

In this review, Boothby et al. summarize some salient advances toward elucidation of the molecular programming of the fate choices and function of B cells in the periphery. They also note unanswered questions that pertain to differences among subsets of B lymphocytes and plasma cells.
Keywords: immunity, lymphocytes, signaling, transcription factors
Abstract
Mature B lymphocytes are crucial components of adaptive immunity, a system essential for the evolutionary fitness of mammals. Adaptive lymphocyte function requires an initially naïve cell to proliferate extensively and its progeny to have the capacity to assume a variety of fates. These include either terminal differentiation (the long-lived plasma cell) or metastable transcriptional reprogramming (germinal center and memory B cells). In this review, we focus principally on the regulation of differentiation and functional diversification of the “B2” subset. An overview is combined with an account of more recent advances, including initial work on mechanisms that eliminate DNA methylation and potential links between intracellular metabolites and chromatin editing.
The adaptive immune system offers paradigms of development in which gene regulation and transitions in cell identity that occur throughout postnatal life are both conceptually appealing and of great importance in human and animal health. The vast universe of microbes with which harmonious relations are needed—or against which defenses must be provided—means that functional diversification even among progeny of a particular clone is a hallmark of lymphocytes. A large and growing body of evidence indicates that developmental transitions impact B-cell function in pathophysiological processes such as metabolism or functioning of the central nervous system, which previously would have been thought of as distinct from immunology. Adaptive immunity, which is mediated by T and B lymphocytes, can be divided into two phases. In the first, populations and subsets of mature resting cells are established. Each group represents a highly diverse set of cells that each displays an individual antigen receptor. These receptors assemble in a combinatorial manner as an essential precondition of developmental progression. This initial phase yields a repertoire of cells that have not been activated or proliferated after their production; these are naïve precursors to multiple fate potentials. A vast trove of findings illuminates the transcriptional regulation and chromatin modifications (for convenience, referred to here as epigenetic) that program developmental progression from common lymphoid progenitors (CLPs) to the establishment of the naïve populations of mature T and B cells (e.g., for review, see Busslinger 2004; Champhekar et al. 2015). Similarly, the process of diversifying subsets of T cells after their activation has been studied and reviewed intensively (Glimcher and Murphy 2000; Fang and Zhu 2017; Henning et al. 2018). Mature B lymphocytes also have the potential to distribute their progeny among several distinct fates or intermediate states after they have encountered a ligand for the B-cell antigen receptor and costimulatory signals. The function of B lymphocytes that has attracted the most attention is their role as precursors to the plasma cells that constitutively secrete immunoglobulins (i.e., antibodies)—both those that are highly antigen-specific and others that are polyreactive or have a broader range of specificities tilted toward recognition of biochemical constituents of micro-organisms. However, there is strong evidence of additional functions for mature cells in the B lineage, some of which even appear to be antibody-independent. This review summarizes some salient advances toward elucidation of the molecular programming of the fate choices and function of B cells in the periphery. In parallel, we note unanswered questions that pertain to differences among subsets of B lymphocytes and plasma cells.
The B lineage in the periphery: B cells and beyond
Fully mature B-cell subtypes include B1 (comprising B1a and B1b) and B2 cells in marginal zone (MZ) and follicular (FO) subsets, but intermediates that are transitional B cells may also influence humoral immunity. A large body of work depicting these events is summarized in Figure 1 (Herzenberg and Herzenberg 1989; Erickson et al. 2002; Martin and Kearney 2002; Dorshkind and Montecino-Rodriguez 2007; Hardy et al. 2007; Allman and Pillai 2008). In adult mammals, B lymphocytes continuously populate the peripheral immune system (Fig. 1) after completing a well-orchestrated developmental process in the bone marrow starting from lymphoid progenitors (CLPs, all-biased lymphoid progenitors [ALPs], and B-cell-biased lymphoid progenitors [BLPs]) (Inlay et al. 2009) beyond the scope of this review. Epigenetic and transcriptional mechanisms that establish B-lineage commitment (Lin et al. 2010; Boller and Grosschedl 2014; Li et al. 2018; Miyai et al. 2018) also govern the maintenance and function of mature B cells. These factors include the helix–loop–helix protein E2A, the early B-cell factor EBF1, and the paired domain transcription factor Pax5 (Nutt and Kee 2007; Treiber et al. 2010; Medvedovic et al. 2011). More recently, ten-eleven translocation (TET) proteins that can erase methylation from DNA to revise epigenetic programming have been found essential for B-cell development (Lio et al. 2016; Orlanski et al. 2016). The establishment of immature B cells is driven by signals generated from successful assembly of a functional B-cell receptor (BCR) and by a first vetting of BCRs to remove autoreactive B cells by receptor editing or death and deletion (Buhl et al. 2000; Melchers et al. 2000; Nemazee and Weigert 2000; Nemazee 2017). On completing this process, these immature IgM+ B cells exit the bone marrow and enter the spleen (part of “the periphery” for lymphocytes), where they mature into microanatomically and phenotypically distinct subsets of mature naive B cells.
Figure 1.
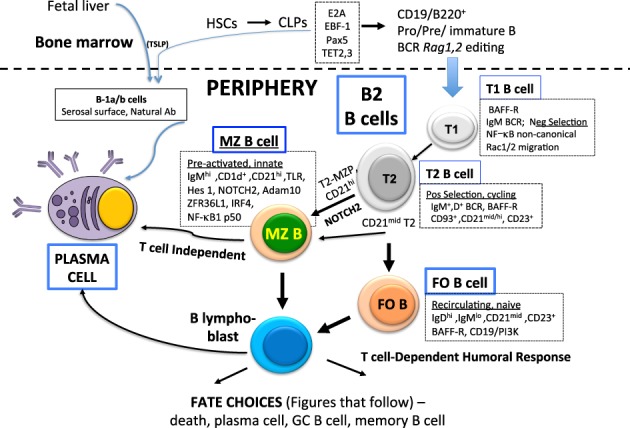
Origins of and fate potential for mature B lymphoid cells. Shown is a simplified outline of the broad array of peripheral B-cell subsets that provide host defense. Two major types of B cells (B1 and B2) populate the peripheral immune system, outside the bone marrow. B1 subtypes, which are outside the scope of this review, are a minor subset that (in mice) is derived largely from fetal liver precursors. In the periphery, they reside principally in serosal spaces such as the peritoneal cavity. The B2 subset constitutes most of the B cells in the peripheral immune system. They emerge from the bone marrow as “immature B cells” via a transcriptional program that is initiated in CLPs. In the periphery, cells with autoreactive BCR are culled from the repertoire at transitional stages, depicted here as T1 and T2, with selected features noted in the text boxes. The small late transitional population displays a range of functions that includes antigen presentation, sensing of innate signals, and immune regulation. These cells are also the precursors for the two major B-cell subsets—FO and MZ—that provide critical effector functions by differentiating into antibody-producing plasma cells. Unique signaling and transcription programs that characterize MZ and FO B cells, some of which are summarized in the text boxes, provide the capacity for immediate and sustained humoral immunity for both innate (T-cell-independent) and T-cell-dependent responses.
The newly emergent B cells mature in the spleen via a multistage transitional process mediated by signals from the BCR and the TNF receptor family member BAFF receptor but also include interactions with myeloid and stromal elements. Transitional B-cell stages have been identified by combined expression levels of surface markers associated with immaturity (CD24, CD93, and AA4.1), BCR isotype, and coreceptor CD21 (complement receptor 2) or CD23 (low-affinity IgE receptor) (Fig. 1; Loder et al. 1999; Allman et al. 2001). In one current classification, CD93 (AA4.1+) IgMhi IgDlow CD21low CD23− B cells are considered to be at transitional stage 1 (T1), and AA4.1+ IgMhi IgDhi CD21med/hi CD23+ identifies T2 B cells. Early stage transitional B cells enter the marginal sinuses and red pulp of the spleen and then—after passing a chemokine- and integrin-mediated checkpoint on survival and migration—pass into the white pulp to colonize a T-cell-rich periarteriolar lymphoid sheath (PALS) (Henderson et al. 2010). Negative selection at this site removes self-reactive B cells (Petro et al. 2002); the remaining T1 B cells acquire IgD and CD23 expression characteristic of T2 B cells. T2 B cells are considered to be positively selected and demonstrate considerable functional diversity for a small subset (∼2% of spleen cells). T2 cells expressing a high level of CD21 (CD21hi) are recognized as precursors of MZ B cells, while T2 CD21mid cells have unique functional properties that include sensing environmental cues and a proliferative capacity that distinguishes them from other transitional B cells (Evans et al. 2007; Meyer-Bahlburg et al. 2008). The T2 CD21mid subset has bimodal potential and can develop into either MZ or FO B cells (Evans et al. 2007; Meyer-Bahlburg et al. 2008). Regulatory functions of B cells, typically dependent on IL-10 production, are often connected to the T2 phenotype. Cells with transitional T2 B-cell properties have been associated with allograft survival, tumor-mediated suppression, and amelioration of autoimmune diseases (Blair et al. 2009; Ganti et al. 2015; Moreau et al. 2015). Thus, the T2 stage appears to be a critically important developmental nexus that determines the fate and function of peripheral B cells. As with several other aspects of diversity in B-cell regulation, interrogating the molecular features with single-cell technologies will be necessary to obtain a better understanding of this small but critical subset.
On completing their maturational transition, a large majority of mature peripheral B cells enter the recirculating pool as naïve B cells (IgDhi IgMlow CD21mid CD23hi) that home to lymphoid follicles of the spleen, lymph nodes, and Peyer's patches. At these sites, B cells organize around a dense network of processes extending from FO dendritic cells (FDCs) as well as other varieties of a specialized stromal cell, the diversity of which is an ongoing area of analysis (Cyster et al. 2000; Rodda et al. 2018). FDCs and additional stromal constituents attract B cells by secreting the chemokine CXCL13, facilitate antigen display, and promote B-cell survival by producing the cytokine BAFF (Allen and Cyster 2008; Wang et al. 2011). B-cell follicles reside adjacent to T-cell zones, an organization that facilitates interactions of FO B cells and activated T helper cells that migrate to the interface of T-cell and B-cell zones after initial activation. Due to their locale, additionally regulated by actions of stromal cell-derived oxysterol on their positioning (Yi et al. 2012), FO B cells are particularly well suited to participate in T-cell-dependent immune responses to protein antigens.
Under homeostatic conditions, B cells are maintained by the action of BAFF—a product of monocytes, DCs, and neutrophils in addition to the stromal and FDC populations already noted—on BAFF receptor and by signaling through the BCR (Lam et al. 1997; Schiemann et al. 2001; Mackay and Browning 2002; Srinivasan et al. 2009). Among B cells, a small residuum (∼10%) of the population survived after elimination of their BCR (Lam et al. 1997). Recent studies use targeted removal of key BCR signaling components to determine whether survival mediated by components of basal BCR signaling differs from those required for development or activation. Initial work showed that mature B cells die off after depletion of the tyrosine kinase Syk, an essential mediator of B-cell activation (Schweighoffer et al. 2013). However, later work noted that about one-fourth of the B-cell repertoire then survived so that Syk was dispensable for survival of many mature B cells (Hobeika et al. 2015). This survival of Syk-deficient B cells was dependent on CD19-dependent phosphatidylinositol 3-kinase (PI3K) signaling as well as BAFF. On similar lines, although Bruton's tyrosine kinase (BTK) is a crucial mediator of BCR and BAFF signals during differentiation, mature B cells from which BTK was conditionally removed survived for >1 mo in the absence of BTK (Nyhoff et al. 2018). However, each of these molecules was necessary for subsequent B-cell activation. Thus, BCR signaling components that are necessary for B-cell development as well as activation may be functionally redundant for maintenance of mature B cells. At the transcriptional level, EBF1 contributes selectively to maintenance of mature B cells in that this critical regulator of early B-cell development was dispensable for survival of recirculating FO B cells yet was essential for MZ and B1 B cells (Gyory et al. 2012; Vilagos et al. 2012). These findings point to a paradigm by which functional differences in mature B-cell subsets are maintained only in part by the signaling and transcription networks that preprogram subsequent effector responses.
A smaller subset (∼5%–7% of mature B cells) resides in a MZ (the outer white pulp of the spleen between the marginal sinus and the red pulp), where they are continuously exposed to blood-borne antigens (for review, see Cerutti et al. 2013). The positioning and retention versus migration of MZ B cells are regulated by sphingosine-1 phosphate and integrin interactions as well as CXCR7 (Lu and Cyster 2002; Wang et al. 2012a; Arnon and Cyster 2014). Upon antigen encounter, MZ B cells are poised for rapid differentiation to plasmablasts and for migration and antigen presentation to T cells. Compared with FO B cells that are primarily involved in T-cell-dependent B-cell responses, MZ B cells have almost 200 differences in gene expression (Kin et al. 2008). Salient distinctions include increased expression of surface immunoglobulin M (IgM), complement receptors (CD35 and CD21), various Toll-like receptors (TLRs; namely, TLR2, TLR3, TLR4, and TLR7) (Rubtsov et al. 2008), and the lipid antigen-presenting molecule CD1d. Development and maintenance of MZ B cells depends on signaling by BCR, BAFF-R, and NOTCH2. Mice lacking expression of NOTCH2, the NOTCH ligand Delta-like 1 (DLL1), or NOTCH signaling components show a dramatic decrease in the number of MZ B cells (for review, see Tanigaki and Honjo 2007). In contrast, constitutive expression of the active NOTCH2 in B cells leads to a marked increase in the number of MZ versus FO B cells (Hampel et al. 2011). In addition to the mechanisms noted above (S1PR, CXCR7, and integrins), the spatial positioning of MZ B is controlled by the transcription factor IRF4 via regulation of Notch2 gene expression (Simonetti et al. 2013). Recently post-translational regulation by the RNA-binding protein ZFP36L1 was found to be essential for maintenance of MZ B cells (Newman et al. 2017). ZFP36L1 controlled a gene expression program in part by limiting expression of the transcription factors KLF2 and IRF8, which are known to enforce the FO B-cell phenotype. MZ B cells are responsible for the antibody response to type 2 thymus-independent (TI-2) antigens, such as polysaccharide from encapsulated bacteria (Fagarasan and Honjo 2000; Martin and Kearney 2000). MZ B cells have innate-like properties using a restricted repertoire of germline-encoded V genes that facilitate multireactive specificities for microbial antigens (Cerutti et al. 2013). These responses are manifested by robust extrafollicular plasmablast formation but not germinal center (GC) formation. Recent studies demonstrate the importance of STAT1 in TLR-mediated differentiation of MZ B cells by its direct regulation of Prdm1 (which encodes Blimp1) as well as protective function (Chen et al. 2016b). Identification and characterization of MZ B cells in humans have been complicated (Weill et al. 2009). Work demonstrating in vitro differentiation of human MZ-like B cells driven by the NOTCH2–DLL1 pathway as well as reduced IgM+ IgD+ CD27+ B cells in NOTCH2 haploinsufficient patients favors the existence of a human counterpart to murine MZ B cells (Descatoire et al. 2014). Further studies will enhance understanding of the functional significance of this subset in humans.
Plasma cells, the major antibody-secreting cells, are derived from B lymphoblasts in several categories of sites (Fig. 2). Plasma cells arise as outputs of GC reactions within the follicles (covered in detail below), after activation of MZ B cells, or in extrafollicular foci. Extrafollicular T-cell-dependent responses can arise when antigen-specific B cells and T cells first interact (MacLennan et al. 2003; Taylor et al. 2012). These appear largely to involve localized short-lived antibody production. T-independent (TI) responses such as those induced by TI-2 antigens with repetitive chemical units also induce extrafollicular responses but do not form GCs. The magnitude of responses can be influenced by affinity of the BCR and by the epitope density of antigen: Increased BCR interactions favor heightened extrafollicular plasmablast formation (Paus et al. 2006), an affinity bias that is less evident for GC-derived plasma cell formation. T cells can also influence the magnitude of the extrafollicular response; depending on the nature of the immunogen driving the response, this effect of T cells is mediated by or independent from IL-21 (Linterman et al. 2010; Lee et al. 2011). Although these extrafollicular responses principally involve germline-encoded BCR and yield low-affinity IgM with only small amounts of switched antibody, they likely provide early host protection during the interval that precedes GC formation. Likewise, MZ B cells exposed to pneumococcus in vivo generate a robust plasma cell response in the marginal sinuses (Martin et al. 2001).
Figure 2.
