

SUMMARY
Inflammation is a risk factor for cancer development. Individuals with preleukemic TET2 mutations manifest clonal hematopoiesis and are at a higher risk of developing leukemia. How inflammatory signals influence the survival of preleukemic hematopoietic stem and progenitor cells (preleukemic-HSPCs) is unclear. We show a rapid increase in the frequency and absolute number of Tet2-KO mature myeloid cells and HSPCs in response to inflammatory stress, which results in enhanced production of inflammatory cytokines, including IL-6, and resistance to apoptosis. IL-6 induces hyperactivation of the Shp2-Stat3 signaling axis, resulting in increased expression of a novel anti-apoptotic lncRNA, Morrbid, in Tet2-KO myeloid cells and HSPCs. Expression of activated Shp2 in HSPCs phenocopies Tet2 loss, with regard to hyperactivation of Stat3 and Morrbid. In vivo, pharmacologic inhibition of Shp2 or Stat3 or genetic loss of Morrbid in Tet2-mutant mice rescues inflammatory stress-induced abnormalities in HSPCs and mature myeloid cells including clonal hematopoiesis.
ETOC paragraph:
Cai et al report that Tet2-deficient hematopoietic stem and progenitor cells manifest hyperactive IL-6/Shp2/Stat3/Morrbid pathway, which promotes cell survival under basal conditions as well as in response to inflammatory stress. Blocking this pathway using anti-inflammatory drugs E3330 and SHP099 or by genetic loss of Morrbid mitigates this response.
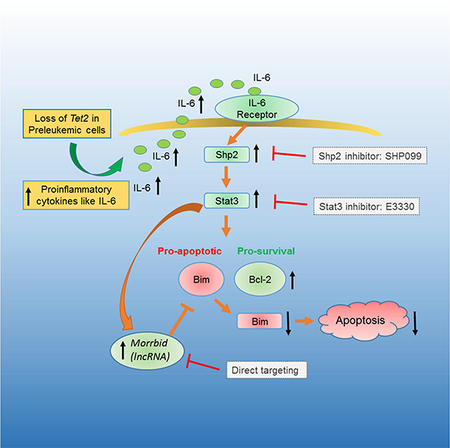
Gaphical Abstract
INTRODUCTION
Myeloid malignancies such as acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CMML), myeloproliferative neoplasia (MPN) and myelodysplastic syndromes (MDS) are considered clonal blood disorders. Hematopoietic stem and progenitor cells (HSPCs) with mutation(s) in AML-related genes such as TET2 or DNMT3A represent what are commonly defined as preleukemic HSPCs (Jan et al., 2012; Shlush and Minden, 2015; Shlush et al., 2014; Sperling et al., 2017). The selection and expansion of preleukemic-HSPC clones precede the development of AML (Abkowitz 2014; Grove and Vassiliou, 2014; Jan et al., 2012). Additionally, preleukemic-HSPCs can transform through serial acquisition of additional somatic mutations over time and contribute to the development of full-blown AML. What is unclear is the nature of environmental signals that might contribute to the “switch” from a preleukemic state to a leukemic state in cells bearing these mutations. In this context, inflammation has been hypothesized to play an essential role, but precisely how inflammatory signals influence the growth, survival, differentiation and the overall engraftment potential of preleukemic-HSPCs is poorly understood.
Older mice carrying loss of function alleles in Tet2 or Dnmt3a manifest an expanded HSPC pool, including a hematopoietic stem cell (HSC)-enriched fraction defined by cell surface markers Lineage-/Sca-1+/c-Kit+ (LSK). Some of these genetically modified mice go on to develop CMML or MPN with modest penetration when aged (Challen et al., 2012; Chu et al., 2012; Ko et al., 2011; Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). The majority of preleukemic mutations on their own are insufficient to cause AML in mice, suggesting that a single mutation among the above-described mutations defines a preleukemic state, and perhaps additional cooperating mutations and/or environmental factors are necessary to provide a more effective selection advantage for preleukemic-HSPCs leading to the development of full-blown leukemia.
Inflammation has been linked to tumor induction and transformation in solid tissues, and has recently been speculated as an enabling characteristic of cancer and its malignancies (Grivennikov et al., 2010; Hanahan and Weinberg, 2011; Mantovani et al., 2008). Inflammation caused by environmental exposure, infection, autoimmunity, or ageing may result in mutations and genomic instability in somatic cells as well as in reprogramming of the tumor microenvironment through regulating angiogenesis and expression of cytokines and chemokines. Considering that both innate and adaptive immune cells are generated from HSPCs and are involved in regulating local as well as whole-body inflammatory processes, the relationship between inflammation and hematopoietic malignancies is likely to be complex. While the influence of inflammatory stress on normal HSPCs has recently gained significant attention, recent studies have begun to address how preleukemic HSPCs respond or contribute to inflammation (Abegunde et al., 2018; Hasselbalch, 2012; Kobayashi et al., 2016; Meisel et al., 2018; Mirantes et al., 2014; Sano et al., 2018; Takizawa et al., 2012; Zhao and Baltimore, 2015). Because HSPCs in adults reside in the bone marrow and are surrounded by mature immune cells, the inflammatory microenvironment is likely to influence the growth and self-renewal of these cells in part by producing pro-inflammatory cytokines and chemokines. In support of this hypothesis are epidemiologic findings demonstrating that infection may act as a trigger for AML development in humans (Kristinsson et al., 2011).
In the present study, we asked whether and how Tet2-KO HSPCs maintain survival advantage during pathological stress by examining how Tet2-KO preleukemic-HSPCs respond to inflammatory stress.
RESULTS
A transient but enhanced neutrophil response to acute inflammatory stress in Tet2-KO mice
Lipopolysaccharide (LPS), a ligand that functions by stimulating the Toll-like receptor 4 (TLR4)/NFκB signaling pathway, is widely used to induce inflammation in mice (Manz and Boettcher, 2014). To assess how Tet2-KO pre-leukemic stem, progenitor and mature cells respond to acute inflammation, we injected LPS in Tet2-KO mice and their wildtype counterparts and followed these mice for 7 days to assess changes in peripheral blood (PB) hematologic parameters (Rodriguez et al., 2009). On day 2 (48 hours) post LPS treatment, a significant increase in white blood cells (WBC) was observed in Tet2-KO mice relative to controls (Figure 1A). The increase in WBCs in these mice was associated with an increase in the absolute number and the frequency of neutrophils (NE), consistent with enhanced “emergency granulopoietic” response (Figure, 1A and 1B) (Manz and Boettcher, 2014). The number of monocytes (MO) was comparable between wildtype and Tet2-KO mice post LPS treatment while eosinophil (EO) and basophil (BA) counts were also elevated like neutrophils in Tet2-KO mice (Figure S1A and data not shown). In addition, the number of lymphocytes (LY), platelets (PLT) and red blood cells (RBC) were comparable between wildtype and Tet2-KO mice (Figure S1A). Although no changes in the histology of bone marrow and spleen was seen upon LPS challenge (Figure S2B), flow cytometry analysis revealed a significant increase in the frequency of Mac1+ myeloid cells in Tet2-KO mice on day 2-post LPS treatment compared to wildtype controls (Figure S1C). Taken together, these data suggest that Tet2-KO mice manifest a transient but significant increase in both number and frequency of neutrophils in response to acute inflammatory stress.
Figure 1. Tet2-KO mice exhibit transient but amplified neutrophil and HSPCs in response to acute inflammatory challenge.

(A) Hematologic changes in the peripheral blood (PB) of LPS-treated wildtype and Tet2-KO mice over a 7-day period. A single dose of LPS (i.p., 0.8 mg/kg) was administered to adult wildtype or Tet2-KO mice (3~4 month old). n=4–10 mice per group, results are pooled from multiple experiments.
(B) LPS induces aberrant emergency granulopoiesis in Tet2-KO mice, revealed by flow cytometry analysis using neutrophil markers Mac1 and Ly6G. n=4 mice per group.
(C) A schematic demonstrating hematopoietic hierarchy.
(D) Representative flow cytometry profiles showing gating strategy and changes in various bone marrow HSPC subsets.
(E) Quantification of the frequency (Freq.) and absolute cell number (No.) of CMP, LSK and HSCs on day 0 and day 2 post LPS treatment in adult wildtype and Tet2-KO mice. n =4 mice per group.
Results are representative of two independent experiments. P value: * P < 0.05, ** P < 0.01, *** P < 0.001. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S1 for additional supporting data.
