

In this study, Bao et al. investigated how transcription is regulated within heterochromatin in fission yeast. They show that overexpressed Epe1 associates with SAGA and recruits SAGA to heterochromatin regions (which leads to an increase in histone acetylation, transcription of repeats, and the disruption of heterochromatin) and that Epe1 recruits SAGA to regulate transcription within heterochromatin when expressed at normal levels.
Keywords: Epe1, SAGA, histone, methylation, heterochromatin, transcription
Abstract
Heterochromatin is a highly condensed form of chromatin that silences gene transcription. Although high levels of transcriptional activities disrupt heterochromatin, transcription of repetitive DNA elements and subsequent processing of the transcripts by the RNAi machinery are required for heterochromatin assembly. In fission yeast, a JmjC domain protein, Epe1, promotes transcription of DNA repeats to facilitate heterochromatin formation, but overexpression of Epe1 leads to heterochromatin defects. However, the molecular function of Epe1 is not well understood. By screening the fission yeast deletion library, we found that heterochromatin defects associated with Epe1 overexpression are alleviated by mutations of the SAGA histone acetyltransferase complex. Overexpressed Epe1 associates with SAGA and recruits SAGA to heterochromatin regions, which leads to increased histone acetylation, transcription of repeats, and the disruption of heterochromatin. At its normal expression levels, Epe1 also associates with SAGA, albeit weakly. Such interaction regulates histone acetylation levels at heterochromatin and promotes transcription of repeats for heterochromatin assembly. Our results also suggest that increases of certain chromatin protein levels, which frequently occur in cancer cells, might strengthen relatively weak interactions to affect the epigenetic landscape.
Eukaryotic genomes contain large amounts of repetitive DNA sequences, which are the preferred sites of heterochromatin formation (Grewal and Jia 2007; Almouzni and Probst 2011). The resulting condensed chromatin state limits the access of the transcription and recombination machinery to restrain the harmful effects of repetitive DNA on genome integrity. Heterochromatin can also spread into neighboring genomic regions, leading to changes in gene expression across large chromosomal domains.
The histones within heterochromatin are usually hypoacetylated but are methylated at histone H3 Lys9 (H3K9me), which recruits heterochromatin protein 1 (HP1) family proteins to compact chromatin (Grewal and Jia 2007; Almouzni and Probst 2011). Therefore, histone deacetylases (HDACs) and histone H3K9 methyltransferases are required for heterochromatin formation, whereas histone H3K9 demethylases and histone acetyltransferases antagonize heterochromatin assembly. Given the importance of heterochromatin in regulating gene expression and genome integrity, it is not surprising that mutations or misregulation of heterochromatin factors have been linked to human diseases. For example, loss of the murine H3K9 methyltransferase SUV39H1/H2 leads to chromosomal instability and increased tumor risk (Peters et al. 2001). On the other hand, histone demethylases are frequently overexpressed in cancer cells (Højfeldt et al. 2013; Johansson et al. 2014). For example, the H3K9 demethylase JMJD2C/GASC1 is amplified in squamous cell carcinoma, breast cancer, and medulloblastoma (Yang et al. 2000; Cloos et al. 2006; Ehrbrecht et al. 2006; Liu et al. 2009).
The fission yeast Schizosaccharomyces pombe has been instrumental in delineating heterochromatin assembly pathways (Grewal and Jia 2007). In this organism, large blocks of heterochromatin are formed at pericentric regions, subtelomeres, and the silent mating-type locus, which share a common repetitive DNA sequence. The formation of heterochromatin at these regions is critically dependent on the histone H3K9 methyltransferase Clr4. The resulting histone H3K9 methylation serves as a signal to recruit HP1 family proteins such as Swi6 and Chp2 (Nakayama et al. 2001; Sadaie et al. 2004). Both Swi6 and Chp2 recruit SHREC, which contains HDAC Clr3 and chromatin remodeling protein Mit1. The combined actions of these two enzymatic activities restrict the access of RNA polymerase II (Pol II), leading to transcriptional gene silencing (Sugiyama et al. 2007; Motamedi et al. 2008). Heterochromatin formation also requires another HDAC, Sir2, which cooperates with SHREC to ensure low histone acetylation levels at heterochromatin (Shankaranarayana et al. 2003; Freeman-Cook et al. 2005; Alper et al. 2013; Buscaino et al. 2013).
Paradoxically, transcription of the DNA repeats is required for heterochromatin assembly. These repeats are transcribed by Pol II, leading to the production of dsRNAs (Djupedal et al. 2005; Kato et al. 2005). Dicer (Dcr1) processes these dsRNAs into siRNAs, which are loaded onto the RNA-induced transcriptional silencing (RITS) complex and guide RITS back to nascent transcripts (Verdel et al. 2004). RITS then recruits the Clr4 complex (CLRC) to initiate H3K9 methylation and heterochromatin assembly at DNA repeats (Zhang et al. 2008; Bayne et al. 2010). Heterochromatic repeats are transcribed mainly during the S phase of the cell cycle, suggesting that the passage of DNA polymerase during DNA replication may disrupt heterochromatin to allow Pol II access (Chen et al. 2008; Kloc et al. 2008). During other stages of the cell cycle, Pol II access to heterochromatin is regulated by the anti-silencing factor Epe1, which is recruited to heterochromatin through its interaction with Swi6 (Zofall and Grewal 2006; Isaac et al. 2007; Trewick et al. 2007). However, how Epe1 functions remains controversial.
Epe1 was first identified as a factor that prevents heterochromatin from expanding outside of its normal boundaries (Ayoub et al. 2003). Loss of Epe1 also results in the formation of ectopic heterochromatin islands and even allows heterochromatin to persist through cell divisions without initiation signals (Zofall et al. 2012; Audergon et al. 2015; Garcia et al. 2015; Ragunathan et al. 2015; Wang et al. 2015). Epe1 contains a JmjC domain, which is commonly the catalytic domain of histone demethylases (Klose et al. 2006). Introducing point mutations within the JmjC domain that are predicted to affect histone demethylase activity, such as the H297A mutation, results in phenotypes similar to epe1Δ (Trewick et al. 2007; Audergon et al. 2015; Ragunathan et al. 2015; Wang et al. 2015), consistent with the idea that Epe1 functions as a H3K9 demethylase. However, no in vitro demethylase activity has been detected for Epe1 (Tsukada et al. 2006), raising the possibility that Epe1 modulates H3K9 methylation indirectly. Indeed, overexpression of the H297A mutant of Epe1 disrupts heterochromatin similarly to overexpression of wild-type Epe1 (Zofall and Grewal 2006; Trewick et al. 2007), demonstrating that Epe1 can affect heterochromatin stability independently from its putative demethylase activity.
