

Abstract
Cortical spreading depression (CSD) is associated with migraine, stroke, and traumatic brain injury, but its mechanisms remain poorly understood. One of the major features of CSD is an hour-long silencing of neuronal activity. Though this silencing has clear ramifications for CSD-associated disease, it has not been fully explained. We used in vivo whole-cell recordings to examine the effects of CSD on layer 2/3 pyramidal neurons in mouse somatosensory cortex and used in vitro recordings to examine their mechanism. We found that CSD caused a reduction in spontaneous synaptic activity and action potential (AP) firing that lasted over an hour. Both pre- and postsynaptic mechanisms contributed to this silencing. Reductions in frequency of postsynaptic potentials were due to a reduction in presynaptic transmitter release probability as well as reduced AP activity. Decreases in postsynaptic potential amplitude were due to an inhibitory shift in the ratio of excitatory and inhibitory postsynaptic currents. This inhibitory shift in turn contributed to the reduced frequency of APs. Thus, distinct but complementary mechanisms generate the long neuronal silence that follows CSD. These cellular changes could contribute to wider network dysfunction in CSD-associated disease, while the pre- and postsynaptic mechanisms offer separate targets for therapy.
Keywords: cortical spreading depression, excitation inhibition balance, somatosensory, whole-cell recording
Introduction
Spreading depolarizations (SD) have been implicated in the pathophysiology of migraine ( Lauritzen 1994 ; Cao et al. 1999 ; Hadjikhani et al. 2001 ; Pietrobon and Moskowitz 2013 ), brain trauma ( Hartings et al. 2009 ; Lauritzen et al. 2011 ), stroke ( Strong et al. 2002 ; Fabricius et al. 2006 ), and subarachnoid hemorrhage ( Dreier 2011 ). Despite their relevance to disease, our understanding of SD and their effects on neuronal function in vivo remain limited.
SD involve synchronous and massive wave-like depolarization of neurons and glia; however, equally prominent is a long-lasting depression of cellular activity that follows the wave ( Leao 1944 ; Sugaya et al. 1975 ; Somjen 2001 ). Cortical spreading depression (CSD), the proposed mechanism of the migraine aura, is actually named after the depression of activity, as it was the first phenotype observed by its discoverer Leão ( Leao 1944 , 1947 ). After more than 70 years, its mechanisms remain unclear.
We examined the intrinsic and synaptic properties of neurons undergoing CSD, in order to understand the mechanisms causing silencing. We used in vivo whole-cell recordings in layer 2/3 pyramidal cells in mouse sensory cortex, supplemented with in vitro recordings to dissect excitatory and inhibitory currents. We found that CSD alters both excitatory and inhibitory input, through both pre- and postsynaptic mechanisms, for at least an hour after passage of the wave. The net result is a strong decrement in synaptic function, which is the likely foundation for the neuronal and network silencing after CSD.
Materials and Methods
All experimental procedures were in accordance with NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Utah.
In Vivo Whole-Cell Recordings
Mice (males; 2–3 months old; C57Bl/6J) were anesthetized by intraperitoneal injection of urethane (0.75 g/kg), supplemented with isoflurane (0.6–0.8%), in a 70/30% nitrogen/oxygen mixture. Vital signs (HR: 470–540 bpm, SpO 2 : 92–98%, respiration: 120–140/min) were monitored (MouseStat, Kent Scientific) throughout the experiment, and maintained within a physiologically normal range. Body temperature was monitored using a rectal probe, and maintained at 36–37°C using a water bath blanket.
An omega-shaped head bar was mounted to the skull, using glue and dental cement and attached to a custom head fixation apparatus. A craniotomy 2.5–3 mm in diameter was made over the somatosensory cortex (hindpaw area: 1 mm caudal to the bregma and 2 mm lateral to the midline). The dura was kept intact, and the craniotomy was filled with 1.5–2% agarose (mixed in saline) in order to keep the cortical surface moist and dampen the movement associated with breathing. Most recordings were obtained using blind-patch technique; however, we added visualized recordings using 2-photon microscopy for confirmation of cell type (see Supplementary Materials ). A patch pipette was filled with intracellular solution containing (in mM; pH = 7.2): 135 K-gluconate, 8 NaCl, 5 EGTA, 10 HEPES, 0.3 GTP, 2 ATP, 7 phosphocreatine. The electrode was placed on the cortical surface with positive pressure of 20 kPa and advanced to a depth of 250–350 µm (layer 2/3). Once the electrode was lowered to the desired cortical depth, pressure was reduced to 4 kPa. In voltage-clamp mode, the pipette was moved in 1 µm steps while monitoring the current response to a 10 mV voltage step. A consistent reduction in response amplitude (∼50%), fluctuation at heartbeat and respiration frequencies, and a sharp increase in pipette resistance indicated cell contact. Positive pressure was removed to zero, and suction was applied if necessary to obtain greater than giga ohm seal resistance recorded through the electrode. A command potential of −70 mV was applied. In this cell-attached configuration, negative pressure was applied to obtain whole-cell access. For selected experiments, using artificial cerebrospinal fluid (ACSF) or potassium–gluconate electrode solution, loose cell-attached recordings (seal resistance <50 MΩ) were obtained by the same procedures. We started recording spontaneous activity immediately after obtaining whole-cell access. We did not observe uninduced CSD in our recordings (i.e., CSD not associated with KCl ejection): frequency of spontaneous postsynaptic potentials (sPSPs) was in a normal range (∼0.3–0.5 Hz) at the start of recordings, in contrast to the significant reductions in PSP frequency seen after CSD (see below). Moreover, in selected recordings with a supplementary field potential electrode, we did not observe CSD induction.
Recordings were performed using thick-wall glass pipettes pulled from borosilicate glass capillaries (OD 1.65 mm, ID 1.2 mm, Garner Glass, Claremont, CA) with a P-87 Flaming-Brown puller (Sutter Instruments, Novato, CA, USA). Patch electrodes of 4–6 MΩ resistance were used (tip size of 3–4 μm). Signals were amplified using a Multiclamp 700B amplifier (Molecular Devices, Union City, CA, USA), sampled at 10 kHz, and low-pass filtered at 2 kHz. Data were acquired and digitized (Digidata-1320A Molecular Devices), stored on a computer, and retrieved with pClamp 8.2/10 software (Molecular Devices).
In Vitro Slice Preparation and Electrophysiology Recordings
Mice (males, 2–3 months old; C57Bl/6J) were anesthetized with 4% isoflurane and killed by rapid decapitation. Brains were removed to ice-cold cutting solution containing (in mM) 220 Sucrose, 3 KCl, 10 MgSO 4 , 1.25 NaH 2 PO 4 , 25 NaHCO 3 , 25 Glucose, 1.3 CaCl 2 . The somatosensory cortex was cut into 350 μm coronal sections using a Leica VT-1000S Vibratome (Leica Microsystems, Wetzlar, Germany). Slices were transferred to a storage chamber maintained at 32–34°C, filled with ACSF containing (in mM) 125 NaCl, 3 KCl, 10 MgSO 4 , 1.25 NaH 2 PO 4 , 25 NaHCO 3 , 25 Glucose, 1.3 CaCl 2 , and saturated with 95% O 2 /5% CO 2 . The slices were rested for >1 h before being transferred to a submerged recording chamber.
To isolate spontaneous excitatory and inhibitory postsynaptic currents (sE/IPSCs), we used an internal solution that was composed of (in mM) 130 CsMeSO 4 ; 3 CsCl; 10 HEPES; 2 MgATP; 0.3 Na 3 GTP; 5 EGTA; 10 Phosphocreatine; 5 QX-314; 8 biocytin (adjusted to pH 7.3 with CsOH, 295 mOsm). Membrane potential (Vm) was clamped at −70 mV to record sEPSCs, and Vm was clamped at +10 mV to record sIPSCs.
Miniature EPSCs and IPSCs (mE/IPSCs) were recorded by adding 1 µM TTX to the bath. To confirm that the EPSCs were AMPA/NMDA receptor-mediated, the slices were bathed in AMPA receptor antagonists (DNQX, 50 µM) at the end of the experiment. To confirm that the IPSCs were GABA A -receptor mediated, the slices were bathed in picrotoxin (100 µM).
The use of Cs and TTX precluded analysis of membrane potential characteristics and neuronal firing patterns. In a subset of experiments, we used potassium gluconate internal solution (identical to in vivo experiments) in current clamp mode to examine these characteristics and record KCl-induced membrane depolarization. We then recorded spontaneous E/IPSCs in voltage-clamp mode.
KCl-Induced Cortical Spreading Depression
In vivo-pressure ejection pulses of 1 M KCl at 1.5 µL/min were applied through a 30-gauge needle onto Layer 1, using a pneumatic drug ejection system for 30–40 s (0.75–1 µL total ejection). In vitro-CSD was induced by pressure ejection (0.5 bar) of 3 M KCl using a pico-spritzer and 0.5 MΩ glass pipette ( Tottene et al. 2009 ).
Data Analysis
In Vivo
We recorded from pyramidal cells, identified by regularly spiking (RS) or intrinsically bursting (IB) phenotypes, and confirmed in selected recordings by 2-photon microscopy with genetically encoded indicators (see Supplementary Materials ). RS and IB cells had similar CSD and post-CSD characteristics so analysis was performed for both groups together. We also identified interneurons, by firing pattern and 2-photon microscopy, but these were not considered for analysis. For additional details on cellular characterization, see Supplementary Materials .
We verified viability of recordings with access (series) resistance. Access resistance was compensated online and was monitored throughout current clamp recordings (typically 24–28 MΩ). If initial access was >50 MΩ and/or values changed >20% during experiment, the recording was not included for further analysis. After CSD, we observed changes in access resistance on the order of 5–10% (8.22 ± 2.46%). Clampfit software (Molecular Devices, Sunnyvale, CA, USA) was used to determine the width at half amplitude of action potentials (APs) and the duration, amplitude, frequency of PSPs. To study AP firing patterns, voltage responses to stepped current injection (−50 to + 500 pA; 1000 ms long square step pulses; frequency–current curve, F–I curve) were recorded. The resting membrane potential (RMP) was the intercept of the fit to the plot of current versus voltage for all nonspiking sweeps. Membrane input resistance ( Rin ) was the slope of the fit to the same plot. Rheobase was the smallest current that evoked an AP. We recorded PSPs at the cells' RMP (approximately−60 mV). Membrane potential variance for each recording was analyzed from 15 upstate and downstate events. CSD-associated depolarization was defined by using the second derivative of the membrane potential trace to localize onset (local maximum in second derivative), maximum depolarization (second derivative zero), and recovery (local minimum in second derivative). The amplitude, frequency, and duration of PSPs in layer 2/3 pyramidal neurons from pre- and post-CSD groups were measured during 180-s periods for each cell ( Wan et al. 1997 ; Halabisky et al. 2010 ) and compared at 2 different times after CSD. To understand immediate and late effects of CSD on intrinsic and synaptic properties, we analyzed data at 5 and 30 min after membrane potential recovered from the depolarization wave. When Vm recovered to baseline, we assigned that time as the zero point from which 5 and 30 min time points were calculated (this is in contrast to calculating timings from the beginning of the wave). For data beyond 60 min after CSD, we were unable to maintain viable recordings (due to access resistance changes >20%, which tended to begin after 40–50 min). For these time points, we recorded from a second cell patched 60–90 min after CSD. In order to control for possible effects of intracellular buffering associated with whole-cell recordings, we also patched cells 5 and 30 min after CSD.
In Vitro
Each PSC was characterized by the following parameters: peak amplitude, duration, rise time constant, and decay tau. All parameters were measured using Clampfit (Molecular Devices, Sunnyvale, CA, USA). The first 25–30 individual nonoverlapping events were selected from 3 min recordings for pre- and post-CSD groups, and then the composite average was calculated. Rise time constant was measured automatically using Clampfit. Decay tau was calculated from double exponential fits (Chebyshev method). Excitatory and inhibitory synaptic charge was analyzed by calculating the area under the curve of individual event. The shift in the balance between excitation and inhibition was analyzed by calculating ratios of sEPSCs to sIPSCs or mEPSCs to mIPSCs, and then comparing between baseline and post-CSD groups. As some of the E/IPSC data was not collected simultaneously, events were randomly assorted to generate a randomized sequence for each current, before and after CSD. Ratios were taken for each sequential event in the randomized sequence, and compared using cumulative distributions.
