

Abstract
For over a century, the structures and reactivities of strained organic compounds have captivated the chemical community. Whereas triple-bond-containing strained intermediates have been well studied, cyclic allenes have received far less attention. Additionally, studies of cyclic allenes that bear heteroatoms in the ring are scarce. We report an experimental and computational study of azacyclic allenes, which features syntheses of stable allene precursors, the mild generation and Diels–Alder trapping of the desired cyclic allenes, and explanations of the observed regio- and diastereoselectivities. Furthermore, we show that stereochemical information can be transferred from an enantioenriched silyl triflate starting material to a Diels–Alder cycloadduct by way of a stereochemically defined azacyclic allene intermediate. These studies demonstrate that heteroatom-containing cyclic allenes, despite previously being overlooked as valuable synthetic intermediates, may be harnessed for the construction of complex molecular scaffolds bearing multiple stereogenic centres.
The chemistry of strained organic compounds has long fascinated the scientific community. Despite once being only scientific curiosities, small rings containing triple bonds can now be used in a host of applications. Breakthroughs in this area include the synthetic chemistry of benzyne (1) and cyclohexyne (2) (Fig. 1a), both of which were once controversial species1. So far, both have been utilized in a variety of synthetic applications spanning ligand synthesis, natural product synthesis, agrochemistry and materials science2–4.
Fig. 1 |. Survey of strained cyclic intermediates and focus of present study.

a, Cyclic alkynes such as benzyne (1) and cyclohexyne (2) were once avoided, but have now been used in many synthetic applications, including ligand synthesis, natural product synthesis, agrochemistry and materials science. 1,2-Cyclohexadiene (3) has been studied computationally and evaluated in cycloaddition reactions since its discovery in 1966, but is underutilized in chemical synthesis. b, Heterocyclic allenes are even less utilized in synthetic applications. Seminal examples of previously generated heterocyclic allenes include compounds 4 –8. c, The present study focuses on the generation and trapping of azacyclic allenes (9) in Diels–Alder and other cycloadditions to furnish products 11 or 12, where A is a carbon, oxygen or nitrogen atom.
Whereas strained cyclic intermediates possessing triple bonds have gained tremendous popularity, the corresponding chemistry of cyclic allenes has remained relatively underdeveloped. The parent allene, 1,2-cyclohexadiene (3, Fig. 1a), was first accessed by Wittig in 19665. Over the subsequent five decades, the field of cyclic allene chemistry has been largely driven by theoretical studies of allene structure and chirality6–11, in addition to studies of cycloaddition mechanisms12. With regard to experiments, various methods to generate cyclic allenes have been developed11,13,14, and subsequently used in cycloaddition methodologies of 1,2-cyclohexadienes15–19. Taken together, these studies suggest the potential synthetic utility of cyclic allene intermediates.
A largely untapped branch of cyclic allene chemistry involves those species that contain a heteroatom. Such reactive intermediates, despite not being used commonly in synthetic chemistry today, were first studied in 1976 (Fig. 1b). In a seminal discovery, Murata and co-workers proposed to have unintentionally accessed cyclic allene 4, an isomer of benzoxepine, using a Doering–Moore–Scattebøl (DMS) ring expansion of a dihalocyclopropane precursor20. Despite the harsh conditions required for this rearrangement (that is, organolithium reagents), a [2+2] cycloadduct was isolated in 33% yield, thus fuelling their hypothesis for the intermediacy of a heterocyclic allene. Led by efforts from the Christl laboratory, reports of the generation of several additional heterocyclic allenes were made in the intervening years, including six-membered cyclic allenes 5–8. Oxacyclic allenes 5 and 6 were first reported by Christl, also using the DMS approach21,22. Subsequently, the groups of Schlosser23 and Caubère24 demonstrated that cyclic allene 6 could also be obtained by the base-mediated dehydrohalogenation of a vinylhalide precursor. With regard to azacyclic allenes, which most closely resemble the chemistry described herein, the generation of 1-aza-3,4-cyclohexadiene (7) by Christl represents a key discovery in the field25. Also generated by the methyllithium-promoted DMS rearrangement, this azacyclic allene underwent Diels–Alder cycloadditions and [2+2] cycloadditions, albeit in modest synthetic efficiency. More recently, efforts were focused on the isomeric 1-aza-2,3-cyclohexadiene. Although this was found to be unstable, N-borylation permitted access to azacyclic allene 8 26. Additional discoveries in this field include the synthesis of a cephalosporinderived cyclic allene formation by Elliott and co-workers27–29 and several studies pertaining to heterocyclic isoarenes7,30–36.
