specific analysis. While sialylated glycans are stabilized by GTOD and can be successfully profiled by MALDI-MS, it is very challenging to assign specific linkages by MS/MS due to quick desialylation, as illustrated in Fig. S10. Fortunately, a linkage-specific assignation of terminal sialic acid could be achieved by combining our GTOD strategy with a mild esterification [48], termed as GTODE which converts α2,6-linked sialic acid to ethyl ester and α2,3-linked sialic acid to lactone under mild room temperature reaction conditions.
As shown in Fig. 5A and B, after GTOD, both 3’-SL and 6’-SL presented only [M]+ peak at m/z 747 without desialylations (Fig. 5A and B, middle panels) whereas desialylations are abundant without GTOD (Fig. 5A and B, upper panels). While this is significant in preventing desialylation, 3’-SL and 6’-SL cannot be discriminated. However, when GTOD was combined with esterification (GTODE) (Fig. 5A and B, lower panels), distinct [M]+ ions at m/z 729 and 775 were produced from 3’-SL and 6’-SL respectively, suggesting lactone formation for 3’-SL-GT with m/z reduction (−ΔDa = 18) and ethyl ester formation for 6’-SL-GT with m/z addition (+ΔDa = 28). These features strongly suggest the potential of using GTODE for distinguishing glycan terminal α2,3-linked sialic acid from α2,6-linked sialic acid. We therefore applied GTODE to sialic acid linkage specification analysis of N-glycans released from human serum glycoprotein (HSG).
Fig. 5.

Positive-ion MS profiles of glycans before and after GTOD or GTODE. (A) MS profiles of 3’-SL before, after GTOD or GTODE; (B) MS profiles of 6’-SL before, after GTOD or GTODE; (C) MS profiles of N-glycan derived from human serum glycoproteins before, after GTOD or GTODE. 3’-sialyllactose (3’-SL), 6’-sialyllactose (6’-SL). HSG N-glycans, human serum glycoprotein N-glycans; Structure formulas: yellow square, N-acetylgalactosamine; blue circle, glucose; yellow circle, galactose; red triangle, fucose; purple diamond, sialic acid (Neu5Ac). * means the [M+K]+ or [M+2Na]+ ionized peaks. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
As presented in Fig. 5C, when treated with GTOD (middle panel) and GTODE (lower panel), cleaner and stronger MALDI-MS profiles and signals of HSG N-glycans were obtained in contrast to that without treatment (upper panel) when detected at the same concentration and MS conditions. Compared with the GTOD-MALDI-MS profile of HSG N-glycans (Fig. 5C, middle panel), m/z values of neutral N-glycans in the GTODE-MALDI-MS profile (Fig. 5C, lower panel) did not change, such as peaks at m/z 1348, 1511, 1576, 1738 and 1901. However, an α2,3-linked sialic acid terminal of N-glycans would yield an 18 Da mass loss and an α2,6-linked sialic acid terminals would yield a 28 Da mass increase. Therefore, the mass changes from sialylated peaks in GTOD-MALDI-MS profile to sialylated peaks in GTODE-MALDI-MS profile can be calculated as n2×28-n1×18, where n1 is the number of α2,3-linked sialic acid and n2 is the number of α2,6-linked sialic acid. For example, peaks at m/z 2045 and 2191 in GTOD-MALDI-MS profile were shifted to m/z 2072 and 2219 in GTODE-MALDI-MS profile, suggesting that these two peaks are α2,6-sialylated glycans. Similarly, all other peaks, including di and tri-sialylated glycans, in the GTODE-MALDI-MS profile can be compared to the GTOD-MALDI-MS profile to provide important information about the terminal sialic acid linkages. Through GTOD strategy, sialic acids residues at HSG N-glycans were stabilized but the linkages specificity could not be determined. After GTODE, not only the sialic acids residues at HSG N-glycans were further stabilized but also the linkage specificity could be well characterized. From HSG N-glycans, we were able to assign specific linkages to 14 glycans with terminal sialic acids moieties within the 1000-3500 Da mass detection range, consistent with data reported in previous works [48,62–64]. Therefor, GTODE allows a one-step profiling of a mixture of glycans and the linkage specific characterization α2,3- and α2,6-linked terminal sialic acids with high fidelity, which will be suitable for high-throughput and robust glycomics analysis of reducing glycans derived from diverse biological specimens.