Interestingly, loss of Epe1 also rescues heterochromatin defects caused by deletions of HDACs such as Clr3 or Sir2 (Ayoub et al. 2003; Aygün et al. 2013; Wang et al. 2013), suggesting that Epe1 counteracts the function of HDACs. However, the exact mechanism by which Epe1 regulates HDACs is unknown. In this study, we found that mutants in the SAGA histone acetyltransferase complex alleviated the effects of Epe1 overexpression on heterochromatin stability. We also found that Epe1 associates with SAGA and recruits SAGA to heterochromatin to promote histone acetylation, which in turn promotes Pol II transcription.
Results
Epe1 overexpression affects heterochromatin integrity
All previous attempts to examine the effects of Epe1 overexpression used plasmid-borne Epe1 (Zofall and Grewal 2006; Trewick et al. 2007). To minimize the effects of potential plasmid copy number variations and facilitate genetic screens with the deletion library, we replaced the endogenous epe1+ promoter with an nmt41 promoter, which can be induced by the removal of thiamine from the growth medium (EMM). We then examined the effects of Epe1 overexpression on heterochromatin integrity by measuring the silencing of reporter genes inserted within the pericentric repeat region (otr:ura4+) or the silent mating-type region (Kint2::ura4+) (Fig. 1A; Allshire et al. 1995; Grewal and Klar 1997). In wild-type cells, the silencing of these reporter genes results in cells that grow weakly on medium without uracil but grow well on medium containing 5-fluoroorotic acid (5-FOA), which is toxic to Ura4-expressing cells. Overexpression of Epe1 leads to defective silencing of the reporter genes, as indicated by increased growth on medium without uracil and decreased growth on 5-FOA-containing medium (Fig. 1B). In addition, ChIP (chromatin immunoprecipitation) analyses showed that H3K9 trimethylation (H3K9me3) and Swi6 levels decrease at the endogenous dh and cenH repeats when Epe1 is overexpressed, accompanied by increased levels of dh and cenH transcripts (Fig. 1B). These results confirm that the silencing defects are due to compromised heterochromatin.
Figure 1.

Overexpression of Epe1 leads to defective gene silencing. (A) Schematic diagram of the reporter genes used. Bars indicate the positions of PCR fragments used in ChIP analyses. (B, left) Tenfold serial dilution analyses of the indicated yeast strains grown on the indicated media to measure the expression of ura4+ reporter genes. (Right) The first two panels show ChIP analyses of H3K9me3 and Swi6 levels at repetitive DNA elements within pericentric (dh) and mating-type regions (cenH), normalized to act1+. The last panel shows quantitative RT–PCR (qRT–PCR) analysis of the dh and cenH transcripts, normalized to act1+.
A genetic screen for suppressors of the effects of Epe1 overexpression on heterochromatin stability
To further understand the mechanism by which Epe1 regulates heterochromatin formation, we performed a screen with the fission yeast deletion library to identify mutations that alleviate silencing defects caused by Epe1 overexpression (Fig. 2A). We constructed a query strain containing the otr::ura4+ reporter and nmt41-epe1+ and crossed it with a mutant library containing ∼3500 nonessential gene deletions. The resulting haploid cells, each containing otr::ura4+, nmt41-epe1+, and a single gene deletion, were grown on medium without thiamine and containing 5-FOA to measure cell growth (Fig. 2B). Mutations of three subunits of the SAGA histone acetyltransferase complex—gcn5Δ, ada3Δ, and tra1Δ (Helmlinger et al. 2008)—were among the top hits of gene deletions that rescued the silencing defects associated with Epe1 overexpression (Supplemental Fig. S1).
Figure 2.

A genetic screen for mutations that alleviate the effects of Epe1 overexpression on heterochromatin integrity. (A) Workflow to introduce otr::ura4+ and nmt41-epe1+ into the deletion library. (B) A representative image of cells grown on medium without thiamine and containing 5-FOA. Each square represents quadruplicates of colonies of the same genotype. The box indicates the position of gcn5Δ. (C, left) Tenfold serial dilution analyses of the indicated yeast strains grown on the indicated media to measure the expression of the otr::ura4+ reporter gene. (Right) The first two panels show ChIP analyses of H3K9me3 and Swi6 levels at pericentric dh repeats, normalized to act1+. The last panel shows qRT–PCR analysis of dh transcript, normalized to act1+. (D) Western blot analysis to measure the levels of HA-tagged Epe1. A nonspecific band served as a loading control. (E) Coimmunoprecipitation analyses of Epe1 and Swi6. The immunoprecipitation was performed with Flag-agarose beads, and Western blot analyses were performed with Flag and Swi6 antibodies. (F) ChIP analyses of HA-Epe1 levels at pericentric dh repeats, normalized to act1+.
To confirm these findings, we constructed an otr::ura4+ nmt41-epe1+ gcn5Δ strain. Serial dilution analyses confirmed that gcn5Δ alleviates silencing defects of otr::ura4+ associated with Epe1 overexpression (Fig. 2C). Moreover, H3K9me3 and Swi6 levels at dh repeats are partially restored in nmt41-epe1+ gcn5Δ cells, accompanied by a reduction in dh transcript levels (Fig. 2C). The rescue is not limited to pericentromeric heterochromatin, as gcn5Δ also alleviated silencing defects of Kint2::ura4+ at the mating-type region when Epe1 is overexpressed (Supplemental Fig. S2).
One possible mechanism by which SAGA mutants alleviate heterochromatin defects caused by Epe1 overexpression is through reducing Epe1 protein levels because the SAGA histone acetyltransferase complex is required for transcriptional regulation of diverse genes (Helmlinger et al. 2008; Wang et al. 2012). Moreover, Epe1 levels at heterochromatin are regulated by the Cul4–Ddb1 E3 ubiquitin ligase (Braun et al. 2011), which might be under the control of SAGA. However, Western blot analyses showed that gcn5Δ has no effect on Epe1 protein levels (Fig. 2D), thus ruling out these possibilities.
It is also possible that gcn5Δ rescues Epe1 overexpression by affecting Epe1 recruitment to heterochromatin. Epe1 interacts with Swi6, and this interaction is required for the localization of Epe1 to heterochromatin (Zofall and Grewal 2006; Isaac et al. 2007; Trewick et al. 2007). Coimmunoprecipitation analysis showed that Epe1 maintains interaction with Swi6 in gcn5Δ cells (Fig. 2E). Moreover, ChIP analyses showed that the levels of Epe1 at pericentric dh repeats even slightly increase in gcn5Δ cells (Fig. 2F). Such an increase could be attributed to increased levels of Swi6 at heterochromatin in these cells. Thus, gcn5Δ does not rescue Epe1 overexpression by affecting its interaction with Swi6 or disrupting the recruitment of Epe1 to heterochromatin.