Acute inflammatory stress results in increased numbers of myeloid progenitors and hematopoietic stem cells in the bone marrow of Tet2-KO mice
Infection induces acute inflammation and modulates hematopoiesis at the level of both hematopoietic stem cells (HSC) and progenitor cells (HPC) to adapt to pathological insult (King and Goodell, 2011; Takizawa et al., 2012). In contrast to steady state hematopoiesis (naïve; hematopoietic hierarchy illustrated in Figure 1C), infection/stress-induced hematopoiesis in the bone marrow (BM) is recognized as “emergency hematopoiesis” (Trumpp et al., 2010; Wilson et al., 2008). Post LPS challenge, we analyzed the LSK and HSC compartments (defined by LSK/CD48-/CD150+) in wildtype and Tet2-KO mice (Kiel et al., 2008). We also analyzed various bone marrow progenitor cell subsets comprising of common myeloid progenitors (CMP, defined by Lin-/Sca-1-/c-Kit+/CD16-/CD34+), granulocyte-macrophage progenitors (GMP, defined by Lin-/Sca-1-/c-Kit+/CD16+/CD34+), and megakaryocyte-erythroid progenitors (MEP, defined by Lin-/Sca-1-/c-Kit+/CD16-/CD34-) in these mice (Figure 1D). We observed a similar drop in the overall BM cellularity in both wildtype and Tet2-KO mice post LPS challenge (Figure 1E). In contrast, LPS treatment resulted in significant differences in the frequencies and absolute number of BM progenitors in Tet2-KO mice relative to controls post LPS challenge. Quantitatively, LPS challenge of Tet2-KO mice resulted in increased number and frequencies of CMPs and GMPs but not MEPs (Figure 1E and data not shown). A significant enrichment in LSK and HSC frequency and numbers was also observed in Tet2-KO mice compared to controls post LPS treatment (Figure 1E). Similarly, Tet2-KO mice exhibit an increase in multipotent progenitors (MPPs, defined by LSK/CD48+/CD150-) and short-term HSCs (ST-HSC, defined by LSK/CD48+/CD150+) post-LPS challenge (Figure S1D). Given that adult (12–16 week old) Tet2-KO mice also manifest increased basal (day 0, before LPS challenge) levels of CMPs, LSKs and HSCs compared to controls, we assessed if juvenile (younger) Tet2-KO mice, which show similar frequency and number of CMPs, LSKs and HSCs as wildtype controls under basal conditions, respond to LPS challenge in a manner similar to older Tet2-KO mice. As shown in Figure S1E, juvenile Tet2-KO mice also exhibit enhanced response to LPS challenge in all the BM progenitor subsets examined including CMPs, LSKs and HSCs compared to controls. Collectively, these data suggest that deficiency of Tet2 in HSPCs primes them to respond to acute inflammatory stress much more efficiently than WT controls.
Differential impact of acute inflammatory stress on the function of normal vs. Tet2-KO hematopoietic stem cells
Recent studies have shown that LPS challenge or bacterial infection in mice not only expands the HSC/LSK population in the BM, but also potentially depletes HSCs and impairs their self-renewing capability (Esplin et al., 2011; Matatall et al., 2016; Rodriguez et al., 2009). We asked whether LPS-challenged Tet2-KO HSPCs also demonstrate impaired stem cell activity after being exposed to an inflammatory stress. To assess this, we first performed a competitive repopulation assay using whole bone marrow cells pre- and post-LPS challenge. The scheme for conducting competitive bone marrow transplantation (cBMT) is shown in Figure 2A. Peripheral blood chimerism revealed that the repopulating activity of CD45.2 donor cells from WT mice on day 2 post LPS treatment (% CD45.2_WT_day 2 in primary recipients) was significantly reduced compared to non LPS treated wildtype donor cells (% CD45.2_WT_day 0 in primary recipients) (% CD45.2_WT_day 2 vs. % CD45.2_WT_day 0, * P<0.05, Figure 2B). In contrast, LPS-stressed Tet2-KO CD45.2 donor cells did not show any decline in repopulating ability on any of the post LPS treatment time points examined (Figures 2B). Further, at every time point post LPS treatment, Tet2-KO CD45.2 bone marrow donor cells demonstrated higher repopulating ability compared to WT CD45.2 donor controls, and the greatest difference was observed on day 2 post LPS treatment (i.e. % CD45.2_WT_Day 2 vs. % CD45.2_Tet2-KO_Day 2, ** P<0.01, Figure 2B and S2A). Secondary cBMT experiments further confirmed that WT CD45.2 donor cells on day 2 post LPS treatment showed a significant decrease in the repopulating activity while Tet2-KO CD45.2 donor cells were resistant to LPS stress and maintained robust engraftment advantage (Figure 2C). Chimerism analysis of various fractions of BM cells including Lin- cells, LSK cells, myeloid cells (labeled by Mac1), B-cells (labeled by CD19) and T-cells (labeled by CD3) in the BM of primary cBMT recipients also demonstrated that the repopulation of Tet2-KO donor cells was higher than controls (Figure S2A, S2B, S2C and S2D).
Figure 2. LPS-stressed Tet2-KO bone marrow cells maintain repopulating advantage.

(A) A schematic describing primary and secondary competitive bone marrow transplantation (cBMT) assay. The age of the mice for donor cells was about 3~4 months old.
(B-C) Tet2-KO bone marrow cells with or without LPS treatment manifest significantly higher engraftment in primary and secondary recipients compared to wildtype controls. (D-F) Identical number of LSK cells were purified from wildtype and Tet2-KO mice pre-and post-LPS treatment and subjected to ex vivo CFU assay (E) and in vivo cBMT assay (F), respectively. Transplant experiments were conducted as described in (D). Data in (B) are from a representative experiment (n=5 recipients for cBMT analysis, mean ± s.e.m.).
Results are representative of two independent experiments. Data in (C) are from a single experiment (n=5 recipients per group, mean ± s.e.m.). Data in (E) is from pooled analysis of two mice per group performed in replicates of 4 (mean ± s.e.m, n=4 replicates). P value: * P < 0.05, ** P < 0.01, *** P < 0.001. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S2 for additional supporting data.
To further compare the repopulating activity of stem cells after LPS-induced inflammatory damage in wildtype and Tet2-KO mice, identical number of LSK cells from pre- and post-LPS treated wildtype and Tet2-KO mice were sorted and subjected to colony forming unit assay (CFU assay) in vitro and BM transplantation assay in vivo (Figure 2D). Because the frequencies of HSCs in the LSK pool were comparable between wildtype and Tet2-KO mice (Figure S2E), the analysis using LSK cells allowed us to assess the functional difference between the two groups with regards to HSCs upon LPS challenge. Consistent with our data utilizing whole bone marrow cells (Figure 2B), purified wildtype LSK cells derived from mice 2 days post LPS treatment showed impaired colony forming ability in vitro and significantly reduced repopulating activity in vivo compared to Tet2-KO LSK cells, which were significantly more resistant to inflammatory insult (Figure 2E and 2F). Taken together, our functional data demonstrate that while the repopulating activity of wildtype HSPCs is significantly impaired in response to LPS-induced inflammatory stress, LPS-treated Tet2-KO HSPCs maintain greater engraftment and are resistant to inflammatory stress.
Hematopoietic stem and progenitor cells lacking Tet2 resist apoptosis in response to acute inflammatory stress
Given our results demonstrating increased frequency, increased number and enhanced engraftment of Tet2-KO HSPCs upon LPS challenge, we sought to determine if Tet2-KO HSPCs compared to wildtype HSPCs manifest differences in apoptosis and/or proliferation upon encountering inflammatory stress. We analyzed apoptosis in HSPCs by Annexin-V and 7-AAD staining followed by flow cytometry analysis post-LPS treatment (Figure 3A). We also determined proliferation by assessing the percentage of Ki67+ HSPCs (Figure S3A). Although we observed only subtle changes in proliferating LSK pool (Figure S3A), remarkable and consistent differences in apoptosis were detected in both Lin-negative and LSK pool between wildtype and Tet2-KO upon LPS challenge (Figure 3A and 3B). Furthermore, the phenotypic observation with regard to reduced apoptosis in Tet2-KO cells post LPS treatment was consistent with changes in the expression of pro-apoptotic genes including Casp1, encoding Caspase 1 and Bcl2l11, encoding Bim, which were significantly reduced in Tet2-KO cells (Figure 3C). Furthermore, pro-survival genes including Bcl2 and Morrbid showed increased expression in Tet2-KO cells in both naïve condition and upon LPS challenge (Figure 3C). Morrbid is a recently identified long non-coding RNA (lncRNA) involved in regulating the expression of Bim and in the survival of myeloid cells including neutrophils; however, the role of Morrbid in the survival of pre-leukemic HSPCs is not known (Kotzin et al., 2016). We hypothesized that the hyperactivation of Morrbid in Tet2-KO HSPCs could be mediated via direct regulation of Morrbid promoter by Tet2 or perhaps via an indirect mechanism involving transcription factors that regulate inflammation. It has been reported that knock-out of a specific promoter region (~800 bp) of Morrbid impairs its activity (Kotzin et al., 2016). Detailed methylation analysis on the ~800-bp promoter region of Morrbid showed no profound difference in either 5-mC or 5-hmC modifications (Figure S3B-E). In contrast, we readily detected hyperactive phospho-Stat3 (pStat3) in Tet2-KO cells by flow cytometry and by western blot analysis (Figure 3E and 3F). Furthermore, we detected increased binding of Stat3 to the Morrbid promoter (Figure 3G). Taken together, these results suggest that Tet2 loss results in survival advantage in preleukemic cells, which is associated with increased expression of Morrbid and activation of Stat3.
Figure 3. Tet2-KO hematopoietic progenitor cells show sustained cell survival and heightened activation of Morrbid in response to acute inflammatory challenge.

(A-B) Level of apoptosis in Lin- cells and LSK cells pre and post LPS treatment as assessed by Annexin-V/7-AAD staining. A, representative flow profile of Annexin/7-AAD staining. B, quantification of apoptosis level.
(C) Differential expression of pro-apoptotic and pro-survival genes in Lin- cells pre- and post-LPS treatment.
(D) A scheme for two possible mechanisms by which Tet2 loss induces hyperactivation of Morrbid: one is through direct regulation of Morrbid promoter and another one is through cytokine-modulated phosphorylation of Stat3 (pStat3), which may bind to Morrbid promoter and activate its expression.
(E-F) Tet2-KO HSPCs maintain elevated expression of pStat3, revealed by flow cytometry (E) and by western blot (F).
(G) Stat3 binds to Morrbid locus revealed by CHIP-qPCR enrichment assay. Shown is relative enrichment in binding (1% of input as unit 1).