4. Conclusions
We have developed a novel approach termed GTOD for on-target derivatization and highly sensitive analysis of neutral and sialylated reducing glycans by MALDI-TOF-MS in positive-ion mode. Through the introduction of Girard's reagent T with a permanent positive charge, the MALDI-MS spectra resolution and peak intensities are greatly improved. Unwanted desialylation during MALDI-MS analysis was significantly suppressed. Importantly, the GTOD-MALDI-MS profiles presents only [M]+ peak without interference from other ion adducts such as [M+H]+, [M+Na]+ and [M+K]+. This results in much cleaner mass spectra and greatly simplified m/z assignment and data interpretation. Specifically, peaks with 16 Da difference, which are often assigned to [M+Na]+ and [M+K]+ adducts, can now be unambiguously assigned, such as glycans with Neu5Ac or Neu5Gc sialylation and fucosylation.
Moreover, the GTOD assay prevents desialylation during MALDI-TOF-MS profiling, allowing for distinct linkage-specific characterization of terminal sialic acids of glycans. When the GTOD approach is combined with esterification, termed GTODE, linkage specific characterization of terminal sialic acids with modifications of α2,3- and/or α2,6-linkages was achieved based on the clean [M]+ profile and distinct sialic acid esterification patterns of different linkages. The sialic acids linkage specificities of N-glycans released from glycoprotein in human serum have been successfully determined.
In summary, the GTOD and GTODE methods are simple, fast, robust and highly reproducible. With all the merits described above, we believe these methods will make significant contribution to glycomics.
Supplementary Material
HIGHLIGHTS.
GTOD greatly increase MALDI-MS peak intensities of free reducing glycans.
Only [M]+ peaks were observed without interference from diverse ion adducts.
GTOD greatly suppresses unwanted desialylation of glycans during MALDI-MS analysis.
Neu5Ac vs. Neu5Gc or fucose vs. hexose in glycans can be assigned unambiguously.
Terminal sialic acids with α2,3- and α2,6-linkages could be assigned through GTODE.
Acknowledgments
This work was funded by National Natural Science Foundation (Nos. 31370804, 21375103, 31670808 and 31300678), the Scientific Research Program Fund for Shannxi Province key laboratory (14JS101) and Shaanxi basic research program of natural science (2016JQ3018). This work was also partially supported by NIH Common Fund Glycoscience (U01GM116254).
Footnotes
Notes
The authors declare no competing financial interest.
Appendix B. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.aca.2018.10.015.
References
- [1].Singh S, Aggarwal A, Bhupathiraju N, Arianna G, Tiwari K, Drain CM, Glycosylated porphyrins, phthalocyanines, and other porphyrinoids for diagnostics and therapeutics, Chem. Rev 115 (2015) 10261–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rao X, Duan X, Mao W, Li X, Li Z, Li Q, Zheng Z, Xu H, Chen M, Wang PG, Wang Y, Shen B, Yi W, O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth, Nat. Commun 6 (2015) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Qin W, Lv PO, Fan XQ, Quan BY, Zhu YT, Qin K, Chen Y, Wang C, Chen X, Quantitative time-resolved chemoproteomics reveals that stable O-GlcNAc regulates box C/D snoRNP biogenesis, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E6749–E6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Stefely JA, Kwiecien NW, Freiberger EC, Richards AL, Jochem A, Rush MJP, Ulbrich A, Robinson KP, Hutchins PD, Veling MT, Guo X, Kemmerer ZA, Connors KJ, Trujillo EA, Sokol J, Marx H, Westphall MS, Hebert AS, Pagliarini DJ, Coon JJ, Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling, Nat. Biotechnol 34 (2016) 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Song X, Ju H, Lasanajak Y, Kudelka MR, Smith DF, Cummings RD, Oxidative release of natural glycans for functional glycomics, Nat. Methods 13 (2016) 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marx V, Metabolism: sweeter paths in glycoscience, Nat. Methods 14 (2017) 667–670. [DOI] [PubMed] [Google Scholar]
- [7].Munkley J, Mills IG, Elliott DJ, The role of glycans in the development and progression of prostate cancer, Nat. Rev. Urol 13 (2016) 324–334. [DOI] [PubMed] [Google Scholar]