The acetyltransferase activity of SAGA is critical for the effects of Epe1 overexpression on heterochromatin stability
SAGA has two enzymatic activities: acetylation and deubiquitination. Gcn5 acts as the catalytic subunit within the acetyltransferase module, which also contains Ada2 and Ada3, whereas Upb8, Sgf73, Sgf11, and Sus1 form the deubiquitination module, with Ubp8 catalyzing the deubiquitination of H2B (Koutelou et al. 2010). We found that mutations in the acetyltransferase module, such as ada2Δ and ada3Δ, alleviate silencing defects associated with Epe1 overexpression (Fig. 3A), but mutations of the deubiquitination module (ubp8Δ and sgf11Δ) or other components such as the SPT module (spt3Δ and spt8Δ) have no effects (Fig. 3B). The effects of these mutations on histone acetylation and ubiquitination were confirmed by Western blot analyses of H3K9 acetylation (H3K9ac) and H2B monoubiquitination (Supplemental Fig. S3).
Figure 3.

The acetyltransferase activity of SAGA is required for Epe1 function. Tenfold serial dilution analyses of the indicated yeast strains grown on the indicated media to measure the expression of the otr::ura4+ reporter gene. (A) The effects of deletion of the SAGA acetyltransferase module. (B) The effects of deletion of the SAGA deubiquitination and SPT modules.
To further examine the role of the histone acetyltransferase activity in regulating the effects of Epe1 overexpression, we obtained a strain containing an E191Q mutation in gcn5+ at its endogenous chromosomal locus. This mutation impairs the enzymatic activity of Gcn5 in vitro and in vivo (Supplemental Fig. S3; Helmlinger et al. 2008). Similar to gcn5Δ, the gcn5-E191Q mutation also alleviates the effects of Epe1 overexpression on the silencing of otr::ura4+ (Fig. 3A), demonstrating that the histone acetyltransferase activity of SAGA is critical for the effects of Epe1 overexpression on heterochromatin stability.
SAGA regulates Epe1 function independently of Epe1's putative demethylase activity
The H297A mutation within the JmjC domain of Epe1 is expected to abolish its putative demethylase activity. Indeed, cells with Epe1-H297A show phenotypes similar to epe1Δ, suggesting that Epe1 functions as a demethylase (Trewick et al. 2007; Audergon et al. 2015; Ragunathan et al. 2015; Wang et al. 2015). However, no in vitro demethylase activity has been detected for Epe1. Therefore, whether the H297A mutation affects Epe1 enzymatic activity is not proven, and it remains possible that this mutation affects Epe1 function in unexpected ways. Nonetheless, we generated nmt41-epe1-H297A at the endogenous epe1+ locus. Overexpression of Epe1-H297A in this context results in silencing defects, as indicated by increased growth on medium without uracil and increased levels of dh transcripts (Supplemental Fig. S4A). However, the silencing defects are milder compared with overexpression of wild-type Epe1, as indicated by relatively robust growth of nmt41-epe1-H297A cells on medium containing 5-FOA even though the mutant is overexpressed at levels similar to wild-type Epe1 (Supplemental Fig. S4A,B). These results suggest that Epe1 likely has demethylase activity-dependent as well as demethylase-independent functions.
Interestingly, when overexpressed from a plasmid, nmt41-epe1-H297A affects heterochromatin similar to the overexpression of wild-type nmt41-epe1+, confirming previous findings (Supplemental Fig. S5A; Trewick et al. 2007 ). We reasoned that the plasmids are present in multiple copies in the cell, leading to higher Epe1 levels and stronger silencing defects. Indeed, when overexpressed from a plasmid, epe1 transcripts levels are about four times higher than those overexpressed from the endogenous chromosomal locus (Supplemental Fig. S4C). Given the more robust silencing defects of plasmid-borne Epe1-H297A on 5-FOA-containing medium, which is our primary assay for heterochromatin silencing, we examined the effects of gcn5Δ on Epe1-H297A overexpression using plasmids.
We found that gcn5Δ rescues silencing defects of plasmid-borne nmt41-epe1+, although the rescue is weaker compared with when Epe1 is overexpressed at the endogenous location. Moreover, gcn5Δ strongly rescues nmt41-epe1-H297A (Supplemental Fig. S5), suggesting that SAGA contributes to Epe1 function mainly independent of Epe1's putative demethylase activity.
SAGA associates with overexpressed Epe1
To further examine the mechanism by which overexpression of Epe1 affects heterochromatin integrity, we generated a Flag-tagged version of Epe1 driven by the nmt41 promoter at the endogenous epe1+ locus and performed affinity purification of overexpressed Flag-Epe1. Interestingly, mass spectrometry analysis of associated proteins identified many components of the SAGA complex (Fig. 4A; Supplemental Tables S1, S2). The association is specific, as no SAGA-specific components were identified in a control purification of cell lysates without any Flag-tagged proteins (Supplemental Table S1) or several mass spectrometry analyses of affinity-purified Flag-Clr4 complex under the same purification conditions (data not shown). Further coimmunoprecipitation analysis confirmed that Flag-Epe1 interacts with Gcn5-myc when Epe1 is overexpressed. Moreover, the interaction was not affected by treatment of cell lysates with ethidium bromide or benzonase, suggesting that Epe1–SAGA interaction is not mediated by DNA/RNA (Fig. 4B; Supplemental Fig. S6). This interaction suggests that Epe1 might recruit SAGA to heterochromatin to affect heterochromatin integrity. Indeed, ChIP analyses showed that Gcn5 is enriched at dh repeats when Epe1 is overexpressed (Fig. 4C).
Figure 4.

Epe1 associates with SAGA. (A) Mass spectrometry analyses of purified protein complexes. The spectral count (left) and the sequence coverage of each protein (right) are indicated. (B) Coimmunoprecipitation analyses of Epe1 and Gcn5. The lysates were treated with benzonase before immunoprecipitation was performed with Flag-agarose beads. Western blot analyses were performed with Flag and myc antibodies. (C) ChIP analyses of Gcn5 levels at pericentric dh repeat, shown as ChIP/input normalized to the no tag control. (D) Tenfold serial dilution analyses of the indicated yeast strains grown on the indicated media to measure the expression of the otr::ura4+ reporter gene.
Interestingly, the majority of SAGA components still interact with Epe1 in gcn5Δ cells (Fig. 4A), suggesting that other components of SAGA mediate the interaction between SAGA and Epe1. We reasoned that if the Epe1–SAGA interaction contributes to the effects of Epe1 overexpression on heterochromatin, then a mutation in a component of SAGA that mediates its interaction with Epe1 would alleviate silencing defects associated with Epe1 overexpression as well. Our genetic screen for suppressors of Epe1 overexpression identified one SAGA subunit deletion that does not affect histone acetyltransferase activity, tra1Δ. We generated a tra1Δ nmt41-epe1+ strain and found that it indeed alleviates the effects of Epe1 overexpression on the silencing of otr::ura4+ (Fig. 4D). Tra1 is required for the recruitment of SAGA to certain gene promoters by transcription activators but has little effect on the composition of the SAGA complex in S. pombe (Koutelou et al. 2010; Helmlinger et al. 2011). To test whether Tra1 mediates the interaction between Epe1 and SAGA, we performed coimmunoprecipitation of Gcn5-myc and overexpressed Flag-Epe1 in a tra1Δ background. Indeed, we found that the interaction between Epe1 and Gcn5 is reduced in tra1Δ cells (Fig. 4B). Moreover, ChIP analysis showed that Gcn5 localization to pericentric heterochromatin is also reduced in tra1Δ cells when Epe1 is overexpressed (Fig. 4C).