Statistics
Paired t -tests were used to compare in vivo intrinsic properties of neurons before and after CSD, where the datasets were found to be normally distributed, as indicated by non-significant Shapiro–Wilk normality test. Unpaired t -tests were used to compare data for >60 min post-CSD data. Paired t -tests were used to compare the interval and amplitude of PSCs between pre- and post-CSD groups. Statistical significance for nonparametric data was determined using the Kolmogoroff–Smirnoff (KS) test. Statistical analyses were performed using SPSS statistics (version 19), GraphPad Prism (version 5.00 for Windows, GraphPad Software), and Matlab (version 7.8.0). Data are expressed as mean ± SEM otherwise stated. For all figures: * P < 0.05; ** P < 0.005.
Results
In Vivo Whole-Cell Recording of CSD
Whole-cell recordings of CSD were made in current clamp mode from 23 pyramidal neurons in Layer 2/3 of mouse somatosensory cortex. Cells had either a regular spiking (RS) or IB phenotype, consistent with prior in vivo whole-cell recordings from this region (RS cells, n = 16 cells, 16 mice; IB cells, n = 7 cells, 7 mice; see Supplementary Fig. 1 for additional characterization). Typical membrane voltage recordings are shown in Figure 1 . Mean RMP for neurons undergoing CSD was −59.55 ± 1.76 mV; mean membrane input resistance was 140.2 ± 4.9 MΩ. After induction of CSD, changes in membrane potential typically began with a relatively small and slow depolarization (“depolarization ramp”; median amplitude: 9.75 mV; median duration: 28.13 s; n = 15 cells, 15 mice; see Fig. 1 and Table 1 ) before the onset of a burst of APs, concurrent with a steep decrease in the membrane potential that heralded the classically defined onset of CSD ( Somjen 2001 ). Membrane potential reached +10 mV (median; range: −7 to +25 mV) at maximum depolarization. The depolarization phase lasted 237.5 s (median; range: 137.6–345.6) followed by recovery of the membrane potential to within 0.5 mV (median; range: −6.7 to +7.4 mV) of pre-CSD baseline values. (Fig. 1 and Table 1 ). As a control, injection of NaCl under identical conditions to KCl did not elicit any changes in the membrane potential ( n = 5 cells, 5 mice).
Figure 1.

In vivo whole-cell recording of CSD. In vivo current clamp recording showing membrane voltage prior to, during, and for 40 min after CSD, induced by focal 1 M KCl ejection >2 mm from recording electrode (induction marked with arrow). Spontaneous synaptic potentials and APs are evident prior to CSD, are completely abolished during and immediately after the depolarization, and return but remain significantly reduced for the duration of the recording. Gaps in recording are during induced hyperpolarization and depolarization current steps to record evoked firing.
Table 1.
Membrane potential changes of Layer 2/3 pyramidal neurons during cortical spreading depression in vivo and in vitro
| Parameters | Median | Range | 25th–75th percentile |
|---|---|---|---|
| In vivo | |||
| Maximum depolarization amplitude (mV) | +10 | −7 to 25 | 4 to 16 |
| Depolarization duration (s) | 237.5 | 137.6 to 345.6 | 203.4 to 301.3 |
| Total amplitude (mV) | 59.11 | 33.29 to 71.17 | 52.67 to 66.68 |
| Depolarization ramp | |||
| Amplitude (mV) | 9.75 | 2.26 to 16.08 | 8.10 to 10.50 |
| Duration (s) | 28.13 | 13.94 to 50.98 | 21.09 to 30.58 |
| Change in Vm after recovery (mV) | 0.5 | −6.7 to + 7.4 | −4.9 to 3.6 |
| In vitro | |||
| Maximum depolarization amplitude (mV) | −10.85 | −54.6 to +1.8 | −33.25 to −16 |
| Depolarization duration (s) | 337.3 | 181.7 to 430 | 201.4 to 371.1 |
| Total amplitude (mV) | 54.15 | 29.30 to 74.50 | 30.60 to 66.35 |
| Change in Vm after recovery (mV) | −0.9 | −8.5 to + 7.3 | −2.78 to 3.68 |
Intrinsic Membrane Properties After CSD In Vivo
We hypothesized that changes in intrinsic neuronal excitability could account for the reduction in activity after CSD. In vitro preparations support the idea of intrinsic changes; however they show mixed effects: decreased membrane input resistance consistent with reduced excitability, but increased AP firing rate to depolarization steps ( Ghadiri et al. 2012 ). We examined intrinsic neuronal excitability after CSD in vivo . Compared with pre-CSD baseline, there was no difference in RMP at 5 min after the end of the depolarization (pre, −56.57 ± 2.87 mV vs. 5 min post-CSD, −55.54 ± 2.56 mV; n = 8 cells, 8 mice; P > 0.05, paired t -test; Fig. 2A ). Interestingly however, at 30 min post-CSD, membrane potential was slightly but significantly depolarized relative to baseline (pre, −59.20 ± 3.35 mV vs. 30 min post-CSD, −48.14 ± 0.97 mV; n = 6 cells, 6 mice; P = 0.02, paired t -test). In order to show that the depolarized membrane potential at 30 min post-CSD was not due to the rundown of whole-cell recordings, we analyzed RMP from cells that we recorded without inducing CSD and found no difference in membrane potential in this group (pre, −60.88 ± 2.19 mV vs. 30 min, −56.11 ± 2.22 mV; paired t -test, P > 0.05; n = 5 cells, 5 mice). Finally, in a separate set of experiments, we recorded >60 min after CSD ( n = 5 cells, 5 mice), from pyramidal cells patched after passage of the wave (holding a cell through and after CSD for that long was not possible in vivo). We found no difference in RMP (pre, −57.45 ± 5.27 mV vs. post-CSD, −57.13 ± 4.28 mV). We concluded that a transient, non-artifactual depolarization of membrane potential occurred tens of minutes after CSD, and subsequently recovered.
Figure 2.

Mild changes in intrinsic membrane properties after CSD in vivo. ( A ) There was a small but significant depolarization in the RMP of Layer 2/3 pyramidal cells 30 min after CSD. (* P = 0.02; paired t -test). Membrane potential before (5 min) and after (60 min) this time point was not significantly different from baseline. Control recordings with no CSD induction (“30 min control”) suggest that this depolarization is not artifactual to the whole-cell preparation. ( B ) There was a significant increase in rheobase 5 min (** P = 0.004, paired t -test) but not 30 or 60 min after CSD. There was no change in input resistance, or in AP frequency to increasing current injection, at any time point after CSD (see text for details). For 5 and 30 min time points after CSD, values were compared for the same neuron before and after CSD; >60 min comparisons were between 2 cells, one patched before and one after CSD in the same animal. 5 min post-CSD: 8 cells, 8 mice; 30 min post-CSD: 6 cells, 6 mice; >60 min data: 2 × 5 cells (5 mice), control data: 5 cells (5 mice).
We found no change in neuronal membrane input resistance at either 5 min, 30 min, or >1 h after CSD (pre, 126.4 ± 6.94 MΩ vs. 5 min post-CSD, 135.0 ± 5.94 MΩ; n = 8 cells, 8 mice; pre, 132.3 ± 7.92 MΩ vs. 30 min post-CSD, 117.3 ± 4.29 MΩ; n = 6 cells, 6 mice; paired t -test, P > 0.05; pre, 130.1 ± 15.11 vs. > 60 min post-CSD, 129.8 ± 11.71; n = 5 cells, 5 mice; unpaired t -test, P > 0.05). In control cells, recorded for 30 min without CSD induction we observed similar characteristics to the values recorded after CSD (pre, 144.7 ± 3.72 MΩ vs. 30 min post, 130.4 ± 5.67 MΩ; paired t -test, P > 0.05; n = 5 cells, 5 mice).
The frequency–current (F–I) curve, measuring the frequency of APs in response to intracellular current pulses, was also analyzed 5 and 30 min post-CSD. There was no change in F–I slope at either time point ( P > 0.05; paired t -test). Rheobase (minimum current required to evoke an AP) was increased at 5 min after CSD, but not at 30 min or > 60 min after CSD (pre-CSD, 162.5 ± 33.74 pA vs. 5 min post-CSD, 293.8 ± 19.9 pA; P = 0.004; n = 8 cells, 8 mice; pre-CSD, 200 ± 54.77 pA vs. 30 min post-CSD, 241.7 ± 27.13 pA; P > 0.05, n = 6 cells, 6 mice; paired t -test; >60 min after CSD: pre, 212.5 ± 85.09 pA vs. post, 162.5 ± 55.43 pA; P > 0.05, n = 5 cells, 5 mice; unpaired t -test; Fig. 2B ). Pearson correlations were computed to determine relationships between rheobase and membrane properties after CSD. There was no significant correlation (vs. RMP: Pearson r = 0.04, P = 0.83; vs. membrane input resistance: Pearson r = 0.23, P = 0.59), indicating that changes in rheobase at 5 min post-CSD did not vary with changes in RMP and membrane input resistance.
Taken together, these data reveal relatively mild changes in intrinsic properties after CSD in vivo, suggesting that synaptic properties might play a greater role in the post-CSD phenotype.
Spontaneous Synaptic Activity After CSD In Vivo
Decreased Amplitude and Frequency of Spontaneous Postsynaptic Potentials
Spontaneous postsynaptic potentials (sPSPs), “upstate events” (spontaneous membrane depolarizations >200 ms long; Timofeev et al. 2000 ; McCormick et al. 2003 ) and APs were routinely observed at baseline prior to CSD (Figs 1 and 3A ). Though they were also observed after CSD, their characteristics were substantially different.
Figure 3.

Significant reductions in spontaneous synaptic activity and AP firing >1 h after CSD in vivo. ( A ) Consecutive membrane voltage traces (30 s) from continuous recordings of sPSPs before, and 5 and 30 min after CSD from the same cell. There is a dramatic reduction in sPSP amplitude and frequency 5 min after CSD, which recovers but remains below baseline 30 min after the event. ( B ) Similar traces comparing a cell patched before CSD to a separate cell patched >60 min after CSD in the same animal. ( C , D ) sPSP amplitude and frequency were significantly reduced 5 and 30 min after CSD when recorded from the same cell. (Histograms above; cumulative plots below. Amplitude: 5 min: P = 2.01 × 10 −12 ; 30 min: P = 2.77 × 10 −8 , 2-sample KS test; Frequency: 5 min: P = 3.78 × 10 −8 ; 30 min: P = 4.59 × 10 −10 , 2-sample KS test; n = 6 cells, 6 mice). Similar changes were seen at 30 min comparing cells recorded before to separate cells recorded 30 min after CSD from the same animal (see Supplementary Fig. 3 ) showing that findings were not due to rundown of whole-cell recordings. ( E ) Recordings comparing cells recorded before with separate cells recorded >60 min after CSD in the same animal: there was no significant difference in sPSP frequency at this time point ( P > 0.05, 5 cells, 5 mice, 2-sample KS test; not shown); however, amplitude of PSPs remained reduced for at least 90 min after CSD ( P = 3.86 × 10 −40 ; 5 cells, 5 mice; 2-sample KS test). ( F ) Box–whisker plot showing no AP firing at 5 min post-CSD and significantly reduced AP firing 30 (upper panel) and >60 min post-CSD (lower panel).
There was a significant reduction in sPSP frequency both 5 and 30 min after the end of the depolarization (5 min: P = 3.78 × 10 −8 ; 30 min: P = 4.59 × 10 −10 ; 2-sample KS test; n = 6 cells, 6 mice; Fig. 3C ). There was also a significant reduction in sPSP amplitude at 5 and 30 min post-CSD (5 min: P = 2.01 × 10 −12 ; 30 min: P = 2.77 × 10 −8 ; 2-sample KS test; Fig. 3D ). Finally, sPSP duration was significantly reduced 5 min post-CSD, returning toward normal values 30 min after CSD (5 min: P = 1.38 × 10 −8 ; 30 min: P = 0.05; KS test; data not shown).