Data in (A) is a representative profile of flow cytometry. Data in (B-G) are from a representative experiment (n=4 mice per group, mean ± s.e.m.). Results are representative of two independent experiments. P value: * P < 0.05, ** P < 0.01, *** P < 0.001. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S3 for additional supporting data.
Tet2-KO mice show enhanced expression of pro-inflammatory cytokines
Acute inflammatory stress can induce an immediate and transient “cytokine storm” to regulate emergency hematopoiesis and granulopoiesis (Manz and Boettcher, 2014). To determine the upstream regulators of Stat3-Morrbid survival pathway and to see if the observed resistance to inflammatory stress in Tet2-KO HSPCs is in part due to dysregulated expression of inflammatory cytokines in Tet2-KO mice, we quantified thirty-one cytokines and chemokines to assess their levels in serum. Fifteen cytokines or chemokines (G-CSF, IL-6, CCL2, CCL4, CXCL1, CCL5, TNFα, CXCL9, CXCL10, IL-10, GM-CSF, IL-1α, IL-1β, M-CSF, IL-2) were found to be stimulated in serum by LPS on day 1 and/or day 2 compared to day 0 in wildtype and Tet2-KO mice (Figure 4A and S4A). However, we consistently observed that loss of Tet2 resulted in a profound increase in serum IL-6 levels on day 0, while a further increase was observed on days 1 and 2 post LPS treatment (Figure 4A). Ccl2, Ccl4, TNFα and CXCL9 were also increased in Tet2-KO mice on day 2 (Figure 4A). G-CSF, one of the most essential cytokines for granulopoiesis, was also slightly but not significantly increased in Tet2-KO mice on day 2 post LPS-treatment compared to wildtype (p=0.06, Figure 4A). Recent studies suggest that LPS can directly induce the production of multiple cytokines including IL-6, IL-1β, GM-CSF and TNFα in wildtype HSPCs (Zhao et al., 2014). We therefore examined to see if Tet2-KO HSPCs also produce these cytokines. IL-6 was expressed more highly by mature BM cells as well as by immature BM cells (HSPCs) lacking Tet2 upon LPS treatment compared to wildtype controls (Figure 4B, 4C, 4D and S4B). TNFα was expressed at a higher level on day 1 in Lin- BM cells derived from Tet2-KO mice (Figure S4C). Expression of IL-1β and GM-CSF was also enhanced by LPS treatment but was comparable between wildtype and Tet2-KO HSPCs (Figure S4D and S4E). QRT-PCR analysis on Lin- BM cells confirmed that Il6 mRNA was significantly elevated on day 0 and days 1 and 2-post LPS treatment in adult Tet2-KO mice relative to controls (Figure 4E). Likewise, in juvenile mice, where serum IL-6 and expression of Il6 were comparable between wildtype and Tet2-KO cells on day 0, LPS challenge resulted in significantly higher expression of Il6 in Lin- cells derived from Tet2-KO mice relative to controls (Figure 4F). Expression of Ccl2 was also increased in the serum and in Lin- cells on day 1 post LPS treatment in Tet2-KO mice relative to controls (Figure 4A, 4E and 4F). Taken together, these data suggest that Tet2-KO preleukemic hematopoietic cells produce elevated pro-inflammatory cytokines including IL-6 upon LPS challenge.
Figure 4. Tet2-KO mice show increased expression of IL-6 in serum and in various bone marrow subsets.

(A) Multiple cytokines/chemokines were increased on day 1 and/or day 2 post- LPS treatment in Tet2-KO mice, compared to wildtype controls.
(B-C) Intracellular flow cytometry analysis (ICFC) of IL-6 expression in total bone marrow cells and in Lin- bone marrow cells pre- and post-LPS treatment.
(D) Expression of IL-6 in indicated bone marrow subsets as assessed by flow cytometry and MFI quantification.
(E-F) QRT-PCR analysis of Il6 and Ccl2 expression in bone marrow Lin- cells derived from adult and juvenile pre- and post-LPS treated wildtype and Tet2-KO mice.
Data in (A) are from a single experiment (n=4 mice per group, mean ± s.e.m.). Data in (B) and (C) are from a representative experiment. Results are representative of two independent experiments. Data in (D), (E) and (F) are from a representative experiment (n= 4 mice per group, mean ± s.e.m.). Results are representative of two independent experiments. P value: * P < 0.05, ** P < 0.01, *** P < 0.001. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S4 for additional supporting data.
A selective Shp2 inhibitor SHP099 or Stat3 inhibitor E3330 blocks IL-6 induced hyperactivaton of Morrbid in vitro
LPS activates canonical TLR4/NFκB signaling, which induces the expression of inflammatory cytokines such as IL-6 to induce emergency hematopoiesis (a schematic of the TLR4/NFκB/IL-6 and IL-6/Shp2/Stat3 signaling pathways is illustrated in Figure 5A) (Lu et al., 2008). Interestingly, multiple components of TLR4/NFκB/IL-6/Stat3 signaling pathway including Tlr4, Nfkbiz, Il6 and Stat3 were elevated in Tet2-KO Linnegative cells with or without LPS challenge, consistent with a heightened inflammatory response in Tet2-KO HSPCs (Figure S5A, S5B and S5C). Given the increased expression of IL-6 in Tet2-KO HSPCs, we asked how IL-6 might contribute to the enhanced survival of Tet2-KO HSPCs. In vitro stimulation of Lin- Tet2-KO cells with IL-6 resulted in reduced apoptosis of these cells compared to controls (Figure 5B). Importantly, the reduced apoptosis of Tet2-KO cells in the presence of IL-6 was associated with increased activation of Stat3 and expression of lncRNA, Morrbid (Figure S5D and 5C). Given the importance of IL-6 in activating Stat3 via Shp2 (Heinrich et al., 2003), we examined the consequence of constitutively active Shp2 expression in Linnegative cells on Morrbid activation. Consistent with IL-6 stimulation of Morrbid in vitro, HSPCs with expression of a constitutive Shp2 isoform, Shp2E76K, also demonstrated reduced apoptosis, enhanced activation of Stat3, increased binding of Stat3 to the Morrbid promoter, increased expression of Morrbid, and reduced expression of Bim; essentially phenocopying the heightened survival advantage of Tet2-KO preleukemic cells (Figure 4D and S5E). Taken together, these gain of function studies further indicate the importance of the IL-6/Shp2/Stat3/Morrbid axis in driving the enhanced survival observed in Tet2-KO Lin- cells.
Figure 5. IL-6 stimulates a Shp2/Stat3/Morrbid-mediated pro-survival pathway in vitro, which is inhibited by pharmacologic inhibitors SHP099 and E3330.

(A) A schematic describing an abbreviated form of canonical TLR4/NFκB/IL-6 and putative IL-6/Stat3/Morrbid signaling pathway.
(B) IL-6 promotes the survival of Lin- cells in liquid culture assay.
(C) IL-6 induces the expression of activation of Morrbid in wildtype and Tet2-KO Lin- cells.
(D) Lin- cells expressing Shp2E76K, a gain-of-function isoform of Shp2, manifest decreased apoptosis, decreased expression of Bim and elevated expression of Morrbid, compared to wildtype controls.
(E and F) E3330 or SHP099 treatment of Tet2-KO Lin-negative bone marrow cells corrects the aberrant colony-forming ability of Tet2-KO cells in primary and secondary replating assay. E3330, 0.5 μM. SHP099, 0.1 μM.
(G) E3330 or SHP099 treatment of Tet2-KO Lin-negative bone marrow cells inhibits IL-6 induced cell survival.
(H) E3330 or SHP099 inhibits Stat3 binding to the Morrbid promoter as revealed by CHIP-qPCR assay. E3330, 0.5 μM. SHP099, 0.1 μM.
Data in (B-H) are from a representative experiment (n=3 to 4 per group, mean ± s.e.m.). Experiments were repeated twice. P value: * P < 0.05, ** P < 0.01, *** P < 0.001. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S5 for additional supporting data.
To further verify the role of Stat3 and Shp2 in regulating the IL-6/Morrbid axis in Tet2-KO Lin-negative cells, we treated the cells with E3330, a pan inhibitor of both NFκB and Stat3 (Cardoso et al., 2012; Jedinak et al., 2011), and SHP099, a novel selective allosteric inhibitor of Shp2 (Chen et al., 2016). E3330 is being advanced in cancer clinical trials as a novel, oral, first-in-class drug in humans to target APE-1 (IND# 125360). SHP099 (TN0155) is also being tested in phase I clinical trial (NCT03114319) for solid tumors with mutations in the receptor tyrosine kinases. We observed a significant inhibition in replating efficiency of Tet2-KO Lin-negative cells relative to controls in the presence of E3330 or SHP099 (Figure 5E and 5F), which was associated with reduced expression of IL-6, reduced activation of Stat3 and enhanced apoptosis (Figure S5F and 5G). Of note, the repressive activity of E3330 and SHP099 exhibited a similar trend in CFU assays in control and Tet2-KO cells, however the repression was more profound in Tet2-KO cells. Consistent with these observations, we also observed a significant reduction in the binding of Stat3 to Morrbid promoter in the presence of these drugs (Figure 5H). Taken together, these data suggest that IL-6, Shp2 and Stat3 are upstream regulators of Morrbid and that E3330 and SHP099 impair the growth of Tet2-KO Lin-negative cells in part by modulating the expression of Morrbid.