- [8].Pinho SS, Reis CA, Glycosylation in cancer: mechanisms and clinical implications, Nat. Rev. Canc 15 (2015) 540–555. [DOI] [PubMed] [Google Scholar]
- [9].An HJ, Kronewitter SR, de Leoz MLA, Lebrilla CB, Glycomics and disease markers, Curr. Opin. Chem. Biol 13 (2009) 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhao R, Liu X, Wang Y, Jie X, Qin R, Qin W, Zhang M, Tai H, Yang C, Li L, Peng P, Shao M, Zhang X, Wu H, Ruan Y, Xu C, Ren S, Gu J, Integrated glycomic analysis of ovarian cancer side population cells, Clin. Proteonomics 13 (2016) 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Griffin ME, Hsieh-Wilson LC, Glycan engineering for cell and developmental biology, Cell Chem. Biol 23 (2016) 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yin B, Gao Y, Chung C.-y, Yang S, Blake E, Stuczynski MC, Tang J, Kildegaard HF, Andersen MR, Zhang H, Betenbaugh MJ, Glycoengineering of Chinese hamster ovary cells for enhanced erythropoietin N-glycan branching and sialylation, Biotechnol. Bioeng 112 (2015) 2343–2351. [DOI] [PubMed] [Google Scholar]
- [13].Law RJ, Law HT, Scurll JM, Scholz R, Santos AS, Shames SR, Deng W, Croxen MA, Li Y, de Hoog CL, van der Heijden J, Foster LJ, Guttman JA, Finlay BB, Quantitative mass spectrometry identifies novel host binding partners for pathogenic escherichia coli type III secretion system effectors, J. Proteome Res 15 (2016) 1613–1622. [DOI] [PubMed] [Google Scholar]
- [14].Yang S, Hu Y, Sokoll L, Zhang H, Simultaneous quantification of N- and O-glycans using a solid-phase method, Nat. Protoc 12 (2017) 1229–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cao L, Diedrich JK, Kulp DW, Pauthner M, He L, Park S-KR, Sok D, Su CY, Delahunty CM, Menis S, Andrabi R, Guenaga J, Georgeson E, Kubitz M, Adachi Y, Burton DR, Schief WR, Yates Iii JR, Paulson JC, Global site-specific N-glycosylation analysis of HIV envelope glycoprotein, Nat. Commun 8 (2017) 14954–14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kailemia MJ, Ruhaak LR, Lebrilla CB, Amster IJ, Oligosaccharide analysis by mass spectrometry: a review of recent developments, Anal. Chem 86 (2014) 196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI, MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics, Nat. Methods 14 (2017) 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Unterieser I, Mischnick P, Labeling of oligosaccharides for quantitative mass spectrometry, Carbohydr. Res. 346 (2011) 68–75. [DOI] [PubMed] [Google Scholar]
- [19].Reinhold V, Zhang H, Hanneman A, Ashline D, Toward a platform for comprehensive glycan sequencing, Mol. Cell. Proteomics 12 (2013) 866–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lu H, Zhang Y, Yang P, Advancements in mass spectrometry-based glyco-proteomics and glycomics, Natl. Sci. Rev 3 (2016) 345–364. [Google Scholar]
- [21].Ruhaak LR, Xu G, Li Q, Goonatilleke E, Lebrilla CB, Mass spectrometry approaches to glycomic and glycoproteomic analyses, Chem. Rev (2018), 10.1021/acs.chemrev.7b00732. Just accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shih CJ, Chen SC, Weng CY, Lai MC, Yang YL, Rapid identification of haloarchaea and methanoarchaea using the matrix assisted laser desorption/ionization time-of-flight mass spectrometry, Sci. Rep 5 (2015) 16326–16337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Harvey DJ, Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: an update for 2009-2010, Mass Spectrom. Rev 34 (2015) 268–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Anumula KR, Advances in fluorescence derivatization methods for high-performance liquid chromatographic analysis of glycoprotein carbohydrates, Anal. Biochem 350 (2006) 1–23. [DOI] [PubMed] [Google Scholar]
- [25].Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M, Glycan labeling strategies and their use in identification and quantification, Anal. Bioanal. Chem 397 (2010) 3457–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harvey DJ, Derivatization of carbohydrates for analysis by chromatography; electrophoresis and mass spectrometry, J. Chromatogr. B 879 (2011) 11y6–1225. [DOI] [PubMed] [Google Scholar]