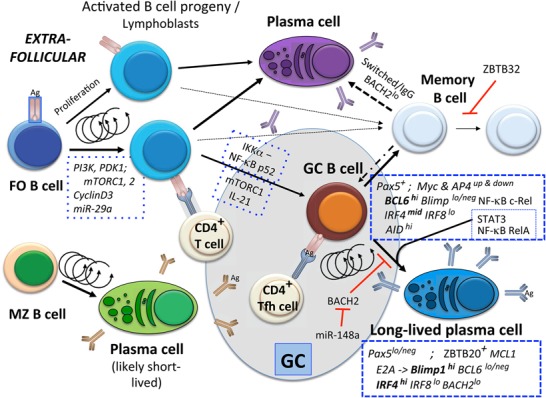
Paths to antibody responses and memory. Simplified cellular progression from FO and MZ B cells to plasma cell differentiation independent from the GC, into memory (Bmem), and via the GC reaction is shown, omitting complexities generated by heavy chain class switching both outside and within the GC. A partial list of molecular regulators, drawn from the text, is shown in boxes enclosed by dashed lines. Multiple rounds of proliferation are shown that are essential for developmental progression, as are indications of some temporal aspects of the extended GC reaction.
Finally, fully matured B-lineage cells or the antibodies that they secrete can exercise major effects on host–tumor interactions and the balance between cancer progression and clearance as well as autoimmunity or tolerance (Gunderson and Coussens 2013; Affara et al. 2014). Precursor–product relationships at the cellular level are not clearly established, but regulatory B-lineage (Breg) cells that secrete IL-10 have been identified (Yanaba et al. 2008; Yoshizaki et al. 2012; Lykken et al. 2015). A plasmablast or plasma cell phenotype has been identified for suppressive cells (Matsumoto et al. 2014) that, in promotion of prostate cancer growth, use lymphotoxin and are IgA+ (Ammirante et al. 2010; Shalapour et al. 2015). Most recently, evidence of a naturally suppressive plasma cell subset expressing the coinhibitory receptor LAG3 and secreting IL-10 in a mouse infection model has emerged (Lino et al. 2018). Other than participation of IL-21 (Yoshizaki et al. 2012) and activated capacity to secrete IL-10 or, more recently, IL-35 (Shen et al. 2014), remarkably little is known about the molecular relationship among the different categories of Breg cells or between them and the B-lineage subsets.
Overview of the basic choice—balancing Blimp1 against BCL6
In general terms, much of the gene expression that frames cellular identity along the B lineage is established and has been modeled through circuit diagrams (Sciammas et al. 2006; Martinez et al. 2012; Nutt et al. 2015). This framework is elaborated on below, particularly with respect to proliferation and the GC, but an outline is as follows (Fig. 2). In the naïve FO B cell, transcription factors BCL6, IRF4, and Blimp1 are at low levels (Bhattacharya et al. 2007; Willis et al. 2014). IRF8 and BACH2 are expressed and antagonize IRF4 and Blimp1, respectively (Carotta et al. 2014; Xu et al. 2015). After activation, IRF4 levels decrease, but, if provided sustained or cognate T-cell help (Tfh), the B lymphoblast may initiate a GC B-cell program in which this transcription factor increases to a level intermediate between that of the GC B cell and the plasma cell (Sciammas et al. 2006; Xu et al. 2015). Alternatively, some B lymphoblast progeny may differentiate directly into antibody-secreting plasma cells. This change of state involves a profound reprogramming in which expression of the transcription factor Pax5—which is essential for pro-B-cell, pre-B-cell, and B-cell identity—is extinguished, while Blimp1 expression achieves high levels (Shaffer et al. 2002; Cobaleda et al. 2007; Medvedovic et al. 2011; Shi et al. 2015). As a consequence, the transcription termination and splicing of antibody heavy chain genes are stably altered so that Ig CH protein is secreted instead of membrane-anchored as an antigen receptor. In addition to loss of the surface BCR, the transition from a Pax5-dependent to an IRF4- and Blimp1-dominated transcriptional landscape reprograms the expression profile for cell surface proteins that trigger signaling or enhance the effective exposure to the prosurvival cytokine APRIL (McCarron et al. 2017). B-cell survival depends on the receptor BAFF-R for survival signals from the stroma-derived cytokine BAFF (Schiemann et al. 2001; Thompson et al. 2001; Mackay and Browning 2002), whereas plasma cells require the related TNF superfamily cytokine APRIL, which then signals through the receptors TACI and BCMA (O'Connor et al. 2004; Bossen et al. 2008; Coquery and Erickson 2012; Ou et al. 2012). These signals and their transcription factor intermediaries lead to altered balances of prosurvival and proapoptotic proteins of the Bcl2 superfamily (e.g., see Lee et al. 2013). Depletion of Mcl1 from GC B cells or plasma cells prevented their persistence (Vikstrom et al. 2010; Clybouw et al. 2011; Peperzak et al. 2013). In contrast, interference with an interaction surface that is essential for the inhibition of proapoptotic BH3-only proteins at the mitochondrial membrane impacted the persistence of Bmem cells and new plasma cell generation but not the continuation of ongoing GC reactions or established long-lived plasma cells (LLPCs) (Carrington et al. 2010). The transition to plasma cell identity appears to be accompanied by a shift in intermediary metabolism in which the dependence on mitochondrial pyruvate import increases (Lam et al. 2016). The high rates of antibody heavy and light chain synthesis, glycosylation, and assembly create a particular demand for activators of the unfolded protein response (UPR) in plasma cells, identified as a requirement for XBP1 (Reimold et al. 2001; Iwakoshi et al. 2003; Shaffer et al. 2004). In addition, some evidence suggests that persistence of plasma cells depends on intact autophagic mechanisms such that loss of function for the autophagy gene Atg7 led to initially higher rates of antibody secretion but a loss of humoral memory due to plasma cell failure over time (Pengo et al. 2013). However, some results indicate that the initial formation of GC B cells, memory, and a primary anti-influenza response does not require the protein ATG5, which, like ATG7, is an essential component of canonical autophagy (Chen et al. 2014, 2015). Elucidation of these issues is of great practical importance in vaccines and autoantibody-mediated disease. However, the fragility and niche dependence of plasma cells, along with challenges in identifying surface markers that distinguish LLPCs versus potentially newer shorter-lived plasma cells, have thus far posed a major barrier to understanding programming in specific subsets of plasma cells in clear biochemical terms.
Signals regulating fate choice
The initial activation of B cells leads to rapid proliferation. The rounds of division, many of which take place before GC formation, appear to be preconditions to all later fates for the B cell and its progeny. Thus, the probabilities of (1) switching or (2) plasma cell differentiation are extremely low prior to a minimum division count (approximately three and six, respectively), after which they increase progressively (Rush et al. 2005; Barwick et al. 2016). Antigen-activated B cells appeared to divide over six to eight times (64–256 potential progeny) before some progeny moved into the GC (Gitlin et al. 2014). The duration of each cell cycle—and thus the frequency and extent of divisions—may be regulated by BCR affinity or the qualities of Tfh received by an activated B cell (Gitlin et al. 2014, 2015). At present, how specific signaling pathways elicited by antigen receptors and costimulation are transduced into altered rates of DNA replication is not fully elucidated.
Nonetheless, triggering and modulation of the signaling pathways downstream from the lipid kinase PI3K is a major impetus to proliferation that is as essential in B cells for antibody responses as it is in earlier development (Fruman et al. 1999; Dil and Marshall 2009; Chiu et al. 2017; Jayachandran et al. 2017). Recent work indicates that the specific Ras isoform R-Ras2 links the BCR and CD19 to PI3K in leading to GC B cells (Mendoza et al. 2018). At a snapshot in time, only a small minority of B cells in the GC light zone (LZ) exhibited evidence of PI3K activation (Dominguez-Sola et al. 2015; Sander et al. 2015; Jellusova et al. 2017). This finding is consistent with a competitive process in which only those cells received a renewed stimulus from Tfh cells, providing cognate help. However, it is possible that the stimulus for the elevated PI3K is elicited just by BCR with CD19 after antigen capture (e.g., from FDCs), and the possibility that the PI3K activation is very transient also cannot be dismissed. Signal relays as well as transcription factors and apoptosis regulators downstream from PI3K transduce the proliferative impulse initiated by PI3K. These include (1) the GTP exchange factor Vav (Doody et al. 2001; Stephenson et al. 2006); (2) PDK1, the main activator of the serine–threonine kinase AKT at T308 (Park et al. 2013; Baracho et al. 2014); (3) mTOR complex 2 (mTORC2), whose phosphorylation of the AKT hydrophobic motif (S473) promotes FoxO1 phosphorylation (Lee et al. 2013); and (4) mTORC1 (Chou et al. 2016; Jones et al. 2016; Raybuck et al. 2018). NF-κB and FoxO1 are regulated directly or indirectly downstream from these signaling complexes (Lee et al. 2005, 2010, 2011, 2012; Park et al. 2013). While not fully worked out specifically in mature B cells, these transcription factors in general regulate proliferation and the proapoptotic and apoptotic genes downstream from mTORC2 (Yusuf et al. 2004; Lee et al. 2013). Within the subset of B cells that exhibited high PI3K activation, the activity of the glycogen synthase kinases GSK3α and GSK3β is another target influencing proliferation and survival (Jellusova et al. 2017).
The calcium–NFAT pathway is one of the signaling and transcription mechanisms most central to lymphocyte activation, differentiation, and function. This pathway plays in the distinction between tolerance and activation (Goodnow et al. 2010). Foundational work provided evidence that calcium signaling without substantial activation of NF-κB or ERK/AP1 characterized the tolerant state in mature B cells chronically stimulated by soluble Ag (Dolmetsch et al. 1997; Healy et al. 1997). The Ca++/NFAT pathway is also active in B cells rendered tolerant to a low-affinity natural self-antigen (insulin) but, in this case, without uncoupling of NF-κB signaling (Acevedo-Suarez et al. 2006). The resulting imbalance in different NFATs maintains tolerance in nonautoimmune B6 mice (Bonami et al. 2014; Williams et al. 2015). However, in genetically autoimmune mice, these B cells are recruited into GCs and produce autoantibody (Wan et al. 2016). This finding provides an important example of how genetic backgrounds change fundamental aspects of the fate potentials of mature B cells. Analyses of calcium signaling in B cells have been invigorated by discovery of Orai1 and stromal interaction molecules (STIM1 and STIM2) along with conditional loss-of-function mutations. Constitutive absence of store-operated calcium entry (SOCE) caused some alterations of lymphocyte differentiation, but, among the B cells that did develop, the capacity to proliferate was substantially reduced (Gwack et al. 2008). The most notable effect and greatest molecular detail point to a function of SOCE in regulating IL-10 production by B cells and their plasmablast or plasma cell progeny (Matsumoto et al. 2011, 2014; Ziegler et al. 2014). A similar heightened IL-10 production is observed in autoreactive B cells functionally silenced by impaired Ca++ signaling (Felton et al. 2018). Lack of STIM protein altered affinity maturation (Mao et al. 2016), implying an effect on GC function. Together, these findings indicate that Ca++/ NFAT signaling likely not only functions in promoting cell-intrinsic proliferation but also may restrain suppressive functions of B lineage cells.
Antibody class choice as a fate
The requirement for proliferation likely arises in part because proliferation facilitates DNA demethylation as well as new DNA methylation, each of which appears to be essential for progressive differentiation after activation of conventional B2 B cells (Barwick et al. 2016, 2018; Orlanski et al. 2016). Two initial decision points for the proliferating B cells are whether the progeny undergo (1) Ig class switching and/or (2) differentiation into plasma cells. Each of these steps can occur without the other either before or after entering the GC and after either activation of the naïve B cell or reactivation of a memory B cell. The switching event influences the fate or status of B cells because of differences in the signal transduction by different classes of BCR (e.g., IgM vs. IgG vs. IgE) (Horikawa et al. 2007; Engels et al. 2009; Liu et al. 2010a,b, 2012; Haniuda et al. 2016; Yang et al. 2016). In addition, the localization of cells switched to these different isotypes and the duration and transcription factor requirements for survival of Bmem cells (e.g., IgM vs. IgG and IgG2a vs. IgA) (Dogan et al. 2009; Pape et al. 2011, 2018; Wang et al. 2012b; Zuccarino-Catania et al. 2014) also highlight class-based molecular and functional distinctions.
Antibody class switching is mediated by activation-induced cytosine deaminase (AID), which catalyzes IgH locus recombination after recruitment to particular germline-encoded switch regions (Muramatsu et al. 1999, 2000; Okazaki et al. 2002; Stavnezer and Schrader 2014). This recruitment is cytokine-regulated and depends on the induction of transcription at specific promoters upstream of quasirepetitive switch regions that precede each isotypic Ig constant region exon (Snapper and Paul 1987; Rothman et al. 1990; Jung et al. 1993; Lee et al. 2001; Stavnezer and Schrader 2014). One barrier to deeper understanding of the molecular events is that it is not known precisely when or where the recombination of a class switching occurs. Since disappearance of the previous constant region and surface expression of the new class are subject to phenotypic lag, this would require direct visualization on a cell-by-cell basis of the cleavage event. In any case, such switches of antibody constant regions affect memory B-cell and plasma cell fates as a consequence of differences in the signaling elicited by engagement of the distinct classes of BCR (IgM, IgG, and IgE) (Horikawa et al. 2007; Engels et al. 2009; Liu et al. 2010a,b; Haniuda et al. 2016; Yang et al. 2016). For example, IL-4 promotes switching to either IgE or IgG1. However, IgG1 humoral memory is durable, whereas IgE-expressing B cells appear to be inefficient at sustained participation in GC reactions, at least in some (but not all) model systems (Talay et al. 2012; Yang et al. 2012; He et al. 2013). Intriguingly, however, some of the data from mouse models may be conditioned by a lack of sufficient Tfh cells (which can provide IL-4) so that antigen-specific Tfh cells are competent to yield sustained IgE in serum, whereas Th2 cells are not (Kobayashi et al. 2017). What the differences are in programming when help for switching to Cε derives from conventional Th2 cells (which might act in extrafollicular switching and plasma cell generation) as compared with Tfh cells or when IgE arises from a secondary switch event (e.g., Cγ1 to Cε) will be important issues for molecular regulation of allergic diseases (He et al. 2013).
The germline CH promoters upstream of switch regions are regulated by cytokine-induced STAT transcription factors, which link cytokines to antibody isotype and additional aspects of the gene expression program. For instance, IFN-γ or IL-27 drive switching to the proinflammatory IgG2c isotype via nuclear induction of STAT1 homodimers and subsequent expression of T-bet (Peng et al. 2002; Yoshimoto et al. 2004; Nguyen et al. 2012; Domeier et al. 2016), and IgG2c+ B cells express CXCR3, a T-bet-dependent chemokine receptor that mediates cellular recruitment to sites of inflammation (Serre et al. 2012). Conversely, IL-4-induced STAT6 inhibits this switch while promoting IgG1 and, at lower efficiency, IgE (Takeda et al. 1996; Linehan et al. 1998; Mao and Stavnezer 2001). In addition to STATs and T-bet, NF-κB regulates the promoters directing transcription from “I” exons upstream of the switch segment constant region pairs that are in the germline DNA of heavy chain genes (Lin and Stavnezer 1996). Moreover, Runx proteins and TGF-β-induced SMAD transcription factors mediate induction of Iα, the transcript for switching to IgA (Cerutti 2008; Watanabe et al. 2010). Finally, chromatin proteins such as Ikaros distinguish between switch regions or isotypes so that it represses the highly proinflammatory or pathogenic IgG2 isotypes but not others (IgG1, etc.) (Sellars et al. 2009). Although it is not clear whether the mechanism is direct (i.e., binding in cis) or indirect, recent evidence indicates that hypoxia-inducible transcription factors (HIFs) exert promoter-specific inhibition of the germline CH transcripts that promote IgG1 and, even more prominently, IgG2c while sparing Iα (Cho et al. 2016). Finally, regulatory RNAs are less explored. Nonetheless, there is evidence that microRNA 146a (miR-146a) promotes switching to Cε (i.e., generation of IgE), perhaps via either increased 14-3-3 or reduced SMAD4 (Li et al. 2017).