Tet2 loss-induced aberrant hematopoietic expansion and emergency granulopoiesis is repressed by E3330 or SHP099
We next asked if SHP099 or E3330 are also effective in mice that mimic Tet2 loss-induced clonal hematopoiesis or early signs of myeloid neoplasia. We first examined the in vivo impact of E3330 and SHP099 treatment on LPS-induced emergency hematopoiesis. A significant decrease in LPS-induced neutrophilia in PB, as well as in the frequency of LSK cells, in BM of Tet2-KO mice was observed as early as 2 days post-LPS treatment in the presence of E3330 or SHP099 treatment, demonstrating that these two drugs possess potent anti-inflammatory activity, not only in vitro but also in vivo (Figure S6A). Following a similar approach as described before (Fuster et al., 2017), we next developed an experimental model of clonal hematopoiesis/clonal expansion by mixing Tet2-KO BM cells (from Tet2 mutant mice, CD45.2) with wildtype cells (from Boy J mice, CD45.1) at a low ratio (1 : 5) prior to transplantation into recipient animals (F1 mice, CD45.2/CD45.1) (Figure 6A). The proportion of Tet2-KO cells consistently expanded with time, and the ratio of Tet2-KO to wildtype cells almost doubled every month (ratio value was calculated by assessing the percentage of CD45.2+ cells to percentage of CD45.1+ cells) (Figure 6B and data not shown). Next, we tested if continuous administration of LPS (mimicking chronic infection), E3330, or SHP099 alters Tet2 loss-induced clonal expansion (procedure scheme illustrated in Figure 6A). As shown in Figure 6B and 6C, stimulation of chimeric mice with LPS further increased the ratio of Tet2-KO to wildtype cells in this model; daily treatment with E3330 or SHP099 significantly repressed the hematopoietic cell expansion of Tet2-KO cells compared to controls. These results demonstrate that Tet2 loss-induced clonal expansion of preleukemic HSPCs is inhibited by the treatment of these mice with E3330 or SHP099.
Figure 6. E3330 and SHP099 treatment represses clonal expansion of Tet2-KO HSPCs and prevents aberrant early signs of MPN in both young and aged Tet2 mutant mice.

(A) A schematic showing the experimental procedure used for generating chimeric mice mimicking clonal expansion of Tet2-KO cells. Tet2-KO bone marrow cells (CD45.2) were mixed with BoyJ bone marrow cells (CD45.1) at a ratio 1 to 5 (100K: 500K). Two months post-transplant, the chimeric mice were subjected to continuous treatment with LPS (0.8 mg/kg, i.p. injection every other day for 1 month) or a daily injection of E3330 or SHP099 (oral gavage, 20 mg/kg for E3330, 50 mg/kg for SHP099 for 3 months). Chimerism in peripheral blood was analyzed monthly at indicated time points.
(B) Clonal expansion of Tet2-KO HSPCs is heightened by continuous administration of LPS.
(C) Clonal expansion of Tet2-KO HSPCs is repressed by daily injection of E3330 or SHP099.
(D) Long-term treatment of juvenile Tet2-KO mice with E3330 or SHP099 results in the rescue of multiple phenotypic defects associated with Tet2 deficiency including frequency of CMPs, LSKs, HSCs, expression of Morrbid, Bim, IL-6 and pStat3 as well as spleen weight.
(E) Short-term treatment of aged Tet2-KO mice with E3330 or SHP099 prevents early hematological signs of CMML in peripheral blood. E3330, 20 mg/kg and SHP099, 50 mg/kg, oral gavage.
Transplant data in (B) and (C) are derived from using 5 recipient mice per group, mean ± s.e.m. 15 doses of LPS were given at 0.8 mg/kg over a period of one month (i.p. injection, every other day). E3330 (20 mg/kg) and SHP099 (50 mg/kg) were administered by oral gavage daily for 12 weeks in (D) or for 2 weeks in (E). Drug treatment studies in (D) and (E) are pooled data from two independent experiments (n=4 to 5 mice per group, mean ± s.e.m.).
P value: * P < 0.05, ** P < 0.01. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S6 for additional supporting data.
In addition to examining the role of these two drugs in chimeric mice demonstrating inhibition of clonal expansion, we also assessed the efficacy of E3330 and SHP099 in a cohort of Tet2-KO mice. Since Tet2-KO mice develop age-dependent CMML-like disease symptoms (Figure S6B), we chose both juvenile (~1 month old) and old Tet2-KO mice (~6 months old) for the drug treatment. Although wildtype control mice were tolerant of the drug treatment and showed no changes in any of the hematologic parameters tested (Figure S6C), a two-month continuous treatment of Tet2-KO mice with E3330 or SHP099 reversed multiple early signs of aberrant HSPC dysregulation associated with Tet2-deficiency, including enhanced frequency of CMPs, LSKs, HSCs and splenomegaly (Figure 6D). Importantly, and consistent with our in vitro observations, expression of IL-6, pStat3, Morrbid and Bim were significantly corrected in Tet2-KO cells treated with E3330 or SHP099 (Figure 6D). In a short-term treatment regimen (14 days) on aged Tet2-KO mice, E3330 and SHP099 failed to reverse the frequency of LSKs and CMPs. However, both these drugs were able to promptly and significantly reduce the neutrophil and white blood cell (WBC) burden, and also restored the anemia including the red blood cell (RBC) and platelet counts in Tet2-KO mice relative to controls (Figure 6E and S6D). In addition, a reduction in spleen weight was observed in SHP099-treated Tet2-KO mice, although it did not reach statistical significance (p=0.09, Figure S6D). Importantly, Serum IL-6 levels were also reduced upon E3330 or SHP099 treatment in Tet2-KO mice (Figure 6E). Taken together, these findings suggest that through targeted inhibition of IL-6/Shp2/Stat3/Morrbid signaling axis, treatment with E3330 or SHP099 offers an anti-inflammatory benefit and rescues clonal hematopoiesis as well as some aspects of Tet2 deficiency-associated HSPC aberrations including CMML-like disease.
Genetic loss of Morrbid in Tet2+/− HSPCs rescues inflammatory stress-induced defects associated with loss of Tet2
Given the observed upregulation of Morrbid via the enhanced binding of Stat3 to the Morrbid gene in Tet2-KO Lin- cells, we next directly tested the requirement for Morrbid in Tet2 loss-induced aberrant hematopoietic phenotype(s), including clonal hematopoiesis and CMML-like disease. We inhibited the expression of Morrbid by knocking-down its expression using a lentivirus in Tet2-KO cells (Figure S7A), or by breeding Morrbid+/− mice to Tet2+/− mice, given that both heterozygous mutants closely mimic the phenotype observed in complete knockout for these genes (Figure 7A) (Kotzin et al., 2016; Quivoron et al., 2011).
Figure 7: Genetic loss of Morrbid represses the growth advantage in Tet2-deficient preleukemic cells.

(A) A schematic showing the experimental procedures for testing genetic requirement of Morrbid in Tet2 deficiency-mediated aberrant hematopoietic phenotypes in naïve mice or LPS-challenged mice, and in ex vivo repopulation assays and in vivo cBMT assays.
(B) Tet2+/−; Morrbid+/− mice fail to manifest amplified “emergency hematopoiesis” seen in Tet2+/− mice upon LPS treatment.
(C) Rescue of splenomegaly in Tet2+/−;Morrbid+/− mice relative to Tet2+/− mice.
(D) Tet2+/− HSPCs with Morrbid haplo-insufficiency corrects the enhanced self-renewing ability of Tet2+/− HSPCs in an in vitro serial replating assay.
(E) Tet2+/− HSPCs with Morrbid haplo-insufficiency rescues the enhanced clonal expansion seen with Tet2+/− HSPCs as assessed in a chimeric cBMT assay.
Data in (B), (C), and (E) are from a representative experiment (n = 4 to 5 mice per group, mean ± s.e.m.). Data in (D) is pooled from two mice performed in replicates of 4. Data in (E) are from 4 to 5 recipient mice for every group at every time point [4 to 16 weeks] (n=4 to 5, mean ± s.e.m.). Similar results were seen in another independent experiment (n=4 to 5 mice per group; analyzed at 8 weeks). P value: * P < 0.05, ** P < 0.01, *** P < 0.001. n.s., not significant. Statistical analysis performed by unpaired, two-tailed Student’s t-test. See also Figure S7 for additional supporting data.
When analyzing the compound mutant mice, loss of Morrbid (Morrbid+/−) mitigated the enhanced emergency granulopoiesis in Tet2+/− mice upon LPS challenge and reversed the splenomegaly normally observed in these mice (Figure 7B and 7C). Similarly, genetic loss of IL-6 or TLR4 (IL-6−/− or TLR4−/−) also reversed LPS-induced heightened emergency granulopoiesis in Tet2-KO mice (Figure S7E and S7F). As shown in Figure S7B and S7C, targeting Morrbid by two distinct shRNAs decreased the replating activity of Tet2-KO cells in a CFU assay in vitro and engraftment in a short-term transplantation assay in vivo. Consistent with these findings, Tet2+/−; Morrbid+/− Lin- cells failed to manifest replating advantage seen in Tet2+/− Lin- cells in a CFU assay in vitro and enhanced repopulating advantage in a cBMT assay in vivo (Figure 7D and 7E). The phenotypic corrections observed in Tet2+/−; Morrbid+/− mice were due in part to increased expression of Bim, which was associated with enhanced apoptosis in both mature myeloid cells and progenitor cells compared to controls (Figure S7D). Taken together, these data demonstrate that Morrbid is an essential mediator of aberrant “emergency hematopoiesis” and clonal hematopoiesis due to loss of Tet2 in HSPCs.