In a separate group of recordings, we recorded from 5 to >60 min after CSD, from cells patched after passage of the event. Here also, there was a significant decrease in sPSP frequency and amplitude in Layer 2/3 neurons when recorded in the 5–30 min period after CSD (frequency: P = 3.10 × 10 −11 , amplitude: P = 1.98 × 10 −24 , 2-sample KS test; n = 5 cells, 5 mice; see Supplementary Fig. 3 A , B ), showing that CSD affects the synaptic network irrespective of the recording conditions, and that rundown of intracellular recordings was unlikely to account for the phenotype.
At >60 min after CSD, sPSP frequency and duration had returned to values indistinguishable from pre-CSD baseline ( P > 0.05, KS test; n = 5 cells, 5 mice; data not shown). However, sPSP amplitude remained lower at >60 min post-CSD ( P = 3.86 × 10 −40 , KS test; Fig. 3B , E ). Taken together, these data show that Layer 2/3 pyramidal neurons after CSD have less frequent, shorter duration sPSPs lasting at least 30 min after the end of the depolarization, and a decrease in amplitude of sPSPs that persists for more than an hour. These findings are consistent with reduced AP firing or release probability of presynaptic neurons; a net inhibitory shift in postsynaptic excitation/inhibition balance; or both.
Decreased Frequency of Spontaneous APs
AP frequency was significantly reduced for tens of minutes after CSD (Fig. 3F ). AP frequency during pre-CSD baseline was 0.21 ± 0.16 Hz (range: 0.01–1.13 Hz), consistent with previous intracellular in vivo recordings that show sparse AP firing rates in Layer 2/3 pyramidal neurons ( Moore and Nelson 1998 ; Zhu and Connors 1999 ; Brecht et al. 2003 ; Petersen et al. 2003 ; Kitamura et al. 2008 ). Five minutes after CSD, we observed no suprathreshold activity in any of the recordings. APs had returned by 30 min after the end of the depolarization (0.04 ± 0.02 Hz, range: 0–0.1 Hz) but remained reduced for more than an hour compared with pre-CSD (pre: 0.65 ± 0.33 Hz vs. >60 min post-CSD: 0.05 ± 0.04 Hz). Thus, CSD caused a massive suppression of suprathreshold activity, with complete silence for at least 5 min after the wave, and persistent reductions lasting at least an hour after passage.
To further examine AP firing after CSD, we performed loose-cell-attached recordings from Layer 2/3 pyramidal neurons ( n = 6 cells, 6 mice). Here again we found reduced firing rates after CSD (pre: 1.92 ± 0.88 Hz vs. post: 0.46 ± 0.23 Hz; P = 0.03, Wilcoxon matched-pairs t -test; Fig. 3F lower panel), suggesting that the reductions observed in whole-cell recordings were not artifactual to the preparation. Taken together, AP data from whole-cell and loose-cell-attached recordings of Layer 2/3 pyramidal cells after CSD suggest that the reduction in frequency of PSPs is due (at least in part) to the reduced activity of excitatory inputs. Conversely, the PSP reduction might account for reduced AP generation.
Decreased Amplitude and Frequency of Upstate Events
At baseline, pyramidal neurons from superficial layers under anesthesia displayed a biphasic membrane potential phenotype, with quiet periods near the RMP (downstates), and periods of depolarization by approximately 20 mV (upstates; Steriade 1997 , 2006 ; Timofeev et al. 2000 ; Lemieux et al. 2015 ). Downstates showed low membrane potential variance (0.54 ± 0.18 mV 2 ) and upstates much larger variance (8.65 ± 2.31 mV 2 ), which is consistent with relative synaptic silence during the downstate, and increased synaptic input during the upstate ( Steriade et al. 2001 ; Hughes and Crunelli 2013 ).
Upstates were significantly different after CSD, showing reduced frequency (at 5 min: P = 0.004; at 30 min: P = 4.91 × 10 −9 ; KS test; n = 6 cells, 6 mice; see Supplementary Fig. 4 A , B ) and amplitude (at 5 min: P = 1.78 × 10 −65 ; at 30 min: P = 6.18 × 10 −71 ; KS test; see Supplementary Fig. 4 A , C ) both 5 and 30 min after passage of the wave. Upstate frequency at >60 min post-CSD had recovered to baseline levels ( P > 0.05 compared with baseline; KS test; data not shown); however, amplitude had not ( P = 1.34 × 10 −28 ; KS test; see Supplementary Fig. 4 D ). Upstates are due to recurrent network activity ( Petersen et al. 2003 ); suppression of these events suggested that CSD effects go beyond local synaptic connections.
Excitatory Synaptic Transmission Onto Pyramidal Cells After CSD In Vitro
Decreased Frequency, Mixed Effects on Amplitude of Spontaneous Excitatory Postsynaptic Currents
Although net neuronal activity is critically determined by the excitatory/inhibitory synaptic inputs a neuron receives, the effects of CSD on excitatory and inhibitory neurotransmission are incompletely understood. Our in vivo recordings were consistent with a reduction in excitatory input, an increase in inhibitory input, or both, on Layer 2/3 pyramidal cells after CSD. We turned to brain slice recordings for the voltage-clamp experiments necessary to dissect excitatory and inhibitory currents.
In voltage-clamp recordings held at −70 mV (cesium internal solution), following KCl administration, we observed a large inward current (duration: 190.4 ± 29.82 s, amplitude: −1.62 ± 0.64 nA; n = 7 cells, 7 mice) that reflected the depolarization wave ( Czeh et al. 1993 ; Aiba and Shuttleworth 2014 ). In recordings holding the cell at +10 mV (the voltage used for recording IPSC's), a large outward current was observed (119.8 ± 35.85 s and 0.94 ± 0.3 nA; n = 7 cells, 7 mice). Membrane potential changes obtained in current clamp mode (potassium gluconate internal solution) from in vitro recordings were consistent with in vivo recordings during CSD (without TTX); membrane potential changes from RMP to the peak potential were similar in magnitude (in vivo: 57.78 ± 2.82 vs. in vitro: 50.59 ± 6.41 mV). Following depolarization, neurons returned to membrane potential of median −0.9 mV (range: −8.5 to 7.3 mV; Table 1 ).
Figure 4A shows sEPSCs (confirmed by elimination with AMPA receptor blockade), recorded from a pyramidal cell held at −70 mV. Frequency of sEPSCs was reduced both 5 and 30 min after the end of the depolarization (5 min: P = 1.62 × 10 −8 , 30 min: P = 2.17 × 10 −15 ; 2-sample KS test; pre: 719 ± 246.8 ms vs. 5 min post: 434.9 ± 115 ms vs. 30 min post: 820.3 ± 237.7 ms; 5 min post-CSD: n = 5 cells, 5 mice and 30 min post-CSD: n = 7 cells, 7 mice; Fig. 4B ). This reduction in sEPSC frequency was also observed in recordings performed using potassium gluconate internal solution (pre vs. 30 min post-CSD: P = 3.53 × 10 −13 ; 2-sample KS test; n = 6 cells, 6 mice; see Supplementary Fig. 5 A ). sEPSC amplitudes were also reduced 5 min after CSD ( P = 3.02 × 10 −4 ; 2-sample KS test; pre vs. post-CSD: −21.91 ± 2.48 pA vs. −25.92 ± 5.15 pA; Fig. 4C ). However, at 30 min amplitudes were increased (pre: −21.91 ± 2.48 pA vs. post: −24.14 ± 3.26 pA; P = 1.72 × 10 −4 , 2-sample KS test; Fig. 4C ). Similar results were obtained with potassium gluconate internal solution (pre vs. 30 min post-CSD: P = 1.42 × 10 −85 ; 2-sample KS test; see Supplementary Fig. 5 B ). Reduction in both frequency and amplitude of sEPSCs at 5 min suggests decreases in presynaptic transmitter release probability, decreased postsynaptic receptor-mediated excitation, or both. The decrease in frequency and increase in amplitude at 30 min are still consistent with reduced presynaptic release, but suggest increased postsynaptic excitation.
Figure 4.
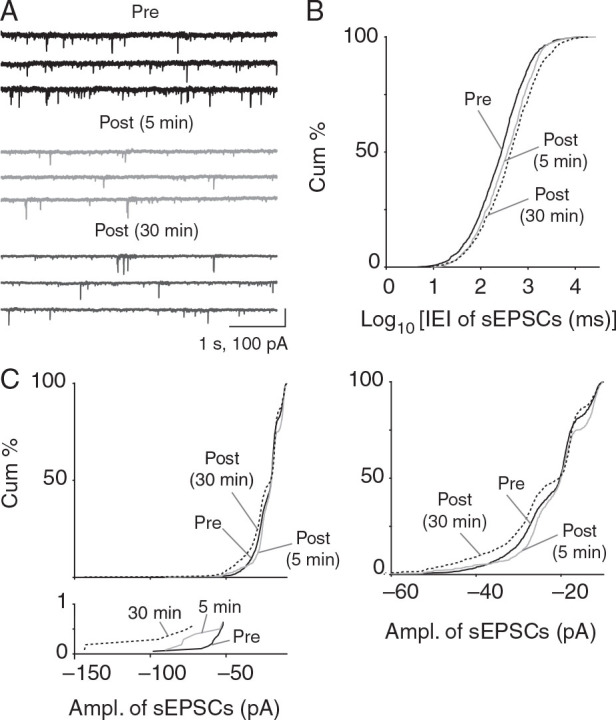
Reduced frequency and mixed effects on sEPSC amplitude after CSD in vitro. ( A ) sEPSCs recorded in Layer 2/3 pyramidal neurons at 5 and 30 mins post-CSD. ( B ) sEPSC frequency is consistently decreased 5 and 30 min after CSD (lengthened interevent interval, rightward shift as shown in cumulative plots; 5 min: P = 1.62 × 10 −8 , 30 min: P = 2.17 × 10 −15 ; 2 sample KS test; 5 min post-CSD, n = 5 cells, 5 mice and 30 min post-CSD, n = 7 cells, 7 mice). ( C ) sEPSC amplitude was decreased at 5 min post-CSD ( P = 3.02 × 10 −4 ; KS test) but increased at 30 min ( P = 1.72 × 10 −4 ; 2-sample KS test; 5 min post-CSD: n = 5 cells, 5 mice and 30 min post-CSD: n = 7 cells, 7 mice). Magnified cumulative plot below shows large amplitude events (greater than −70 pA) that were only evident in post-CSD groups. Magnified cumulative plot on right including only less than −60 pA events shows clear rightward shift (smaller amplitudes) at 5 min and leftward shift (larger amplitudes) 30 min after CSD.
Decreased Frequency, Mixed Effects on Amplitude of Miniature Excitatory Postsynaptic Currents
To assess the relative contribution of miniature events to the decreased sEPSC frequency observed after CSD, we recorded in the presence of 1 µM TTX. TTX treatment also gave the opportunity to examine the effect of sodium channel blockade on the CSD phenotype . We found that TTX significantly altered CSD characteristics ( n = 7 cells, 7 mice; Fig. 5A ). Compared with CSD without TTX, both amplitude and duration of depolarization were significantly reduced (amplitude: 54.15 mV, range: 29.30–74.50 mV vs. TTX CSD: 13.40 mV, range: 10.40–42.40; duration: 337.3 s, range: 181.7–430.0 vs. TTX CSD: 211.2 s, range: 22.04–370.1; see Table 1 ). When we compared mEPSCs obtained after CSD induced under TTX, we found no significant differences in their amplitude or frequency either 5 or 30 min post-CSD ( P > 0.05, 2-sample KS test; n = 4 cells, 4 mice; data not shown). We concluded that an event induced by KCl in the presence of TTX is phenotypically different than the wave without TTX, and that the lack of changes in mEPSCs might be a result of the much smaller depolarizations we observed.