With the aforementioned advances in mind, we sought to provide an advance that would enable the field of heterocyclic allene chemistry to enter mainstream synthesis. Notably, all previous examples of heterocyclic allene generation require harsh, strongly basic reaction conditions and, as a consequence, often suffer from modest reaction yields. Additionally, no studies of functionalized heterocyclic allene precursors, either for the sake of synthetic utility or for the investigation of reactivity and selectivity, have been reported. As such, we sought to prepare compounds of the type 9 (Fig. 1c). If intermediates 9 could be generated under mild conditions, they would provide a valuable tool to assemble stereochemically complex derivatives of the medicinally privileged piperidine scaffold. Functionalized piperidines are the most common heterocycle seen in medicines37, so new strategies for their synthesis are highly sought. Moreover, the study of substituted azacyclic allenes would provide opportunities to understand and modulate various aspects of regio- and stereoselectivities using a blend of computations and experiments. Computational studies of azacyclic allenes 9 have not been reported.
We now report (1) the syntheses of several silyl triflate precursors to azacyclic allene intermediates; (2) the mild generation and Diels–Alder trapping of the desired cyclic allenes (9 + 10 → 11 + 12, Fig. 1c) to give decorated piperidine products, including some bearing quaternary stereocentres; (3) the first examples of [3+ 2] cycloadditions of any heterocyclic allene; (4) computational studies, including application of the distortion–interaction model, to explain experimentally observed selectivities. Moreover, we demonstrate that stereochemical information can be transferred from an enantioenriched silyl triflate starting material to Diels–Alder cycloadducts by way of a stereochemically defined azacyclic allene intermediate.
Results and discussion
Computational analysis of azacyclic allene structures.
Figure 2 presents a comparison of linear allene 13 and azacyclic allene 14. was ω B97XD deemed the most appropriate density functional theory (DFT) method for the cycloaddition studies (described in the ‘DFT calculations’ section), and was used to optimize the ground-state geometries of interest. The linear allene C=C bond length is 1.31 Å in 13. As expected, the allene π orbitals are orthogonal but degenerate. In comparison, the C= C bond length of azacyclic allene 14 is slightly longer (1.32 Å), with the internal angle at the central allene carbon being 133°, rather than 180°, as a result of the ring constraint. The allene π orbitals are no longer perfectly orthogonal or degenerate, and the allene CH bonds are twisted out of plane, by 42° and 37°, respectively. Azacyclic allene 14 possesses 27.3 kcal mol−1 of strain energy (Supplementary Part II-C) due to its distortion from linearity, an attribute that facilitates cycloaddition reactions.
Fig. 2 |. Comparison of geometry-optimized structures of allenes 13 and 14 (ωB97XD/6–31G(d)).

a, The π orbitals of 13 are orthogonal and the compound has a linear geometry. b, Azacyclic allene 14 is bent and possesses an internal angle of 133° at the central allene carbon. For the geometryoptimized structure of compound 14, the CO2Me is omitted for clarity.
Syntheses of azacyclic allene precursors.
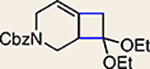
With the ultimate aim of accessing azacyclic allenes 25–28, we developed a divergent synthetic route to silyl triflates 19, 20, 23 and 24, stemming from common intermediate 16 (Fig. 3). 4-Methoxypyridine (15) was elaborated to compound 16 using a straightforward sequence, analogous to that used to prepare the known trimethylsilyl derivative38. Subsequent 1,4-reduction proceeded smoothly using l-selectride to give an intermediate lithium enolate. Whereas acidic workup furnished silyl ketone 17, trapping with methyl iodide led to α-methylated ketone 18. Subsequent triflation of 17 and 18 delivered silyl triflates 19 and 20, respectively. Alternatively, intermediates 17 and 18 could be elaborated to esters 21 and 22, respectively, following deprotonation and quenching with Mander’s reagent. Enols 21 and 22 were readily triflated using sodium hydride and triflic anhydride, thus furnishing the corresponding silyl triflates 23 and 24. It should be noted that, by preparing four silyl triflates, we hoped to probe substituent effects on regio- and diastereoselectivities. Silyl triflate 19 would serve as a precursor to the parent azacyclic allene 25. Allenes 26 and 27, optimistically accessible from silyl triflates 20 and 23, respectively, each bear one additional substituent relative to the parent allene 25, but with varying electronic properties (methyl versus ester). Finally, fully substituted allene 28 would be derived from silyl triflate 24.
Fig. 3 |. Syntheses of silyl triflates 19, 20, 23 and 24.

Common intermediate 16 was accessed in two steps from 15 and carried forward to ketones 17 and 18. A deprotonation/triflation sequence then delivered 19 and 20, whereas a two-step sequence involving α-carboxymethylation and triflation afforded silyl triflates 23 and 24. Silyl triflates 19, 20, 23 and 24 would serve as precursors to allenes 25–28, respectively. Colours are used to highlight the key substituents that influence regioselectivities in cycloaddition reactions (yellow = electron-donating methyl group; blue = electron-deficient ester group). Cbz, carboxybenzyl; LDA, lithium diisopropylamide; KHMDS, potassium bis(trimethylsilyl)amide; Comins’ reagent, N-(5-chloro-2-pyridyl)bis(trifluoromethanesulfonimide); Mander’s reagent, methyl cyanoformate; Tf2O, trifluoromethanesulfonic anhydride.