- [27].Zhou H, Hanneman AJ, Chasteen ND, Reinhold VN, Anomalous N-glycan structures with an internal fucose branched to GlcA and GlcN residues isolated from a mollusk shell-forming fluid, J. Proteome Res 12 (2013) 4547–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Honda S, Akao E, Suzuki S, Okuda M, Kakehi K, Nakamura J, High-performance liquid chromatography of reducing carbohydrates as strongly ultraviolet-absorbing and electrochemically sensitive 1-phenyl-3-methyl5-pyrazolone derivatives, Anal. Biochem 180 (1989) 351–357. [DOI] [PubMed] [Google Scholar]
- [29].Palaniappan KK, Bertozzi CR, Chemical glycoproteomics, Chem. Rev 116 (2016) 14277–14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Krasnova L, Wong CH, Understanding the chemistry and biology of glycosylation with glycan synthesis, Annu. Rev. Biochem 85 (2016) 599–630. [DOI] [PubMed] [Google Scholar]
- [31].Jiang K, Aloor A, Qu J, Xiao C, Wu Z, Ma C, Zhang L, Wang PG, Rapid and sensitive MALDI MS analysis of oligosaccharides by using 2-hydrazinopyrimidine as a derivative reagent and co-matrix, Anal. Bioanal. Chem 409 (2017) 421–429. [DOI] [PubMed] [Google Scholar]
- [32].Dirksen A, Dawson PE, Rapid oxime and hydrazone ligations with aromatic aldehydes for biomolecular labeling, Bioconjug. Chem 19 (2008) 2543–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kaneshiro K, Watanabe M, Terasawa K, Uchimura H, Fukuyama Y, Iwamoto S, Sato T-A, Shimizu K, Tsujimoto G, Tanaka K, Rapid quantitative profiling of N-glycan by the glycan-labeling method using 3-aminoquinoline/α-cyano-4-hydroxycinnamic acid, Anal. Chem 84 (2012) 7146–7151. [DOI] [PubMed] [Google Scholar]
- [34].Lattová E, Chen VC, Varma S, Bezabeh T, Perreault H, Matrix-assisted laser desorption/ionization on-target method for the investigation of oligosaccha-rides and glycosylation sites in glycopeptides and glycoproteins, Rapid Commun. Mass Spectrom 21 (2007) 1644–1650. [DOI] [PubMed] [Google Scholar]
- [35].Lattova E, Perreault H, Labelling saccharides with phenylhydrazine for electrospray and matrix-assisted laser desorption-ionization mass spectrometry, J. Chromatogr. B 793 (2003) 167–179. [DOI] [PubMed] [Google Scholar]
- [36].Lattova E, Perreault H, Profiling of N-linked oligosaccharides using phenylhydrazine derivatization and mass spectrometry, J. Chromatogr. A 1016 (2003) 71–87. [DOI] [PubMed] [Google Scholar]
- [37].Varki A, Sialic acids in human health and disease, Trends Mol. Med 14 (2008) 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Walker HG, Gee M, McCready RM, Complete methylation of reducing carbohydrates, J. Org. Chem 27 (1962) 2100–2102. [Google Scholar]
- [39].Yang S, Zhang H, Glycomic analysis of glycans released from glycoproteins using chemical immobilization and mass spectrometry, Curr Protoc Chem Biol 6 (2014) 191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bereman MS, Comins DL, Muddiman DC, Increasing the hydrophobicity and electrospray response of glycans through derivatization with novel cationic hydrazides, Chem. Commun 46 (2010) 237–239. [DOI] [PubMed] [Google Scholar]
- [41].Erika L, Hélène P, The usefulness of hydrazine derivatives for mass spectrometric analysis of carbohydrates, Mass Spectrom. Rev 32 (2013) 366–385. [DOI] [PubMed] [Google Scholar]
- [42].Wang CJ, Wu ZY, Yuan JB, Wang B, Zhang P, Zhang Y, Wang ZF, Huang LJ, Simplified quantitative glycomics using the stable isotope label Girard's reagent P by electrospray ionization mass spectrometry, J. Proteome Res 13 (2014) 372–384. [DOI] [PubMed] [Google Scholar]
- [43].Gil GC, Kim YG, Kim BG, A relative and absolute quantification of neutral N-linked oligosaccharides using modification with carboxymethyl trimethylammonium hydrazide and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, Anal. Biochem 379 (2008) 45–59. [DOI] [PubMed] [Google Scholar]
- [44].Naven TJP, Harvey DJ, Cationic derivatization of oligosaccharides with Girard's T reagent for improved performance in matrix-assisted laser desorption/ionization and electrospray Rapid Commun, Mass Spectrom. 10 (1996) 829–834. [Google Scholar]
- [45].Zhang Y, Wang C, Liu Y, Yao W, Sun Y, Zhang P, Huang LJ, Wang ZF, Fluorescein-5-thiosemicarbazide (FTSC) labeling for fluorescent imaging of pectin-derived oligogalacturonic acid transported in living cells by confocal microscopy, Eur, Food Res. Technol. 239 (2014) 1–9. [Google Scholar]