Figure 5.
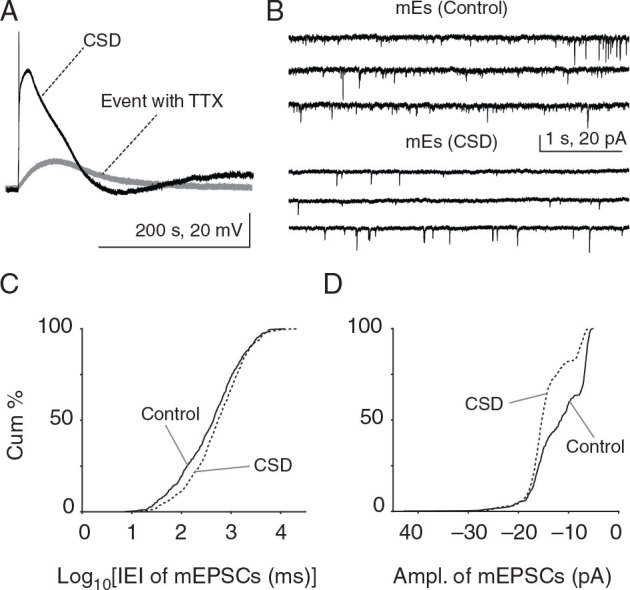
Reduced frequency and increased amplitude of mEPSCs after CSD in vitro. ( A ) Whole-cell recordings were made at −70 mV in current clamp mode to record membrane potential changes during CSD. Typical CSD depolarization of a Layer 2/3 pyramidal cell showing significantly reduced membrane potential changes during KCl-induced depolarization with TTX treatment (50.59 ± 6.41 mV vs. event with TTX: 20.27 ± 4.91 mV). There was no change in mEPSC frequency or amplitude after CSD induced in the presence of TTX ( P > 0.05, 2-sample KS test; n = 4 cells, 4 mice; data not shown). ( B ) mEPSCs from separate post-CSD and control groups in different slices from the same animal, where TTX was delivered after CSD (or sham CSD—NaCl ejection—in control groups). ( C , D ) There was a significant decrease in mEPSC frequency (rightward shift in cumulative plot; P = 1.7 × 10 −4 , 2-sample KS test; control: n = 4 cells, 4 mice and CSD: n = 6 cells, 6 mice) and a significant increase in mEPSC amplitude 30 min post-CSD (leftward shift toward larger amplitudes; P = 1.31 × 10 −31 ; KS test; control: n = 4 cells, 4 mice and CSD: n = 6 cells, 6 mice) after “intact” CSD.
The significant effect of TTX on CSD characteristics prompted a modification of experimental protocol. Rather than recording events in the same cell during a potentially unrepresentative CSD event, we compared miniature events from 2 populations of cells, with TTX added either after CSD or after control recordings without CSD. Under these conditions, with “intact” CSD waves, we observed significant changes in miniature events 30 min after the end of the depolarization (Fig. 5B ). (Because it took >5 min to wash on TTX, 5 min post-CSD recordings were not possible for these experiments.) There was a significant decrease in the frequency (intervals: control: 607.7 ± 176.9 ms vs. post-CSD: 1274 ± 562.9 ms; P = 1.7 × 10 −4 , 2-sample KS test; control: n = 4 cells, 4 mice and CSD: n = 6 cells, 6 mice; Fig. 5C ) and increase in the amplitude (control: −12.37 ± 2.37 pA vs. post-CSD: −15.15 ± 2.15 pA; P = 1.31 × 10 −31 ; KS test; Fig. 5D ) of mEPSCs recorded from Layer 2/3 pyramidal neurons at 30 min post-CSD.
Taken together, the in vitro data on EPSCs suggest that a decrease in excitatory presynaptic activity contributes to the decrease in frequency of sPSPs and APs we observed in vivo . However, the increased amplitude of s- and mEPSCs would be expected to generate an increase in sPSP amplitudes, rather than the decrease we observed in vivo. We hypothesized that the increase in EPSCs would have to be met by a larger increase in IPSCs to generate this phenotype.
Inhibitory Synaptic Transmission Onto Pyramidal Cells After CSD In Vitro
Decreased Frequency, Increased Amplitude of Spontaneous Inhibitory Postsynaptic Currents
The reduction in spontaneous synaptic activity we observed after CSD could be due to reduced excitatory input, increased inhibitory input, or both. EPSC recordings were consistent with reduced presynaptic excitatory input, but a larger excitatory postsynaptic response. To examine the effect of inhibitory inputs, we measured changes in sIPSCs and mIPSCs (confirmed at the end of each experiment with GABA blockade).
Similar to sEPSCs, the frequency of sIPSCs was reduced at 5 and 30 min post-CSD (intervals: pre: 403.2 ± 126 ms vs. 5 min post-CSD: 726 ± 166.4 ms vs. 30 min post-CSD: 604.9 ± 127.5 ms; 5 min: P = 3.13 × 10 −28 and 30 min: P = 0.09; KS test; 5 min post-CSD: n = 8 cells, 8 mice and 30 min post-CSD: n = 9 cells, 9 mice; Fig. 6A , B ). In contrast to the mixed effects seen with sEPSCs, sIPSCs showed an increase in amplitude both 5 and 30 min after CSD (pre: 23.09 ± 2.72 pA vs. 5 min post-CSD: 34.91 ± 3.69 pA vs. 30 min post-CSD: 32.17 ± 4.33 pA; 5 min: P = 1.67 × 10 −64 and 30 min: P = 4.67 × 10 −20 ; KS test; Fig. 6C ).
Figure 6.
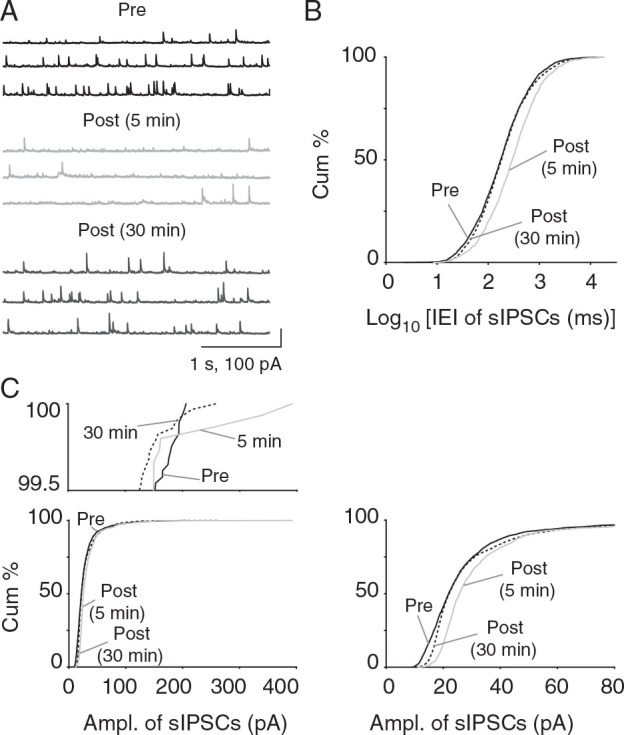
Reduced frequency and consistently increased amplitude of sIPSCs after CSD in vitro. ( A ) Voltage-clamp recordings of sIPSCs from Layer 2/3 pyramidal cells after CSD. Note the increase in amplitude of IPSCs post-CSD. ( B ) There was a significant reduction in sIPSC frequency 5 min post-CSD (rightward shift towards longer intervals; P = 3.13 × 10 −28 ; 2-sample KS test; 5 min post-CSD: n = 8 cells, 8 mice and 30 min post-CSD: n = 9 cells, 9 mice) but not 30 min after CSD ( P = 0.09; 2-sample KS test). ( C ) Consistent increase in sIPSC amplitude 5 and 30 min after CSD (5 min post-CSD: P = 1.67 × 10 −64 , 30 min post-CSD: P = 4.67 × 10 −20 ; KS test; 5 min post-CSD: n = 8 cells, 8 mice and 30 min post-CSD: n = 9 cells, 9 mice). Magnified cumulative plot above shows larger amplitude events observed only in post-CSD group. Magnified cumulative plot to right shows expansion of histogram for lower amplitude range—rightward shift indicates larger EPSC amplitude for 5 and 30 min post-CSD groups.
Decreased Frequency, Increased Amplitude of Miniature Inhibitory Postsynaptic Currents
Next, we examined the effects of CSD on AP-independent inhibitory synaptic transmission (control: n = 5 cells, 5 mice and CSD: n = 6 cells, 6 mice). Similar to mEPSCs, mIPSC frequency was significantly reduced in the 30 min post-CSD group ( P = 2.34 × 10 −58 ; KS test; Fig. 7A , B ), and mIPSC amplitude was significantly increased (control vs. CSD: 15.53 ± 2.6 vs. 18.31 ± 1.65 pA; P = 1.80 × 10 −99 ; KS test, Fig. 7C ).
Figure 7.
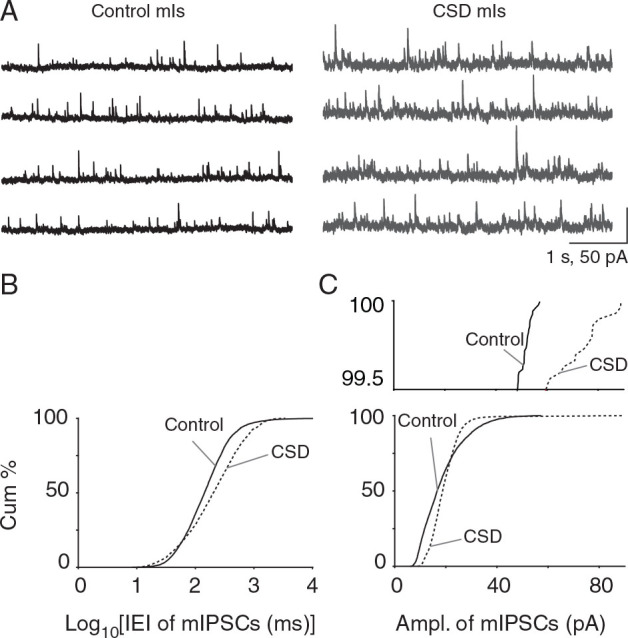
Reduced frequency and increased amplitude of mIPSCs after CSD in vitro. ( A ) Recordings of mIPSCs from control and CSD groups. As with mEPSCs, TTX was delivered after CSD or sham. ( B ) There was a significant decrease in mIPSC frequency ( P = 2.34 × 10 −58 , KS-test; control: n = 5 cells, 5 mice and CSD: n = 6 cells, 6 mice). ( C ) A significant increase in mIPSC amplitude 30 min after CSD ( P = 1.80 × 10 −99 ; KS test). Magnified cumulative plot above shows larger than 40 pA IPSC events evident in post-CSD groups but not sham.
We interpreted the decrease in s- and mIPSC frequency as evidence of decreased presynaptic interneuronal AP firing or reduction in transmitter release probability at inhibitory synapses—this could be expected to mitigate the effects of s/mEPSC reductions, though presumably not enough to change the depression of sPSPs observed in vivo. The consistent increase in amplitude of s/mIPSCs at both 5 and 30 min could be expected to oppose the increased s/mEPSC amplitude and contribute to the overall decrement in sPSP amplitude after CSD. However, which effect is dominant ultimately depends on the relative strength of excitation and inhibition. We approached this question in 2 ways:
Increased Decay Constant of Miniature Postsynaptic Currents
mE/IPSC kinetics can be used to assess changes excitatory and inhibitory synaptic strength at both pre- and postsynaptic levels ( Otis et al. 1994 ; Jones and Westbrook 1997 ; Turrigiano et al. 1998 ; Postlethwaite et al. 2007 ). We calculated rise and decay time constants for mE- and IPSCs before and after CSD. We saw no change in rise and decay time constants for mEPSCs after CSD (rise time constant, control: 0.35 ± 0.08 ms vs. CSD: 0.52 ± 0.16 ms; decay time constant, control: 3.35 ± 0.7 ms vs. CSD: 4.0 ± 0.78 ms; both P > 0.05, unpaired t -test). In contrast to mEPSCs, while there was no difference in mIPSC rise time constants after CSD (control: 0.29 ± 0.05 ms vs. CSD: 0.82 ± 0.23 ms; P > 0.05, unpaired t -test), there was a significant increase in decay time constants (7.55 ± 0.61 and 11.18 ± 0.47 ms in control and CSD neurons, respectively; P = 0.001, unpaired t -test). This is evidence of an increased postsynaptic inhibitory response, not seen for excitation, which could help explain the large decrement in sPSP amplitude after CSD in vivo.