Scope of methodology.
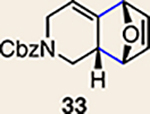
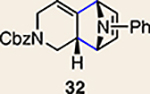
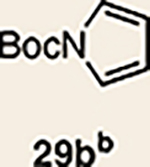
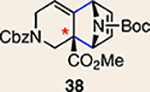
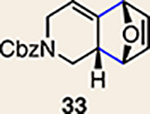
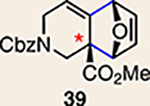
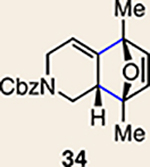
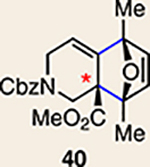
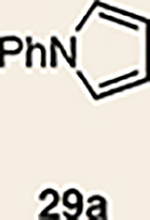
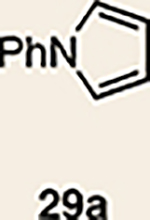
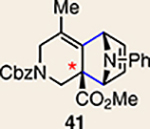
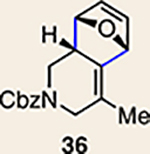
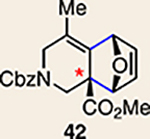
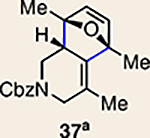
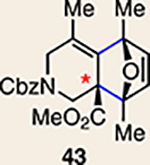
With the necessary silyl triflates in hand, we sought to generate and trap azacyclic allenes 25–28 in Diels–Alder reactions (Table 1). This idea was realized by implementing a simple experimental protocol, wherein CsF was added to a mixture of a given silyl triflate and a particular diene in acetonitrile at ambient temperature, to ultimately afford a variety of cycloadducts. For example, we found that unsubstituted allene 25 could be generated and trapped by N-phenylpyrrole (29a) to furnish [2.2.1]-azabicycle 32 in 77% yield and 4.7:1 d.r. (Table 1, entry 1). Similarly, oxa-bicycles 33 and 34 were accessed by trapping of 25 with furans 30 and 31, respectively (entries 2 and 3). In all cases, the endo products were formed preferentially. We also generated methyl-substituted allene 26, which was found to undergo Diels–Alder trapping with 29a, 30 and 31 (entries 4–6, respectively). In each case, cycloaddition occurred regioselectively on the olefin positioned distal to the methyl group, again with notable levels of diastereoselectivity. By switching to allene 27, bearing an electron-withdrawing ester in place of the methyl group in 26, we observed a switch in regioselectivity. As shown by the formation of 38–40, cycloaddition occurred on the olefin proximal to the ester substituent (entries 7–9, respectively). It should be noted that the trapping experiments of allene 27 allow for the formation of quaternary centres in a controlled manner. Finally, we examined cycloadditions between fully substituted allene 28 and dienes 29a, 30 and 31. As seen in entries 10–12, the cycloadditions occurred smoothly, giving rise to cycloadducts 41–43 in 93–95% yield and > 20:1 d.r. Consistent with the regioselectivites seen in the cases of allenes 26 and 27, cycloadditions involving allene 28 occur proximal to the ester and distal to the methyl group. Moreover, the trapping of 28 in Diels–Alder cycloadditions allows for the efficient assembly of highly functionalized piperidine scaffolds bearing quaternary stereocentres.
Table 1.