While the germline CH promoters for “sterile CH transcripts” are crucial determinants of switch targeting, the DNA breaks and recombination require AID (product of the Aicda gene). PI3Kδ, one isoform of the PI3K catalytic subunit, was found to repress AID expression (Omori et al. 2006) even though PI3K, mTORC1, and mTORC2 activities are crucial for proliferation and later formation of GCs (Lee et al. 2013; Dominguez-Sola et al. 2015; Sander et al. 2015; Jones et al. 2016; Chiu et al. 2017; Raybuck et al. 2018). In part, this finding appeared to be mediated through FoxO1 downstream from AKT kinase (Dengler et al. 2008; Dominguez-Sola et al. 2015; Sander et al. 2015; Inoue et al. 2017), but an important element was that this PI3K isoform regulated Blimp1, whose expression would block the program leading to Aicda. In part, these findings illustrate the complexities of population analyses with highly dynamic cells and the ultimate need for single-cell analyses of mRNA, protein, and even enzyme activities. AID is active in both extrafollicular and, at higher levels, GC B cells. Moreover, the cytosine deaminase activity, whose function is to introduce both point mutations and DNA strand breaks, creates inherent risks of death and oncogenic transformation (Daniel and Nussenzweig 2013; Casellas et al. 2016). As such, a large array of transcription factors and other regulators has been tied to determination of AID levels. The E-box-binding E2A factors and their negative regulators, Id proteins (Gonda et al. 2003; Sayegh et al. 2003), as well as BCL6, Batf, NF-κB, YY1, and the homeobox protein HoxC4 are among transcription factors, likely as direct regulators of Aicda gene expression (Park et al. 2009; Pavri et al. 2010; Basso et al. 2012; Pone et al. 2012; Zaprazna and Atchison 2012; Ise et al. 2018). In addition, AID protein levels, activity, and retention on chromatin likely also are regulated (Pavri et al. 2010; Sun et al. 2013; Zan and Casali 2013), and microRNAs such as miR-155 are also modulators of Aicda mRNA (Teng et al. 2008; Basso et al. 2012; Bouamar et al. 2015). Analyses of the impact of hypoxia and HIFs on switching indicated that AID (which, from the outset, was identified as IL-4-stimulated) (Muramatsu et al. 1999) is regulated in a manner dependent on the cytokine milieu and that hypoxia inhibited AID expression in the presence of IFN-γ, whereas it had no effect in IgA-promoting conditions (TGF-β and retinoic acid) (Cho et al. 2016). However, it remains to be determined whether there is any direct effect of HIF on Aicda or whether, instead, the result is due to sensing of the activity of mTORC1. Nonetheless, the work taken together evokes models in which there is an interplay among nutrient microenvironment, cytokine milieu, and B-cell sensors in a manner whereby the local conditions adjust AID activity (Keim et al. 2013) and contribute to the balance of fates that emerge.
Once a B cell differentiates into a plasma cell, the localization of LLPC populations tracks with their antibody class. As such, both the regulation of the promoters that precede the quasirepetitive Ig CH switch regions and that of AID expression are important determinants of B-cell fates. IgA plasma cells are more likely to be at mucosal sites (gut and respiratory tract), whereas IgG LLPCs are in the marrow (Moser et al. 2006; Kometani and Kurosaki 2015). Based on clinical parameters, it is almost certain that there are IgE LLPCs in allergic disease patients. Although typically rare, IgE plasma cells can be detected in mucosa-associated lymphoid tissues (Chvatchko et al. 1996; Eckl-Dorna and Niederberger 2013). Distinct homing receptors as well as patterns of where plasma cells producing the different antibody classes arise likely account for the differences in localization, but the transcriptional regulatory patterns that create and maintain these differences remain to be determined.
GCs—sources of enhanced affinity, memory, and plasma cell durability
If an activated B lymphoblast acquires suitably persistent T-cell help, decreases expression of the G-protein-coupled receptor EBI2 (Pereira et al. 2009), and avoids rapid differentiation into a plasmablast with high IRF4 and Blimp1 and lower IRF8, it may enter or contribute to the establishment of a GC reaction (Fig. 3). This specialized microanatomic niche within the overall follicle involves a mutually reinforcing program in which both B and CD4 T cells express progressively higher concentrations of the repressor BCL6 (Zhang et al. 2017). The cytokine IL-21 is essential for the gene expression program of the GC B cell as well as for generation of plasma cells from the follicle (Ozaki et al. 2002; Yoon et al. 2009; Bessa et al. 2010; Linterman et al. 2010; Zotos et al. 2010; Berglund et al. 2013). As an example of flexibility in immunity, however, extrafollicular antibody responses can appear IL-21-dependent or IL-21-independent. For instance, an extrafollicular IgG2c response to Salmonella was not affected by the absence of this cytokine (Linterman et al. 2010). As noted above, the GC program involves superinduction of AID, which is essential for introduction of new somatic mutations targeted to the immunoglobulin locus (Muramatsu et al. 2000). GC B-cell biology hinges on a capacity for iterative cycling of the B cells between LZs—rich in FDCs laden with antigens (potential BCR ligands) and Tfh cells—and dark zones (DZs) in which B cells have resumed proliferation (Allen et al. 2007; Victora et al. 2010; Victora and Nussenzweig 2012). Gene regulation and expression profiles as well as the analyses of them are made more complex by this cyclical migration and by an admixture of cells in the LZ with low or briefly high PI3K activity (Dominguez-Sola et al. 2015; Sander et al. 2015; Jellusova et al. 2017). Moreover, recent work with a sensor to identify cells on their way toward death indicated that substantial portions of a GC B-cell pool likely are set to undergo apoptosis within hours (Mayer et al. 2017). These findings indicate that dynamically expressed factors that are PI3K-regulated may be at very different levels in a majority of B cells that will not contribute after affinity selection from the minority that ultimately establishes humoral memory. Moreover, B-lineage-restricted loss-of-function analyses indicate that mTORC1 promotes the expression of both Bcl6 and Irf4 mRNA and protein (Raybuck et al. 2018) as one of the mechanisms by which this branch of PI3K signaling regulates the GC program. In any case, genetic approaches extend earlier findings with the immune suppressant rapamycin to indicate that both mTORC1 (Cho et al. 2016; Jones et al. 2016; Ersching et al. 2017; Raybuck et al. 2018) and mTORC2 (Lee et al. 2013) complexes are essential in B cells for a GC reaction.
Figure 3.
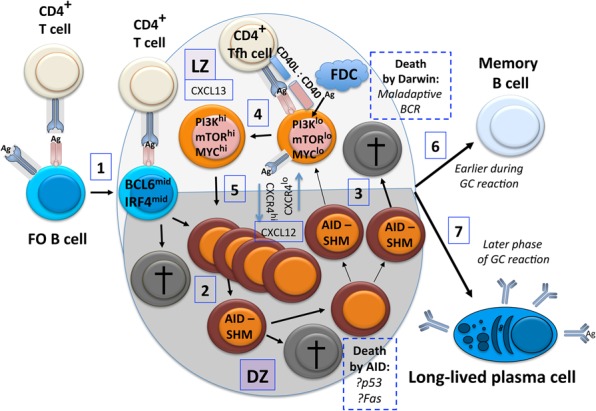
Entry, exit, life, and death in the light–dark cycle of a GC. After an activated B lymphoblast encounters a helper T cell (best of all, one whose T-cell receptor [TCR] recognizes a peptide derived from the same antigenic protein to which the B cell responded; referred to as “cognate” help), an initial T–B conjugate may form. (1) These cells may then move into the developing GC, in part via CXCR5 responding to a CXCL13 gradient along with decreased expression of the 7α,25-dihydroxycholesterol-binding G-protein-coupled receptor EBI2. (2) The B lymphoblast continues proliferation, although, in the DZ, death rates of its progeny (gray cells) may approach 50% and are due in large part to AID, likely involving its generation of genomic stress (genotoxicity). The high AID expression and replicating DNA allow targeting of the cytosine deaminase and somatic hypermutation (SHM) that yields sequence variants of the BCR expressed on the B-cell clone that entered the GC. (3) As a timed program of DZ residence runs out and signaling by the chemokine receptor CXCR4 falls, CXCR5-directed chemotaxis leads GC B cells from the DZ to the LZ. In the LZ, mutated BCR specificity may lead to apoptosis from either a failure to compete for antigen and Tfh or generation of an autoreactive receptor. (4) However, some clones will have BCRs that succeed in the competition to capture the antigen or immune complex from FDCs in the LZ, internalizing and processing the captured protein for display of peptides in MHC-II. These successful cells will be reinvigorated and activated by the antigen and help from Tfh (IL-21 and CD40 ligand), leading to a short burst of higher PI3K activity and c-Myc. (5) These reactivated GC B cells can migrate back to the DZ, thereby fostering both greater numbers of progeny from more proliferation and the potential to increase BCR affinity for antigens after further SHM. (6) Relatively earlier in the GC reaction, some GC B cells may convert to a quiescent state and take on the characteristics of memory B cells. (7) At later times, the output of cells shifting from the GC phenotype to become plasma cells increases.
Further complicating analyses, intravital labeling found dense patches of hypoxic cells in most but not all GCs (Cho et al. 2016; Jellusova et al. 2017). The hypoxia appeared mostly but not exclusively to colocalize with FDCs (i.e., the LZ) (Cho et al. 2016). In addition, HIF-1α stabilization was increased in the GC, which could be due to either antigen activation or hypoxia. As noted above, AID creates danger for GC B cells by activating p53 and through the risk of unresolved damage. Indeed, analyses of dying B cells in vivo suggest a substantial degree of death due to AID in the DZ (Zaheen et al. 2009; Mayer et al. 2017). As such, its level and activity likely need tight control. As noted above, increased HIF lowered AID expression; in addition, sustained HIF elevation appears to attenuate mTORC1 activity, and partial reductions in mTORC1 reduced both Aicda gene expression and the frequency of somatic hypermutation (SHM) in GC phenotype B cells (Cho et al. 2016; Raybuck et al. 2018). Parallel work has provided evidence that dynamic adjustments in mTOR activity are mechanistically involved in the deceleration of B-cell proliferation in the DZ (Ersching et al. 2017). Ultimately, this competitive fitness process involves both survival and extent of proliferation and leads to increased affinity for Ag over the affinity profile of the BCR on B cells that initiate a response to immunization.
Long-term humoral memory—embodied in both LLPCs and functional Bmem cells—can arise from extrafollicular reactions or after TI proliferation (Bortnick et al. 2012; Taylor et al. 2012), but the efficiency of generating such cells is enhanced by participation in the GC reaction. Although the capacity of an individual B cell to see both fates may depend on its BCR (Taylor et al. 2015), each GC—often arising at disparate times after initiation of the immune reaction—likely can yield a population of B-cell progeny that includes Bmem, short-lived plasmablasts, and LLPCs. What, then, are the mechanisms that govern the fate of GC B cells and progeny that survive trial by selection? Some points are crucial to underscore prior to consideration of signals and factors. A first consideration is that, across time, the nonsynchronous population of GCs tends to be programmed toward a greater production of memory cells during an earlier phase and then transition later to a greater probability of producing bone marrow plasma cells (BMPCs) (Weisel et al. 2016). The second is that most immune responses will elicit a mix of extrafollicular plasma cells that produce antigen-binding antibodies. These antibodies likely have two immediate impacts on the GC. One is that they will form immune complexes that can be captured and, in a controlled process, displayed by the FDC of the GC LZ (Wu et al. 2008; Heesters et al. 2014) and thereby enhance capture by B cells that thereby acquire epitopes to display in competing for Tfh. However, if the concentration and qualities of antibody reach some threshold, they can eliminate the supply of antigen needed for BCR stimulation and Ag presentation (Zhang et al. 2013b). Inasmuch as BCR stimulation is central to the physiology, it seems likely that this influence of soluble antibody contributes to signaling the shift from Bmem (early) to BMPC (later) bias in normal physiology. Moreover, it is likely that the precise nature of signaling from higher-affinity receptors—selected after random SHM—differs from the preimmune BCR population (Ise et al. 2018). Below, the evidence is reviewed for a few signals or transcription factors that mediate qualitative discrimination, for instance, in which the loss-of-function mutant selectively impairs one fate. In parallel, however, a major impact likely lies in the integration of a balance between a number of signals and DNA-binding transcription regulators. Nonetheless, it is striking how little is established clearly about molecular mechanisms of signaling or transcription that transduce the temporal information and the cell surface receptor stimulation (the BCR complex and CD40) in ways that account for alternative fates: quiescence as memory versus progression to plasma cell differentiation.
Humoral memory—LLPCs and the Bmem limb
As noted above, one limb of humoral memory involves the constitutive secretion of antibodies from LLPCs that were elicited by an immune experience. The molecular programming of durability and persistence of LLPCs is basically that of the survival niches, anti-apoptotic programs, and management of the UPR (discussed above). With clearance of the antigen, the spectrum of antibody qualities will not change much, and yet many microbes generate variants of the functional proteins that they display. Perhaps for this reason, a second limb of humoral memory involves the generation of quiescent antigen-experienced B-cell populations that returned to a quiescent state while retaining a capacity for reactivation (McHeyzer-Williams et al. 2012; Weisel and Shlomchik 2017; Inoue et al. 2018). The restimulated Bmem cell can then either rapidly differentiate into a new plasma cell or participate again in a GC reaction, thereby creating a new round of somatic mutation, affinity maturation, and dynamic allocation of progeny to memory or plasma cell fates (McHeyzer-Williams et al. 2012; Weisel and Shlomchik 2017; Inoue et al. 2018).
As is typical of immunity, complexity and a diversity of processes are features of the Bmem state. Thus, in addition to the canonical Bmem cell that is class-switched (has deleted the μ−δ heavy chain coding DNA that was in cis to its clonal VDJ region) and derived from a GC reaction, an extrafollicular pathway also generates antibody-secreting cells as well as B-cell memory with unswitched (IgM+) Bmem cells (Toyama et al. 2002; Inamine et al. 2005; Dogan et al. 2009; Pape et al. 2011; Kaji et al. 2012; Taylor et al. 2012). Recall antibody responses then draw on the pools of Bmem cells that are IgM+ or isotype-switched to improve antibody levels and qualities when an antigen similar to that 10 immunity is encountered. Certainly, B-cell memory can arise despite disruption of the GC, but an important unresolved area is that of the relative contributions of different types of antibody and Bmem cells (IgM vs. switched, GC vs. others, and CD80/PD-L2+/+ vs. CD80/PD-L2−/−) (Zuccarino-Catania et al. 2014) to neutralization and long-term recall. Some work suggests the IgM+ Bmem is vital (Yates et al. 2013; Perez-Shibayama et al. 2014); other mechanistic studies along with clinical (human) data (e.g., conjugate vs. nonconjugate vaccines and broadly neutralizing antibody vs. influenza and HIV) provide evidence supporting a major role of GC-derived memory (Corti and Lanzavecchia 2013; Klein et al. 2013). Even with the further refinement of classification (Zuccarino-Catania et al. 2014), the molecular mechanisms within B-lineage cells that regulate the formation and maintenance of Bmem cell populations and the strength of recall antibody responses are far less elucidated than those of most other B-lineage states.