DISCUSSION
In this study, we examined the impact of inflammation on the function of pre-leukemic stem and progenitor cells lacking the enzyme Tet2. TET2 is frequently mutated in patients with myeloid malignancies and these mutations are also present in normal individuals who lack signs of AML but demonstrate clonal hematopoiesis (Busque et al., 2012; Genovese et al., 2014; Jaiswal et al., 2014). Some of these individuals develop full-blown hematologic malignancies later on in life. It is however unclear what the risk factors are in these individuals, which push them to progress from clonal hematopoiesis to full-blown leukemia. In solid tumors, infection/inflammation contribute to colon cancer (Bernstein et al., 2001; Newman et al., 2001). Here, we have examined how acute inflammation changes the behavior of hematopoietic stem and progenitor cells in a mouse model of pre-leukemic stem and progenitor cells that shows signs of clonal hematopoiesis. Specifically, we studied if Tet2-KO pre-leukemic stem and progenitor cells themselves produce inflammatory cytokines and how they respond to an inflammatory challenge. Our findings demonstrate that Tet2-KO pre-leukemic stem and progenitor cells show enhanced response to inflammatory stimuli within the more primitive and mature stem/progenitor cell compartment of the BM. Tet2-KO HSPCs show sustained survival in response to inflammation. Serum IL-6 levels, as well as the expression of IL-6 is significantly upregulated in Tet2-KO HSPCs with or without LPS stimulation. Functionally, Tet2-KO HSCs demonstrate resistance to inflammation-induced damage. Pharmacologically, an anti-inflammatory drug E3330 or a SHP2 inhibitor, SHP099, normalizes inflammation-induced emergency hematopoiesis, represses aberrant hematopoietic phenotypes in the bone marrow of young Tet2-KO mice, and reverses the imbalance of WBC/RBCs in aged Tet2-KO mice. Mechanistically, hyperactive Morrbid, in part contributes, to the aberrant growth advantage caused by Tet2 loss. Taken together, these findings suggest that Tet2-KO pre-leukemic HSPCs are powered with a selection advantage under conditions of inflammation-induced stress. The growth advantage seen in Tet2-KO pre-leukemic stem cells is likely due to elevated NFκB/IL-6/Stat3/Morrbid signaling in both mature (supplying IL-6) and immature cells (supplying and responding to IL-6), which likely forms a feed-forward loop to promote myeloid malignancies with age (Figure S7G).
TET2 catalyzes the 5-hydroxylation of 5-methylcytosine (5-mc) to 5-hydroxymethylcytosine (5-hmc) and is an essential epigenetic regulator of the genome (Shih et al., 2012). TET2 is a tumor suppressor in myeloid and solid cancer (Ko et al., 2010; Lian et al., 2012). Although studies have shown that Tet2-deficient (Tet2−/− or Tet2+/−) LSK/HSC cells have increased self-renewal activity, the underlying molecular mechanisms that control this process are largely unknown. During the course of our studies, Zhang et al, utilized Tet2-KO dendritic cells and macrophages and showed that, upon LPS treatment, Tet2-KO mature immune cells produce higher levels of IL-6 and fail to resolve inflammation as efficiently as wildtype cells, implicating an important role of Tet2 in regulating inflammation (Zhang et al., 2015). Jaiswal et al and Fuster et al also reported that Tet2 loss results in aberrant inflammation and clonal hematopoietic expansion, which further promotes myeloid cell-driven diseases such as cardiovascular disease (Fuster et al., 2017; Jaiswal et al., 2017). In the current study, we observed higher levels of IL-6 in the serum and in HSPCs lacking Tet2 upon LPS challenge. Surprisingly, we also found that adult and older naïve Tet2-KO mice also possess increased expression of IL-6 in serum and in HSPCs. This observation has not been appreciated before and strongly suggests that Tet2-KO HSPCs perpetuate a baseline high level of inflammation, which may be one of the mechanism(s) involved in driving clonal hematopoiesis (Figure S7G).
IL-6 is one of the major pro-inflammatory cytokine circulating in the blood and also functions locally (Hunter and Jones, 2015). IL-6 can be synthesized in large amounts in response to LPS challenge. In addition to playing an essential role in regulating immunity, IL-6 can also regulate hematopoietic cell development and leukemia transformation. Recent studies utilizing a mouse model of chronic myeloid leukemia (CML) showed that leukemia in this model is dependent on increased levels of inflammatory cytokine IL-6, and that blocking IL-6/IL6R signaling prevents CML development (Reynaud et al., 2011; Welner et al., 2015). These observations and our present findings support a persuasive hypothesis that increased levels of the pro-inflammatory cytokine IL-6 is an essential trigger of CMML-like disease observed in Tet2-KO mice with age (Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). In addition to IL-6, other cytokines such as IFNα, IFNγ, IL-1α, IL-1β, and TNFα can also directly activate HSCs (Baldridge et al., 2010; Essers et al., 2009; Pietras et al., 2014; Pietras et al., 2016; Sujer, 2014). As we only tested the levels of intracellular IL-6, IL-1β, TNFα and GM-CSF at defined time points, we cannot rule out the possibility that the presence of these additional cytokines may also contribute to the observed phenotype seen in Tet2-KO mice. Interestingly, we detected increased expression of TLR4 in Tet2-KO HSPCs (Figure S5C). Meisel et al have recently reported increased expression of IL6Rα in Tet2-KO HSPCs (Meisel et al., 2018). These observations may provide further explanation as to why Tet2-deficient (Tet2−/− or Tet2+/−) preleukemic cells are primed and more sensitive to LPS and IL-6 signaling compared to wildtype cells.
While Bim−/− mice manifest increased myeloid cells in peripheral blood (Bouillet et al., 1999), Morrbid negatively regulates Bcl2l12 (encoding Bim) expression, and Morrbid+/− or Morrbid−/− mice manifest decreased myeloid cells in peripheral blood (Kotzin et al., 2016). We show that shRNA-mediated knockdown of Morrbid or genetic loss of Morrbid in the setting of Tet2-deficiency failed to undergo aberrant hematopoietic clonal expansion; and that Tet2+/−; Morrbid+/− HSPCs failed to manifest amplified “emergency granulopoiesis” upon LPS challenge. Likewise, when a cohort of young Tet2-KO mice were treated with E3330 or SHP099, recovery of Bim and repression of IL-6 was observed, suggesting that these two drugs largely function via the NFκB/IL-6/Stat3/Morrbid pathway for inducing the disease phenotype associated with Tet2 loss.
It is generally accepted that cancer, including solid tumors and leukemia, undergo an evolutionary process relying on adaptive advantages of acquired somatic mutations (intrinsic factors) with fitness for niche selection (extrinsic factors), with similar bioecological principles as indicated in Darwinian natural selection (Greaves and Maley, 2012). A recent study showed that once lymphocyte progenitors are primed with constitutive expression of oncogenes such as BCR-ABL, NRASV12 or Myc, progenitor cells have a selection advantage in age-induced inflammatory niche and transform into leukemia (Henry et al., 2015). By conducting primary and secondary cBMT assays, we show that Tet2-KO HSPCs always outperform wildtype control cells. Essentially, when wildtype donor cells are isolated from their endogenous microenvironment on day 2-post LPS treatment, they lose their normal repopulating activity in recipient mice. In contrast, and in agreement with the finding by Ko et al (Ko et al., 2011), even though the primary cBMT only received half the number of Tet2-KO bone marrow donor cells, the recipient animals still developed early signs of MPN or CMML, strongly indicating that Tet2-deficiency induces growth and repopulating advantage to HSCs.
Based on the results from our study along with what others have shown, we have created a schematic that summarizes our findings (Figure S7G). Our data suggest that loss of Tet2 results in multiple changes in the level of key molecules including IL-6 and Morrbid, which may render the self-renewal, differentiation, survival and clonal evolution of mutant HSCs to include myeloid skewing and development of MPN or CMML like disease with age. Given the hyperactivation of the IL-6/Shp2/Stat3/Morrbid pathway in Tet2-KO cells, we employed small molecule inhibitors, E3330 and SHP099, to determine their impact on emergency hematopoiesis. Our results show that both E3330 and SHP099 can effectively repress LPS-induced emergency granulopoiesis and LSK expansion. More importantly, our results show that E3330 and SHP099 treatment prevents aberrant hematopoietic phenotypes in bone marrow of young Tet2-KO mice and reverses neutrophil counts and serum IL-6 levels in aged naïve Tet2-KO mice, suggesting that these drugs, through their specific inhibition of Ape1-NFκB activation or Shp2-Stat3 activation, might provide an anti-inflammatory benefit in AML patients bearing TET2 mutations and in normal healthy individuals bearing TET2 mutations and clonal hematopoiesis. Given that emerging evidence suggests that inflammation very likely plays a causative role in the pathology of MPN (Fleischman, 2015; Koschmieder et al., 2016), testing these drugs in other pre-leukemic models may be of clinical benefit.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Reuben Kapur (rkapur@iupui.edu).
EXPERIMENTAL MODEL
Mice
All mice were bred and maintained under specified pathogen-free (SPF) conditions in the animal facility at Indiana University School of Medicine with 12-hr light/dark cycle and were provided food and water ad libitum. The Institutional Animal Care and Use Committee (IACUC) at Indiana University School of Medicine approved experiments with mice. Tet2-deficient mice (Tet2−/− or Tet2+/−, CD45.2) and Morrbid-deficient mice (Morrbid+/−, CD45.2) are on C57BL/6 genetic background and have been described previously (Kotzin et al., 2016; Li et al., 2011). Tet2+/− mice were bred with Morrbid+/− mice to generate Tet2+/−; Morrbid+/− mice and all the controls. Wildtype C57BL/6 (CD45.2) mice were purchased from The Jackson Laboratory and used as controls. BoyJ mice (CD45.1) were purchased from The Jackson Laboratory for transplantation and chimerism analysis. The Shp2E76K mice have been described (Xu et al., 2011). TLR4−/− and IL6−/− were purchased from Jackson Laboratory (Cat #007727; Cat #002650). Whenever possible littermates were used as controls for all experiments. The age of mice has been indicated specifically for certain analysis in Figure 1 to Figure 7. In general, juvenile mice were between 1~2 months of age, adult mice between 3~4 months of age and older mice between 6~12 months of age. Mice of both sexes were used for experiments. Mice were not mated or used in previous procedure before experimentation.