Scope of Diels-Alder cycloadditions of azacyclic aliene intermediates 25–28
| Entry | Allene | Diene | Products (yield, d.r.) | Entry | Allene | Diene | Products (yield, d.r.) |
|---|---|---|---|---|---|---|---|
| 1 | 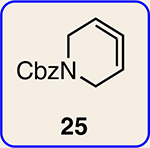 |
 |
 |
7 | 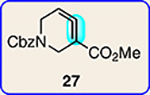 |
 |
 |
| 2 | 25 | 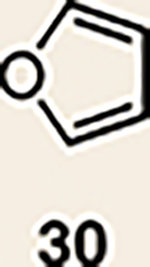 |
 |
8 | 27 | 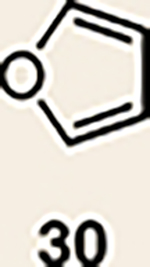 |
 |
| 3 | 25 | 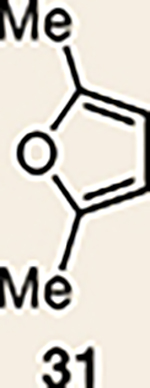 |
 |
9 | 27 | 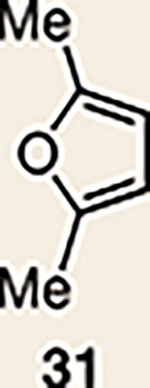 |
 |
| 4 | 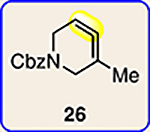 |
 |
 |
10 | 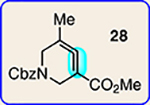 |
 |
 |
| 5 | 26 | 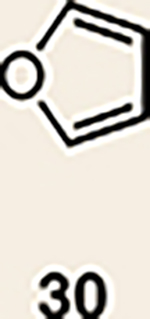 |
 |
11 | 28 | 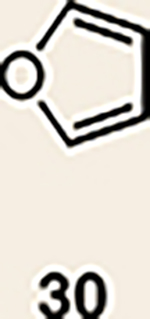 |
 |
| 6 | 26 | 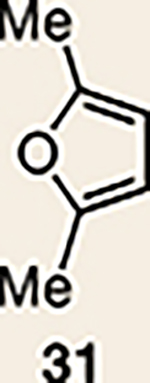 |
 |
12 | 28 | 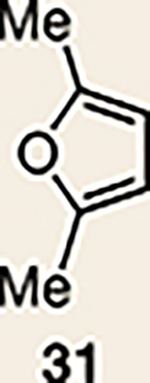 |
 |
Conditions unless otherwise stated: silyl triflate substrate (1.0equiv.), diene (5.0–10.0equiv.), CsF (5.0equiv.), acetonitrile (0.1 M) at 23°C. Yields shown reflect the average of two isolation experiments.
The regioisomer of 37 was also observed (∼20% yield), resulting from endo cycloaddition on the more substituted olefin of allene 26.
Pyrrole 29b was used in place of pyrrole 29a, as the cycloaddition of 29a with 27 proceeded in low yield for reasons that are not presently understood. Highlighted bonds indicate which double bond of the cyclic allene undergoes cycloaddition, with colours corresponding to the methyl and ester group substituents depicted in Fig. 3. Cbz, carboxybenzyl; A, methyl group or hydrogen atom; X, oxygen atom, NBoc or NPh; Boc, ferf-butyloxy carbonyl.
Several features of the allene generation and trapping experiments should be emphasized. (1) In the absence of CsF, no reaction occurs, even at elevated temperature, which is suggestive of allene formation (as opposed to Diels–Alder cycloaddition, followed by silyl triflate elimination). (2) All reactions take place at room temperature, thus allowing for azacyclic allene generation under exceptionally mild reaction conditions. (3) For all reactions with the exception of the formation of 37, the opposite regioisomer was not observed. (4) This methodology provides a facile, metal-free means to access decorated piperidines, by formation of two new bonds and three stereocentres, with high levels of diastereoselectivity. (5) The substituents on the azacyclic allene have a profound effect on modulating regioselectivities in the cycloaddition reactions.
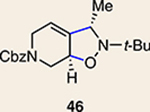
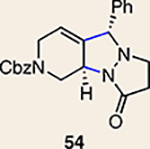
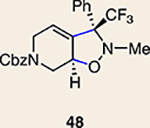
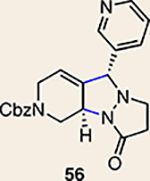
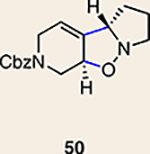
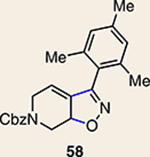
Although not the focus of the current study, it should be noted that azacyclic allene 25 could also be trapped in [3+2] and [2+2] cycloadditions (Table 2). For example, an assortment of nitrones can be employed as 1,3-dipoles. Whereas earlier studies of 1,2-cyclohexadiene nitrone cycloadditions were performed optimally at elevated temperatures to accelerate reaction rates18, in the case of azacyclic allene 25, reactions proceeded efficiently at ambient temperature (see Supplementary Information, Part I, Section C for reaction times). Moreover, comparable selectivities were seen in control experiments performed at 50 °C. Use of aldehyde-derived nitrone 45 provided isoxazolidine 46 (Table 2, entry 1), whereas trapping with ketone-derived nitrone 47 furnished 48, bearing a tertiary centre and trifluoromethyl group (entry 2). When cyclic nitrones 49 and 51 were utilized, tri- and tetracyclic products 50 and 52 were obtained, respectively (entries 3 and 4). Additionally, trapping of the allene intermediate with azomethine imines19 53 and 55 gave the corresponding pyrazolidine products 54 and 56 (entries 5 and 6). Of note, 56 contains three distinct nitrogen-containing heterocycles: a piperidine, a pyrazolidine and a pyridine ring. Nitrile oxide19 57 was also employed as a trapping agent and gave rise to isoxazoline 58 in 81% yield. These reactions (entries 1–7) represent the first [3+ 2] cycloadditions of heterocyclic allenes. Finally, we attempted to utilize olefin 59 in a [2+2] cycloaddition. This reaction proceeded smoothly to deliver cyclobutane derivative 60 in 78% yield as a single isomer. Collectively, the trapping experiments of azacyclic allenes shown in Tables 1 and 2 demonstrate their utility in the rapid generation of stereochemically rich hetererocycles.