Profiling studies with mouse and human B cells have identified limited sets of mRNA at different levels in Bmem cells compared with naïve and/or GC B cells (Klein et al. 2003; Luckey et al. 2006; Haining et al. 2008), with at least a few gene products in common in the studies. Recent work with DZ and LZ GC B cells suggests that expression of the chemokine receptor CCR6 may foreshadow development into Bmem (Suan et al. 2017). In this latter state, salient features of these quiescent cells appear to be shared with memory CD8 T cells and include a stem-like signature (Luckey et al. 2006). Compared with the body of work on T cells, however, relatively little is known about how signal transduction or transcriptional mechanisms regulate the phases of memory: resolution of proliferation and initiation of quiescence, formation, and then persistence of various memory B classes. Long-term recall antibody responses in some models—and likely in humans—require the adapter protein SAP in T cells to promote a longer residence of B cells in GCs, which would also entail longer periods of costimulation via CD40 and of signaling through integrins (Crotty et al. 2003; Qi et al. 2008; Cannons et al. 2010). Although the role of CD40 is complex because its overstimulation can drive excess plasmablast fate and foreclose on others, this receptor on B cells is vital for both the GC reaction and memory formation (Banchereau et al. 1994; Foy et al. 1994; Gray et al. 1994; Laman et al. 1996; Erickson et al. 2002; Basso et al. 2004). IL-4, IL-21, and their receptors on B cells also are crucial (Ozaki et al. 2002; Glatman Zaretsky et al. 2009; King and Mohrs 2009). Based on conditional loss-of-function experiments, it is inferred that a tonic, presumably antigen-independent signal from the BCR complex (including its invariant accessory chains) acts to maintain the resting Bmem population in a manner that can be rescued via PI3K, as is the case for the bulk of B cells (Lam et al. 1997; Kraus et al. 2004; Srinivasan et al. 2009). Downstream from the BCR, an Ig Cγ1-Cre transgene for post-switch termination of PLC-γ1 reduced memory phenotype (CD38hi GL7neg CD138lo) B cells and recall antibody responses (Hikida et al. 2009). An analogous strategy for elimination of Syk gene expression well after immunization showed that this tyrosine kinase initiated survival signals for the Ag-specific Bmem population (Ackermann et al. 2015). In addition, the signal transducers Grb2 and the small GTPase DOCK8 are vital for GC formation and ultimate humoral memory (Randall et al. 2009; Jang et al. 2011). Of note, PI3K and mTOR are downstream from these signal relays (Pearce and Pearce 2013), and IgG1 has means of recruiting Grb2 that differ from IgM (Engels et al. 2014). Ultimately, the signals that promote memory appear to converge on anti-apoptotic members of the Bcl-2 superfamily (Klein et al. 2003; Carrington et al. 2010). Consistent with a status proposed to be poised at the brink of apoptosis, use of an Aicda (AID)-Cre transgene to deplete Mcl-1 eliminated Ag-specific memory phenotype B cells and recall antibody responses (Vikstrom et al. 2010; Clybouw et al. 2011). Intriguingly, Mcl-1 protein stability is maintained by a mechanism involving activity of intermediary metabolism in model cells (Maurer et al. 2006; Zhao et al. 2007; Coloff et al. 2011; Boller and Grosschedl 2014). These findings suggest that even in the relatively quiescent state, the programming of metabolism in Bmem cells may contribute to their persistence, as likely is the case for B cells’ capacity for canonical autophagy (Chen et al. 2014, 2015).
Regulation of the gene expression programs—transcription factors and RNAs
One of the challenges to a clear understanding of the regulation of mature B cells’ fates and the allocation of progeny cells among them is that most of the molecular mechanisms (signal transduction, transcriptional controls, and regulatory RNA) are essential at an earlier stage of development. With an abnormal developmental history or one that is not clearly established, conclusions are particularly tenuous or provisional. Nonetheless, analyses of the fates of mature B cells have reaped the benefits of conditional loss of function in recent years. Examples of the challenges include the sequence of transcriptional activators of initial B-lineage differentiation, proceeding from E2A proteins to EBF1 to Pax5. Each of these portions of a coordinated transcriptional program is essential at the early stages of ontogeny, and their absence eliminated normal mature B cells (for review, see Busslinger and Tarakhovsky 2014). These proteins upstream of Pax5 have been eliminated using constitutive Cre expression driven by a Cd23 transgene that acts only as full maturation of B cells is occurring in the periphery (Kwon et al. 2008; Gyory et al. 2012; Vilagos et al. 2012). Along with analogous work (Gloury et al. 2016), it is clear that these factors initiating Pax5 expression and an epigenetic network that maintains B-cell identity become dispensable for its maintenance, but E2A and EBF are crucial for steady-state homeostasis of a normal B-cell population. In addition, these factors appear important for establishment of GCs and plasma cell output after immunization.
As discussed above, regulated rates of proliferation and survival are central to the mechanisms by which the outputs from B-lineage cells are achieved; i.e., GC physiology, the capacity to switch effector class of antibody, and generation of plasma cells. Major checkpoint regulation is exercised by cyclin D in G1 and cyclin E at the initiation of S. One insight is that after activation, most cyclin D expression in FO B cells appears to be that of D2 and D3 (Cato et al. 2011), encoded by Ccnd2 and Ccnd3 genes, respectively. When a constitutive loss-of-function mutation for Cyclin D3 (Ccnd3−/−) was in the bone marrow progenitors used to reconstitute the B lineage in B-cell-deficient µMT mice, the resultant population could initiate early GC stages, but the response then failed to mature (Cato et al. 2011). This defect resulted in an initial impairment of affinity maturation but not in total IgM or IgG1, suggesting that cyclin D3 was functionally redundant with D2 for extrafollicular progression through switching and plasma cell differentiation but not in the initial GC. Intriguingly, parallel work with whole-body Ccnd3−/− mice (in which T-cell development is perturbed) found that this initial block to affinity maturation was alleviated progressively during two recall immunizations (Peled et al. 2010). These findings suggest that the capacity to form Bmem cells of the subset with a higher probability of entering secondary GC was enhanced. However, further work to elucidate regulation of cycling in primary and recall GCs will be needed, since the effect measured for increased Tfh using the DEC205 BCR knock-in model was on rates of replication fork progression during S phase (Gitlin et al. 2015). Moreover, mechanistic evidence on how a selected higher BCR affinity for Ag is then linked to DNA polymerization rates will be of mechanistic interest. Certain microRNAs present an emerging area in which there are tantalizing clues to new mechanisms that participate in modulating the strength of BCR signaling or cell cycling. For instance, miR-28 has been identified as a GC-specific microRNA that restrains proliferation, class switching, and the cellular products of the GC; i.e., Bmem and plasma cells (Bartolome-Izquierdo et al. 2017).
Temporally regulated levels of the proto-oncogene protein c-Myc, a central regulator of proliferation and apoptosis, are another crucial feature of GC physiology (Calado et al. 2012; Dominguez-Sola et al. 2012). Although originally Myc levels appeared lower for the GC as a whole than in activated lymphoblasts, recent work indicates that protein levels are in fact high in a smaller subset of GC B cells (Calado et al. 2012; Dominguez-Sola et al. 2012). These appear to be LZ cells in a limited period after reactivation through BCR stimulation followed by a pulse of help from Tfh cells. In part, the increased Myc may represent a coordinated induction that involves the expected collaboration of BCR and CD40 (Luo et al. 2018). Nonetheless, the conundrum remained that proliferation was greatest in the DZ, in most of which the Myc levels had declined. This apparent paradox may be resolved at least in part by Myc induction of the transcription regulator AP4 (Chou et al. 2016). This latter bound to similar sites and promoted metabolic pathways similar to a function of Myc, suggesting that a functional relay by the later transcription factor after Tfh restimulated B cells moved to the DZ. As noted, a major function of Myc in licensing proliferation is that it assists in reorganization of nutrient uptake and programming levels of gene expression for numerous pathways of intermediary metabolism. However, the spectrum of Myc target genes depends on the level of Myc and broadens with increasing concentration so as to include new pathways (Ji et al. 2011; Le et al. 2012). Experiments manipulating Myc levels provide results indicating that this proto-oncogene can function as a timer of proliferative duration in a manner dependent on its concentration and balances apoptosis (at high Myc with insufficient anti-apoptotic rescue factors) against maintenance of a proliferating state (Heinzel et al. 2017). Below a certain threshold level of Myc, lymphoblasts would exit cell cycle. This model is compatible with previous work noting a timed program for transition from blasts (DZ) to LZ centrocyte status for B cells (Bannard et al. 2013). Therefore, an issue to be addressed for B cells in humoral immunity is one of dose response—and the levels of Myc specifically—in the cells that progress or exit the GC reaction to die, enter relative quiescence as memory cells, or keep up some proliferation as plasmablasts. Inasmuch as precedents more analyzed in T cells rather than B cells provide indications that mTORC1 supports c-Myc stability (Verbist et al. 2016), it is possible that such a timer mechanism contributes to the observation that smaller B cells tend to exit cycle and the GC DZ (Ersching et al. 2017). With regard to timer mechanisms and BCR affinities in the GC (Bannard et al. 2013; Shinnakasu et al. 2016; Weisel et al. 2016), one model might be that early in the GC reaction, B lymphoblasts more readily reduce Myc and AP4 below the threshold of proliferation and exit into memory prior to signals sufficient to induce plasma cell commitment.
A central determinant of the branch points of GC B-cell status, memory fate, and plasma cell differentiation is the balance between BCL6 and Blimp1 along with the level of IRF4. At the first level, BCL6 expression and Blimp1 expression are mutually antagonistic so that each directly inhibits the other. The concentrations of IRF4 and its competition with IRF8 appear to be pivotal (Sciammas et al. 2006; Ochiai et al. 2013; Carotta et al. 2014; Xu et al. 2015). With that being the case, IRF4 is regulated in part by turnover mechanisms so that Cbl ubiquitin ligases target the transcription factor for degradation (Li et al. 2018). This regulatory mechanism presents another model for mediating cooperation of BCR stimulation with CD40 signaling, as Cbl expression was found to be highest in GC B cells of the LZ and enhanced by these stimuli (Li et al. 2018). Thus, GC B cells that capture Ag from FDCs and are stimulated by CD40L of Tfh cells would keep the level of IRF4 in check and thereby decrease the likelihood that they would initiate conversion into plasma cells (Sciammas et al. 2011; Martinez et al. 2012; Nutt et al. 2015). In principle, this mechanism could fit with the temporal sequence in which GC-derived plasma cells emerge later than Bmem (Weisel et al. 2016), at a time when the effective availability of antigen may be on the wane (Zhang et al. 2013b). In addition, the functional capacity of Blimp1 is opposed by the transcriptional cofactor BACH2 (Kometani et al. 2013; Huang et al. 2014), which itself can be modulated by mTORC1 (Tamahara et al. 2017). This antagonism influences the capacity of an activating BCR signal to send a B cell to the plasmablast fate (Kometani et al. 2013; Shinnakasu et al. 2016). Differences in BCR signaling qualities between IgM and IgG translate to a difference in BACH2 level and are likely a major mechanism causing class-switched Bmem cells to be less likely to re-enter a GC reaction than IgM memory. Since the stoichiometric balance and functions of BCL6, IRF4, Blimp1, and BACH2 are so central to whether a lymphoblast reaches a tipping point into plasma cell differentiation, it makes sense at a system level that these parameters be targeted for tuning by secondary factors. For instance, the microRNA miR-148a promotes the plasma cell fate by decreasing mRNA encoding BACH2 and an inhibitor of IRF4, MITF (Porstner et al. 2015). Although their mechanisms remain to be established, yet further microRNAs (miR-29a and miR-217) appear to enhance or restrain the GC reaction and its outputs (de Yebenes et al. 2014; van Nieuwenhuijze et al. 2017), and almost nothing is yet known about long noncoding RNA that can regulate gene expression.
NF-κB/Rel dimers represent a major transcription factor family downstream from the BCR, CD40, and PI3K (Hayden and Ghosh 2012). In line with the requirement for tonic signaling through PI3K to maintain B cells in the periphery (Srinivasan et al. 2009), canonical NF-κB signaling through IKK2 (IKKβ) is essential for B-cell survival (Derudder et al. 2016). In addition, mTORC2 and activity of at least the noncanonical (RelB:p52) NF-κB pathway appear to be required (Claudio et al. 2002; Lee et al. 2013). After B-cell activation, both the canonical (RelA:p50 and c-Rel:p50) and noncanonical NF-κB pathways and complexes support normal GC function in promoting affinity-matured antibodies (Zarnegar et al. 2004; Mills et al. 2007; Heise et al. 2014; De Silva et al. 2016). However, specific deletions of c-Rel versus RelA revealed a critical distinction: Whereas c-Rel was required for maintenance of GC, the distinction of the LZs and DZs, and metabolic activity, RelA was not. Instead, RelA promoted Blimp1 expression so that depletion of RelA from mature B cells undermined their ability to develop into plasma cells (Heise et al. 2014). Importantly, these findings place RelA upstream of expression of the Prdm1 gene that encodes Blimp1; what remains to be determined is whether the absence of RelA impaired the formation or maintenance of Bmem cells, either IgM or GC-derived (high-affinity and class-switched). Beyond these important findings, receptor activation of IκB kinases (IKKs) and other work with specific loss of function show evidence of regulatory finesse of the pathways. Thus, point mutations in IKKα, the apical activator of noncanonical NF-κB via p52 generation, eliminated GC induction but not the extrafollicular antibody response, and, in aging mice, absence of p50/NFκB1 led to deregulated IL-6 production by B cells, driving a breach of tolerance (Mills et al. 2007; de Valle et al. 2016).
The importance of IL-21 and proinflammatory functions of IL-6 (each discussed above) calls attention to STAT3, which is activated by receptors for these cytokines. Apart from its functions in other cell types, B-cell-intrinsic STAT3 is established as crucial for the late stages of differentiation that yield antibody production (Reljic et al. 2000; Fornek et al. 2006; Ding et al. 2016). Several other transcription factors also contribute. Various endogenously generated metabolites as well as environmental toxicants such as dioxins can engage the aryl hydrocarbon receptor (AHR), and recent work shows that AHR restrains class switching and plasma cell differentiation (Vaidyanathan et al. 2017), as can a dioxin (Zhang et al. 2013a). However, much remains to be determined about the relationships between such findings in vivo or as related to endogenously generated AHR ligands. Similarly, there have been exciting findings about the mechanisms by which a family of zinc finger BTB domain-containing ZBTB transcription factors preferentially limit the Bmem subset and duration of recall responses (ZBTB32) (Wang and Bhattacharya 2014) or promote the persistence of adjuvant-specific but not extrafollicular TLR-driven LLPCs (ZBTB20) (Jash et al. 2016; for review, see Zhu et al. 2018).
Epigenetic regulation of peripheral B cells
Specialized transcriptional programs of functionally and developmentally distinct cell types are reflected in their epigenetic landscapes. Genome-scale maps of chromatin accessibility, post-translational histone modifications, DNA methylation, and transcription factor-binding sites have revealed dedicated sets of cis-regulatory elements for a diverse number of cell types and developmental states. The transcriptional activities that separate naïve B cells into functionally distinct subsets of plasma cells, GC B cells, or Bmem cells involve extensive epigenetic reprogramming, ultimately resulting in unique DNA methylation and chromatin signatures of terminally differentiated cells. The establishment of these signatures relies on a highly ordered stepwise process of gene regulation.
In developing B cells, the hematopoietic master regulator PU.1 collaborates with B-lineage-specific transcription factors E2A, EBF, Oct-2, and NF-κB at an exclusive set of genomic sites marked by H3K4 monomethylation and H3K27 acetylation, which are indicators of open chromatin and have been correlated with enhancer activity (Heinz et al. 2010). Both PU.1 and EBF1 have been shown to act as pioneer factors that initiate nucleosome repositioning and provide a permissive chromatin state for secondary transcription factors to bind at enhancers of key genes during subsequent stages of differentiation (Mercer et al. 2011; Li et al. 2018). These two factors interact with and possibly recruit TET2 to target loci, which facilitates further chromatin accessibility and DNA demethylation through oxidation of 5-methylcytosine (Lio et al. 2016). The molecular mechanics of this process are not well understood, but replication may play an important role in passive dilution of 5-hydroxymethylcytosine (Otani et al. 2013). Indeed, the impact of TET2 and TET3 loss is most profound in highly replicating cells, blocking the pro-B-cell-to-pre-B-cell transition (Lio et al. 2016; Orlanski et al. 2016), which may be due to impaired regulation of IRF4/IRF8 and inhibited IgK ρε arrangement (for review, see Tsagaratou et al. 2017). However, a more robust understanding of the functions of the metabolite- and vitamin C-regulated TET dioxygenases (Sasidharan Nair et al. 2016; Yue et al. 2016) in mature B cells and humoral immunity will hinge on use of later-acting and conditional Cre transgenes.
Consistent with other models of differentiation, targeted DNA hypomethylation of plasma cells occurs at specific cis-regulatory sites and is associated with binding of stage-specific transcription factors such as AP-1 or IRF, which depends on the number of divisions the cells have undergone (Barwick et al. 2016). Unlike plasma cells, GC B cells experience global DNA demethylation of heterochromatic and repetitive elements, while polycomb-repressed regions become hypermethylated. Likewise, the transition from naïve to GC cells involves loss of methylation and, to a lesser extent, gain of methylation for thousands of genomic loci that are enriched near genes targeted for SHM. AID has been shown to be responsible for the majority of the changes observed during this transition (Dominguez et al. 2015).
Maintenance of methylation by the DNA methyltransferase DNMT1 is critical for GC generation, and genetic and pharmacological inhibition of DNMT1 skews differentiation of naïve B cells, leading to loss of GC cells and increased plasma cell numbers. In contrast, de novo methylation by DNMT3a/3b is not required for normal B-cell development and does not effect gene expression or chromatin accessibility in GC cells. Upon antigen stimulation, however, loss of DNMT3 produces abnormal plasma cell responses to immunization (Barwick et al. 2018). This may be explained by misregulation of genes resulting from failure to methylate regions surrounding genes that are normally down-regulated during B-cell development.