Inhibitory Shift in Synaptic Current Balance
Next, we examined the balance of excitation and inhibition by comparing the ratio of both amplitude and charge for s- and mE/IPSCs. sEPSC/sIPSC amplitude ratio was significantly reduced post-CSD ( P = 2.59 × 10 −24 ; 2-sample KS test; n = 5 cells, 5 mice; Fig. 8A ). mEPSC/mIPSC amplitude ratio was similarly reduced (2.51 × 10 −12 ; KS test; n = 4–6 cells, 4–6 mice; Fig. 8B ). However, it is possible that our amplitude-based analysis, which is based on individual thresholded events, missed counting smaller events that might contribute substantially to excitation/inhibition balance. We used charge analysis, which computes area under the current trace, to control for this possibility. Here again both sEPSC/sIPSC ( P = 0.004; KS test, n = 5 cells, 5 mice; Fig. 8C ) and mEPSC/mIPSC ratios ( P = 1.08 × 10 −17 ; 2-sample KS test, n = 4–6 cells, 4–6 mice; Fig. 8D ) were significantly reduced after CSD. We concluded that there was a significant increase in relative inhibitory synaptic strength after CSD, likely explaining the persistent reduction of sPSP amplitude observed in vivo.
Figure 8.
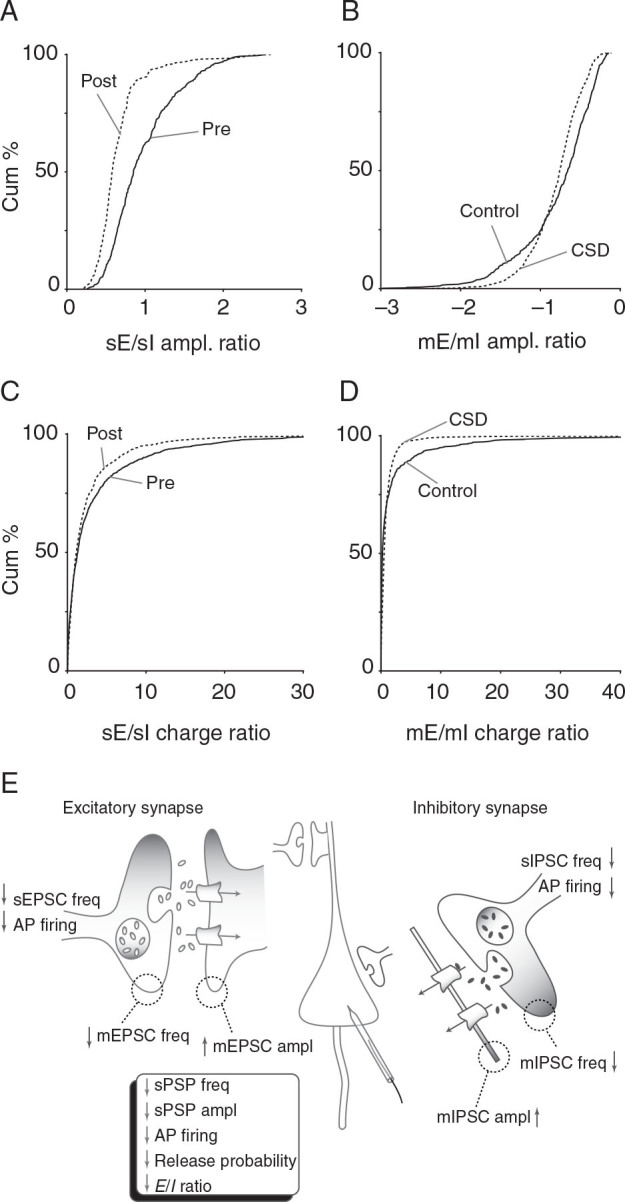
Inhibitory shift in excitation/inhibition ratio after CSD. Different measures of excitation/inhibition balance all show inhibitory shift 30 min after CSD. ( A ) Decrease in ratio of sEPSC to sIPSC amplitude ( P = 2.59 × 10 −24 , KS-test) in post-CSD group. ( B ) Decrease in ratio mEPSC to mIPSC amplitude ( P = 2.51 × 10 −12 , 2-sample KS test). ( C ) Decrease in ratio of sEPSC to sIPSC charge ( P = 0.004, KS test). ( D ) Decrease in ratio of mEPSC to mIPSC charge ( P = 1.08 × 10 −17 ; 2-sample KS test) . ( E ) Schematic shows summary of intrinsic and synaptic changes 30 min after CSD. Box summarizes overall findings. Presynaptic decrease in AP firing and release probability combine with a postsynaptic inhibitory shift in excitation/inhibition ratio to generate the sustained depression in neuronal activity observed after CSD.
Discussion
Silencing of Neuronal Activity After CSD
A signature feature of CSD—discovered before the depolarization itself—is the profound silencing of spontaneous neuronal activity after passage of the wave (in vivo: Leao 1944 ; Grafstein 1956 ; Marshall 1959 ; Sugaya et al. 1975 ; in vitro :Jing et al. 1991 ; Takano et al. 2007 ; Aiba and Shuttleworth 2012 ). This feature is of clear importance to all disorders affected by CSD and indeed all SD in general ( Dreier 2011 ; Theriot et al. 2012 ), yet its mechanisms are only beginning to be understood. Our principal finding is that CSD induces distinct but complementary pre- and postsynaptic changes that combine to explain this long-lasting suppression (Fig. 8E ).
Intrinsic Membrane Properties Are Unlikely to Explain Post-CSD Silencing
A possible explanation for the pronounced decrement in spontaneous neuronal activity after CSD is a change in intrinsic membrane properties. We did observe significant changes in these properties, though overall they were less dramatic than synaptic changes (Fig. 2 ).
Resting Membrane Potential Depolarization
RMP was mildly but significantly depolarized 30 min post-CSD. A depolarized membrane potential could explain the reduced amplitude of sPSPs that also occurred 30 min after CSD, due to a reduced driving force for excitation and increased driving force for inhibition. However, we observed a similar but larger reduction in sPSP amplitude 5 min post-CSD, with no concomitant change in RMP. We tested for a correlation between PSP amplitude and membrane potential found none (post-CSD 5 min, Pearson r = 0.58, P = 0.31; post-CSD 30 min, Pearson r = −0.22, P = 0.73).
We thus consider it unlikely that RMP changes played a major role in the suppression of spontaneous neuronal activity. However, the RMP changes may provide an explanation for a second extracellular negative direct current shift that occurs tens of minutes after CSD ( Chang et al. 2010 ); compare Figure 1 with Figure 4a of Chang et al.). We had speculated that these changes could be due a variety of factors including tissue impedance and astrocytic or vascular depolarization, in addition to neuronal membrane depolarization. Though our findings do not rule out these other causes, they confirm neuronal depolarization as part of the mechanism of the second direct current shift.
The ultimate causes of depolarized membrane potential, sometime after passage of the CSD wave, are unclear. Depolarized RMP could indicate deficiency in energy metabolism, as membrane potential is ultimately ATP dependent ( Calabresi et al. 1995 ; Balestrino et al. 1999 ), and long-lasting changes in neurovascular coupling and metabolism occur after CSD ( Piilgaard and Lauritzen 2009 ; Chang et al. 2010 ). Another possible explanation for depolarized RMP after CSD is downregulation of potassium leak conductance ( Hille 2001 ). Because we saw no changes in RMP following control saline ejection, and access resistance was maintained in all of our 30 min recordings, we do not suspect the RMP changes are artifactual to our whole-cell recording conditions.
Increase in Rheobase
Changes in spontaneous neuronal activity after CSD could also be caused by decreases in membrane input resistance. We observed a change in rheobase 5 min after CSD that might be reflective of a decreased membrane input resistance. We did not detect any significant changes in membrane input resistance; indeed there was a trend for increase at 5 min post-CSD (pre, 126.4 ± 6.94 vs. 5 min post-CSD, 135.0 ± 5.94 MΩ). However our recordings of membrane input resistance may reflect changes at the soma and not at the level of dendrites where most excitatory synapses are located ( Bernander et al. 1994 ). A shunting mechanism underlying CSD-induced depression of sPSPs thus cannot be ruled out at 5 min; however it is less likely to explain the changes we observed at 30 min and >60 min after the event.
Significant changes in intrinsic membrane properties would be expected to change the probability of AP generation, and thus the slope of the frequency–current (F–I) curve. However, we saw no significant changes in F–I characteristics at any of the post-CSD time points. This is consistent with observations that APs are energetically less expensive than synaptic potentials ( Alle et al. 2009 ; Belanger et al. 2011 ), and thus might be expected to recover faster after CSD. Interestingly, our in vivo F–I curve results diverge from in vitro studies, where increases in evoked firing (as well as reduced membrane input resistance) were observed ( Ghadiri et al. 2012 ). Taken together, our data reveal relatively mild changes in neuronal intrinsic properties after CSD that do not appear to explain the long-term synaptic silencing we observed.
Changes in Synaptic Properties Account for Post-CSD Silencing
CSD Decreases Presynaptic Release Probability
The major changes we observed in vivo after CSD were decreases in the frequency and amplitude of synaptic activity, and a (likely concomitant) decrease in AP firing. Though these changes could have explanations based on altered intrinsic membrane properties, our data suggest that this is unlikely. We thus examined excitatory and inhibitory synaptic properties in greater depth at the in vitro level . In contrast to intrinsic properties, we found that changes in excitatory and inhibitory synaptic transmission explained the post-CSD phenotype well.
Frequency of synaptic events is primarily controlled by presynaptic factors such as the number of synaptic terminals, the activity of presynaptic neurons, or the probability of vesicle release from the terminal ( Kerchner and Nicoll 2008 ). Though CSD causes transient dendritic beading during passage of the wave ( Takano et al. 2007 ), there is no evidence that CSD causes changes in presynaptic terminal number, so we focused on the latter 2 factors.
Both whole-cell and loose-cell-attached recordings in vivo showed a significant decrease in AP firing after CSD (see Fig. 3F ). This clearly shows a decrease in net neuronal excitability, which we ultimately attribute to an inhibitory shift in excitation/inhibition balance (see below). Though we only recorded after CSD from pyramidal cells, our in vitro data show that both sEPSC and sIPSC frequency are reduced after CSD. Thus, CSD likely affects the sub- and supra-threshold activity of both excitatory and inhibitory neurons.