METHOD DETAILS
In vivo treatments
Lipopolysaccharide (LPS) was purchased from Sigma (Cat # L8643) and dissolved in sterile phosphate-buffered saline (PBS) prior to being administered to mice (single dose; 0.8 mg/kg, i.p.). E3330, also called APX3330 was synthesized as described (Jiang et al., 2010; Nyland et al., 2010). E3330 was dissolved in Cremophor: EtOH (1:1) (Cremophor was purchased from Sigma, Cat # C5135) for stock solution generation and diluted in PBS prior to use for pre-LPS treatment or post-LPS treatment (20 mg/kg, twice a day, i.p. for Figure S6A; 20 mg/kg, one dose per day, daily by gavage for experiments in Figure 6C, 6D, 6E and Figure S6C and S6D). SHP099 (provided by Novartis), was dissolved in 0.5% Methylcellulose (Sigma, Cat# M0262) and 0.1% Tween-80 (Fisher Scientific, Cat #BP338–500) and given to animals by gavage (daily, 50 mg/kg) for all the in vivo treatments. Male and female mice between 3–4 weeks as juvenile mice, or 8–16 weeks of age as adult mice, or 6~8 month of age as aged mice, were used for indicated experiments.
Hematological cell counts
Hematologic parameters in peripheral blood (PB, from tail bleeding) were analyzed by an automated cell counter machine (Drew Hemavet 950). Total bone marrow (BM) cells were harvested from two femurs and two tibias and kept on ice or in refrigerator and stored in sterile blocking buffer containing 2% rat-serum prior to analysis. BM cellularity (viable cell counts) was analyzed by an automated cell counter (Beckman the Vi-CELL™ Cell Counter for Cell Viability Analyzer).
Flow cytometry
Non-lysed BM cells were used for analysis of erythroid lineage progenitor cells (Ter119 and CD71 staining). Remaining flow cytometry analysis was performed on lysed bone marrow cells (Lysis Buffer, BD, Cat # 555899). Antibodies against Ter119, Mac1, Gr1, B220, CD3, CD4 and CD8 were used for detecting mature cells (Lineage labeling). Progenitor cells were labeled and analyzed using the indicated antibodies. Antibody labeled BM cells were analyzed using a BD FACS-CANTO II machine with a two-laser and six-filter configuration. The properly compensated flow data were analyzed by Flow Jo software (V10.2). Events plotting, calculation of frequency, mean fluorescence intensity (MFI), and histogram overlays were analyzed by Flow Jo software. A panel of PE-conjugated antibodies against TER-119, Gr1, Mac1, B220, CD3, CD4 and CD8a surface antigens were mixed as a cocktail for lineage labeling (Lin-PE cocktail). A panel of antibodies containing Lin-PE cocktail, c-Kit APC, Sca-1 APC/Cy7, CD150 PE/Cy5 and CD48 PE/Cy7 were used for LSK/HSC labeling. A panel of antibodies containing Lin-PE cocktail, c-Kit APC, Sca-1 APC/Cy7, CD127 PE/Cy5, CD16/32 PE/Cy7 and CD34 FITC were used for CMP/GMP/MEP/CLP labeling. A panel of antibodies containing Lin-PE cocktail, c-Kit APC, Sca-1 APC/Cy7, Annexin FITC and 7-AAD were used for apoptosis labeling. For detecting the expression of TLR4 in mature cells, TLR4 PE/Cy7 was mixed with Mac1 PE, B220 APC and CD3 PE. For detecting the expression of TLR4 in HSPCs, TLR4 PE/Cy7 was mixed with Lin-PE cocktail, c-Kit APC, Sca-1 APC/Cy7 and CD150 PE/Cy5. For chimerism analysis in mature cells, CD45.2 PerCP/Cy5.5 and CD45.1 PE/Cy7 were mixed with Mac1 PE, B220 APC and CD3 PE. For chimerism analysis in HSPCs, CD45.2 PerCP/Cy5.5 and CD45.1 PE/Cy7 were mixed with Lin-PE cocktail, c-Kit APC, Sca-1 APC/Cy7 and CD150 PE/Cy5. Mac1 PE and Ly6G FITC were mixed for neutrophil labeling. Intracelluar flow cytometry (ICFC) was performed to detect the expression of Bim, GM-CSF, IL-6, IL-1β, Ki-67, NFκB, phospho-Stat3, and TNFα. Briefly, freshly prepared bone marrow cells were pre-stained by using cell surface antibodies for mature cells or for HSPCs and then fixed with BD Cytofix and washed using BD Cytoperm three times. The pre-stained cells were then restained with the appropriate indicated antibodies. Staining with an Annexin-V and 7-AAD kit (BioLegend, Cat # 640922) was performed according to the manufacturer’s instruction for apoptosis analysis, along with labeling of LSK cells.
A full list of antibodies is provided in KEY RESOURCES TABLE.
Multiplex cytokine assays
Serum samples were prepared from PB (tail-bleeding) and diluted in sterile PBS (1 to 2 dilution). Thirty-one cytokines or chemokines were quantified by multiplex immunoassay with a BioPlex 200 instrument (Eve Technologies, Mouse Cytokine Array/Chemokine Array 31-Plex, Cat # MD31).
Isolation of Lin-negative BM cells, LSK cells and qRT-PCR assays
Lin- BM cells (~1 × 106) were purified by an EasySep™ Mouse Hematopoietic Progenitor Cell Isolation Kit (StemCell, Cat # 19856) according to manufacturer’s instruction. LSK cells were purified from Lin- negative BM cells by staining the cells with antibodies against c-Kit and Sca-1 followed by sorting them (Fluorescence-activated cell sorting (FACS) (BD FACSARIA). Total RNA was extracted from Lin- cells using RNeasy Mini Kit (Qiagen, Cat # 74104) according to the manufacturer’s instructions. Isolated RNA was quantified by spectrophotometer and RNA concentrations were normalized. cDNA was synthesized by SuperScript II Reverse Transcriptase (ThermoFisher Scientific, Cat # 18064014). Resulting cDNA was analyzed by SYBR Green master mix (Life Technologies, Cat # 4385612) with indicated primers on a ViiA7 Real-Time PCR instrument. Expression of Actin-b was used as an internal control (Forward, 5’-GACGGCCAGGTCATCACTATTG-3’ and Reverse 5’-AGGAAGGCTGGAAAAGAGCC-3’) for calculating fold changes of indicated genes. The information of qRT-PCR primers for Morrbid and IL-6 is provided in KEY RESOURCES TABLE. See also Table Supplement-1 for a list of additional qRT-PCR primers.
CFU assay
Bone marrow Lin- negative cells or LSK cells were isolated as described above and platted in 24-well plate (10K cells per 0.5 mL medium per well) using MethoCult™ GF M3434 (Stem Cell). E3330 and SHP099 were dissolved in DMSO as inhibitors. Colonies were counted after 7-days of culture.
In vitro suspension culture with IL-6 and drugs
Bone marrow Lin- negative cells were isolated as described above and cultured in basal medium (Gibco IMDM containing 2% FBS) or with IL-6 as indicated in Figure 6 and S6. Drugs E3330 or SHP099 were added as indicated. After cultured for 36~48 hours, cells were washed by PBS and analyzed by flow cytometry or by qRT-PCR.
CHIP-qPCR assay
BM Lin-negative cells were used to extract chromatin DNA using MAGnify™ Chromatin Immunoprecipitation System (ThermoFisher, Cat # 492024) according to the manufacturer’s instruction. CHIP purified chromatin DNA and input DNA were normalized to identical concentration for qPCR validation and enrichment analysis (1% enrichment of input level was defined as unit 1). Anti-Stat3 (Cell Signaling Technologies, #9319) was used for chromatin precipitation. Primers for CHIP-qPCR analysis are listed in KEY RESOURCES TABLE.
Analysis of 5mC and 5hmC level on the Morrbid promoter
Genomic DNA from Lin-negative cells derived from wild type and Tet2-KO mice was purified by Thermo Scientific Genomic DNA Purification Kit (Cat #K0512). For analyzing 5mC level of the indicated CCGG site by MspI and HpaI enzymes, the DNA samples were pretreated by Qiagen EpiTect Bisulfite Kits (Cat #59104) for bisulfite conversion, followed by MspI or HapI digestion. The digested products were used as templates for quantitative PCR (qPCR) analysis and/or DNA gel running by the indicated primers targeting the promoter region (oligos used for the ~800-bp promoter are included in KEY RESOURCES TABLE). For analyzing 5hmC level of the indicated CCGG site, the DNA was pretreated by Zymo Quest 5-hmC Detection Kit for 5ghmC conversion, followed by Msp digestion (Cat #D5415). Similarly the digested products were used as templates for quantitative PCR (qPCR) analysis and/or DNA gel running by the indicated primers targeting the promoter region. For analyzing the indicated CpG sites 1 to 4 by bisulfite sequencing PCR (BSP), the DNA samples were pretreated by Qiagen EpiTect Bisulfite Kit (Cat #59104) for bisulfite conversion and then amplified by indicated primers (oligos used for the 4 CpG sites are included in Supplemental Table-4). The amplified products were cloned into T-vector (Promega, Cat #A3610). Twenty-four proper clones were randomly selected for sequencing analysis to determine the methylation level on the CpG sites. 5mC level (percentage) is measured by the percentage of “C-T” conversion.