Despite these observations, fewer regions gain methylation and lose chromatin accessibility compared with the number of regions that lose methylation and gain accessibility during plasma or B-cell differentiation (Scharer et al. 2018). In order to repress gene expression programs of alternate cell fates, transcription factors such as Blimp-1 in plasma cells or BCL6 in B cells recruit repressive Polycomb protein EZH2 to target promoters and enhancers (Shaffer et al. 2002; Beguelin et al. 2016; Guo et al. 2018). EZH2, the histone methyltransferase responsible for trimethylation of H3K27 in nucleosomes at target sites, is required for GC formation, indicating that this repressive histone modification is functionally significant. Because of technical limitations and the variegation of B cells even within a single GC, much remains to be determined about the marks within Ag-specific cells of different fates and at key junctions on the paths toward developing plasma cells versus Bmem as alternative fates for the mature B cell.
Closing thoughts
In this review and status report, a theme of how there are distinctions between subtypes of cell (for instance, LLPCs that secrete different antibody classes or arise under different conditions/natures of adjuvant or Tfh) and in the current pioneering phase of the molecular basis for developing and programming these differences remains to be explored. This emphasis was chosen in part because of the underlying physiology of immunity and in part because the fast-evolving frontier of single-cell technologies may be approaching the robustness (rigor and reproducibility) to make these questions ripe for elucidation. A contention is that answers will be valuable in seeking to develop universal or broadly neutralizing vaccines to certain viruses (influenza and HIV) in outbred and diverse populations. Moreover, key information about the genes and molecular mechanisms that account for sustained breaches of tolerance in human autoimmune diseases mediated by pathological antibodies is still needed. Mapping sequence variants in noncoding regions has become an area of intense interest because of their potential to affect gene regulatory circuits. In fact, genome-wide associations studies (GWAS) have demonstrated that a substantial portion of common disease- and trait-associated SNPs (single-nucleotide polymorphisms) is located in noncoding functional regulatory elements. Pairing GWAS with functional genomic data reveals that disease risk variants not only are enriched in regulatory DNA but also localize primarily within cell type-specific enhancers of disease-relevant tissues (Maurano et al. 2012). Recent studies have probed relationships between molecular phenotypes such as gene expression, chromatin accessibility, or DNA methylation status of cis-acting regulatory sites and immune disease loci, finding a strong connection between noncoding genetic variation and epigenetic regulation (Farh et al. 2015; Koues et al. 2015; Chen et al. 2016a). The obvious implication of this relationship is that sequence variation may alter the stability of transcription factor occupancy, thus altering the epigenetic status and regulatory output of the enhancer. However, developing conclusive data that identify direct cause and effect relationships remains another challenge for the future.
Acknowledgments
We first acknowledge that the scope and extent of papers and mechanisms exceeds what could fit in one review and apologize to authors of work omitted from this synopsis of the fate of mature B cells. We gratefully acknowledge P. Basso, A. Raybuck, K. Stengel, and our peer reviewers for helpful comments from close readings of the manuscripts; P. Basso for building blocks of great assistance in preparing figures; and National Institutes of Health support of the time invested in creating the manuscript (R01 AI113292 and HL106812 to M.R.B., K22 CA184308 to E.H., and R01 AI051448 to J.W.T.).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.320192.118.
References
- Acevedo-Suarez CA, Kilkenny DM, Reich MB, Thomas JW. 2006. Impaired intracellular calcium mobilization and NFATc1 availability in tolerant anti-insulin B cells. J Immunol 177: 2234–2241. 10.4049/jimmunol.177.4.2234 [DOI] [PubMed] [Google Scholar]
- Ackermann JA, Nys J, Schweighoffer E, McCleary S, Smithers N, Tybulewicz VL. 2015. Syk tyrosine kinase is critical for B cell antibody responses and memory B cell survival. J Immunol 194: 4650–4656. 10.4049/jimmunol.1500461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, Bergsland E, Steinhoff M, Li Y, Gong Q, et al. 2014. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 25: 809–821. 10.1016/j.ccr.2014.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Cyster JG. 2008. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol 20: 14–25. 10.1016/j.smim.2007.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Okada T, Tang HL, Cyster JG. 2007. Imaging of germinal center selection events during affinity maturation. Science 315: 528–531. 10.1126/science.1136736 [DOI] [PubMed] [Google Scholar]
- Allman D, Pillai S. 2008. Peripheral B cell subsets. Curr Opin Immunol 20: 149–157. 10.1016/j.coi.2008.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. 2001. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol 167: 6834–6840. 10.4049/jimmunol.167.12.6834 [DOI] [PubMed] [Google Scholar]
- Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. 2010. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 464: 302–305. 10.1038/nature08782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon TI, Cyster JG. 2014. Blood, sphingosine-1-phosphate and lymphocyte migration dynamics in the spleen. Curr Top Microbiol Immunol 378: 107–128. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Bazan F, Blanchard D, Brière F, Galizzi JP, van Kooten C, Liu YJ, Rousset F, Saeland S. 1994. The CD40 antigen and its ligand. Annu Rev Immunol 12: 881–926. 10.1146/annurev.iy.12.040194.004313 [DOI] [PubMed] [Google Scholar]
- Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. 2013. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity 39: 912–924. 10.1016/j.immuni.2013.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracho GV, Cato MH, Zhu Z, Jaren OR, Hobeika E, Reth M, Rickert RC. 2014. PDK1 regulates B cell differentiation and homeostasis. Proc Natl Acad Sci 111: 9573–9578. 10.1073/pnas.1314562111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome-Izquierdo N, de Yebenes VG, Alvarez-Prado AF, Mur SM, Lopez Del Olmo JA, Roa S, Vazquez J, Ramiro AR. 2017. miR-28 regulates the germinal center reaction and blocks tumor growth in preclinical models of non-Hodgkin lymphoma. Blood 129: 2408–2419. 10.1182/blood-2016-08-731166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwick BG, Scharer CD, Bally APR, Boss JM. 2016. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol 17: 1216–1225. 10.1038/ni.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR, Bally APR, Kohlmeier JE, Boss JM. 2018. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun 9: 1900 10.1038/s41467-018-04234-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Klein U, Niu H, Stolovitzky GA, Tu Y, Califano A, Cattoretti G, Dalla-Favera R. 2004. Tracking CD40 signaling during germinal center development. Blood 104: 4088–4096. 10.1182/blood-2003-12-4291 [DOI] [PubMed] [Google Scholar]
- Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, Dalla-Favera R. 2012. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med 209: 2455–2465. 10.1084/jem.20121387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, et al. 2016. EZH2 and BCL6 cooperate to assemble CBX8–BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell 30: 197–213. 10.1016/j.ccell.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, Boisson-Dupuis S, Wong M, Adelstein S, Arkwright PD, et al. 2013. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood 122: 3940–3950. 10.1182/blood-2013-06-506865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessa J, Kopf M, Bachmann MF. 2010. Cutting edge: IL-21 and TLR signaling regulate germinal center responses in a B cell-intrinsic manner. J Immunol 184: 4615–4619. 10.4049/jimmunol.0903949 [DOI] [PubMed] [Google Scholar]
- Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, Weissman IL. 2007. Transcriptional profiling of antigen-dependent murine B cell differentiation and memory formation. J Immunol 179: 6808–6819. 10.4049/jimmunol.179.10.6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair PA, Chavez-Rueda KA, Evans JG, Shlomchik MJ, Eddaoudi A, Isenberg DA, Ehrenstein MR, Mauri C. 2009. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. J Immunol 182: 3492–3502. 10.4049/jimmunol.0803052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boller S, Grosschedl R. 2014. The regulatory network of B-cell differentiation: a focused view of early B-cell factor 1 function. Immunol Rev 261: 102–115. 10.1111/imr.12206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonami RH, Wolfle WT, Thomas JW, Kendall PL. 2014. NFATc2 (NFAT1) assists BCR-mediated anergy in anti-insulin B cells. Mol Immunol 62: 321–328. 10.1016/j.molimm.2014.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortnick A, Chernova I, Quinn WJ III, Mugnier M, Cancro MP, Allman D. 2012. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J Immunol 188: 5389–5396. 10.4049/jimmunol.1102808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, Scott ML, Maquelin A, Belnoue E, Siegrist CA, et al. 2008. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 111: 1004–1012. 10.1182/blood-2007-09-110874 [DOI] [PubMed] [Google Scholar]
- Bouamar H, Jiang D, Wang L, Lin AP, Ortega M, Aguiar RC. 2015. MicroRNA 155 control of p53 activity is context dependent and mediated by Aicda and Socs1. Mol Cell Biol 35: 1329–1340. 10.1128/MCB.01446-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl AM, Nemazee D, Cambier JC, Rickert R, Hertz M. 2000. B-cell antigen receptor competence regulates B-lymphocyte selection and survival. Immunol Rev 176: 141–153. 10.1034/j.1600-065X.2000.00613.x [DOI] [PubMed] [Google Scholar]
- Busslinger M. 2004. Transcriptional control of early B cell development. Annu Rev Immunol 22: 55–79. 10.1146/annurev.immunol.22.012703.104807 [DOI] [PubMed] [Google Scholar]
- Busslinger M, Tarakhovsky A. 2014. Epigenetic control of immunity. Cold Spring Harb Perspect Biol 6: a024174 10.1101/cshperspect.a024174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, de Alboran IM, Janz M, Rodig S, Rajewsky K. 2012. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol 13: 1092–1100. 10.1038/ni.2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, Wakeland EK, Germain RN, Schwartzberg PL. 2010. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity 32: 253–265. 10.1016/j.immuni.2010.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotta S, Willis SN, Hasbold J, Inouye M, Pang SH, Emslie D, Light A, Chopin M, Shi W, Wang H, et al. 2014. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med 211: 2169–2181. 10.1084/jem.20140425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington EM, Vikstrom IB, Light A, Sutherland RM, Londrigan SL, Mason KD, Huang DC, Lew AM, Tarlinton DM. 2010. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci 107: 10967–10971. 10.1073/pnas.1005256107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. 2016. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol 16: 164–176. 10.1038/nri.2016.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cato MH, Chintalapati SK, Yau IW, Omori SA, Rickert RC. 2011. Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol Cell Biol 31: 127–137. 10.1128/MCB.00650-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A. 2008. The regulation of IgA class switching. Nat Rev Immunol 8: 421–434. 10.1038/nri2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A, Cols M, Puga I. 2013. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol 13: 118–132. 10.1038/nri3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champhekar A, Damle SS, Freedman G, Carotta S, Nutt SL, Rothenberg EV. 2015. Regulation of early T-lineage gene expression and developmental progression by the progenitor cell transcription factor PU.1. Genes Dev 29: 832–848. 10.1101/gad.259879.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, Corry DB, Kheradmand F, Wang J. 2014. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med 20: 503–510. 10.1038/nm.3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Kodali S, Jang A, Kuai L, Wang J. 2015. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol 194: 2607–2615. 10.4049/jimmunol.1403001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martin D, Watt S, Yan Y, Kundu K, Ecker S, et al. 2016a. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167: 1398–1414.e24. 10.1016/j.cell.2016.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TT, Tsai MH, Kung JT, Lin KI, Decker T, Lee CK. 2016b. STAT1 regulates marginal zone B cell differentiation in response to inflammation and infection with blood-borne bacteria. J Exp Med 213: 3025–3039. 10.1084/jem.20151620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu H, Mallya S, Nguyen P, Mai A, Jackson LV, Winkler DG, DiNitto JP, Brophy EE, McGovern K, Kutok JL, et al. 2017. The selective phosphoinoside-3-kinase p110δ inhibitor IPI-3063 potently suppresses B cell survival, proliferation, and differentiation. Front Immunol 8: 747 10.3389/fimmu.2017.00747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, Thomas JW, Hiebert S, Haase VH, Boothby MR. 2016. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 537: 234–238. 10.1038/nature19334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C, Verbaro DJ, Tonc E, Holmgren M, Cella M, Colonna M, Bhattacharya D, Egawa T. 2016. The transcription factor AP4 mediates resolution of chronic viral infection through amplification of germinal center B cell responses. Immunity 45: 570–582. 10.1016/j.immuni.2016.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvatchko Y, Kosco-Vilbois MH, Herren S, Lefort J, Bonnefoy JY. 1996. Germinal center formation and local immunoglobulin E (IgE) production in the lung after an airway antigenic challenge. J Exp Med 184: 2353–2360. 10.1084/jem.184.6.2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E, Brown K, Park S, Wang H, Siebenlist U. 2002. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat Immunol 3: 958–965. 10.1038/ni842 [DOI] [PubMed] [Google Scholar]
- Clybouw C, Fischer S, Auffredou MT, Hugues P, Alexia C, Bouillet P, Raphael M, Leca G, Strasser A, Tarlinton DM, et al. 2011. Regulation of memory B-cell survival by the BH3-only protein Puma. Blood 118: 4120–4128. 10.1182/blood-2011-04-347096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M. 2007. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 449: 473–477. 10.1038/nature06159 [DOI] [PubMed] [Google Scholar]
- Coloff JL, Macintyre AN, Nichols AG, Liu T, Gallo CA, Plas DR, Rathmell JC. 2011. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res 71: 5204–5213. 10.1158/0008-5472.CAN-10-4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquery CM, Erickson LD. 2012. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit Rev Immunol 32: 287–305. 10.1615/CritRevImmunol.v32.i4.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D, Lanzavecchia A. 2013. Broadly neutralizing antiviral antibodies. Annu Rev Immunol 31: 705–742. 10.1146/annurev-immunol-032712-095916 [DOI] [PubMed] [Google Scholar]
- Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. 2003. SAP is required for generating long-term humoral immunity. Nature 421: 282–287. 10.1038/nature01318 [DOI] [PubMed] [Google Scholar]
- Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, Ngo VN. 2000. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev 176: 181–193. 10.1034/j.1600-065X.2000.00618.x [DOI] [PubMed] [Google Scholar]
- Daniel JA, Nussenzweig A. 2013. The AID-induced DNA damage response in chromatin. Mol Cell 50: 309–321. 10.1016/j.molcel.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, Rickert RC. 2008. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol 9: 1388–1398. 10.1038/ni.1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derudder E, Herzog S, Labi V, Yasuda T, Köchert K, Janz M, Villunger A, Schmidt-Supprian M, Rajewsky K. 2016. Canonical NF-κB signaling is uniquely required for the long-term persistence of functional mature B cells. Proc Natl Acad Sci 113: 5065–5070. 10.1073/pnas.1604529113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descatoire M, Weller S, Irtan S, Sarnacki S, Feuillard J, Storck S, Guiochon-Mantel A, Bouligand J, Morali A, Cohen J, et al. 2014. Identification of a human splenic marginal zone B cell precursor with NOTCH2-dependent differentiation properties. J Exp Med 211: 987–1000. 10.1084/jem.20132203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva NS, Anderson MM, Carette A, Silva K, Heise N, Bhagat G, Klein U. 2016. Transcription factors of the alternative NF-κB pathway are required for germinal center B-cell development. Proc Natl Acad Sci 113: 9063–9068. 10.1073/pnas.1602728113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Valle E, Grigoriadis G, O'Reilly LA, Willis SN, Maxwell MJ, Corcoran LM, Tsantikos E, Cornish JK, Fairfax KA, Vasanthakumar A, et al. 2016. NFκB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J Exp Med 213: 621–641. 10.1084/jem.20151182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Yebenes VG, Bartolome-Izquierdo N, Nogales-Cadenas R, Perez-Duran P, Mur SM, Martinez N, Di Lisio L, Robbiani DF, Pascual-Montano A, Canamero M, et al. 2014. miR-217 is an oncogene that enhances the germinal center reaction. Blood 124: 229–239. 10.1182/blood-2013-12-543611 [DOI] [PubMed] [Google Scholar]