Our data also show that CSD affects presynaptic release probability. Both spontaneous and miniature E- and IPSC frequency was significantly reduced 30 min after wave passage. sE/IPSC events can be due to both AP-dependent and -independent processes, but mE/IPSCs are AP independent, and thus provide evidence for an effect on presynaptic release machinery rather than just global cellular excitability. A potential candidate for presynaptic activity suppression is adenosine, which acts at presynaptic A1 receptors to inhibit glutamate release at excitatory terminals ( Yoon and Rothman 1991 ; Prince and Stevens 1992 ; Dunwiddie and Masino 2001 ), by inhibiting calcium influx through voltage-gated channels ( Yawo and Chuhma 1993 ; Wu and Saggau 1994 ), or by interfering with phosphorylation of presynaptic proteins ( Scanziani et al. 1992 ; Thompson et al. 1993 ). Indeed adenosine is released during and after CSD ( Kaku et al. 1994 ; Lindquist and Shuttleworth 2012 , 2014 ), and is responsible for evoked PSP suppression during and a few minutes after the passage of the wave ( Lindquist and Shuttleworth 2012 ). However, the duration of adenosine increase after CSD is on the order of 5–10 min unless metabolism is impaired ( Lindquist and Shuttleworth 2014 ); thus, purinergic activity is less likely to explain our findings at 30 min or >60 min after wave passage. Additionally, there is evidence that adenosine does not affect inhibitory neurotransmitter release ( Yoon and Rothman 1991 ; Prince and Stevens 1992 ). Activity at GABA B ( Laviv et al. 2010 ) and cannabinoid receptors ( McAllister and Glass 2002 ; Chevaleyre et al. 2006 ) could cause decreases in release probability, but one might expect similar release kinetics to adenosine—that is, associated primarily with the wave itself, not the tonic release necessary to exert effects 30 min after wave passage. Depletion of transmitter pools is a possible mechanism for decreased excitatory and inhibitory release probability, but repletion of these pools is very rapid compared with the time scale of post-CSD suppression (even reserve pools replete within a few minutes; Rizzoli and Betz 2005 ) so it is unlikely to account for the long-lasting effects we observe. Finally, it is possible that persistent disruptions in ionic homeostasis, membrane polarization, and metabolic activity could contribute to reduced transmitter release probability. There is ample evidence of metabolic compromise after CSD, mainly associated with altered neurovascular coupling ( Scheckenbach et al. 2006 ; Piilgaard and Lauritzen 2009 ; Chang et al. 2010 ). However, these changes are associated with tissue ( Chang et al. 2010 ) and cellular (Figs 1 and 2 ) depolarization, which one might expect to increase release probability.
At the 30 min time scale after CSD, it is quite possible that Hebbian plasticity mechanisms are operant [homeostatic plasticity changes are thought to occur on longer timescales, in response to longer-duration stimuli ( Turrigiano 2012 )]. It is possible that the reduced release probability we observe represents presynaptic long-term depression (LTD) induced by CSD. We have observed changes consistent with LTD in evoked sensory activity after CSD ( Theriot et al. 2012 ). Moreover, presynaptic LTD is known to be induced by presynaptic NMDA receptor activation ( Atwood et al. 2014 ), which occurs during CSD ( Zhou et al. 2013 ). Interestingly, though purinergic and cannabinoid effects are typically thought of as reversible, there is some evidence that A1 and CB1 receptor-mediated activity can also cause LTD ( Heifets and Castillo 2009 ; Atwood et al. 2014 ). Adenosine ( Lindquist and Shuttleworth 2012 ) and possibly cannabinoids ( Kazemi et al. 2012 ) released during CSD thus might not have to be tonically active to exert the long-term effects we observe ( Castillo 2012 ). Finally, given the significant astrocytic depolarization that accompanies CSD ( Sugaya et al. 1975 ) and the role of astrocytes in both excitatory and inhibitory plasticity ( De et al. 2015 ; Haydon and Nedergaard 2015 ), the potential role of gliotransmission also needs to be considered. Whatever the underlying stimulus, our data clearly show that CSD reduces presynaptic release probability, through a combination of reduced AP firing and effects on presynaptic release machinery. We speculate that the latter is due to presynaptic LTD; the former is likely due to increased inhibitory tone (see below).
CSD Causes Inhibitory Shift in Excitation/Inhibition Ratio
After CSD in vivo , we observed a significant decrease in the amplitude of spontaneous PSPs that lasted beyond the maximum duration of our experiments (>90 min). In vitro experiments showed an increase in the amplitude of both spontaneous and miniature E- and IPSCs, but the IPSC changes were temporally more consistent, and were of greater amplitude, leading to a shift in the excitation/inhibition ratio toward inhibition (see Fig. 8 ). We believe this increase in inhibitory tone, rather than a change in intrinsic properties, is responsible for the large decrement in AP firing after CSD. Reduced AP firing, like PSP amplitude changes, lasted at least an hour after the event. We believe the increased inhibitory tone also explains the >1 h decrement in the amplitude of upstates, as these events are ultimately determined by the balance of network-based excitation and inhibition impinging on a cell ( Wilson 2008 ). The change in upstate phenotypes is relevant, because it is evidence that CSD exerts effect on network characteristics, in addition to cellular excitability.
The amplitude of PSCs is determined largely by postsynaptic mechanisms, most prominently the number and conductance of postsynaptic receptors ( Jonas et al. 1993 ; Perkel and Nicoll 1993 ). However, presynaptic mechanisms can also contribute, via increases in vesicle size or release synchrony, if postsynaptic receptors are not saturated ( Frerking et al. 1995 ; Forti et al. 1997 ; Liu et al. 1999 ). A reduced number of larger presynaptic release events could provide a single unifying explanation for the decreased frequency and increased amplitude we observed in both spontaneous and miniature E- and IPSCs after CSD.
To determine whether pre- or postsynaptic mechanisms were involved in post-CSD synaptic amplitude changes, we examined the amplitude distribution and kinetics of mPSCs. In both control and post-CSD groups, for both mE- and mIPSCS, amplitude histograms were unimodal, with no significant difference in shape. In the event of increased vesicle synchrony, a multipeaked histogram would have been expected ( Bekkers et al. 1990 ; Edwards et al. 1990 ; Ulrich and Luscher 1993 ). Synchronous multivesicular release also produces a prolonged rise time constant because of temporal jitter in the time between vesicle releases ( Otis et al. 1994 ; Raghavachari and Lisman 2004 ; Lisman et al. 2007 ). We observed no significant differences in rise time between control and post-CSD mE- or mIPSCs. Thus, it appears unlikely that synchronous multiquantal release is responsible for increased amplitude of mPSCs after CSD. Changes in quantal/vesicular size are very unlikely in our preparations: they are typically associated with increases rather than decreases in PSC frequency ( Gao et al. 1998 ; El-Husseini et al. 2000 ), and to our knowledge they have not been demonstrated on the time scale we observed. Thus, postsynaptic rather than presynaptic mechanisms likely account for the changes in post-CSD synaptic amplitude.
Both receptor insertion and increased channel conductance could contribute to a postsynaptic increase in current amplitude. Unfortunately, the nature of our data did not allow for the nonstationary noise analysis that could dissect these contributions ( Otis et al. 1994 ). In epilepsy models, which arguably have conditions most resembling CSD, (GABA) receptor insertion rather than increased channel conductance is observed ( Nusser et al. 1998 ), though this certainly does not rule out conductance changes in CSD. The insertion and removal of AMPA receptors in either Hebbian or homeostatic plasticity paradigms is well known ( Malenka and Bear 2004 ; Shepherd and Huganir 2007 ; Turrigiano 2012 ). Here again, we favor Hebbian plasticity mechanisms over homeostatic plasticity, due to the time course over which we observed the changes. Consistent with this, BDNF, which is known to be released after CSD ( Kawahara et al. 1997 ) promotes postsynaptic potentiation within minutes after high-frequency ( Jia et al. 2010 ) or weak ( Kovalchuk et al. 2002 ) synaptic stimulation. What is interesting is that in the case of CSD, excitatory and inhibitory synaptic activity moves in the same direction (i.e., increased amplitude in both, albeit a significantly larger change in inhibition). Opposite activity for excitation versus inhibition is more typically observed in plasticity paradigms ( O'Brien et al. 1998 ; Turrigiano et al. 1998 ; Malenka and Bear 2004 ; Shepherd and Huganir 2007 ). We suspect that the diffuse depolarization of CSD resulted in potentiation of both excitatory and inhibitory synapses.
A remaining question is how synaptic inhibition was favored over excitation. Interneurons differ in structure, intrinsic excitability, and plasticity mechanisms from pyramidal cells. It is not difficult to envision differential effects of CSD on the 2 cell types. It is also possible that the location of inhibitory synapses contributed to a greater net inhibition—though there is great diversity, inhibitory synapses tend to be located where they can exert a stronger influence on excitability than excitatory synapses ( Markram et al. 2004 ). Diffuse potentiation of all excitatory and inhibitory synapses on a cell could thus lead to a net inhibitory shift in tone. Though the inhibitory shift appears to “make sense” from a homeostatic perspective, its specific mechanisms await future work.
Cellular Electrophysiology of CSD in In Vivo
The principal purpose of our experiments was to examine the intrinsic and synaptic effects of CSD; however, our recordings also allow us to examine the wave itself and compare our observations with others made with complementary techniques. In 2 pioneering papers, Higashida et al. ( 1974 ) and Sugaya et al. ( 1975 ) used sharp electrodes to record from both neurons and astrocytes during CSD in anesthetized cats. Sharp and whole-cell recordings each have advantages and disadvantages. Sharp recordings minimally perturb the intracellular ionic environment; however, they introduce a large membrane leak current that can reduce the cell's membrane input resistance ( Spruston and Johnston 1992 ; Li et al. 2004 ). Whole-cell recordings induce minimal leak current and have greater signal-to-noise for synaptic events, but dialyze the intracellular compartment ( Karmazinova and Lacinova 2010 ). It is thus instructive to consider sharp and whole-cell recordings together. In addition, given the rarity of in vivo single cell recordings, it is important to compare both in vivo techniques with in vitro work.
Comparing our work with Higashida et al. ( 1974 ) and Sugaya et al. ( 1975 ), we found larger changes in membrane potential: 33–72 mV baseline to peak potential versus 20–41 mV in their sharp recordings ( Higashida et al. 1974 ). Our observations are closer in depolarization amplitude to recordings from brain slices, where control over membrane variables is presumably tighter than in vivo ( Snow et al. 1983 ; Czeh et al. 1993 ; Muller and Somjen 2000 ; Gniel and Martin 2010 ; also see Table 1 ). Interestingly, sharp recordings from brain slices also show a relatively large amplitude (∼40 mV) depolarization ( Mody et al. 1987 ; Ghadiri et al. 2012 ). The smaller depolarization values obtained with in vivo sharp recordings may be due to the membrane leakage associated with this type of recording, which might be expected to be larger in vivo ( Sugaya et al. 1975 ) than in vitro ( Snow et al. 1983 ; Mody et al. 1987 ; Ghadiri et al. 2012 ). On the other hand, the larger depolarization values obtained in whole-cell recordings could be due to internal solution composition (e.g., by generating a larger driving force for depolarization). It is also noteworthy that only 3 studies of CSD-Higashida et al. ( 1974 ), Snow et al. ( 1983 ), and Ghadiri et al. ( 2012 )-measured true transmembrane voltage by placing the ground electrode adjacent to the recorded cell and thus accounted for the large changes in extracellular ionic concentration that accompany the wave ( Somjen 2001 ). Snow et al. ( 1983 ) and Ghadiri et al. ( 2012 ) observed a large (>40 mV) depolarization.
We also observed a high incidence of positive membrane potential at peak depolarization (Fig. 1 and Table 1 ), which is relatively infrequently reported in prior in vivo and in vitro work ( Higashida et al. 1974 ; Sugaya et al. 1975 ; Snow et al. 1983 ; Gniel and Martin 2010 ; Ghadiri et al. 2012 ). This again could be due to the composition of our internal solution, though it did not vary significantly from other whole-cell recordings (e.g., Gniel and Martin 2010 ; the other compared studies are all sharp recordings). Another possible confounding factor could be incompletely corrected junction potential, though we had no difficulty correcting for junction potential in vivo or in vitro, and we verified in 3 separate in vivo recordings that the correction (Pipette Offset) was maintained after breaking out of whole-cell recording (data not shown).
It is possible that positive deflections during CSD are simply under-recognized. The 3 studies that account for transmembrane voltage, in vivo ( Higashida et al. 1974 ) and in vitro ( Snow et al. 1983 ; Ghadiri et al. 2012 ) all show positive membrane potential deflections. In vivo, 4/5 recorded neurons showed positive deflections; the incidence was lower in the slice studies—1/11 neurons in Snow et al. ( 1983 ) and reported “sometimes' in Ghadiri et al. ( 2012 ). Interestingly, we see a similar difference between our in vivo recordings (+10 mV positive deflections; 13/15 cells with positive deflections) and our in vitro recordings (maximum +1.8 mV; 2/8 positive deflections). It should be noted that an excursion to positive values during CSD is not problematic from a biophysical point of view—APs routinely enter the positive range, and modeling studies of CSD also generate positive values ( Wei et al. 2014 ). Ultimately the endpoint of CSD depends on the driving forces and conductances for transmembrane charge flow—there is no constraint that prevents these flows generating positive membrane potentials.