Table 2.
[3+2] and [2+2] cycloadditions of azacyclic aliene intermediate 25
| Entry | Trapping agent | Products (yield, d.r.) | Entry | Trapping agent | Products (yield, d.r.) |
|---|---|---|---|---|---|
| 1 | 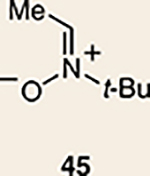 |
 |
5 | 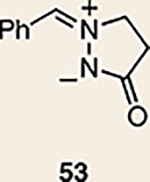 |
 |
| 2 | 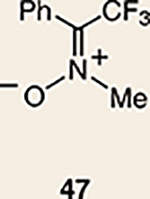 |
 |
6 | 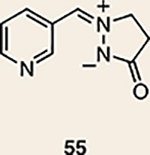 |
 |
| 3 | 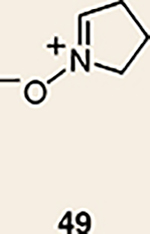 |
 |
7 | 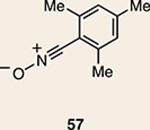 |
 |
| 4 | 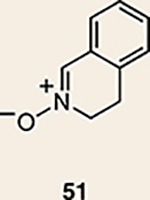 |
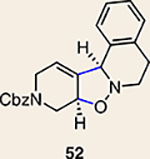 |
8 |  |
 |
Conditions unless otherwise stated: silyl triflate 19 (1.0equiv.), trapping agent (1.2–5.0equiv.), CsF (5.0equiv.), acetonitrile (0.1 M) at 23°C. Yields shown reflect the average of two isolation experiments. Cbz, carboxybenzyl; A, carbon substituent; B, carbon, nitrogen or oxygen substituent.
DFT calculations.
To learn about the nature of the mechanism (concerted or stepwise) and to understand how the methyl and ester groups guide selectivities, DFT calculations were performed for the Diels–Alder reactions of allenes 61 and 62 (carbomethoxy was used as a surrogate for the larger carboxybenzyl (Cbz) group to simplify computations) reacting with furan (30) (Fig. 4). We considered pathways leading to all regio- and stereoisomers via three possible scenarios: concerted cycloaddition, stepwise zwitterionic cycloaddition and stepwise diradical cycloaddition. B3LYP was previously used to study the mechanism of Diels–Alder cycloadditions with 1,2-cyclohexadiene. However, Brinck and co-workers have noted a tendency for B3LYP to overestimate the asynchronicity of highly asynchronous transition states and to ultimately favour stepwise pathways39; their recent benchmark study demonstrates that ω B97XD performs better than B3LYP for highly asynchronous Diels–Alder cycloadditions and has been shown to reproduce transition state geometries obtained using a high accuracy method, CCSD(T). A variety of methods and basis sets were evaluated for this study, and ω B97XD/6–311+ G(d,p)/ SMD(MeCN) was selected as the computational method of choice (Supplementary Part II-B).
Fig. 4 |. Computations provide insight into regio- and diastereoselectivities.

a, Reaction of furan (30) with allenes 61 and 62 to give endo products was examined computationally (ω B97XD/6–311+ G(d,p)/SMD(MeCN)). The ΔG‡ and ΔE‡ values determined were consistent with experimental regiochemical observations. A distortion/ interaction analysis was performed, where the ΔE‡ was broken down into the ΔE‡ of distortion and the ΔE‡ of interaction. The ΔE‡ of interaction was found to be a significant contributor to the difference in ΔE‡ values for both allenes 61 and 62. b, Optimized endo transition states leading to the observed major regioisomers (ωB97XD/6–31G(d)). c, Orbital coefficients for 61 and 62 and depiction of LUMOs (HF/6–31G). Highlighted bonds indicate which double bond of the cyclic allene undergoes cycloaddition, with colours corresponding to the methyl and ester group substituents depicted in Fig. 3. Pink shading emphasizes that the magnitude of the ΔΔE‡ of interaction is significant and leads to the observed regiochemistry.