Overexpressed Epe1 recruits SAGA to acetylate histones at heterochromatin regions
Epe1 promotes the localization of Pol II to heterochromatin (Zofall and Grewal 2006). Given that histone acetylation is frequently associated with active transcription (Pokholok et al. 2005), a plausible hypothesis is that overexpressed Epe1 recruits SAGA to heterochromatin to acetylate histones to promote Pol II-mediated transcription, which could disrupt heterochromatin. SAGA acetylates a number of lysines on histones, including H3K9 and H3K14 (Nugent et al. 2010). ChIP analyses showed that both H3K9ac and H3K14ac levels at pericentric dh repeats increase when Epe1 is overexpressed (Fig. 5A,B). Consistent with the idea that Gcn5 mediates these acetylation events, H3K9ac and H3K14ac levels are reduced in gcn5Δ nmt41-epe1+ cells (Fig. 5A,B). Furthermore, ChIP analysis with an antibody against Pol II CTD (C-terminal domain) phosphorylated at Ser2, which represents the elongating form of Pol II, showed that when Epe1 is overexpressed, Pol II levels increase at heterochromatin but reduce in gcn5Δ nmt41-epe1+ cells (Fig. 5C). The Pol II ChIP data are consistent with dh transcripts levels, which increase when Epe1 is overexpressed and decrease in gcn5Δ nmt41-epe1+ cells (Fig. 2C). Altogether, these results support the idea that overexpressed Epe1 recruits SAGA to heterochromatin to promote histone acetylation and transcription of the underlying repeats, leading to heterochromatin defects.
Figure 5.

SAGA regulates histone acetylation and Pol II access at heterochromatin when Epe1 is overexpressed. (A–C) ChIP analyses of the levels of H3K9ac, H3K14ac, and the Ser2 phosphorylated form (Ser2P) of Pol II at the pericentric dh repeat, shown as ChIP/input normalized to wild type.
SAGA interacts with Epe1 at normal expression levels
To examine the interaction between endogenous Epe1 and SAGA, we obtained a strain that carries myc-tagged Spt7, which is a component of the SAGA complex, at the endogenous chromosome locus (Helmlinger et al. 2008). We detected a specific interaction between Epe1-Flag and Spt7-myc under benzonase treatment to disrupt interaction mediated by nucleic acids. (Fig. 6A). Moreover, the interaction is reduced in tra1Δ cells, consistent with the idea that Tra1 mediates the interaction between Epe1 and SAGA. However, we note that the interaction is much weaker compared with overexpressed Epe1. ChIP analyses indicated that both Gcn5 and Spt7 are localized at heterochromatin, and their levels are reduced in epe1Δ cells (Fig. 6B), consistent with the idea that Epe1 recruits SAGA to heterochromatin.
Figure 6.

SAGA counteracts the effects of HDAC Sir2. (A) Coimmunoprecipitation analyses of Epe1-Flag and Spt7-myc. The lysates were treated with benzonase before immunoprecipitation was performed with Flag-agarose beads. Western blot analyses were performed with Flag and myc antibodies. (B,D,E,F) ChIP analyses of SAGA components, H3K9ac, H3K14ac, and Pol II Ser2P at the pericentric dh repeat, shown as ChIP/input normalized to wild type. (C) Tenfold serial dilution analyses of the indicated yeast strains grown on the indicated media to measure the expression of the imr::ura4+ reporter gene. (G) qRT–PCR analysis of the dh transcript, normalized to act1+.
SAGA counteracts HDAC Sir2 to regulate heterochromatin stability
Epe1 counteracts HDACs Clr3 and Sir2 in heterochromatin assembly (Ayoub et al. 2003; Zofall and Grewal 2006; Wang et al. 2013). Both sir2Δ and clr3Δ result in strong silencing defects of the imr::ura4+ reporter inserted at pericentric heterochromatin, but the defects are alleviated in sir2Δ epe1Δ and clr3Δ epe1Δ cells (Fig. 6C; Supplemental Fig. S7). We found that sir2Δ gcn5Δ partially restores silencing of imr::ura4+, although gcn5Δ clr3Δ does not (Fig. 6C; Supplemental Fig. S7). ChIP analyses showed that both H3K9ac and H3K14ac and Ser2 phosphorylated form (Ser2P) of Pol II levels at dh repeats increase in sir2Δ cells yet decrease in sir2Δ gcn5Δ cells (Fig. 6D–F). Consistent with these data, dh transcript levels also increase in sir2Δ cells and decrease in sir2Δ gcn5Δ cells (Fig. 6G). ChIP analysis also showed that H3K9ac and H3K14ac levels at dh repeats increase in clr3Δ cells yet decrease in clr3Δ gcn5Δ cells (Supplemental Fig. S8) even though gcn5Δ could not rescue the silencing defects of clr3Δ (Supplemental Fig. S7), suggesting that Clr3 might deacetylate other histone residues important for heterochromatin formation that are not acetylated by SAGA. Alternatively, the acetylation levels in gcn5Δ clr3Δ might still be above the threshold for proper heterochromatin formation.
We also found that epe1-H297A partially rescues the silencing of imr::ura4+ in sir2Δ cells and that epe1-H297A gcn5Δ completely rescues silencing defects of sir2Δ, similar to epe1Δ cells (Supplemental Fig. S7). These results demonstrate that when Epe1 is expressed at normal levels, it may function as both a demethylase and a recruiter of SAGA to counteract the effects of HDAC Sir2 at heterochromatin to promote transcription of repeats.
Discussions
Although heterochromatin represses transcription, the formation of heterochromatin at repetitive DNA elements requires transcription of repeats. These transcripts serve as scaffolds for the recruitment of chromatin-modifying activities and a source for the production of siRNAs. Multiple mechanisms have evolved to promote transcription within heterochromatin. For example, in plants, the transcription of the repeats is mediated by two specialized RNA polymerases: Pol IV, which initiates small RNA biogenesis, and Pol V, which generates scaffold transcripts for the recruitment of chromatin factors (Haag and Pikaard 2011). However, Pol II transcribes the repeats in other organisms, indicating the existence of special mechanisms to overcome the repressive effects of heterochromatin. For example, in flies, the HP1 homolog Rhino recruits a transcription factor Moonshiner to heterochromatin to initiate Pol II-dependent transcription of the underlying repeats (Andersen et al. 2017). In fission yeast, Swi6 recruits the JmjC domain protein Epe1, which promotes Pol II-mediated transcription of repeats (Zofall and Grewal 2006). However, the mechanisms of Epe1 function are unknown and controversial.