Duration of depolarization, at approximately 3.5 min in Layer 2/3 (see Fig. 1 ), was longer than that observed by Higashida et al. and Sugaya et al. [2–2.5 min at 1000 μm depth ( Higashida et al. 1974 ) and at the cortical surface ( Sugaya et al. 1975 )] and also longer than that observed in most (∼1.5 min in hippocampus: Snow et al. 1983 ; Mody et al. 1987 ), but not all (∼3–6 min in layer 5 somatosensory cortex: Gniel and Martin 2010 ; Ghadiri et al. 2012 ) brain slice recordings. It is possible that anesthetic variables (isoflurane/urethane vs. pentobarbital) and species (mouse vs. cat) account for some of the in vivo differences. It is also possible that differences between our recordings and others are due to the cell and cortical layer recorded. CSD durations from most ( Snow et al. 1983 ; Mody et al. 1987 ) but not all ( Gniel and Martin 2010 ; Ghadiri et al. 2012 ) slice CSD recordings are from hippocampus. Both amplitude and duration of CSD vary by hippocampal and cortical layer ( Herreras and Somjen 1993 ; Gniel and Martin 2010 ). At least for Layer 2/3 of sensory cortex, the CSD characteristics of 2 populations of pyramidal cells were fairly consistent: in our recordings from RS and IB cells, we did not observe any significant differences between depolarization duration, number of APs firing, and peak amplitude changes during depolarization (see Supplementary Materials ).
Conclusion
In conclusion, we show that a long-lasting reduction in synaptic activity underlies the suppression of spontaneous neuronal activity after CSD, and we elucidate the mechanisms of this reduction. A decrease in PSP frequency is mediated by a reduction in presynaptic release probability and decreased presynaptic AP firing. A decrease in PSP amplitude and AP firing is caused by an inhibitory shift in postsynaptic excitation/inhibition ratio. These pre- and postsynaptic mechanisms of post-CSD suppression may explain the effects CSD exerts on circuits of disease, and they offer specific targets for intervention.
Supplementary Material
Supplementary material can be found here. .
Funding
This study was supported by the National Institutes of Health (R01 NS085413, R21 NS083010) and the Department of Defense (CDMRP PR 100060, PR 130373).
Supplementary Material
Notes
We thank Drs William Shuttleworth, Jeremy Theriot, and Jay Spampanato for valuable comments and suggestions. Conflict of Interest : None declared.
References
- Aiba I , Shuttleworth CW . 2012. . Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons . J Physiol . 590 : 5877 – 5893 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba I , Shuttleworth CW . 2014. . Characterization of inhibitory GABA-A receptor activation during spreading depolarization in brain slice . PLoS One . 9 : e110849 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alle H , Roth A , Geiger JR . 2009. . Energy-efficient action potentials in hippocampal mossy fibers . Science . 325 : 1405 – 1408 . [DOI] [PubMed] [Google Scholar]
- Atwood BK , Lovinger DM , Mathur BN . 2014. . Presynaptic long-term depression mediated by Gi/o-coupled receptors . Trends Neurosci . 37 : 663 – 673 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M , Young J , Aitken P . 1999. . Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices . Brain Res . 838 : 37 – 44 . [DOI] [PubMed] [Google Scholar]
- Bekkers JM , Richerson GB , Stevens CF . 1990. . Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices . Proc Natl Acad Sci USA . 87 : 5359 – 5362 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M , Allaman I , Magistretti PJ . 2011. . Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation . Cell Metab . 14 : 724 – 738 . [DOI] [PubMed] [Google Scholar]
- Bernander O , Koch C , Douglas RJ . 1994. . Amplification and linearization of distal synaptic input to cortical pyramidal cells . J Neurophysiol . 72 : 2743 – 2753 . [DOI] [PubMed] [Google Scholar]
- Brecht M , Roth A , Sakmann B . 2003. . Dynamic receptive fields of reconstructed pyramidal cells in layers 3 and 2 of rat somatosensory barrel cortex . J Physiol . 553 : 243 – 265 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P , Pisani A , Mercuri NB , Bernardi G . 1995. . Hypoxia-induced electrical changes in striatal neurons . J Cereb Blood Flow Metab . 15 : 1141 – 1145 . [DOI] [PubMed] [Google Scholar]
- Cao Y , Welch KM , Aurora S , Vikingstad EM . 1999. . Functional MRI-BOLD of visually triggered headache in patients with migraine . Arch Neurol . 56 : 548 – 554 . [DOI] [PubMed] [Google Scholar]
- Castillo PE . 2012. . Presynaptic LTP and LTD of excitatory and inhibitory synapses . Cold Spring Harb Perspect Biol . 4 : a005728 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC , Shook LL , Biag J , Nguyen EN , Toga AW , Charles AC , Brennan KC . 2010. . Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression . Brain . 133 : 996 – 1012 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V , Takahashi KA , Castillo PE . 2006. . Endocannabinoid-mediated synaptic plasticity in the CNS . Annu Rev Neurosci . 29 : 37 – 76 . [DOI] [PubMed] [Google Scholar]
- Czeh G , Aitken PG , Somjen GG . 1993. . Membrane currents in CA1 pyramidal cells during spreading depression (SD) and SD-like hypoxic depolarization . Brain Res . 632 : 195 – 208 . [DOI] [PubMed] [Google Scholar]
- De PM , Brunel N , Volterra A . 2015. . Astrocytes: orchestrating synaptic plasticity? Neuroscience . 10.1016/j.neuroscience.2015.04.001 . [DOI] [PubMed] [Google Scholar]
- Dreier JP . 2011. . The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease . Nat Med . 17 : 439 – 447 . [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV , Masino SA . 2001. . The role and regulation of adenosine in the central nervous system . Annu Rev Neurosci . 24 : 31 – 55 . [DOI] [PubMed] [Google Scholar]
- Edwards FA , Konnerth A , Sakmann B . 1990. . Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study . J Physiol . 430 : 213 – 249 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE , Schnell E , Chetkovich DM , Nicoll RA , Bredt DS . 2000. . PSD-95 involvement in maturation of excitatory synapses . Science . 290 : 1364 – 1368 . [PubMed] [Google Scholar]
- Fabricius M , Fuhr S , Bhatia R , Boutelle M , Hashemi P , Strong AJ , Lauritzen M . 2006. . Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex . Brain . 129 : 778 – 790 . [DOI] [PubMed] [Google Scholar]
- Forti L , Bossi M , Bergamaschi A , Villa A , Malgaroli A . 1997. . Loose-patch recordings of single quanta at individual hippocampal synapses . Nature . 388 : 874 – 878 . [DOI] [PubMed] [Google Scholar]
- Frerking M , Borges S , Wilson M . 1995. . Variation in GABA mini amplitude is the consequence of variation in transmitter concentration . Neuron . 15 : 885 – 895 . [DOI] [PubMed] [Google Scholar]
- Gao BX , Cheng G , Ziskind-Conhaim L . 1998. . Development of spontaneous synaptic transmission in the rat spinal cord . J Neurophysiol . 79 : 2277 – 2287 . [DOI] [PubMed] [Google Scholar]
- Ghadiri MK , Kozian M , Ghaffarian N , Stummer W , Kazemi H , Speckmann EJ , Gorji A . 2012. . Sequential changes in neuronal activity in single neocortical neurons after spreading depression . Cephalalgia . 32 : 116 – 124 . [DOI] [PubMed] [Google Scholar]
- Gniel HM , Martin RL . 2010. . Changes in membrane potential and the intracellular calcium concentration during CSD and OGD in layer V and layer II/III mouse cortical neurons . J Neurophysiol . 104 : 3203 – 3212 . [DOI] [PubMed] [Google Scholar]
- Grafstein B . 1956. . Mechanism of spreading cortical depression . J Neurophysiol . 19 : 154 – 171 . [DOI] [PubMed] [Google Scholar]
- Hadjikhani N , Sanchez Del RM , Wu O , Schwartz D , Bakker D , Fischl B , Kwong KK , Cutrer FM , Rosen BR , Tootell RB et al. . 2001. . Mechanisms of migraine aura revealed by functional MRI in human visual cortex . Proc Natl Acad Sci USA . 98 : 4687 – 4692 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabisky B , Parada I , Buckmaster PS , Prince DA . 2010. . Excitatory input onto hilar somatostatin interneurons is increased in a chronic model of epilepsy . J Neurophysiol . 104 : 2214 – 2223 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA , Strong AJ , Fabricius M , Manning A , Bhatia R , Dreier JP , Mazzeo AT , Tortella FC , Bullock MR . 2009. . Spreading depolarizations and late secondary insults after traumatic brain injury . J Neurotrauma . 26 : 1857 – 1866 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG , Nedergaard M . 2015. . How do astrocytes participate in neural plasticity? Cold Spring Harb Perspect Biol . 7 : a020438 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD , Castillo PE . 2009. . Endocannabinoid signaling and long-term synaptic plasticity . Annu Rev Physiol . 71 : 283 – 306 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreras O , Somjen GG . 1993. . Propagation of spreading depression among dendrites and somata of the same cell population . Brain Res . 610 : 276 – 282 . [DOI] [PubMed] [Google Scholar]
- Higashida H , Mitarai G , Watanabe S . 1974. . A comparative study of membrane potential changes in neurons and neuroglial cells during spreading depression in the rabbit . Brain Res . 65 : 411 – 425 . [DOI] [PubMed] [Google Scholar]
- Hille B . 2001. . Ion channels of excitable membranes . Sunderland: ( MA: ): Sinauer Associates, Inc; . [Google Scholar]
- Hughes S , Crunelli V . 2013. . UP states rise from the depths . Nat Neurosci . 16 : 115 – 117 . [DOI] [PubMed] [Google Scholar]
- Jia Y , Gall CM , Lynch G . 2010. . Presynaptic BDNF promotes postsynaptic long-term potentiation in the dorsal striatum . J Neurosci . 30 : 14440 – 14445 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing J , Aitken PG , Somjen GG . 1991. . Lasting neuron depression induced by high potassium and its prevention by low calcium and NMDA receptor blockade . Brain Res . 557 : 177 – 183 . [DOI] [PubMed] [Google Scholar]
- Jonas P , Major G , Sakmann B . 1993. . Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus . J Physiol . 472 : 615 – 663 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV , Westbrook GL . 1997. . Shaping of IPSCs by endogenous calcineurin activity . J Neurosci . 17 : 7626 – 7633 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku T , Hada J , Hayashi Y . 1994. . Endogenous adenosine exerts inhibitory effects upon the development of spreading depression and glutamate release induced by microdialysis with high K+ in rat hippocampus . Brain Res . 658 : 39 – 48 . [DOI] [PubMed] [Google Scholar]
- Karmazinova M , Lacinova L . 2010. . Measurement of cellular excitability by whole cell patch clamp technique . Physiol Res . 59(Suppl. 1) : S1 – S7 . [DOI] [PubMed] [Google Scholar]
- Kawahara N , Croll SD , Wiegand SJ , Klatzo I . 1997. . Cortical spreading depression induces long-term alterations of BDNF levels in cortex and hippocampus distinct from lesion effects: implications for ischemic tolerance . Neurosci Res . 29 : 37 – 47 . [DOI] [PubMed] [Google Scholar]
- Kazemi H , Rahgozar M , Speckmann EJ , Gorji A . 2012. . Effect of cannabinoid receptor activation on spreading depression . Iran J Basic Med Sci . 15 : 926 – 936 . [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA , Nicoll RA . 2008. . Silent synapses and the emergence of a postsynaptic mechanism for LTP . Nat Rev Neurosci . 9 : 813 – 825 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K , Judkewitz B , Kano M , Denk W , Hausser M . 2008. . Targeted patch-clamp recordings and single-cell electroporation of unlabeled neurons in vivo . Nat Methods . 5 : 61 – 67 . [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y , Hanse E , Kafitz KW , Konnerth A . 2002. . Postsynaptic induction of BDNF-mediated long-term potentiation . Science . 295 : 1729 – 1734 . [DOI] [PubMed] [Google Scholar]