Our computations reveal that pathways leading to endo products are most energetically favourable, consistent with experimental results, and proceed in an asynchronous concerted fashion. Key results are summarized in Fig. 4a (see Supplementary Part II-D and E for analyses of pathways leading to exo products). The ΔΔG‡ for the two competing endo pathways for the Diels–Alder reaction of methyl-substituted allene 61 and furan (30) was calculated to be –1.5 kcal mol−1, with the cycloaddition occurring on the olefin distal to the methyl group. This correlates well with the experimental result shown in Table 1 (entry 5). To understand the origin of regioselectivity, we performed a distortion/interaction activation strain analysis40. Thus, ΔE‡ was calculated for the endo pathways leading to the two possible regioisomers, and ΔE‡ was further broken down into its components, the distortion energy (ΔEdist‡, an energetic cost associated with the structural distortion of the reactants) and the interaction energy (ΔEint‡‡, an energetic benefit resulting from favourable orbital interactions). Whereas the ΔEdist‡ was found to be comparable for the transition states leading to the major and minor regioisomers (ΔΔEdist‡ = –0.4 kcal mol−1), ΔEint‡ was more favourable in the transition state leading to the major regioisomer (ΔΔEint‡ = –1.3 kcal mol−1). This more favourable ΔEint‡ results from more stabilizing orbital interactions between the lowest unoccupied molecular orbital (LUMO) of allene 61 and the highest occupied molecular orbital (HOMO) of furan (30) in TS1 (Fig. 4b), which leads to the major observed regioisomer. That is, the LUMO is more concentrated on the less substituted double bond of the allene.
The corresponding analysis was performed for the competing endo pathways for the Diels–Alder reaction of ester-substituted allene 62 and furan (30). ΔΔG‡ and ΔΔE‡ were calculated to be –4.9 and –4.7 kcal mol−1, respectively, with the cycloaddition occurring on the olefin proximal to the ester, consistent with experimental findings (Table 1, entry 8). ΔEdist‡ was again found to be comparable for the transition states leading to the major and minor regioisomers (ΔΔEdist‡ = 0.7 kcal mol−1, favouring the minor pathway), but ΔEint‡ was much more favourable in the transition state leading to the major regioisomer (ΔΔEint‡ = –5.4 kcal mol−1). More favourable orbital interactions in TS2 (Fig. 4b) occur following interaction with the LUMO, now concentrated on the ester-substituted double bond. As shown in Fig. 4b, the major pathway (TS2) is highly asynchronous, with initial bond formation occurring on the central allene carbon.
To better understand the favourable electronic interactions that led to the observed regioselectivities, Hartree-Fock molecular orbitals were calculated for allenes 61 and 62. The aforementioned helical LUMOs of 61 and 62 are depicted (Fig. 4c), along with the pz orbital coefficients for both termini of the allene, which project in the direction at which bond formation occurs. In the case of 61, the less substituted terminus possesses a larger coefficient in the LUMO, which correlates to the regioselectivity we observe in experiments involving Cbz derivative 26. On the other hand, for ester 62, the larger orbital coefficient resides on the more substituted allene terminus, which is also consistent with our experimental results involving allene 27. The slight charge separation in the transition state also contributes to the better stabilization of those transition states leading to the observed products.
Enantioenriched silyl triflates and transfer-of-chirality studies.
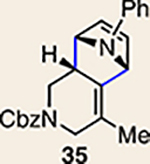
We were also intrigued by the idea of accessing an enantioenriched cycloadduct by intercepting an enantioenriched heterocyclic allene intermediate. Key precedent for this idea stems from the studies of Christl and Engels, who accessed a highly enantioenriched cycloadduct from in situ generated 1-phenyl-1,2-cyclohexadiene using a low-temperature Skattebøl rearrangement, albeit in low yield15,41. As an alternative approach, we hypothesized that the silyl triflate precursors to heterocyclic allenes could be utilized in enantioenriched form. To this end, we separated gram quantities of the enantiomers of ketones 17 and 18 and elaborated them to enantioenriched silyl triflates 20 and 23, respectively. The chiral separations were achieved using preparative chiral supercritical fluid chromatography (SFC), either by batch processing or direct purification of gram quantities, an approach also used for gram-scale purifications in industrial settings42. As shown in Fig. 5, we have found that the success of this strategy is highly dependent on the nature of the heterocyclic allene substituent. When employing optically enriched ester-containing silyl triflate (+)-23 in the Diels–Alder cycloaddition with pyrrole 29b or furans 30 or 31, adducts 38–40 were obtained as expected, but in racemic form (Fig. 5a). In contrast, when enantioenriched methyl-substituted silyl triflate (+)-20 was utilized in the corresponding Diels–Alder reactions, the expected cycloadducts (+)-35, (–)-36 and (+)-37 were obtained in 81–98% e.e. (Fig. 5b). As noted earlier in Table 1, cycloadduct 37 is obtained as a mixture of regioisomers in the cycloaddition between allene 32 and dimethylfuran (31). Interestingly, the formation of the minor regioisomer in this reaction also proceeded without significant stereochemical erosion to give the corresponding cycloadduct (+)-63 bearing vicinal tetrasubstituted stereocentres in 97% e.e.
Fig. 5 |. Attempted transfer of stereochemical information from silyl triflates to cycloadducts via azacyclic allene intermediates.