Epe1 protein levels are controlled by Cul4–Ddb1-mediated ubiquitylation and subsequent degradation by the proteasome (Braun et al. 2011). Compromising Cul4–Ddb1 or overexpression of Epe1 leads to elevated transcription of repeats and heterochromatin defects (Zofall and Grewal 2006; Trewick et al. 2007; Braun et al. 2011), suggesting that a tight control of Epe1 levels is essential for promoting transcription within heterochromatin without disrupting heterochromatin structure. We found that when Epe1 is expressed at normal levels, it weakly associates with SAGA. SAGA counteracts HDAC Sir2 to promote histone acetylation and Pol II-mediated transcription of repeats, generating sufficient amounts of transcripts for RNAi-mediated heterochromatin assembly without destabilizing heterochromatin (Fig. 7). When Epe1 is overexpressed, it recruits higher levels of SAGA to heterochromatin. This in turn leads to high levels of histone acetylation and Pol II transcription, which disrupt heterochromatin.
Figure 7.
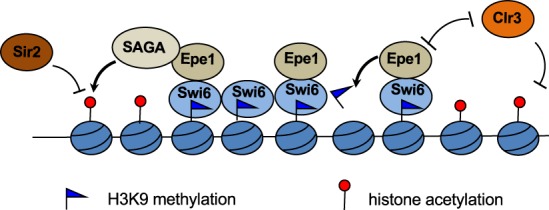
Model for the function of Epe1 at heterochromatin. Epe1 demethylates H3K9, competes with HDAC Clr3 for binding to Swi6, and recruits SAGA to counteract HDAC Sir2. The combined activities lead to higher levels of histone acetylation at heterochromatin, which promotes transcription of the underlying DNA repeats.
We noticed that when overexpressed from its endogenous chromosome location, the silencing defects in nmt41-epe1+ cells is stronger than those in nmt41-epe1-H297A cells, indicating that Epe1's putative demethylase activity also contributes to the effects of Epe1 overexpression on heterochromatin. On the other hand, when expressed at higher levels through a multicopy number plasmid, nmt41-epe1-H297A caused silencing defects similar to those in nmt41-epe1+. However, gcn5Δ alleviated silencing defects in nmt41-epe1-H297A better than those in nmt41-epe1+, suggesting the SAGA regulates Epe1 function independently from Epe1's putative histone demethylase activity, although we could not rule out the possibility that SAGA also regulates Epe1's putative demethylase activity. We found that neither recombinant Gcn5 nor purified SAGA complex acetylates recombinant Epe1 in an in vitro acetyltransferase assay (Supplemental Fig. S9). It remains possible that SAGA regulates Epe1 demethylase activity by acetylating histones to provide a better substrate for Epe1. However, we were unable to test such possibilities given the difficulties in detecting Epe1 enzymatic activity in vitro (Tsukada et al. 2006; Zofall and Grewal 2006; Trewick et al. 2007).
Similar to epe1Δ, gcn5Δ partially rescues sir2Δ. However, gcn5Δ does not phenocopy epe1Δ in many other assays. For example, unlike epe1Δ, we did not observe significant heterochromatin spreading into a reporter gene inserted near pericentric heterochromatin (IRC1::ura4+) in gcn5Δ cells (data not shown). It should be noted that in budding yeast, SAGA components are required for the proper formation of heterochromatin boundaries (Kamata et al. 2014, 2016). The difference in the requirement of SAGA for heterochromatin boundary function in fission yeast and budding yeast could be due to the different chromatin machineries involved in the formation of heterochromatin, with the fission yeast requiring histone H3K9 methylation and HP1 proteins for heterochromatin assembly, and SAGA functions in boundary formation might be substituted by other histone acetyltransferases such as Mst1 (Wang et al. 2013). In addition, unlike epe1Δ, gcn5Δ does not rescue the defects of RNAi mutants in pericentric heterochromatin function (Trewick et al. 2007; Reddy et al. 2011). This discrepancy could be due to other functions of Epe1, such as its putative histone demethylase activity (Trewick et al. 2007; Audergon et al. 2015; Ragunathan et al. 2015). Consistent with this idea, the epe1-H297A gcn5Δ behaved similarly to epe1Δ in suppressing sir2Δ, suggesting that the putative demethylase activity and SAGA recruitment are independent functions of Epe1 (Supplemental Fig. S7). We also found that, unlike epe1Δ, gcn5Δ could not rescue the silencing defects associated with clr3Δ. Moreover, neither epe1-H297A nor epe1-H297A gcn5Δ could rescue clr3Δ (Supplemental Fig. S7). These results can be explained by the fact that Epe1 directly competes with Clr3 for localization to heterochromatin (Shimada et al. 2009). Therefore, our results are consistent with those of others, showing that Epe1 functions through multiple mechanisms to regulate heterochromatin formation: SAGA recruitment, competition with Clr3 for binding to Swi6, and possibly histone demethylation (Fig. 7). While the putative demethylase activity is important for the function of Epe1, the SAGA–Epe1 interaction also plays important roles in heterochromatin regulation.
While our data suggest a direct role of Epe1 in the recruitment of SAGA to heterochromatin to regulate transcription of repeats, SAGA might regulate heterochromatin assembly through additional mechanisms. So far, we ruled out the effects of SAGA mutations on Epe1 expression levels, Epe1–Swi6 interaction, and Epe1 localization to heterochromatin (Fig. 2). SAGA regulates diverse cellular processes. In budding yeast, the gcn5 mutant delays the cell cycle, and cells accumulate at the G2/M phase (Zhang et al. 1998; Burgess et al. 2010), raising the possibility that cell cycle delay indirectly alleviates the effects of Epe1 overexpression. In fission yeast, cdr1Δ and cdr2Δ cause cell cycle delay at G2/M (Breeding et al. 1998). We found that these two mutants (which were confirmed to be correct in the strains) did not suppress Epe1 overexpression in our genetic screen. Furthermore, serial dilution analyses of cdr1Δ nmt41-epe1+ and cdr2Δ nmt41-epe1+ strains showed that cdr1Δ and cdr2Δ could not rescue Epe1 overexpression (Supplemental Fig. S10). Therefore, it is unlikely that the rescue of Epe1 overexpression observed in SAGA mutants is through misregulation of cell cycle progression.
We showed previously that Epe1 associates with bromodomain protein Bdf2, which is required for the formation of proper heterochromatin boundaries (Wang et al. 2013). Consistent with this finding, our mass spectrometry analysis of protein associated with overexpressed Epe1 also contains Bdf2 (Supplemental Table S1). Moreover, ChIP analysis showed that Bdf2 levels are higher at pericentric regions when Epe1 is overexpressed (Supplemental Fig. S11A), consistent with the fact Epe1 associates with Bdf2. However, bdf2Δ does not rescue silencing defects caused by Epe1 overexpression (Supplemental Fig. S11B). Therefore, although Bdf2 is recruited to heterochromatin by overexpressed Epe1, it does not contribute to the silencing defects caused by Epe1 overexpression. This might be due to the ability of histone acetylation to directly regulate transcription machinery without help from bromodomain proteins.