Competitive bone marrow transplantation (cBMT)
B6.SJL-Ptprca Pepcb/Boy (BoyJ, CD45.1) mice were purchased from The Jackson Laboratory. Recipient animals (F1, CD45.2/CD45.1) were lethally irradiated (700 cGy plus 400 cGy) one day prior to transplantation (intravenous tail injection) of donor cells. For primary cBMT, CD45.2 donor BM cells from naïve or LPS-treated mice were mixed with BoyJ CD45.1 competitor BM donor cells (with an equal number of viable total cells, 500K: 500K). For secondary cBMT, donor BM cells from primary cBMT recipients were mixed with BoyJ CD45.1 competitor BM cells (with an equal number of viable total cells). For LSK cell engraftment, 2000 LSK cells from LPS treated or control mice were mixed with 500,000 BoyJ CD45.1 supporting cells (2K: 500K). For generating chimeric mice mimicking hematopoietic clonal expansion, Tet2-KO donor cells and BoyJ donor cells were mixed at a ratio of 1: 5 (100K: 500K). For assessing the repopulating advantage of Tet2+/−; Morrbid+/− cells, bone marrow donor cells from Tet2+/−; Morrbid+/− mice and all other controls were equally mixed with BoyJ donor cells respectively (500K: 500K). Chimerism analysis for progressive engraftment was analyzed on PB samples monthly (every 4-week interval) post BM transplantation.
shRNA generation and approach for knocking down the expression of Morrbid
Empty vector for pGreen shRNA cloning and shRNA-containing vectors for knocking-down Morrbid, shRNA1 and shRNA2 have been described before (Kotzin et al., 2016). Lentivirus supernatants were generated by transfecting 293T cells with an envelope plasmid (Addgene: #12260), packaging plasmid (Addgene: #12260), and the shRNA-containing vector or empty vector (1ug: 3ug: 6ug) per 10-cm dish. Virus soup was collected on day 1, day 2 and day 3 after transfection, combined, filtered and enriched by high-speed centrifugation (10 × 103 g). Lin- cells from wildtype or Tet2-KO mice were freshly isolated and pre-cultured over-night at 37°C in 6-well plate (1× 106 per mL) with medium containing mIL-3 (10 ng/mL), mIL-6 (5 ng/mL) and mSCF (50 ng/mL) (all are from PeproTech). Cell number was readjusted to make them equal at equal in 2 mL at 2.0 × 106 per well in a 6-well plate and spinfected for 2 hours with 5 ug/mL polybrene. After overnight culture, the transfection efficiency was determined by flow cytometry for GFP expression. Approximately 60–70% infection efficiency was achieved in most cases. For CFU assays, infected cells were purified by FACS. For cBMT assays, infected cells were washed three times in PBS, diluted and mixed with BoyJ cells (100K: 500K) prior to transplantation. Cells targeted by Morrbid-shRNA or empty vector were monitored by examining the GFP+% in the CD45.2 pool.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experimental procedures on Tet2-KO samples were always run in parallel with wildtype controls (sex and age matched littermate controls when possible) for observing experiment variabilities. Analysis of grouped data was not blinded and no samples were excluded. Aged Tet2-KO mice were randomized into two groups for treatment with E3330 or vehicle, SHP099 or vehicle (Figure 6). P value was calculated using an unpaired t-test for comparing means of two groups (GraphPad Prism 6.0). Error bars indicate the standard error of mean (s.e.m.).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tet2−/− (Tet2-KO), Tet2+/− | Li et al., 2011 | N/A |
| Mouse: Morrbid+/− | Kotzin et al., 2016 | N/A |
| Mouse: Shp2E76K | Xu et al., 2011 | N/A |
| Mouse: Tlr4-KO | Jackson Laboratory | Cat #007727 |
| Mouse: IL6-KO | Jackson Laboratory | Cat #002650 |
| Mouse: C57/B6 | Jackson Laboratory | Cat #000664 |
| Mouse: BoyJ | Jackson Laboratory | Cat #002014 |
| Critical Commercial Assays | ||
| Multiplex cytokine assays | Eve Technologies | #MD31 |
| CFU assays | StemCell | #M3434 |
| EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit | StemCell | #19856 |
| Rneasy Mini Kit | Qiagen | #74104 |
| SuperScript II Reverse Transcriptase | Fisher Scientific | #18064014 |
| SYBR Green master mix | Life Technologies | #4385612 |
| MAGnify Chromatin Immunoprecipitation System | Fisher Scientific | #492024 |
| Genomic DNA purification Kit | Fisher Scientific | #K0512 |
| EpiTect Bisulfite Kit | Qiagen | #59104 |
| Quest 5-hmC Detection Kit | Zymo | #5415 |
| MspI | NEB | R0106S |
| HpaI | NEB | R0105S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS | Sigma | #L8643 |
| Cremophor | Sigma | #C5135 |
| Methylcellulose | Sigma | #M0262 |
| Tween-80 | Fisher Scientific | #BP338–500 |
| E3330 | Kelley Laboratory | N/A |
| SHP099 | Mohseni Laboratory (Novartis) | N/A |
| Lysis Buffer | BD | #555899 |
| Cytofix/Cytoperm Kit | BD | #554714 |
| mIL-3 peptide | PeproTech | #213–13 |
| mIL-6 peptide | PeproTech | #216–16 |
| mSCF peptide | PeproTech | #250–03 |
| Antibodies for Western Blot or CHIP assays | ||
| phospho-Stat3 | Cell Signaling Tech | #9145 |
| Stat3 | Cell Signaling Tech | #9139 |
| Antibodies for Flow Cytometry | ||
| TER-119, PE | BioLegend | #116208 |
| Gr1, PE | BioLegend | #108408 |
| Mac1, PE | BioLegend | #101208 |
| B220, PE | BioLegend | #103208 |
| CD3, PE | BioLegend | #100206 |
| CD4, PE | BioLegend | #116006 |
| CD8a, PE | BioLegend | #100708 |
| c-Kit, APC | BioLegend | #105812 |
| Sca-1, APC/Cy7 | BioLegend | #108126 |
| CD150, PE/Cy5 | BioLegend | #115912 |
| CD48, PE/Cy7 | BioLegend | #103424 |
| CD127, PE/Cy5 | BioLegend | #135016 |
| CD16/32, PE/Cy7 | bioLegend | #101318 |
| CD34, FITC | eBioscience | #11–0341-85 |
| Annexin V, FITC | BioLegend | #640906 |
| 7-AAD | BioLegend | #79993 |
| TLR4, PE/Cy7 | BioLegend | #145408 |
| IL-6, AF488 | BD | #561363 |
| TNFa, AF488 | BioLegend | #506315 |
| IL-1b, FITC | eBioscience | #11–7114-80 |
| GM-CSF, FITC | BioLegend | #505403 |
| Ki67, FITC | BioLegend | #652410 |
| NFkB1 (p50), PE | Cell Signaling Tech | #24961 |
| phospho-Stat3, AF488 | Cell Signaling Tech | #4323 |
| Bim mAb (C34C5), AF488 | Cell Signaling Tech | #94805S |
| CD45.2, PerCP/Cy5.5 | BioLegend | #109928 |
| CD45.1, PE/Cy7 | BioLegend | #110730 |
| Ly-6G, FITC | BioLegend | #127606 |
| Oligonucleotides | ||
| Part-I: Key oligonucleotides for qRT-PCR, See also Table Supplement-1 for information of additional oligonucleotides for qRT-PCR | ||
| Morrbid, Forward TCTGAGAATGAGGGGACTGG | Kotzin, J. J. et al., Nature, 2016 | N/A |
| Morrbid, Reverse TGTGCTGTGAAGATCCCAAG | Kotzin, J. J. et al., Nature, 2016 | N/A |
| II6, Forward AGTTGCCTTCTTGGGACTGA | Inoue, S. et al., Cancer Cell, 2016 | N/A |
| II6, Reverse TCCACGATTTCCCAGAGAAC | Inoue, S. et al., Cancer Cell, 2016 | N/A |
| Part-II: Oligonucleotides for CHIP-qPCR | ||
| Promoter of Morrbid (~130 bp), Forward AGCACGAGTCATCTGGTTCC | Kotzin, J. J. et al., Nature, 2016 | N/A |
| Promoter of Morrbid (~130 bp), Reverse ACCCAGTCCCCTCATTCTCA | Kotzin, J. J. et al., Nature, 2016 | N/A |
| Part-III: Oligonucleotides for 5mC/5hmC analysis | ||
| Promoter of Morrbid (~800 bp), Forward ACC CCC AAG TCT CCTA ACCA | Kotzin, J. J. et al., Nature, 2016 | N/A |
| Promoter of Morrbid (~800 bp), Reverse GTT CAA CCT CAG TGC CCAGT | Kotzin, J. J. et al., Nature, 2016 | |
| Promoter Region with 4 CpG island sites, Forward ATTTAAGGTTTGGGAAGTTGTTTTT | This paper | N/A |
| Promoter Region with 4 CpG island sites, Reverse CAAACACCTCAATCTTCATTATCACTA | This paper | |
| Software and Algorithms | ||
| FlowJo | FlowJo | V10.2 |
| Prism | GraphPad Software | V6.0 |
| Adobe Illustrator | Adobe | CC-2015 |
Highlights:
Tet2-KO HSPCs maintain repopulation advantage post-acute inflammatory insult
Hyperactivation of IL-6/Shp2/Stat3/Morrbid and reduced apoptosis in Tet2-KO HPSCs
Anti-inflammation drugs E3330 or SHP099 inhibit survival advantage of Tet2-KO HSPCs
Morrbid+/− counters emergency granulopoiesis and clonal hematopoiesis in Tet2+/−
ACKNOWLEDGMENTS
We thank Dr. Mingjiang Xu for providing Tet2-KO mice and Dr. Peilin Ma for helping us with CHIP-qPCR assays. We thank our colleagues for technical support, for critically reading our manuscript and their suggestions to improve the manuscript. This work was supported in part by grants from National Institutes of Health (R01HL077177 to RK, R01CA173852 to R.K and R01CA134777 to RK) and by funds to RK from the Leukemia and Lymphoma Society (LLS) and from the Riley Children’s Foundation. ZC is supported by T32HL007910. We would also like to thank Ms. Tracy Winkle for her administrative support.