a, Use of enantioenriched silyl triflate (+)-23 led to the anticipated bicycles 38–40, but in racemic form. b, In contrast, use of enantioenriched methyl-substituted silyl triflate (+)-20 furnished cycloadducts (+)-35, (–)-36 and (+)-37 with transfer of stereochemical information. c, The racemization barrier for allene 62 (ωB97XD/6–311+ G(d,p)/SMD(MeCN)) was calculated to be low, whereas the racemization barrier for allene 63 (ωB97XD/6–311+ G(d,p)/SMD(MeCN)) was calculated to be high, thus accounting for the observed enantiospecific cycloadditions. Colours are used to highlight the key substituents that influence regioselectivities in cycloaddition reactions (yellow = electron-donating methyl group; blue = electron-deficient ester group). Cbz, carboxybenzyl; A, methyl group or hydrogen atom; X, oxygen atom, NBoc or NPh; Boc, tert-butyloxy carbonyl.
Calculations were performed to assess the racemization barriers for allenes 62 and 61 to determine whether the substituent had an effect on the barrier for racemization of the chiral allenoate intermediate generated from the corresponding silyl triflate. Previous theoretical studies on the racemization of 1,2-cyclohexadiene (3) by Johnson and co-workers showed that the racemization of 1,2-cyclohexadiene (3) occurs via a diradical transition state6. As shown in Fig. 5c, the racemization barrier for estercontaining allene 62 was determined to be only 14.1 kcal mol−1 and occurs via a diradical transition state. In the case of methylsubstituted allene 61, the barrier for racemization was calculated to be 16.4 kcal mol−1, which is consistent with faster racemization of 62 than 61, a result of greater stabilization of the diradical transition state from 62. As such, we hypothesize that allene 62 undergoes racemization faster than cycloaddition, thus accounting for the formation of racemic 38–40 (Fig. 5a). With racemization being disfavourable, the Diels–Alder reactions to give 35 –37 proceed with significant transfer of stereochemical information (Fig. 5b). At present, we cannot rule out the possibility of racemization occurring after desilylation, but before allene formation. Nonetheless, these results demonstrate that heterocyclic allenes may be employed as building blocks for enantioenriched products, while further showcasing the key role of substituents in governing selectivities in reactions of heterocyclic allenes.
We have performed an experimental and computational study of unusual azacyclic allene intermediates. Our study includes syntheses of stable allene precursors, which in turn can be used to access the desired azacyclic allenes in situ under mild reaction conditions. Diels–Alder trapping of the allene intermediates provides an array of functionalized piperidines bearing multiple stereogenic centres, including quaternary centres in some cases. DFT studies show that computations correctly predict the observed diastereoselectivities, with cycloadditions occurring through concerted asynchronous endo transition states. A detailed distortion/interaction analysis explains the origins of the observed regiochemistry of the Diels–Alder cycloadditions. Finally, by assessing enantioenriched silyl triflates, we found that stereochemical information can be transferred from a silyl triflate starting material to Diels–Alder cycloadducts, provided the allene is appropriately substituted.
The structure and reactivity of strained organic compounds has intrigued the chemical community for over a century. However, cyclic allenes have received relatively little attention, especially in comparison to cyclic alkynes. Our present study demonstrates that strained azacyclic allenes, although largely neglected, serve as valuable building blocks for the construction of complex molecular scaffolds bearing multiple stereogenic centres.
Supplementary Material
Acknowledgments
The authors acknowledge the NIH-NIGMS (R01 GM090007 to N.K.G., R01 GM109078 to K.N.H. and F32 GM122245 to E.R.D.), the National Science Foundation (NSF; CHE-1361104 to K.N.H. and DGE-1144087 to M.M.Y.), the University of California, Los Angeles, the UCLA Cota Robles Fellowship Program (M.R.) and the Chemistry–Biology Interface training program (J.S.B., USPHS National Research Service Award 5T32GM008496–20) for financial support. Pier Champagne is acknowledged for computational assistance. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631). Computations were performed with resources made available from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575), as well as the UCLA Institute of Digital Research and Education (IDRE).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41557-018-0080-1.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims inpublished maps and institutional affiliations.
Data availability. Data supporting the findings of this study are available in the Supplementary Information or from the corresponding author upon request. The Supplementary Information contains full details on the synthesis and characterization of compounds, as well as details of computational studies.