- Lauritzen M . 1994. . Pathophysiology of the migraine aura. The spreading depression theory . Brain . 117(Pt 1) : 199 – 210 . [DOI] [PubMed] [Google Scholar]
- Lauritzen M , Dreier JP , Fabricius M , Hartings JA , Graf R , Strong AJ . 2011. . Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury . J Cereb Blood Flow Metab . 31 : 17 – 35 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviv T , Riven I , Dolev I , Vertkin I , Balana B , Slesinger PA , Slutsky I . 2010. . Basal GABA regulates GABA(B)R conformation and release probability at single hippocampal synapses . Neuron . 67 : 253 – 267 . [DOI] [PubMed] [Google Scholar]
- Leao AA . 1944. . Spreading depression of activity in the cerebral cortex . J Neurophysiol . 7 : 359 – 390 . [DOI] [PubMed] [Google Scholar]
- Leao AA . 1947. . Further observations on the spreading depression of activity in the cerebral cortex . J Neurophysiol . 10 : 409 – 414 . [DOI] [PubMed] [Google Scholar]
- Lemieux M , Chauvette S , Timofeev I . 2015. . Neocortical inhibitory activities and long-range afferents contribute to the synchronous onset of silent states of the neocortical slow oscillation . J Neurophysiol . 113 : 768 – 779 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WC , Soffe SR , Roberts A . 2004. . A direct comparison of whole cell patch and sharp electrodes by simultaneous recording from single spinal neurons in frog tadpoles . J Neurophysiol . 92 : 380 – 386 . [DOI] [PubMed] [Google Scholar]
- Lindquist BE , Shuttleworth CW . 2012. . Adenosine receptor activation is responsible for prolonged depression of synaptic transmission after spreading depolarization in brain slices . Neuroscience . 223 : 365 – 376 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist BE , Shuttleworth CW . 2014. . Spreading depolarization-induced adenosine accumulation reflects metabolic status in vitro and in vivo . J Cereb Blood Flow Metab . 34 : 1779 – 1790 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE , Raghavachari S , Tsien RW . 2007. . The sequence of events that underlie quantal transmission at central glutamatergic synapses . Nat Rev Neurosci . 8 : 597 – 609 . [DOI] [PubMed] [Google Scholar]
- Liu G , Choi S , Tsien RW . 1999. . Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices . Neuron . 22 : 395 – 409 . [DOI] [PubMed] [Google Scholar]
- Malenka RC , Bear MF . 2004. . LTP and LTD: an embarrassment of riches . Neuron . 44 : 5 – 21 . [DOI] [PubMed] [Google Scholar]
- Markram H , Toledo-Rodriguez M , Wang Y , Gupta A , Silberberg G , Wu C . 2004. . Interneurons of the neocortical inhibitory system . Nat Rev Neurosci . 5 : 793 – 807 . [DOI] [PubMed] [Google Scholar]
- Marshall WH . 1959. . Spreading cortical depression of Leao . Physiol Rev . 39 : 239 – 279 . [DOI] [PubMed] [Google Scholar]
- McAllister SD , Glass M . 2002. . CB(1) and CB(2) receptor-mediated signalling: a focus on endocannabinoids . Prostaglandins Leukot Essent Fatty Acids . 66 : 161 – 171 . [DOI] [PubMed] [Google Scholar]
- McCormick DA , Shu Y , Hasenstaub A , Sanchez-Vives M , Badoual M , Bal T . 2003. . Persistent cortical activity: mechanisms of generation and effects on neuronal excitability . Cereb Cortex . 13 : 1219 – 1231 . [DOI] [PubMed] [Google Scholar]
- Mody I , Lambert JD , Heinemann U . 1987. . Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices . J Neurophysiol . 57 : 869 – 888 . [DOI] [PubMed] [Google Scholar]
- Moore CI , Nelson SB . 1998. . Spatio-temporal subthreshold receptive fields in the vibrissa representation of rat primary somatosensory cortex . J Neurophysiol . 80 : 2882 – 2892 . [DOI] [PubMed] [Google Scholar]
- Muller M , Somjen GG . 2000. . Na(+) dependence and the role of glutamate receptors and Na(+) channels in ion fluxes during hypoxia of rat hippocampal slices . J Neurophysiol . 84 : 1869 – 1880 . [DOI] [PubMed] [Google Scholar]
- Nusser Z , Hajos N , Somogyi P , Mody I . 1998. . Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses . Nature . 395 : 172 – 177 . [DOI] [PubMed] [Google Scholar]
- O'Brien RJ , Lau LF , Huganir RL . 1998. . Molecular mechanisms of glutamate receptor clustering at excitatory synapses . Curr Opin Neurobiol . 8 : 364 – 369 . [DOI] [PubMed] [Google Scholar]
- Otis TS , De KY , Mody I . 1994. . Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents . Proc Natl Acad Sci USA . 91 : 7698 – 7702 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkel DJ , Nicoll RA . 1993. . Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation . J Physiol . 471 : 481 – 500 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC , Hahn TT , Mehta M , Grinvald A , Sakmann B . 2003. . Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex . Proc Natl Acad Sci USA . 100 : 13638 – 13643 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D , Moskowitz MA . 2013. . Pathophysiology of migraine . Annu Rev Physiol . 75 : 365 – 391 . [DOI] [PubMed] [Google Scholar]
- Piilgaard H , Lauritzen M . 2009. . Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex . J Cereb Blood Flow Metab . 29 : 1517 – 1527 . [DOI] [PubMed] [Google Scholar]
- Postlethwaite M , Hennig MH , Steinert JR , Graham BP , Forsythe ID . 2007. . Acceleration of AMPA receptor kinetics underlies temperature-dependent changes in synaptic strength at the rat calyx of Held . J Physiol . 579 : 69 – 84 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA , Stevens CF . 1992. . Adenosine decreases neurotransmitter release at central synapses . Proc Natl Acad Sci USA . 89 : 8586 – 8590 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavachari S , Lisman JE . 2004. . Properties of quantal transmission at CA1 synapses . J Neurophysiol . 92 : 2456 – 2467 . [DOI] [PubMed] [Google Scholar]
- Rizzoli SO , Betz WJ . 2005. . Synaptic vesicle pools . Nat Rev Neurosci . 6 : 57 – 69 . [DOI] [PubMed] [Google Scholar]
- Scanziani M , Capogna M , Gahwiler BH , Thompson SM . 1992. . Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus . Neuron . 9 : 919 – 927 . [DOI] [PubMed] [Google Scholar]
- Scheckenbach KE , Dreier JP , Dirnagl U , Lindauer U . 2006. . Impaired cerebrovascular reactivity after cortical spreading depression in rats: Restoration by nitric oxide or cGMP . Exp Neurol . 202 : 449 – 455 . [DOI] [PubMed] [Google Scholar]
- Shepherd JD , Huganir RL . 2007. . The cell biology of synaptic plasticity: AMPA receptor trafficking . Annu Rev Cell Dev Biol . 23 : 613 – 643 . [DOI] [PubMed] [Google Scholar]
- Snow RW , Taylor CP , Dudek FE . 1983. . Electrophysiological and optical changes in slices of rat hippocampus during spreading depression . J Neurophysiol . 50 : 561 – 572 . [DOI] [PubMed] [Google Scholar]
- Somjen GG . 2001. . Mechanisms of spreading depression and hypoxic spreading depression-like depolarization . Physiol Rev . 81 : 1065 – 1096 . [DOI] [PubMed] [Google Scholar]
- Spruston N , Johnston D . 1992. . Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons . J Neurophysiol . 67 : 508 – 529 . [DOI] [PubMed] [Google Scholar]
- Steriade M . 2006. . Grouping of brain rhythms in corticothalamic systems . Neuroscience . 137 : 1087 – 1106 . [DOI] [PubMed] [Google Scholar]
- Steriade M . 1997. . Synchronized activities of coupled oscillators in the cerebral cortex and thalamus at different levels of vigilance . Cereb Cortex . 7 : 583 – 604 . [DOI] [PubMed] [Google Scholar]
- Steriade M , Timofeev I , Grenier F . 2001. . Natural waking and sleep states: a view from inside neocortical neurons . J Neurophysiol . 85 : 1969 – 1985 . [DOI] [PubMed] [Google Scholar]
- Strong AJ , Fabricius M , Boutelle MG , Hibbins SJ , Hopwood SE , Jones R , Parkin MC , Lauritzen M . 2002. . Spreading and synchronous depressions of cortical activity in acutely injured human brain . Stroke . 33 : 2738 – 2743 . [DOI] [PubMed] [Google Scholar]
- Sugaya E , Takato M , Noda Y . 1975. . Neuronal and glial activity during spreading depression in cerebral cortex of cat . J Neurophysiol . 38 : 822 – 841 . [DOI] [PubMed] [Google Scholar]
- Takano T , Tian GF , Peng W , Lou N , Lovatt D , Hansen AJ , Kasischke KA , Nedergaard M . 2007. . Cortical spreading depression causes and coincides with tissue hypoxia . Nat Neurosci . 10 : 754 – 762 . [DOI] [PubMed] [Google Scholar]
- Theriot JJ , Toga AW , Prakash N , Ju YS , Brennan KC . 2012. . Cortical sensory plasticity in a model of migraine with aura . J Neurosci . 32 : 15252 – 15261 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM , Capogna M , Scanziani M . 1993. . Presynaptic inhibition in the hippocampus . Trends Neurosci . 16 : 222 – 227 . [DOI] [PubMed] [Google Scholar]
- Timofeev I , Grenier F , Steriade M . 2000. . Impact of intrinsic properties and synaptic factors on the activity of neocortical networks in vivo . J Physiol Paris . 94 : 343 – 355 . [DOI] [PubMed] [Google Scholar]
- Tottene A , Conti R , Fabbro A , Vecchia D , Shapovalova M , Santello M , van den Maagdenberg AM , Ferrari MD , Pietrobon D . 2009. . Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice . Neuron . 61 : 762 – 773 . [DOI] [PubMed] [Google Scholar]
- Turrigiano G . 2012. . Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function . Cold Spring Harb Perspect Biol . 4 : a005736 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG , Leslie KR , Desai NS , Rutherford LC , Nelson SB . 1998. . Activity-dependent scaling of quantal amplitude in neocortical neurons . Nature . 391 : 892 – 896 . [DOI] [PubMed] [Google Scholar]
- Ulrich D , Luscher HR . 1993. . Miniature excitatory synaptic currents corrected for dendritic cable properties reveal quantal size and variance . J Neurophysiol . 69 : 1769 – 1773 . [DOI] [PubMed] [Google Scholar]
- Wan Q , Xiong ZG , Man HY , Ackerley CA , Braunton J , Lu WY , Becker LE , MacDonald JF , Wang YT . 1997. . Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin . Nature . 388 : 686 – 690 . [DOI] [PubMed] [Google Scholar]
- Wei Y , Ullah G , Schiff SJ . 2014. . Unification of neuronal spikes, seizures, and spreading depression . J Neurosci . 34 : 11733 – 11743 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C . 2008. . Up and down states . Scholarpedia . 3 : 1410 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG , Saggau P . 1994. . Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus . Neuron . 12 : 1139 – 1148 . [DOI] [PubMed] [Google Scholar]
- Yawo H , Chuhma N . 1993. . Preferential inhibition of omega-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors . Nature . 365 : 256 – 258 . [DOI] [PubMed] [Google Scholar]
- Yoon KW , Rothman SM . 1991. . Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus . J Neurosci . 11 : 1375 – 1380 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N , Rungta RL , Malik A , Han H , Wu DC , MacVicar BA . 2013. . Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression . J Cereb Blood Flow Metab . 33 : 1582 – 1594 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ , Connors BW . 1999. . Intrinsic firing patterns and whisker-evoked synaptic responses of neurons in the rat barrel cortex . J Neurophysiol . 81 : 1171 – 1183 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.