- Dil N, Marshall AJ. 2009. Role of phosphoinositide 3-kinase p110δ in TLR4- and TLR9-mediated B cell cytokine production and differentiation. Mol Immunol 46: 1970–1978. 10.1016/j.molimm.2009.03.010 [DOI] [PubMed] [Google Scholar]
- Ding C, Chen X, Dascani P, Hu X, Bolli R, Zhang HG, McLeish KR, Yan J. 2016. STAT3 signaling in B cells is critical for germinal center maintenance and contributes to the pathogenesis of murine models of lupus. J Immunol 196: 4477–4486. 10.4049/jimmunol.1502043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, Reynaud CA, Weill JC. 2009. Multiple layers of B cell memory with different effector functions. Nat Immunol 10: 1292–1299. 10.1038/ni.1814 [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. 1997. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386: 855–858. 10.1038/386855a0 [DOI] [PubMed] [Google Scholar]
- Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, Cooper TK, Kitamura D, Rahman ZS. 2016. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med 213: 715–732. 10.1084/jem.20151722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez PM, Teater M, Chambwe N, Kormaksson M, Redmond D, Ishii J, Vuong B, Chaudhuri J, Melnick A, Vasanthakumar A, et al. 2015. DNA methylation dynamics of germinal center B cells are mediated by AID. Cell Rep 12: 2086–2098. 10.1016/j.celrep.2015.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, Dalla-Favera R. 2012. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 13: 1083–1091. 10.1038/ni.2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, Dalla-Favera R. 2015. The FOXO1 transcription factor instructs the germinal center dark zone program. Immunity 43: 1064–1074. 10.1016/j.immuni.2015.10.015 [DOI] [PubMed] [Google Scholar]
- Doody GM, Bell SE, Vigorito E, Clayton E, McAdam S, Tooze R, Fernandez C, Lee IJ, Turner M. 2001. Signal transduction through Vav-2 participates in humoral immune responses and B cell maturation. Nat Immunol 2: 542–547. 10.1038/88748 [DOI] [PubMed] [Google Scholar]
- Dorshkind K, Montecino-Rodriguez E. 2007. Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nat Rev Immunol 7: 213–219. 10.1038/nri2019 [DOI] [PubMed] [Google Scholar]
- Eckl-Dorna J, Niederberger V. 2013. What is the source of serum allergen-specific IgE? Curr Allergy Asthma Rep 13: 281–287. 10.1007/s11882-013-0348-x [DOI] [PubMed] [Google Scholar]
- Engels N, König LM, Heemann C, Lutz J, Tsubata T, Griep S, Schrader V, Wienands J. 2009. Recruitment of the cytoplasmic adaptor Grb2 to surface IgG and IgE provides antigen receptor-intrinsic costimulation to class-switched B cells. Nat Immunol 10: 1018–1025. 10.1038/ni.1764 [DOI] [PubMed] [Google Scholar]
- Engels N, Konig LM, Schulze W, Radtke D, Vanshylla K, Lutz J, Winkler TH, Nitsche L, Wienands J. 2014. The immunoglobulin tail tyrosine motif upgrades memory-type BCRs by incorporating a Grb2–Btk signalling module. Nat Commun 5: 5456 10.1038/ncomms6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson LD, Durell BG, Vogel LA, O'Connor BP, Cascalho M, Yasui T, Kikutani H, Noelle RJ. 2002. Short-circuiting long-lived humoral immunity by the heightened engagement of CD40. J Clin Invest 109: 613–620. 10.1172/JCI0214110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, Dominguez-Sola D, Sabatini DM, Victora GD. 2017. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity 46: 1045–1058.e6. 10.1016/j.immuni.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, Mauri C. 2007. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol 178: 7868–7878. 10.4049/jimmunol.178.12.7868 [DOI] [PubMed] [Google Scholar]
- Fagarasan S, Honjo T. 2000. T-Independent immune response: new aspects of B cell biology. Science 290: 89–92. 10.1126/science.290.5489.89 [DOI] [PubMed] [Google Scholar]
- Fang D, Zhu J. 2017. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J Exp Med 214: 1861–1876. 10.1084/jem.20170494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. 2015. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518: 337–343. 10.1038/nature13835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton JL, Maseda D, Bonami RH, Hulbert C, Thomas JW. 2018. Anti-insulin B cells are poised for antigen presentation in type 1 diabetes. J Immunol 201: 861–873. 10.4049/jimmunol.1701717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornek JL, Tygrett LT, Waldschmidt TJ, Poli V, Rickert RC, Kansas GS. 2006. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood 107: 1085–1091. 10.1182/blood-2005-07-2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, Noelle RJ. 1994. gp39–CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med 180: 157–163. 10.1084/jem.180.1.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, Cantley LC. 1999. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science 283: 393–397. 10.1126/science.283.5400.393 [DOI] [PubMed] [Google Scholar]
- Ganti SN, Albershardt TC, Iritani BM, Ruddell A. 2015. Regulatory B cells preferentially accumulate in tumor-draining lymph nodes and promote tumor growth. Sci Rep 5: 12255 10.1038/srep12255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin AD, Shulman Z, Nussenzweig MC. 2014. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509: 637–640. 10.1038/nature13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJ, Koren A, Nussenzweig MC. 2015. HUMORAL IMMUNITY. T cell help controls the speed of the cell cycle in germinal center B cells. Science 349: 643–646. 10.1126/science.aac4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatman Zaretsky A, Taylor JJ, King IL, Marshall FA, Mohrs M, Pearce EJ. 2009. T follicular helper cells differentiate from Th2 cells in response to helminth antigens. J Exp Med 206: 991–999. 10.1084/jem.20090303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH, Murphy KM. 2000. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev 14: 1693–1711. [PubMed] [Google Scholar]
- Gloury R, Zotos D, Zuidscherwoude M, Masson F, Liao Y, Hasbold J, Corcoran LM, Hodgkin PD, Belz GT, Shi W, et al. 2016. Dynamic changes in Id3 and E-protein activity orchestrate germinal center and plasma cell development. J Exp Med 213: 1095–1111. 10.1084/jem.20152003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, Yokota Y, Shimizu A. 2003. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med 198: 1427–1437. 10.1084/jem.20030802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. 2010. Control systems and decision making for antibody production. Nat Immunol 11: 681–688. 10.1038/ni.1900 [DOI] [PubMed] [Google Scholar]
- Gray D, Dullforce P, Jainandunsing S. 1994. Memory B cell development but not germinal center formation is impaired by in vivo blockade of CD40–CD40 ligand interaction. J Exp Med 180: 141–155. 10.1084/jem.180.1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson AJ, Coussens LM. 2013. B cells and their mediators as targets for therapy in solid tumors. Exp Cell Res 319: 1644–1649. 10.1016/j.yexcr.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM, Scharer CD. 2018. EZH2 Represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol 200: 1039–1052. 10.4049/jimmunol.1701470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, et al. 2008. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol 28: 5209–5222. 10.1128/MCB.00360-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyory I, Boller S, Nechanitzky R, Mandel E, Pott S, Liu E, Grosschedl R. 2012. Transcription factor Ebf1 regulates differentiation stage-specific signaling, proliferation, and survival of B cells. Genes Dev 26: 668–682. 10.1101/gad.187328.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haining WN, Ebert BL, Subrmanian A, Wherry EJ, Eichbaum Q, Evans JW, Mak R, Rivoli S, Pretz J, Angelosanto J, et al. 2008. Identification of an evolutionarily conserved transcriptional signature of CD8 memory differentiation that is shared by T and B cells. J Immunol 181: 1859–1868. 10.4049/jimmunol.181.3.1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel F, Ehrenberg S, Hojer C, Draeseke A, Marschall-Schroter G, Kuhn R, Mack B, Gires O, Vahl CJ, Schmidt-Supprian M, et al. 2011. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood 118: 6321–6331. 10.1182/blood-2010-12-325944 [DOI] [PubMed] [Google Scholar]
- Haniuda K, Fukao S, Kodama T, Hasegawa H, Kitamura D. 2016. Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol 17: 1109–1117. 10.1038/ni.3508 [DOI] [PubMed] [Google Scholar]
- Hardy RR, Kincade PW, Dorshkind K. 2007. The protean nature of cells in the B lymphocyte lineage. Immunity 26: 703–714. 10.1016/j.immuni.2007.05.013 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. 2012. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26: 203–234. 10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He JS, Meyer-Hermann M, Xiangying D, Zuan LY, Jones LA, Ramakrishna L, de Vries VC, Dolpady J, Aina H, Joseph S, et al. 2013. The distinctive germinal center phase of IgE+ B lymphocytes limits their contribution to the classical memory response. J Exp Med 210: 2755–2771. 10.1084/jem.20131539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, Goodnow CC. 1997. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 6: 419–428. 10.1016/S1074-7613(00)80285-X [DOI] [PubMed] [Google Scholar]
- Heesters BA, Myers RC, Carroll MC. 2014. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol 14: 495–504. 10.1038/nri3689 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel S, Binh Giang T, Kan A, Marchingo JM, Lye BK, Corcoran LM, Hodgkin PD. 2017. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat Immunol 18: 96–103. 10.1038/ni.3598 [DOI] [PubMed] [Google Scholar]
- Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, Klein U. 2014. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J Exp Med 211: 2103–2118. 10.1084/jem.20132613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson RB, Grys K, Vehlow A, de Bettignies C, Zachacz A, Henley T, Turner M, Batista F, Tybulewicz VL. 2010. A novel Rac-dependent checkpoint in B cell development controls entry into the splenic white pulp and cell survival. J Exp Med 207: 837–853. 10.1084/jem.20091489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning AN, Roychoudhuri R, Restifo NP. 2018. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol 18: 340–356. 10.1038/nri.2017.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA, Herzenberg LA. 1989. Toward a layered immune system. Cell 59: 953–954. 10.1016/0092-8674(89)90748-4 [DOI] [PubMed] [Google Scholar]
- Hikida M, Casola S, Takahashi N, Kaji T, Takemori T, Rajewsky K, Kurosaki T. 2009. PLC-γ2 is essential for formation and maintenance of memory B cells. J Exp Med 206: 681–689. 10.1084/jem.20082100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Levit-Zerdoun E, Anastasopoulou V, Pohlmeyer R, Altmeier S, Alsadeq A, Dobenecker MW, Pelanda R, Reth M. 2015. CD19 and BAFF-R can signal to promote B-cell survival in the absence of Syk. EMBO J 34: 925–939. 10.15252/embj.201489732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa K, Martin SW, Pogue SL, Silver K, Peng K, Takatsu K, Goodnow CC. 2007. Enhancement and suppression of signaling by the conserved tail of IgG memory-type B cell antigen receptors. J Exp Med 204: 759–769. 10.1084/jem.20061923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Geng H, Boss I, Wang L, Melnick A. 2014. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood 123: 1012–1020. 10.1182/blood-2013-07-518605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamine A, Takahashi Y, Baba N, Miyake K, Tokuhisa T, Takemori T, Abe R. 2005. Two waves of memory B-cell generation in the primary immune response. Int Immunol 17: 581–589. 10.1093/intimm/dxh241 [DOI] [PubMed] [Google Scholar]
- Inlay MA, Bhattacharya D, Sahoo D, Serwold T, Seita J, Karsunky H, Plevritis SK, Dill DL, Weissman IL. 2009. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev 23: 2376–2381. 10.1101/gad.1836009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Shinnakasu R, Ise W, Kawai C, Egawa T, Kurosaki T. 2017. The transcription factor Foxo1 controls germinal center B cell proliferation in response to T cell help. J Exp Med 214: 1181–1198. 10.1084/jem.20161263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Moran I, Shinnakasu R, Phan TG, Kurosaki T. 2018. Generation of memory B cells and their reactivation. Immunol Rev 283: 138–149. 10.1111/imr.12640 [DOI] [PubMed] [Google Scholar]
- Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T, et al. 2018. T follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity 48: 702–715.e4. 10.1016/j.immuni.2018.03.027 [DOI] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4: 321–329. 10.1038/ni907 [DOI] [PubMed] [Google Scholar]
- Jang IK, Cronshaw DG, Xie LK, Fang G, Zhang J, Oh H, Fu YX, Gu H, Zou Y. 2011. Growth-factor receptor-bound protein-2 (Grb2) signaling in B cells controls lymphoid follicle organization and germinal center reaction. Proc Natl Acad Sci 108: 7926–7931. 10.1073/pnas.1016451108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jash A, Wang Y, Weisel FJ, Scharer CD, Boss JM, Shlomchik MJ, Bhattacharya D. 2016. ZBTB32 restricts the duration of memory B cell recall responses. J Immunol 197: 1159–1168. 10.4049/jimmunol.1600882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayachandran N, Landego I, Hou S, Alessi DR, Marshall AJ. 2017. B-cell-intrinsic function of TAPP adaptors in controlling germinal center responses and autoantibody production in mice. Eur J Immunol 47: 280–290. 10.1002/eji.201646596 [DOI] [PubMed] [Google Scholar]
- Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung CR, Chen C, Richardson AD, Conner EM, Benschop RJ, Woodgett JR, et al. 2017. Gsk3 is a metabolic checkpoint regulator in B cells. Nat Immunol 18: 303–312. 10.1038/ni.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, et al. 2011. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One 6: e26057 10.1371/journal.pone.0026057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, Allman D. 2016. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest 126: 4250–4261. 10.1172/JCI86504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Rajewsky K, Radbruch A. 1993. Shutdown of class switch recombination by deletion of a switch region control element. Science 259: 984–987. 10.1126/science.8438159 [DOI] [PubMed] [Google Scholar]
- Kaji T, Ishige A, Hikida M, Taka J, Hijikata A, Kubo M, Nagashima T, Takahashi Y, Kurosaki T, Okada M, et al. 2012. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J Exp Med 209: 2079–2097. 10.1084/jem.20120127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim C, Kazadi D, Rothschild G, Basu U. 2013. Regulation of AID, the B-cell genome mutator. Genes Dev 27: 1–17. 10.1101/gad.200014.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin NW, Crawford DM, Liu J, Behrens TW, Kearney JF. 2008. DNA microarray gene expression profile of marginal zone versus follicular B cells and idiotype positive marginal zone B cells before and after immunization with Streptococcus pneumoniae. J Immunol 180: 6663–6674. 10.4049/jimmunol.180.10.6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IL, Mohrs M. 2009. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J Exp Med 206: 1001–1007. 10.1084/jem.20090313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J Jr, Miljkovic V, Cattoretti G, Califano A, Dalla-Favera R. 2003. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci 100: 2639–2644. 10.1073/pnas.0437996100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pancera M, Zhou T, Incesu RB, Fu BZ, et al. 2013. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153: 126–138. 10.1016/j.cell.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Iijima K, Dent AL, Kita H. 2017. Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol 139: 300–313.e7. 10.1016/j.jaci.2016.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kometani K, Kurosaki T. 2015. Differentiation and maintenance of long-lived plasma cells. Curr Opin Immunol 33: 64–69. 10.1016/j.coi.2015.01.017 [DOI] [PubMed] [Google Scholar]
- Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, Furukawa K, Koseki H, Takemori T, Kurosaki T. 2013. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39: 136–147. 10.1016/j.immuni.2013.06.011 [DOI] [PubMed] [Google Scholar]
- Koues OI, Kowalewski RA, Chang LW, Pyfrom SC, Schmidt JA, Luo H, Sandoval LE, Hughes TB, Bednarski JJ, Cashen AF, et al. 2015. Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma. Immunity 42: 186–198. 10.1016/j.immuni.2014.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. 2004. Survival of resting mature B lymphocytes depends on BCR signaling via the Igα/β heterodimer. Cell 117: 787–800. 10.1016/j.cell.2004.05.014 [DOI] [PubMed] [Google Scholar]
- Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, Busslinger M. 2008. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 28: 751–762. 10.1016/j.immuni.2008.04.014 [DOI] [PubMed] [Google Scholar]
- Lam KP, Kühn R, Rajewsky K. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90: 1073–1083. 10.1016/S0092-8674(00)80373-6 [DOI] [PubMed] [Google Scholar]
- Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, McCommis KS, Fahrmann J, Pizzato HA, Nunley RM, et al. 2016. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity 45: 60–73. 10.1016/j.immuni.2016.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman JD, Claassen E, Noelle RJ. 1996. Functions of CD40 and its ligand, gp39 (CD40L). Crit Rev Immunol 16: 59–108. 10.1615/CritRevImmunol.v16.i1.40 [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. 2012. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15: 110–121. 10.1016/j.cmet.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CG, Kinoshita K, Arudchandran A, Cerritelli SM, Crouch RJ, Honjo T. 2001. Quantitative regulation of class switch recombination by switch region transcription. J Exp Med 194: 365–374. 10.1084/jem.194.3.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, D'Acquisto F, Hayden MS, Shim JH, Ghosh S. 2005. PDK1 nucleates T cell receptor-induced signaling complex for NF-κB activation. Science 308: 114–118. 10.1126/science.1107107 [DOI] [PubMed] [Google Scholar]
- Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. 2010. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32: 743–753. 10.1016/j.immuni.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, et al. 2011. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med 208: 1377–1388. 10.1084/jem.20102065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park DS, Potter R, Chen J, Volanakis E, Boothby M. 2012. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J Exp Med 209: 713–728. 10.1084/jem.20111470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Heffington L, Jellusova J, Nam KT, Raybuck A, Cho SH, Thomas JW, Rickert RC, Boothby M. 2013. Requirement for Rictor in homeostasis and function of mature B lymphoid cells. Blood 122: 2369–2379. 10.1182/blood-2013-01-477505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Huang Y, Huang YY, Kuang YS, Wei YJ, Xiang L, Zhang XJ, Jia ZC, Jiang S, Li JY, et al. 2017. MicroRNA-146a promotes IgE class switch in B cells via upregulating 14-3-3σ expression. Mol Immunol 92: 180–189. 10.1016/j.molimm.2017.10.023 [DOI] [PubMed] [Google Scholar]
- Li R, Cauchy P, Ramamoorthy S, Boller S, Chavez L, Grosschedl R. 2018. Dynamic EBF1 occupancy directs sequential epigenetic and transcriptional events in B-cell programming. Genes Dev 32: 96–111. 10.1101/gad.309583.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Stavnezer J. 1996. Activation of NF-κB/Rel by CD40 engagement induces the mouse germ line immunoglobulin Cγ1 promoter. Mol Cell Biol 16: 4591–4603. 10.1128/MCB.16.9.4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. 2010. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635–643. 10.1038/ni.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan LA, Warren WD, Thompson PA, Grusby MJ, Berton MT. 1998. STAT6 is required for IL-4-induced germline Ig gene transcription and switch recombination. J Immunol 161: 302–310. [PubMed] [Google Scholar]
- Lino AC, Dang VD, Lampropoulou V, Welle A, Joedicke J, Pohar J, Simon Q, Thalmensi J, Baures A, Fluhler V, et al. 2018. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity 49: 120–133.e9. 10.1016/j.immuni.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. 2010. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med 207: 353–363. 10.1084/jem.20091738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio CW, Zhang J, González-Avalos E, Hogan PG, Chang X, Rao A. 2016. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 5: e18290 10.7554/eLife.18290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Meckel T, Tolar P, Sohn HW, Pierce SK. 2010a. Antigen affinity discrimination is an intrinsic function of the B cell receptor. J Exp Med 207: 1095–1111. 10.1084/jem.20092123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Meckel T, Tolar P, Sohn HW, Pierce SK. 2010b. Intrinsic properties of immunoglobulin IgG1 isotype-switched B cell receptors promote microclustering and the initiation of signaling. Immunity 32: 778–789. 10.1016/j.immuni.2010.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chen E, Zhao XW, Wan ZP, Gao YR, Davey A, Huang E, Zhang L, Crocetti J, Sandoval G, et al. 2012. The scaffolding protein synapse-associated protein 97 is required for enhanced signaling through isotype-switched IgG memory B cell receptors. Sci Signal 5: ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. 1999. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med 190: 75–90. 10.1084/jem.190.1.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Cyster JG. 2002. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science 297: 409–412. 10.1126/science.1071632 [DOI] [PubMed] [Google Scholar]
- Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, Mathis D. 2006. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci 103: 3304–3309. 10.1073/pnas.0511137103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Weisel F, Shlomchik MJ. 2018. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity 48: 313–326.e5. 10.1016/j.immuni.2018.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykken JM, Candando KM, Tedder TF. 2015. Regulatory B10 cell development and function. Int Immunol 27: 471–477. 10.1093/intimm/dxv046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F, Browning JL. 2002. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol 2: 465–475. 10.1038/nri844 [DOI] [PubMed] [Google Scholar]
- MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, Cook MC, Vinuesa CG. 2003. Extrafollicular antibody responses. Immunol Rev 194: 8–18. 10.1034/j.1600-065X.2003.00058.x [DOI] [PubMed] [Google Scholar]
- Mao CS, Stavnezer J. 2001. Differential regulation of mouse germline Igγ1 and ε promoters by IL-4 and CD40. J Immunol 167: 1522–1534. 10.4049/jimmunol.167.3.1522 [DOI] [PubMed] [Google Scholar]
- Mao X, Zhang J, Han Y, Luan C, Hu Y, Hao Z, Chen M. 2016. Deficient for endoplasmic reticulum calcium sensors Stim1 and Stim2 affects aberrant antibody affinity maturation in B cells. Oncotarget 7: 60885–60895. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martin F, Kearney JF. 2000. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a ‘natural immune memory’. Immunol Rev 175: 70–79. 10.1111/j.1600-065X.2000.imr017515.x [DOI] [PubMed] [Google Scholar]
- Martin F, Kearney JF. 2002. Marginal-zone B cells. Nat Rev Immunol 2: 323–335. 10.1038/nri799 [DOI] [PubMed] [Google Scholar]
- Martin F, Oliver AM, Kearney JF. 2001. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 14: 617–629. 10.1016/S1074-7613(01)00129-7 [DOI] [PubMed] [Google Scholar]
- Martinez MR, Corradin A, Klein U, Alvarez MJ, Toffolo GM, di Camillo B, Califano A, Stolovitzky GA. 2012. Quantitative modeling of the terminal differentiation of B cells and mechanisms of lymphomagenesis. Proc Natl Acad Sci 109: 2672–2677. 10.1073/pnas.1113019109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. 2011. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity 34: 703–714. 10.1016/j.immuni.2011.03.016 [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, Kallies A, Nutt SL, Sakaguchi S, Takeda K, et al. 2014. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 41: 1040–1051. 10.1016/j.immuni.2014.10.016 [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. 2006. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21: 749–760. 10.1016/j.molcel.2006.02.009 [DOI] [PubMed] [Google Scholar]
- Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, et al. 2017. The microanatomic segregation of selection by apoptosis in the germinal center. Science 358: eaao2602 10.1126/science.aao2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron MJ, Park PW, Fooksman DR. 2017. CD138 mediates selection of mature plasma cells by regulating their survival. Blood 129: 2749–2759. 10.1182/blood-2017-01-761643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. 2012. Molecular programming of B cell memory. Nat Rev Immunol 12: 24–34. 10.1038/nri3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, Busslinger M. 2011. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol 111: 179–206. 10.1016/B978-0-12-385991-4.00005-2 [DOI] [PubMed] [Google Scholar]
- Melchers F, ten Boekel E, Seidl T, Kong XC, Yamagami T, Onishi K, Shimizu T, Rolink AG, Andersson J. 2000. Repertoire selection by pre-B-cell receptors and B-cell receptors, and genetic control of B-cell development from immature to mature B cells. Immunol Rev 175: 33–46. 10.1111/j.1600-065X.2000.imr017510.x [DOI] [PubMed] [Google Scholar]
- Mendoza P, Martinez-Martin N, Bovolenta ER, Reyes-Garau D, Hernansanz-Agustin P, Delgado P, Diaz-Munoz MD, Oeste CL, Fernandez-Pisonero I, Castellano E, et al. 2018. R-Ras2 is required for germinal center formation to aid B cells during energetically demanding processes. Sci Signal 11: eaal1506 10.1126/scisignal.aal1506 [DOI] [PubMed] [Google Scholar]
- Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T, Glass CK, Murre C. 2011. Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity 35: 413–425. 10.1016/j.immuni.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Bahlburg A, Andrews SF, Yu KO, Porcelli SA, Rawlings DJ. 2008. Characterization of a late transitional B cell population highly sensitive to BAFF-mediated homeostatic proliferation. J Exp Med 205: 155–168. 10.1084/jem.20071088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills DM, Bonizzi G, Karin M, Rickert RC. 2007. Regulation of late B cell differentiation by intrinsic IKKα-dependent signals. Proc Natl Acad Sci 104: 6359–6364. 10.1073/pnas.0700296104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyai T, Takano J, Endo TA, Kawakami E, Agata Y, Motomura Y, Kubo M, Kashima Y, Suzuki Y, Kawamoto H, et al. 2018. Three-step transcriptional priming that drives the commitment of multipotent progenitors toward B cells. Genes Dev 32: 112–126. 10.1101/gad.309575.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau A, Blair PA, Chai JG, Ratnasothy K, Stolarczyk E, Alhabbab R, Rackham CL, Jones PM, Smyth L, Elgueta R, et al. 2015. Transitional-2 B cells acquire regulatory function during tolerance induction and contribute to allograft survival. Eur J Immunol 45: 843–853. 10.1002/eji.201445082 [DOI] [PubMed] [Google Scholar]
- Moser K, Tokoyoda K, Radbruch A, MacLennan I, Manz RA. 2006. Stromal niches, plasma cell differentiation and survival. Curr Opin Immunol 18: 265–270. 10.1016/j.coi.2006.03.004 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 274: 18470–18476. 10.1074/jbc.274.26.18470 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563. 10.1016/S0092-8674(00)00078-7 [DOI] [PubMed] [Google Scholar]
- Nemazee D. 2017. Mechanisms of central tolerance for B cells. Nat Rev Immunol 17: 281–294. 10.1038/nri.2017.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazee D, Weigert M. 2000. Revising B cell receptors. J Exp Med 191: 1813–1818. 10.1084/jem.191.11.1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman R, Ahlfors H, Saveliev A, Galloway A, Hodson DJ, Williams R, Besra GS, Cook CN, Cunningham AF, Bell SE, et al. 2017. Maintenance of the marginal-zone B cell compartment specifically requires the RNA-binding protein ZFP36L1. Nat Immunol 18: 683–693. 10.1038/ni.3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HV, Mouly E, Chemin K, Luinaud R, Despres R, Fermand JP, Arnulf B, Bories JC. 2012. The Ets-1 transcription factor is required for Stat1-mediated T-bet expression and IgG2a class switching in mouse B cells. Blood 119: 4174–4181. 10.1182/blood-2011-09-378182 [DOI] [PubMed] [Google Scholar]
- Nutt SL, Kee BL. 2007. The transcriptional regulation of B cell lineage commitment. Immunity 26: 715–725. 10.1016/j.immuni.2007.05.010 [DOI] [PubMed] [Google Scholar]
- Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. 2015. The generation of antibody-secreting plasma cells. Nat Rev Immunol 15: 160–171. 10.1038/nri3795 [DOI] [PubMed] [Google Scholar]
- Nyhoff LE, Clark ES, Barron BL, Bonami RH, Khan WN, Kendall PL. 2018. Bruton's tyrosine kinase is not essential for B cell survival beyond early developmental stages. J Immunol 200: 2352–2361. 10.4049/jimmunol.1701489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, Chong AS, Klein U, Dinner AR, Singh H, et al. 2013. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38: 918–929. 10.1016/j.immuni.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ. 2004. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 199: 91–98. 10.1084/jem.20031330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki IM, Kinoshita K, Muramatsu M, Yoshikawa K, Honjo T. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature 416: 340–345. 10.1038/nature727 [DOI] [PubMed] [Google Scholar]
- Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, Rickert RC. 2006. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 25: 545–557. 10.1016/j.immuni.2006.08.015 [DOI] [PubMed] [Google Scholar]
- Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin-Klein R, Koralov SB, Skversky Y, Rajewsky K, Cedar H, et al. 2016. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Natl Acad Sci 113: 5018–5023. 10.1073/pnas.1604365113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani J, Kimura H, Sharif J, Endo TA, Mishima Y, Kawakami T, Koseki H, Shirakawa M, Suetake I, Tajima S. 2013. Cell cycle-dependent turnover of 5-hydroxymethyl cytosine in mouse embryonic stem cells. PLoS One 8: e82961 10.1371/journal.pone.0082961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X, Xu S, Lam KP. 2012. Deficiency in TNFRSF13B (TACI) expands T-follicular helper and germinal center B cells via increased ICOS-ligand expression but impairs plasma cell survival. Proc Natl Acad Sci 109: 15401–15406. 10.1073/pnas.1200386109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC III, Liu C, Schwartzberg PL, Leonard WJ. 2002. A critical role for IL-21 in regulating immunoglobulin production. Science 298: 1630–1634. 10.1126/science.1077002 [DOI] [PubMed] [Google Scholar]
- Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. 2011. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331: 1203–1207. 10.1126/science.1201730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape KA, Maul RW, Dileepan T, Paustian AS, Gearhart PJ, Jenkins MK. 2018. Naive B cells with high-avidity germline-encoded antigen receptors produce persistent IgM+ and transient IgG+ memory B cells. Immunity 48: 1135–1143.e4. 10.1016/j.immuni.2018.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, Xu Z, Mai T, Casali P. 2009. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol 10: 540–550. 10.1038/ni.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Long M, Kang JA, Kim WS, Lee CR, Im SH, Strickland I, Schulze-Luehrmann J, Hayden MS, Ghosh S. 2013. The kinase PDK1 is essential for B-cell receptor mediated survival signaling. PLoS One 8: e55378 10.1371/journal.pone.0055378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. 2006. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med 203: 1081–1091. 10.1084/jem.20060087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, et al. 2010. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143: 122–133. 10.1016/j.cell.2010.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Pearce EJ. 2013. Metabolic pathways in immune cell activation and quiescence. Immunity 38: 633–643. 10.1016/j.immuni.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JP, Kelly LM, Xu Y, Cyster JG. 2009. EBI2 mediates B cell segregation between the outer and centre follicle. Nature 460: 1122–1126. 10.1038/nature08226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled JU, Yu JJ, Venkatesh J, Bi E, Ding BB, Krupski-Downs M, Shaknovich R, Sicinski P, Diamond B, Scharff MD, et al. 2010. Requirement for cyclin D3 in germinal center formation and function. Cell Res 20: 631–646. 10.1038/cr.2010.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Szabo SJ, Glimcher LH. 2002. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci 99: 5545–5550. 10.1073/pnas.082114899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, Fagioli C, Merlini A, Mariani E, Pasqualetto E, et al. 2013. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 14: 298–305. 10.1038/ni.2524 [DOI] [PubMed] [Google Scholar]
- Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Coquery CM, Erickson LD, Fairfax K, Mackay F, Strasser A, et al. 2013. Mcl-1 is essential for the survival of plasma cells. Nat Immunol 14: 290–297. 10.1038/ni.2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Shibayama C, Gil-Cruz C, Pastelin-Palacios R, Cervantes-Barragan L, Hisaki E, Chai Q, Onder L, Scandella E, Regen T, Waisman A, et al. 2014. IFN-γ-producing CD4+ T cells promote generation of protective germinal center-derived IgM+ B cell memory against Salmonella Typhi. J Immunol 192: 5192–5200. 10.4049/jimmunol.1302526 [DOI] [PubMed] [Google Scholar]
- Petro JB, Gerstein RM, Lowe J, Carter RS, Shinners N, Khan WN. 2002. Transitional type 1 and 2 B lymphocyte subsets are differentially responsive to antigen receptor signaling. J Biol Chem 277: 48009–48019. 10.1074/jbc.M200305200 [DOI] [PubMed] [Google Scholar]
- Pone EJ, Xu Z, White CA, Zan H, Casali P. 2012. B cell TLRs and induction of immunoglobulin class-switch DNA recombination. Front Biosci (Landmark Ed) 17: 2594–2615. 10.2741/4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstner M, Winkelmann R, Daum P, Schmid J, Pracht K, Corte-Real J, Schreiber S, Haftmann C, Brandl A, Mashreghi MF, et al. 2015. miR-148a promotes plasma cell differentiation and targets the germinal center transcription factors Mitf and Bach2. Eur J Immunol 45: 1206–1215. 10.1002/eji.201444637 [DOI] [PubMed] [Google Scholar]
- Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. 2008. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455: 764–769. 10.1038/nature07345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall KL, Lambe T, Johnson AL, Treanor B, Kucharska E, Domaschenz H, Whittle B, Tze LE, Enders A, Crockford TL, et al. 2009. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol 10: 1283–1291. 10.1038/ni.1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck AL, Cho SH, Li J, Rogers MC, Lee K, Williams CL, Shlomchik M, Thomas JW, Chen J, Williams JV, et al. 2018. B cell-intrinsic mTORC1 promotes germinal center-defining transcription factor gene expression, somatic hypermutation, and memory B cell generation in humoral immunity. J Immunol 200: 2627–2639. 10.4049/jimmunol.1701321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. 2001. Plasma cell differentiation requires the transcription factor XBP-1. Nature 412: 300–307. 10.1038/35085509 [DOI] [PubMed] [Google Scholar]