Based on sequence homology, Epe1 belongs to the KDM2 family of histone demethylases (Klose et al. 2006). Like Epe1, mammalian KDM2A, which demethylates H3K36, also associates with HP1 proteins and localizes to heterochromatin regions. However, unlike Epe1, KDM2A represses transcription within heterochromatin rather than promoting transcription (Frescas et al. 2008).
JmjC domain demethylases have been shown to have functions independent of their enzymatic activities. For example, JmjD1A, a H3K9 demethylase, interacts with the SWI/SNF complex to mediate long-range chromatin interaction to activate gene expression (Abe et al. 2015). In addition, KDM2B recruits Polycomb-repressive complex 1 (PRC1) to CpG islands through its CxxC-ZF domain (He et al. 2013). Furthermore, Drosophila histone demethylase KDM4A has nonenzymatic roles in controlling heterochromatin integrity and position effect variegation (PEV) (Colmenares et al. 2017). All of these findings highlight that, like Epe1, other JmjC domain proteins also function through mechanisms that are independent of their demethylase activity.
Histone H3K9 demethylases are frequently overexpressed in cancer cells (Højfeldt et al. 2013; Johansson et al. 2014). While the changes in their levels are expected to alter the epigenetic landscape of these cancer cells through histone demethylation, it is also possible that the overexpression of proteins enhances their interactions with other chromatin regulators. Such enhanced interactions might also contribute to changes in epigenetic landscape during tumorigenesis. Therefore, it would be interesting to examine whether mammalian H3K9 demethylases would significantly change their protein interactions when overexpressed.
Materials and methods
Fission yeast strains and genetic analyses
Yeast strains containing nmt41-HA-epe1, nmt41-Flag-epe1, Gcn5-myc, and Gcn5-Flag were generated by a PCR-based module method (Bahler et al. 1998). Deletion strains such as ada2Δ, ada3Δ, tra1Δ, ubp8Δ, sgf11Δ, spt3Δ, spt8Δ, cdr1Δ, and cdr2Δ were derived from the Bioneer deletion library, and the absence of the gene-coding regions was confirmed by PCR analyses. Plasmid-borne nmt41-epe1+ and nmt41-epe1-H297A were constructed by cloning the Epe1 ORF into the pREP41-MHN vector and were transformed into yeast cells by electroporation. All other strains were constructed by genetic crosses. A list of yeast strains used is in Supplemental Table S3. For serial dilution plating assays, 10-fold dilutions of a mid-log-phase culture were plated on the indicated media and grown for 3–4 d at 30°C.
Screen for suppressors of Epe1 overexpression
The query strain (nmt41-epe1+-natMX6 otr:ura4+-hphMX6) was crossed with a library of strains that contain individual gene deletions marked with kanMX6 cassette using a Singer RoToR HDA pinning robot as described previously (Roguev et al. 2007). The desired haploid progenies, which contain nmt41-epe1+-natMX6 otr:ura4+-hphMX6 and a single gene deletion, were selected and pinned first onto EMM plates for 1 d to induce nmt41 expression and subsequently pinned onto EMM plates supplemented with 100 µg/mL FOA to measure growth.
ChIP analyses
ChIP experiments were performed as described previously (Shan et al. 2016). The antibodies used were H3K9me3 (Active Motif, 39161), M2 Flag (Sigma, A2220), H3K9ac (Upstate Biotechnology, 07-352), H3K14ac (Upstate Biotechnology, 07-353), and H3K9me2 (Abcam, 115159). HA antibody was a kind gift from Dr. Michael Keogh. Pol II Ser2P antibody was a kind gift from Dr. James Manley. DNA serial dilutions were used as templates to generate a standard curve of amplification for each pair of primers, and the relative concentration of target sequence was calculated accordingly. An act1 fragment was used as reference to calculate the enrichment of ChIP over whole-cell extract for H3K9me3, Swi6, and Bdf2. For all other ChIP experiments, ChIP/input at the specified locus was used to calculate enrichment levels. A list of DNA oligos used is in Supplemental Table S4.
RNA analyses
RNA was extracted from log-growth phase yeast cultures using MasterPure yeast RNA purification kit (Epicentre). RT-qPCR analyses were performed with Power SYBR Green RNA-to-CT one-step kit (Thermo Fisher Scientific) in a StepOne Plus real-time PCR system (Applied Biosystems). RNA serial dilutions were used as templates to generate the standard curve of amplification for each pair of primers, and the relative concentration of target sequence was calculated accordingly. An act1 fragment served as a reference to normalize the concentrations of samples. The concentration of each target in wild type was arbitrarily set to 1 and served as a reference for other samples. A list of DNA oligos used is in Supplemental Table S4.
Coimmunoprecipitation, Western blotting, and mass spectrometry analysis
Immunoprecipitation of Flag-tagged Epe1 was performed as described previously (Wang et al. 2016). Briefly, 2 L of exponentially growing cells was harvested and washed first with PBS buffer and then with 2× HC buffer (300 mM HEPES-KOH at pH 7.6, 1 mM EDTA, 100 mM KCl, 20% glycerol, 5 mM 2-mercaptoethanol, 0.1% NP40, protease inhibitor cocktail [Roche]). Cells were then pushed through a syringe into liquid nitrogen to create flash-frozen cell balls. The frozen cells were blended using a household blender in the presence of dry ice. After the dry ice sublimed, the lysates were resuspended in 1× HC buffer with 250 mM KCl and incubated for 30 min on a rotator at 4°C. The lysate was cleared by centrifugation at 20,000g for 1 h. The supernatant was incubated with M2 Flag-agarose beads (Sigma, A2220) overnight. For mass spectrometry analysis, the beads were washed eight times with 1× HC containing 250 mM KCl. Bound proteins were eluted with 200 µg/mL 3xFlag peptides followed by TCA precipitation. MudPIT (multidimensional protein identification technology) mass spectrometry analysis was performed as described previously (Wang et al. 2014). For coimmunoprecipitation experiments, the beads were washed four times with 1× HEMN containing 100 mM KCl. For benzonase treatment, 250 U of benzonase and 1.5 mM MgCl2 were added to the lysate and incubated together with Flag beads for 2 h at 4°C. The total and immunoprecipitated portions were resolved by SDS-PAGE followed by Western blot analysis with Myc (Santa Cruz Biotechnology, A14) and Flag (Sigma, F7425) antibodies. Another antibody used for Western blot was HA (Roche, 3F10).