- [46].Zhang Y, Yuan J, Song J, Wang ZF, Huang LJ, An efficient method for selectively in situ imaging and quantifying the expression of sialylated glycoproteins on living cells, Glycobiology 23 (2012) 643–653. [DOI] [PubMed] [Google Scholar]
- [47].Zhang Y, Sun Y, Wang ZF, Huang LJ, Fluorescein-5-thiosemicarbazide as a probe for directly imaging of mucin-type O-linked glycoprotein within living cells, Glycoconj. J 29 (2012) 445–452. [DOI] [PubMed] [Google Scholar]
- [48].Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M, High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification, Anal. Chem 86 (2014) 5784–5793. [DOI] [PubMed] [Google Scholar]
- [49].Kunz C, Rudloff S, Baier W, Klein N, Strobel S, Oligosaccharides in human milk: structural, functional, and metabolic aspects, Annu. Rev. Nutr 20 (2000) 699–722. [DOI] [PubMed] [Google Scholar]
- [50].Yuan J, Wang C, Sun Y, Huang L, Wang Z, Nonreductive chemical release of intact N-glycans for subsequent labeling and analysis by mass spectrometry, Anal. Biochem 462 (2014) 1–9. [DOI] [PubMed] [Google Scholar]
- [51].Yang S, Li Y, Shah P, Zhang H, Glycomic analysis using glycoprotein immobilization for glycan extraction, Anal. Chem 85 (2013) 5555–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Song X, Smith DF, Cummings RD, Nonenzymatic release of free reducing glycans from glycosphingolipids, Anal. Biochem 429 (2012) 82–87. [DOI] [PubMed] [Google Scholar]
- [53].Naven TJP, Harvey DJ, Cationic derivatization of oligosaccharides with Girard's T reagent for improved performance in matrix-assisted laser desorption/ionization and electrospray mass spectrometry, Rapid Commun. Mass Spectrom. 10 (1996) 829–834. [Google Scholar]
- [54].Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M, Glycan labeling strategies and their use in identification and quantification, Anal. Bioanal. Chem 397 (2010) 3457–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Walker SH, Lilley LM, Enamorado MF, Comins DL, Muddiman DC, Hydrophobic derivatization of N-linked glycans for increased ion abundance in electrospray ionization mass spectrometry, J. Am. Soc. Mass Spectrom 22 (2011) 1309–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tong W, Han H, Song Z, Ma C, Pan Y, Zhang Y, Qin W, Qian X, Chemical derivatization with a polycyclic aromatic hydrocarbon for highly sensitive detection of N-linked glycans using MALDI-TOF MS, Anal. Methods 4 (2012) 3531–3535. [Google Scholar]
- [57].Walker SH, Budhathoki-Uprety J, Novak BM, Muddiman DC, Stable-isotope labeled hydrophobic hydrazide reagents for the relative quantification of N-linked glycans by electrospray ionization mass spectrometry, Anal. Chem 83 (2011) 6738–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bereman MS, Comins DL, Muddiman DC, Increasing the hydrophobicity and electrospray response of glycans through derivatization with novel cationic hydrazides, Chem. Commun 46 (2010) 237–239. [DOI] [PubMed] [Google Scholar]
- [59].Nwosu CC, Aldredge DL, Lee H, Lerno LA, Zivkovic AM, German JB, Lebrilla CB, Comparison of the human and bovine milk N-glycome via high-performance microfluidic chip liquid chromatography and tandem mass spectrometry, J. Proteome Res 11 (2012) 2912–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ninonuevo MR, Park Y, Yin H, Zhang J, Ward RE, Clowers BH, German JB, Freeman SL, Killeen K, Grimm R, Lebrilla CB, A strategy for annotating the human milk glycome, J. Agric. Food Chem 54 (2006) 7471–7480. [DOI] [PubMed] [Google Scholar]
- [61].de Haan N, Reiding KR, Haberger M, Reusch D, Falck D, Wuhrer M, Linkage-specific sialic acid derivatization for MALDI-TOF-MS profiling of IgG glycopeptides, Anal. Chem 87 (2015) 8284–8291. [DOI] [PubMed] [Google Scholar]
- [62].Knezevic A, Bones J, Kracun SK, Gornik O, Rudd PM, Lauc G, High throughput plasma N-glycome profiling using multiplexed labelling and UPLC with fluorescence detection, Analyst 136 (2011) 4670–4673. [DOI] [PubMed] [Google Scholar]
- [63].Stumpo KA, Reinhold VN, The N-glycome of human plasma, J. Proteome Res 9 (2010) 4823–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Arnold JN, Saldova R, Hamid UMA, Rudd PM, Evaluation of the serum N-linked glycome for the diagnosis of cancer and chronic inflammation, Proteomics 8 (2008) 3284–3293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.