- Reljic R, Wagner SD, Peakman LJ, Fearon DT. 2000. Suppression of signal transducer and activator of transcription 3-dependent B lymphocyte terminal differentiation by BCL-6. J Exp Med 192: 1841–1848. 10.1084/jem.192.12.1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda LB, Lu E, Bennett ML, Sokol CL, Wang X, Luther SA, Barres BA, Luster AD, Ye CJ, Cyster JG. 2018. Single-cell RNA sequencing of lymph node stromal cells reveals niche-associated heterogeneity. Immunity 48: 1014–1028.e6. 10.1016/j.immuni.2018.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman P, Chen YY, Lutzker S, Li SC, Stewart V, Coffman R, Alt FW. 1990. Structure and expression of germ line immunoglobulin heavy-chain ε transcripts: interleukin-4 plus lipopolysaccharide-directed switching to C ε. Mol Cell Biol 10: 1672–1679. 10.1128/MCB.10.4.1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Swanson CL, Troy S, Strauch P, Pelanda R, Torres RM. 2008. TLR agonists promote marginal zone B cell activation and facilitate T-dependent IgM responses. J Immunol 180: 3882–3888. 10.4049/jimmunol.180.6.3882 [DOI] [PubMed] [Google Scholar]
- Rush JS, Liu M, Odegard VH, Unniraman S, Schatz DG. 2005. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci 102: 13242–13247. 10.1073/pnas.0502779102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, Li S, Imami K, Selbach M, Di Virgilio M, et al. 2015. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity 43: 1075–1086. 10.1016/j.immuni.2015.10.021 [DOI] [PubMed] [Google Scholar]
- Sasidharan Nair V, Song MH, Oh KI. 2016. Vitamin C facilitates demethylation of the Foxp3 enhancer in a Tet-dependent manner. J Immunol 196: 2119–2131. 10.4049/jimmunol.1502352 [DOI] [PubMed] [Google Scholar]
- Sayegh CE, Quong MW, Agata Y, Murre C. 2003. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol 4: 586–593. 10.1038/ni923 [DOI] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Guo M, Bally APR, Boss JM. 2018. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat Commun 9: 1698 10.1038/s41467-018-04125-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. 2001. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science 293: 2111–2114. 10.1126/science.1061964 [DOI] [PubMed] [Google Scholar]
- Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, Tybulewicz VL. 2013. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 38: 475–488. 10.1016/j.immuni.2012.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. 2006. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25: 225–236. 10.1016/j.immuni.2006.07.009 [DOI] [PubMed] [Google Scholar]
- Sciammas R, Li Y, Warmflash A, Song Y, Dinner AR, Singh H. 2011. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol Syst Biol 7: 495 10.1038/msb.2011.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellars M, Reina-San-Martin B, Kastner P, Chan S. 2009. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med 206: 1073–1087. 10.1084/jem.20082311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre K, Cunningham AF, Coughlan RE, Lino AC, Rot A, Hub E, Moser K, Manz R, Ferraro A, Bird R, et al. 2012. CD8 T cells induce T-bet-dependent migration toward CXCR3 ligands by differentiated B cells produced during responses to alum-protein vaccines. Blood 120: 4552–4559. 10.1182/blood-2012-03-417733 [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, et al. 2002. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17: 51–62. 10.1016/S1074-7613(02)00335-7 [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, et al. 2004. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21: 81–93. 10.1016/j.immuni.2004.06.010 [DOI] [PubMed] [Google Scholar]
- Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, Willimsky G, Ammirante M, Strasner A, Hansel DE, et al. 2015. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521: 94–98. 10.1038/nature14395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Roch T, Lampropoulou V, O'Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, et al. 2014. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507: 366–370. 10.1038/nature12979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, Corcoran LM. 2015. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol 16: 663–673. 10.1038/ni.3154 [DOI] [PubMed] [Google Scholar]
- Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, Kurosaki T. 2016. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17: 861–869. 10.1038/ni.3460 [DOI] [PubMed] [Google Scholar]
- Simonetti G, Carette A, Silva K, Wang H, De Silva NS, Heise N, Siebel CW, Shlomchik MJ, Klein U. 2013. IRF4 controls the positioning of mature B cells in the lymphoid microenvironments by regulating NOTCH2 expression and activity. J Exp Med 210: 2887–2902. 10.1084/jem.20131026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper CM, Paul WE. 1987. Interferon-γ and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 236: 944–947. 10.1126/science.3107127 [DOI] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. 2009. PI3 kinase signals BCR-dependent mature B cell survival. Cell 139: 573–586. 10.1016/j.cell.2009.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Schrader CE. 2014. IgH chain class switch recombination: mechanism and regulation. J Immunol 193: 5370–5378. 10.4049/jimmunol.1401849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson LM, Miletic AV, Kloeppel T, Kusin S, Swat W. 2006. Vav proteins regulate the plasma cell program and secretory Ig production. J Immunol 177: 8620–8625. 10.4049/jimmunol.177.12.8620 [DOI] [PubMed] [Google Scholar]
- Suan D, Krautler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, Avery DT, Young C, Statham A, Elliott M, et al. 2017. CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light-zone location and predominant low antigen affinity. Immunity 47: 1142–1153.e4. 10.1016/j.immuni.2017.11.022 [DOI] [PubMed] [Google Scholar]
- Sun J, Keim CD, Wang J, Kazadi D, Oliver PM, Rabadan R, Basu U. 2013. E3-ubiquitin ligase Nedd4 determines the fate of AID-associated RNA polymerase II in B cells. Genes Dev 27: 1821–1833. 10.1101/gad.210211.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. 1996. Essential role of Stat6 in IL-4 signalling. Nature 380: 627–630. 10.1038/380627a0 [DOI] [PubMed] [Google Scholar]
- Talay O, Yan D, Brightbill HD, Straney EE, Zhou M, Ladi E, Lee WP, Egen JG, Austin CD, Xu M, et al. 2012. IgE+ memory B cells and plasma cells generated through a germinal-center pathway. Nat Immunol 13: 396–404. 10.1038/ni.2256 [DOI] [PubMed] [Google Scholar]
- Tamahara T, Ochiai K, Muto A, Kato Y, Sax N, Matsumoto M, Koseki T, Igarashi K. 2017. The mTOR–Bach2 cascade controls cell cycle and class switch recombination during B cell differentiation. Mol Cell Biol 37: e00418-17 10.1128/MCB.00418-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigaki K, Honjo T. 2007. Regulation of lymphocyte development by Notch signaling. Nat Immunol 8: 451–456. 10.1038/ni1453 [DOI] [PubMed] [Google Scholar]
- Taylor JJ, Pape KA, Jenkins MK. 2012. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med 209: 597–606. 10.1084/jem.20111696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JJ, Pape KA, Steach HR, Jenkins MK. 2015. Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naive B cell. Science 347: 784–787. 10.1126/science.aaa1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. 2008. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 28: 621–629. 10.1016/j.immuni.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, et al. 2001. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 293: 2108–2111. 10.1126/science.1061965 [DOI] [PubMed] [Google Scholar]
- Toyama H, Okada S, Hatano M, Takahashi Y, Takeda N, Ichii H, Takemori T, Kuroda Y, Tokuhisa T. 2002. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 17: 329–339. 10.1016/S1074-7613(02)00387-4 [DOI] [PubMed] [Google Scholar]
- Treiber T, Mandel EM, Pott S, Györy I, Firner S, Liu ET, Grosschedl R. 2010. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription- independent poising of chromatin. Immunity 32: 714–725. 10.1016/j.immuni.2010.04.013 [DOI] [PubMed] [Google Scholar]
- Tsagaratou A, Lio CJ, Yue X, Rao A. 2017. TET methylcytosine oxidases in T cell and B cell development and function. Front Immunol 8: 220 10.3389/fimmu.2017.00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan B, Chaudhuri A, Yewdell WT, Angeletti D, Yen WF, Wheatley AK, Bradfield CA, McDermott AB, Yewdell JW, Rudensky AY, et al. 2017. The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J Exp Med 214: 197–208. 10.1084/jem.20160789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nieuwenhuijze A, Dooley J, Humblet-Baron S, Sreenivasan J, Koenders M, Schlenner SM, Linterman M, Liston A. 2017. Defective germinal center B-cell response and reduced arthritic pathology in microRNA-29a-deficient mice. Cell Mol Life Sci 74: 2095–2106. 10.1007/s00018-017-2456-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbist KC, Guy CS, Milasta S, Liedmann S, Kamiński MM, Wang R, Green DR. 2016. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 532: 389–393. 10.1038/nature17442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC. 2012. Germinal centers. Annu Rev Immunol 30: 429–457. 10.1146/annurev-immunol-020711-075032 [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. 2010. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143: 592–605. 10.1016/j.cell.2010.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikstrom I, Carotta S, Luthje K, Peperzak V, Jost PJ, Glaser S, Busslinger M, Bouillet P, Strasser A, Nutt SL, et al. 2010. Mcl-1 is essential for germinal center formation and B cell memory. Science 330: 1095–1099. 10.1126/science.1191793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilagos B, Hoffmann M, Souabni A, Sun Q, Werner B, Medvedovic J, Bilic I, Minnich M, Axelsson E, Jaritz M, et al. 2012. Essential role of EBF1 in the generation and function of distinct mature B cell types. J Exp Med 209: 775–792. 10.1084/jem.20112422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Thomas JW, Unanue ER. 2016. Class-switched anti-insulin antibodies originate from unconventional antigen presentation in multiple lymphoid sites. J Exp Med 213: 967–978. 10.1084/jem.20151869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bhattacharya D. 2014. Adjuvant-specific regulation of long-term antibody responses by ZBTB20. J Exp Med 211: 841–856. 10.1084/jem.20131821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, Cyster JG. 2011. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med 208: 2497–2510. 10.1084/jem.20111449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Beaty N, Chen S, Qi CF, Masiuk M, Shin DM, Morse HC III. 2012a. The CXCR7 chemokine receptor promotes B-cell retention in the splenic marginal zone and serves as a sink for CXCL12. Blood 119: 465–468. 10.1182/blood-2011-03-343608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, McHeyzer-Williams MG. 2012b. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORα. Nat Immunol 13: 604–611. 10.1038/ni.2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Sugai M, Nambu Y, Osato M, Hayashi T, Kawaguchi M, Komori T, Ito Y, Shimizu A. 2010. Requirement for Runx proteins in IgA class switching acting downstream of TGF-β1 and retinoic acid signaling. J Immunol 184: 2785–2792. 10.4049/jimmunol.0901823 [DOI] [PubMed] [Google Scholar]
- Weill JC, Weller S, Reynaud CA. 2009. Human marginal zone B cells. Annu Rev Immunol 27: 267–285. 10.1146/annurev.immunol.021908.132607 [DOI] [PubMed] [Google Scholar]
- Weisel F, Shlomchik M. 2017. Memory B cells of mice and humans. Annu Rev Immunol 35: 255–284. 10.1146/annurev-immunol-041015-055531 [DOI] [PubMed] [Google Scholar]
- Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. 2016. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 44: 116–130. 10.1016/j.immuni.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JM, Bonami RH, Hulbert C, Thomas JW. 2015. Reversing tolerance in isotype switch-competent anti-insulin B lymphocytes. J Immunol 195: 853–864. 10.4049/jimmunol.1403114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Good-Jacobson KL, Curtis J, Light A, Tellier J, Shi W, Smyth GK, Tarlinton DM, Belz GT, Corcoran LM, et al. 2014. Transcription factor IRF4 regulates germinal center cell formation through a B cell-intrinsic mechanism. J Immunol 192: 3200–3206. 10.4049/jimmunol.1303216 [DOI] [PubMed] [Google Scholar]
- Wu Y, Sukumar S, El Shikh ME, Best AM, Szakal AK, Tew JG. 2008. Immune complex-bearing follicular dendritic cells deliver a late antigenic signal that promotes somatic hypermutation. J Immunol 180: 281–290. 10.4049/jimmunol.180.1.281 [DOI] [PubMed] [Google Scholar]
- Xu H, Chaudhri VK, Wu Z, Biliouris K, Dienger-Stambaugh K, Rochman Y, Singh H. 2015. Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat Immunol 16: 1274–1281. 10.1038/ni.3287 [DOI] [PubMed] [Google Scholar]
- Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. 2008. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 28: 639–650. 10.1016/j.immuni.2008.03.017 [DOI] [PubMed] [Google Scholar]
- Yang Z, Sullivan BM, Allen CD. 2012. Fluorescent in vivo detection reveals that IgE+ B cells are restrained by an intrinsic cell fate predisposition. Immunity 36: 857–872. 10.1016/j.immuni.2012.02.009 [DOI] [PubMed] [Google Scholar]
- Yang Z, Robinson MJ, Chen X, Smith GA, Taunton J, Liu W, Allen CD. 2016. Regulation of B cell fate by chronic activity of the IgE B cell receptor. Elife 5: e21238 10.7554/eLife.21238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Racine R, McBride KM, Winslow GM. 2013. T cell-dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J Immunol 191: 1240–1249. 10.4049/jimmunol.1300062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, Gustafsson JA, Russell DW, Cyster JG. 2012. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity 37: 535–548. 10.1016/j.immuni.2012.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Zhang X, Berner P, Choi YS. 2009. IL-21 and IL-10 have redundant roles but differential capacities at different stages of plasma cell generation from human germinal center B cells. J Leukoc Biol 86: 1311–1318. 10.1189/jlb.0409268 [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Okada K, Morishima N, Kamiya S, Owaki T, Asakawa M, Iwakura Y, Fukai F, Mizuguchi J. 2004. Induction of IgG2a class switching in B cells by IL-27. J Immunol 173: 2479–2485. 10.4049/jimmunol.173.4.2479 [DOI] [PubMed] [Google Scholar]
- Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, Spolski R, Poe JC, Leonard WJ, Tedder TF. 2012. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 491: 264–268. 10.1038/nature11501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda-Martinez JA, Lio CW, Li X, Huang Y, Vijayanand P, et al. 2016. Control of Foxp3 stability through modulation of TET activity. J Exp Med 213: 377–397. 10.1084/jem.20151438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. 2004. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood 104: 784–787. 10.1182/blood-2003-09-3071 [DOI] [PubMed] [Google Scholar]
- Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A. 2009. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood 14: 547–554. 10.1182/blood-2009-03-211763 [DOI] [PubMed] [Google Scholar]
- Zan H, Casali P. 2013. Regulation of Aicda expression and AID activity. Autoimmunity 46: 83–101. 10.3109/08916934.2012.749244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaprazna K, Atchison ML. 2012. YY1 controls immunoglobulin class switch recombination and nuclear activation-induced deaminase levels. Mol Cell Biol 32: 1542–1554. 10.1128/MCB.05989-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar B, He JQ, Oganesyan G, Hoffmann A, Baltimore D, Cheng G. 2004. Unique CD40-mediated biological program in B cell activation requires both type 1 and type 2 NF-κB activation pathways. Proc Natl Acad Sci 101: 8108–8113. 10.1073/pnas.0402629101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Kline DE, Bhattacharya S, Crawford RB, Conolly RB, Thomas RS, Andersen ME, Kaminski NE. 2013a. All-or-none suppression of B cell terminal differentiation by environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 268: 17–26. 10.1016/j.taap.2013.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, Young SP, Reynolds A, Falciani F, Waisman A, et al. 2013b. Germinal center B cells govern their own fate via antibody feedback. J Exp Med 210: 457–464. 10.1084/jem.20120150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TT, Gonzalez DG, Cote CM, Kerfoot SM, Deng S, Cheng Y, Magari M, Haberman AM. 2017. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. Elife 6: e19552 10.7554/eLife.19552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, et al. 2007. Glycogen synthase kinase 3α and 3β mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol 27: 4328–4339. 10.1128/MCB.00153-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Chen G, Zhao Y, Gao XM, Wang J. 2018. Regulation of the development and function of B cells by ZBTB transcription factors. Front Immunol 9: 580 10.3389/fimmu.2018.00580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler S, Gartner K, Scheuermann U, Zoeller T, Hantzschmann J, Over B, Foermer S, Heeg K, Bekeredjian-Ding I. 2014. Ca2+-related signaling events influence TLR9-induced IL-10 secretion in human B cells. Eur J Immunol 44: 1285–1298. 10.1002/eji.201343994 [DOI] [PubMed] [Google Scholar]
- Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner KM, Smyth MJ, et al. 2010. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med 207: 365–378. 10.1084/jem.20091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, Shlomchik MJ. 2014. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15: 631–637. 10.1038/ni.2914 [DOI] [PMC free article] [PubMed] [Google Scholar]