Acetyltransferase assay
HeLa histone octamers or recombinant GST-Epe1 were incubated with recombinant GST-Gcn5 or Flag-Gcn5 complex purified from yeast cells in the presence of 3H-labeled acetyl-CoA in histone acetyltransferase reaction buffer (50 mM Tris at pH 8.0, 0.1 mM EDTA, 1 mM DTT, 50 mM KCl) for 1 h at 30°C. The reactions were then subjected to SDS-PAGE. The gel was stained with Coomassie and dried. The dried gel was fluorographed with EN3HANCE (PerkinElmer).
Supplementary Material
Acknowledgments
We thank Jiyong Wang for technical assistance, Alok Nimgaonkar for assistance with validating results of the SGA screen, Fred Winston for yeast strains, Michael Keogh for the HA antibody, James Manley for Pol II antibody, and Arjun Srivatsa and Tasneem Ebrahim for critical reading of the manuscript. This work was supported by National Institutes of Health grants R01-GM085145 and R35-GM126910 to S.J., the National Center for Research Resources (P41-RR011823), and the National Institute of General Medical Sciences (P41-GM103533).
Author contributions: K.B., C.-M.S., and S.J. designed the project, performed experiments, and interpreted data. J.M. and J.Y. performed mass spectrometry analyses. K.B and S.J. wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.318030.118.
References
- Abe Y, Rozqie R, Matsumura Y, Kawamura T, Nakaki R, Tsurutani Y, Tanimura-Inagaki K, Shiono A, Magoori K, Nakamura K, et al. 2015. JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nat Commun 6: 7052 10.1038/ncomms8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, Nimmo ER, Ekwall K, Javerzat JP, Cranston G. 1995. Mutations derepressing silent centromeric domains in fission yeast disrupt chromosome segregation. Genes Dev 9: 218–233. 10.1101/gad.9.2.218 [DOI] [PubMed] [Google Scholar]
- Almouzni G, Probst AV. 2011. Heterochromatin maintenance and establishment: lessons from the mouse pericentromere. Nucleus 2: 332–338. 10.4161/nucl.2.5.17707 [DOI] [PubMed] [Google Scholar]
- Alper BJ, Job G, Yadav RK, Shanker S, Lowe BR, Partridge JF. 2013. Sir2 is required for Clr4 to initiate centromeric heterochromatin assembly in fission yeast. EMBO J 32: 2321–2335. 10.1038/emboj.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PR, Tirian L, Vunjak M, Brennecke J. 2017. A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 549: 54–59. 10.1038/nature23482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audergon PNCB, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, Allshire RC. 2015. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348: 132–135. 10.1126/science.1260638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygün O, Mehta S, Grewal SIS. 2013. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat Struct Mol Biol 20: 547–554. 10.1038/nsmb.2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Noma K-i, Isaac S, Kahan T, Grewal SIS, Cohen A. 2003. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol Cell Biol 23: 4356–4370. 10.1128/MCB.23.12.4356-4370.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A III, Steever AB, Wach A, Philippsen P, Pringle JR. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14: 943–951. [DOI] [PubMed] [Google Scholar]
- Bayne EH, White SA, Kagansky A, Bijos DA, Sanchez-Pulido L, Hoe KL, Kim DU, Park HO, Ponting CP, Rappsilber J, et al. 2010. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 140: 666–677. 10.1016/j.cell.2010.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. 2011. The Cul4–Ddb1Cdt2 ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144: 41–54. 10.1016/j.cell.2010.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeding CS, Hudson J, Balasubramanian MK, Hemmingsen SM, Young PG, Gould KL. 1998. The cdr2+ gene encodes a regulator of G2/M progression and cytokinesis in Schizosaccharomyces pombe. Mol Biol Cell 9: 3399–3415. 10.1091/mbc.9.12.3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, Zhou H, Han J, Zhang Z. 2010. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell 37: 469–480. 10.1016/j.molcel.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscaino A, Lejeune E, Audergon P, Hamilton G, Pidoux A, Allshire RC. 2013. Distinct roles for Sir2 and RNAi in centromeric heterochromatin nucleation, spreading and maintenance. EMBO J 32: 1250–1264. 10.1038/emboj.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SIS. 2008. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451: 734–737. 10.1038/nature06561 [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. 2006. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442: 307–311. 10.1038/nature04837 [DOI] [PubMed] [Google Scholar]
- Colmenares SU, Swenson JM, Langley SA, Kennedy C, Costes SV, Karpen GH. 2017. Drosophila histone demethylase KDM4A has enzymatic and non-enzymatic roles in controlling heterochromatin integrity. Dev Cell 42: 156–169.e5. 10.1016/j.devcel.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djupedal I, Portoso M, Spåhr H, Bonilla C, Gustafsson CM, Allshire RC, Ekwall K. 2005. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev 19: 2301–2306. 10.1101/gad.344205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrbrecht A, Muller U, Wolter M, Hoischen A, Koch A, Radlwimmer B, Actor B, Mincheva A, Pietsch T, Lichter P, et al. 2006. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol 208: 554–563. 10.1002/path.1925 [DOI] [PubMed] [Google Scholar]
- Freeman-Cook LL, Gomez EB, Spedale EJ, Marlett J, Forsburg SL, Pillus L, Laurenson P. 2005. Conserved locus-specific silencing functions of Schizosaccharomyces pombe sir2+. Genetics 169: 1243–1260. 10.1534/genetics.104.032714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, Guardavaccaro D, Kuchay SM, Kato H, Poleshko A, Basrur V, Elenitoba-Johnson KS, Katz RA, Pagano M. 2008. KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 7: 3539–3547. 10.4161/cc.7.22.7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JF, Al-Sady B, Madhani HD. 2015. Intrinsic toxicity of unchecked heterochromatin spread is suppressed by redundant chromatin boundary functions in Schizosaccharomyces pombe. G3 (Bethesda) 5: 1453–1461. 10.1534/g3.115.018663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, Jia S. 2007. Heterochromatin revisited. Nat Rev Genet 8: 35–46. 10.1038/nrg2008 [DOI] [PubMed] [Google Scholar]
- Grewal SI, Klar AJ. 1997. A recombinationally repressed region between mat2 and mat3 loci shares homology to centromeric repeats and regulates directionality of mating-type switching in fission yeast. Genetics 146: 1221–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag JR, Pikaard CS. 2011. Multisubunit RNA polymerases IV and V: purveyors of non-coding RNA for plant gene silencing. Nat Rev Mol Cell Biol 12: 483–492. 10.1038/nrm3152 [DOI] [PubMed] [Google Scholar]
- He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y. 2013. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat Cell Biol 15: 373–384. 10.1038/ncb2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Marguerat S, Villen J, Gygi SP, Bahler J, Winston F. 2008. The S. pombe SAGA complex controls the switch from proliferation to sexual differentiation through the opposing roles of its subunits Gcn5 and Spt8. Genes Dev 22: 3184–3195. 10.1101/gad.1719908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Marguerat S, Villén J, Swaney DL, Gygi SP, Bähler J, Winston F. 2011. Tra1 has specific regulatory roles, rather than global functions, within the SAGA co-activator complex. EMBO J 30: 2843–2852. 10.1038/emboj.2011.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Højfeldt JW, Agger K, Helin K. 2013. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov 12: 917–930. 10.1038/nrd4154 [DOI] [PubMed] [Google Scholar]