Footnotes
DECLARATION OF INTERESTS
Dr. Mark R. Kelley has licensed E3330 (APX3330) through Indiana University Research and Technology Corporation to Apexian Pharmaceuticals. Apexian Pharmaceuticals had neither control nor oversight of the studies, interpretation, or presentation of the data in this manuscript. Morvarid Mohseni is an employee of Novartis Institutes of Biomedical Research. Other authors declare no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven Figures and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abegunde SO, Buckstein R, Wells RA, and Rauh MJ (2018). An inflammatory environment containing TNFalpha favors Tet2 mutant clonal hematopoiesis. Experimental Hematology 59, 60–65. [DOI] [PubMed] [Google Scholar]
- Abkowitz JL (2014). Clone Wars — The Emergence of Neoplastic Blood-Cell Clones with Aging. New England Journal of Medicine 371, 2523–2525. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein CN, Blanchard JF, Kliewer E, and Wajda A (2001). Cancer risk in patients with inflammatory bowel disease. Cancer 91, 854–862. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, and Strasser A (1999). Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286, 1735–1738. [DOI] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 44, 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y, Maitra A, Kelley MR, and Fishel ML (2012). APE1/Ref-1 Regulates STAT3 Transcriptional Activity and APE1/Ref-1–STAT3 Dual-Targeting Effectively Inhibits Pancreatic Cancer Cell Survival. PLOS ONE 7, e47462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2012). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH-T, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- Chu SH, Heiser D, Li L, Kaplan I, Collector M, Huso D, Sharkis Saul J., Civin C, and Small D (2012). FLT3-ITD Knockin Impairs Hematopoietic Stem Cell Quiescence/Homeostasis, Leading to Myeloproliferative Neoplasm. Cell Stem Cell 11, 346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, Humphrey MB, Yang Q, Borghesi LA, and Kincade PW (2011). Chronic Exposure to a TLR Ligand Injures Hematopoietic Stem Cells. The Journal of Immunology 186, 5367–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, and Trumpp A (2009). IFN[agr] activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908. [DOI] [PubMed] [Google Scholar]
- Fleischman AG (2015). Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediators of Inflammation 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. New England Journal of Medicine 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, and Maley CC (2012). Clonal evolution in cancer. Nature 481, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, and Karin M (2010). Immunity, Inflammation, and Cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove CS, and Vassiliou GS (2014). Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Disease Models and Mechanisms 7, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg Robert A. (2011). Hallmarks of Cancer: The Next Generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hasselbalch HC (2012). Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 119, 3219–3225. [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, MÜLler-Newen G, and Schaper F (2003). Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochemical Journal 374, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Casás-Selves M, Kim J, Zaberezhnyy V, Aghili L, Daniel AE, Jimenez L, Azam T, McNamee EN, Clambey ET, et al. (2015). Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. The Journal of Clinical Investigation 125, 4666–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CA, and Jones SA (2015). IL-6 as a keystone cytokine in health and disease. Nat Immunol 16, 448–457. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, and Majeti R (2012). Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Science Translational Medicine 4, 149ra118–149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedinak A, Dudhgaonkar S, Kelley MR, and Sliva D (2011). Apurinic/Apyrimidinic Endonuclease 1 Regulates Inflammatory Response in Macrophages. Anticancer Research 31, 379–385. [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Sandusky GE, Kelley MR, and Fishel ML (2010). Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Investigation 28, 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2008). SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- King KY, and Goodell MA (2011). Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol 11, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, and Rao A (2011). Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proceedings of the National Academy of Sciences 108, 14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. (2010). Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Suda T, and Takubo K (2016). How hematopoietic stem/progenitors and their niche sense and respond to infectious stress. Experimental Hematology 44, 92–100. [DOI] [PubMed] [Google Scholar]
- Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ, Jeryczynski G, Gisslinger H, Jutzi JS, Pahl HL, et al. (2016). Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia 30, 1018–1024. [DOI] [PubMed] [Google Scholar]
- Kotzin JJ, Spencer SP, McCright SJ, Kumar DBU, Collet MA, Mowel WK, Elliott EN, Uyar A, Makiya MA, Dunagin MC, et al. (2016). The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature 537, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristinsson SY, Björkholm M, Hultcrantz M, Derolf ÅR, Landgren O, and Goldin LR (2011). Chronic Immune Stimulation Might Act As a Trigger for the Development of Acute Myeloid Leukemia or Myelodysplastic Syndromes. Journal of Clinical Oncology 29, 2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai C-L, Wang J, Zhang W, Petersen BE, Yang F-C, and Xu M (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Christine G., Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, et al. (2012). Loss of 5-Hydroxymethylcytosine Is an Epigenetic Hallmark of Melanoma. Cell 150, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y-C, Yeh W-C, and Ohashi PS (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, and Balkwill F (2008). Cancer-related inflammation. Nature 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Manz MG, and Boettcher S (2014). Emergency granulopoiesis. Nat Rev Immunol 14, 302–314. [DOI] [PubMed] [Google Scholar]
- Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, and King KY (2016). Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Reports 17, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, Pierre JF, Ernest JD, Galipeau HJ, Thuille N, et al. (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirantes C, Passegué E, and Pietras EM (2014). Pro-inflammatory cytokines: Emerging players regulating HSC function in normal and diseased hematopoiesis. Experimental Cell Research 329, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa Maria E., Vasanthakumar A, Patel J, Zhao X, et al. (2011). Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 20, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JV, Kosaka T, Sheppard BJ, Fox JG, and Schauer DB (2001). Bacterial Infection Promotes Colon Tumorigenesis in Apc Min/+ Mice. Journal of Infectious Diseases 184, 227–230. [DOI] [PubMed] [Google Scholar]
- Nyland RL, Luo M, Kelley MR, and Borch RF (2010). Design and Synthesis of Novel Quinone Inhibitors Targeted to the Redox Function of Apurinic/Apyrimidinic Endonuclease 1/Redox Enhancing Factor-1 (Ape1/Ref-1). J Med Chem 53, 1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, and Passegué E (2014). Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. The Journal of Experimental Medicine 211, 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner J-M, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronné L, Della Valle V, Lopez Cécile K., Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern M-H, et al. (2011). TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 20, 25–38. [DOI] [PubMed] [Google Scholar]
- Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, Sala-Torra O, Radich Jerald P., and Passegué E. (2011). IL-6 Controls Leukemic Multipotent Progenitor Cell Fate and Contributes to Chronic Myelogenous Leukemia Development. Cancer Cell 20, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S, Chora A, Goumnerov B, Mumaw C, Goebel WS, Fernandez L, Baydoun H, HogenEsch H, Dombkowski DM, Karlewicz CA, et al. (2009). Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood 114, 4064–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. (2018). Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. Journal of the American College of Cardiology 71, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Abdel-Wahab O, Patel JP, and Levine RL (2012). The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12, 599–612. [DOI] [PubMed] [Google Scholar]
- Shlush LI, and Minden MD (2015). Preleukemia: the normal side of cancer. Current Opinion in Hematology 22, 77–84. [DOI] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. (2014). Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling AS, Gibson CJ, and Ebert BL (2017). The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 17, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujer S (2014). Lipopolysaccharide-induced activation of haematopoietic stem cells in vivo PhD Dissertation. [Google Scholar]
- Takizawa H, Boettcher S, and Manz MG (2012). Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 119, 2991–3002. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Essers M, and Wilson A (2010). Awakening dormant haematopoietic stem cells. Nat Rev Immunol 10, 201–209. [DOI] [PubMed] [Google Scholar]
- Welner Robert S., Amabile G, Bararia D, Czibere A, Yang H, Zhang H, Pontes Lorena Lobo De F., Ye M, Levantini E, Di Ruscio A, et al. (2015). Treatment of Chronic Myelogenous Leukemia by Blocking Cytokine Alterations Found in Normal Stem and Progenitor Cells. Cancer Cell 27, 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Xu D, Liu X, Yu W-M, Meyerson HJ, Guo C, Gerson SL, and Qu C-K (2011). Non–lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase <em>Ptpn11</em> (Shp2) on malignant transformation of hematopoietic cells. The Journal of Experimental Medicine 208, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. (2015). Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JL, and Baltimore D (2015). Regulation of stress-induced hematopoiesis. Current Opinion in Hematology 22, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Jimmy L., Ma C, O’Connell Ryan M., Mehta A, DiLoreto R, Heath James R., and Baltimore D. (2014). Conversion of Danger Signals into Cytokine Signals by Hematopoietic Stem and Progenitor Cells for Regulation of Stress-Induced Hematopoiesis. Cell Stem Cell 14, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.