- Isaac S, Walfridsson J, Zohar T, Lazar D, Kahan T, Ekwall K, Cohen A. 2007. Interaction of epe1 with the heterochromatin assembly pathway in Schizosaccharomyces pombe. Genetics 175: 1549–1560. 10.1534/genetics.106.068684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, Oppermann U. 2014. The roles of Jumonji-type oxygenases in human disease. Epigenomics 6: 89–120. 10.2217/epi.13.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata K, Goswami G, Kashio S, Urano T, Nakagawa R, Uchida H, Oki M. 2014. The N-terminus and Tudor domains of Sgf29 are important for its heterochromatin boundary formation function. J Biochem 155: 159–171. 10.1093/jb/mvt108 [DOI] [PubMed] [Google Scholar]
- Kamata K, Shinmyozu K, Nakayama JI, Hatashita M, Uchida H, Oki M. 2016. Four domains of Ada1 form a heterochromatin boundary through different mechanisms. Genes Cells 21: 1125–1136. 10.1111/gtc.12421 [DOI] [PubMed] [Google Scholar]
- Kato H, Goto D, Martienssen RA, Urano T, Furukawa K, Murakami Y. 2005. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science 309: 467–469. 10.1126/science.1114955 [DOI] [PubMed] [Google Scholar]
- Kloc A, Zaratiegui M, Nora E, Martienssen R. 2008. RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol 18: 490–495. 10.1016/j.cub.2008.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Kallin EM, Zhang Y. 2006. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 7: 715–727. 10.1038/nrg1945 [DOI] [PubMed] [Google Scholar]
- Koutelou E, Hirsch CL, Dent SYR. 2010. Multiple faces of the SAGA complex. Curr Opin Cell Biol 22: 374–382. 10.1016/j.ceb.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, Yang ZQ. 2009. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28: 4491–4500. 10.1038/onc.2009.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi MR, Hong EJ, Li X, Gerber S, Denison C, Gygi S, Moazed D. 2008. HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell 32: 778–790. 10.1016/j.molcel.2008.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113. 10.1126/science.1060118 [DOI] [PubMed] [Google Scholar]
- Nugent RL, Johnsson A, Fleharty B, Gogol M, Xue-Franzén Y, Seidel C, Wright AP, Forsburg SL. 2010. Expression profiling of S. pombe acetyltransferase mutants identifies redundant pathways of gene regulation. BMC Genomics 11: 59 10.1186/1471-2164-11-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, et al. 2001. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337. 10.1016/S0092-8674(01)00542-6 [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Tong IL, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122: 517–527. 10.1016/j.cell.2005.06.026 [DOI] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, Moazed D. 2015. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348: 1258699 10.1126/science.1258699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy BD, Wang Y, Niu L, Higuchi EC, Marguerat SB, Bahler J, Smith GR, Jia S. 2011. Elimination of a specific histone H3K14 acetyltransferase complex bypasses the RNAi pathway to regulate pericentric heterochromatin functions. Genes Dev 25: 214–219. 10.1101/gad.1993611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, Wiren M, Weissman JS, Krogan NJ. 2007. High-throughput genetic interaction mapping in the fission yeast Schizosaccharomyces pombe. Nat Methods 4: 861–866. 10.1038/nmeth1098 [DOI] [PubMed] [Google Scholar]
- Sadaie M, Iida T, Urano T, Nakayama J. 2004. A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J 23: 3825–3835. 10.1038/sj.emboj.7600401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan CM, Wang J, Xu K, Chen H, Yue JX, Andrews S, Moresco JJ, Yates JR, Nagy PL, Tong L, et al. 2016. A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. eLife 5: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaranarayana GD, Motamedi MR, Moazed D, Grewal SIS. 2003. Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr Biol 13: 1240–1246. 10.1016/S0960-9822(03)00489-5 [DOI] [PubMed] [Google Scholar]
- Shimada A, Dohke K, Sadaie M, Shinmyozu K, Nakayama J-I, Urano T, Murakami Y. 2009. Phosphorylation of Swi6/HP1 regulates transcriptional gene silencing at heterochromatin. Genes Dev 23: 18–23. 10.1101/gad.1708009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, Grewal SIS. 2007. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128: 491–504. 10.1016/j.cell.2006.12.035 [DOI] [PubMed] [Google Scholar]
- Trewick SC, Minc E, Antonelli R, Urano T, Allshire RC. 2007. The JmjC domain protein Epe1 prevents unregulated assembly and disassembly of heterochromatin. EMBO J 26: 4670–4682. 10.1038/sj.emboj.7601892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y-i, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. 2006. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439: 811–816. 10.1038/nature04433 [DOI] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SIS, Moazed D. 2004. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303: 672–676. 10.1126/science.1093686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kallgren SP, Reddy BD, Kuntz K, López-Maury L, Thompson J, Watt S, Ma C, Hou H, Shi Y, et al. 2012. Histone H3 lysine 14 acetylation is required for activation of a DNA damage checkpoint in fission yeast. J Biol Chem 287: 4386–4393. 10.1074/jbc.M111.329417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tadeo X, Hou H, Tu PG, Thompson J, Yates JR, Jia S. 2013. Epe1 recruits BET family bromodomain protein Bdf2 to establish heterochromatin boundaries. Genes Dev 27: 1886–1902. 10.1101/gad.221010.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tadeo X, Hou H, Andrews S, Moresco JJ, Yates JR, Nagy PL, Jia S. 2014. Tls1 regulates splicing of shelterin components to control telomeric heterochromatin assembly and telomere length. Nucleic Acids Res 42: 11419–11432. 10.1093/nar/gku842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Reddy BD, Jia S. 2015. Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. eLife 2015: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cohen AL, Letian A, Tadeo X, Moresco JJ, Liu J, Yates JR, Qiao F, Jia S. 2016. The proper connection between shelterin components is required for telomeric heterochromatin assembly. Genes Dev 30: 827–839. 10.1101/gad.266718.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. 2000. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res 60: 4735–4739. [PubMed] [Google Scholar]
- Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY. 1998. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J 17: 3155–3167. 10.1093/emboj/17.11.3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, Grewal SI. 2008. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol 15: 381–388. 10.1038/nsmb.1406 [DOI] [PubMed] [Google Scholar]
- Zofall M, Grewal SIS. 2006. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol Cell 22: 681–692. 10.1016/j.molcel.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Zofall M, Yamanaka S, Reyes-Turcu FE, Zhang K, Rubin C, Grewal SIS. 2012. RNA elimination machinery targeting meiotic mRNAs promotes facultative heterochromatin formation. Science 335: 96–100. 10.1126/science.1211651 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.