References
- 1.Heaney H The benzyne and related intermediates. Chem. Rev 62, 81–97 (1962). [Google Scholar]
- 2.Tadross PM & Stoltz BM A comprehensive history of arynes in natural product total synthesis. Chem. Rev 112, 3550–3577 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Dubrovskiy AV, Markina NA & Larock RC Use of benzynes for the synthesis of heterocycles. Org. Biomol. Chem 11, 191–218 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Goetz AE, Shah TK & Garg NK Pyridynes and indolynes as building blocks for functionalized heterocycles and natural products. Chem. Commun 51, 34–45 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Wittig G & Fritze P On the intermediate occurrence of 1,2-cyclohexadiene. Angew. Chem. Int Ed 5, 846 (1966). [Google Scholar]
- 6.Angus RO, Schmidt MW & Johnson RP Small-ring cyclic cumulenes: theoretical studies of the structure and barrier to inversion in cyclic allenes. J. Am. Chem. Soc 107, 532–537 (1985). [Google Scholar]
- 7.Engels B, Schöneboom JC, Münster AF, Groetsch S & Christl M Computational assessment of electronic structures of cyclohexa-1,2,4-triene, 1-oxacyclohexa-2,3,5-triene (3δ 2-pyran), their benzo derivatives, and cyclohexa-1,2-diene. An experimental approach to 3δ2-pyran. J. Am. Chem. Soc 124, 287–297 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Hänninen MM, Peuronen A & Tuononen HM Do extremely bent allenes exist? Chem. Eur. J 15, 7287–7291 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Daoust KJ et al. Strain estimates for small-ring cyclic allenes and butatrienes. J. Org. Chem 71, 5708–5714 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Johnson RP Strained cyclic cumulenes. Chem. Rev 89, 1111–1124 (1989). [Google Scholar]
- 11.Wentrup C, Gross G, Maquestiau A & Flammang R 1,2-Cyclohexadiene. Angew. Chem. Int. Ed. Engl 22, 542–543 (1983). [Google Scholar]
- 12.Nendel M, Tolbert LM, Herring LE, Islam MN & Houk KN Strained allenes as dienophiles in the Diels–Alder reaction: an experimental and computational study. J. Org. Chem 64, 976–983 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Moore WR & Moser WR The reaction of 6,6-dibromobicyclo[3.1.0]hexane with methyllithium. Evidence for the generation of 1,2-cyclohexadiene and 2,2′ -dicyclohexenylene. J. Am. Chem. Soc 92, 5469–5474 (1970). [Google Scholar]
- 14.Quintana I, Peña D, Pérez D & Guitián E Generation and reactivity of 1,2-cyclohexadiene under mild reaction conditions. Eur. J. Org. Chem 2009, 5519–5524 (2009). [Google Scholar]
- 15.Christl M, Fischer H, Arnone M & Engels B 1-Phenyl-1,2-cyclohexadiene: astoundingly high enantioselectivities on generation in a Doering–Moore–Skattebøl reaction and interception by activated olefins. Chem. Eur. J 15, 11266–11272 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Bottini AT, Hilton LL & Plott J Relative reactivities of 1,2-cyclohexadiene with conjugated dienes and styrene. Tetrahedron 31, 1997–2001 (1975). [Google Scholar]
- 17.Bottini AT, Corson FP, Fitzgerald R & Frost KA II Reactions of 1-halocyclohexenes and methyl substituted 1-halocyclohexenes with potassium t-butoxide. Tetrahedron 28, 4883–4904 (1972). [Google Scholar]
- 18.Barber JS et al. Nitrone cycloadditions of 1,2-cyclohexadiene. J. Am. Chem. Soc 138, 2512–2515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lofstrand VA & West FG Efficient trapping of 1,2-cyclohexadienes with 1,3-dipoles. Chem. Eur. J 22, 10763–10767 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Uyegaki M, Ito S, Sugihara Y & Murata I 1-Benzoxepin and its valence isomers, 4,5-benz-3-oxatricyclo[4.1.0.02,7]heptene and 3,4-benz-2-oxabicyclo[3.2.1]hepta-3,6-diene. Tetrahedron Lett 49, 4473–4476 (1976). [Google Scholar]
- 21.Schreck M & Christl M Generation and interception of 1-oxo-3,4-cyclohexadiene. Angew. Chem. Int. Ed 26, 690–692 (1987). [Google Scholar]
- 22.Christl M & Braun M Friesetzung und Abfangreaktionen von 1-oxa-2,3-cyclohexadien. Chem. Ber 122, 1939–1946 (1989). [Google Scholar]
- 23.Ruzziconi R, Naruse Y & Schlosser M 1-Oxa-2,3-cyclohexadiene (‘2H-isopyran’): a strained heterocyclic allene undergoing cycloaddition reactions with characteristic typo-, regio-, and stereoselectivities. Tetrahedron 47, 4603–4610 (1991). [Google Scholar]
- 24.Jamart-Grégoire B, Mercier-Girardot S, Ianelli S, Nardelli M & Caubère P Aggregative activation and heterocyclic chemistry. II Nucleophilic condensation of ketone enolates on dehydrodihydropyran generated by complex bases. Tetrahedron 51, 1973–1984 (1995). [Google Scholar]
- 25.Christl M, Braun M, Wolz E & Wagner W 1-Phenyl-1-aza-3,4-cyclohexadien, das erste Isodihydropyridin: ertzeugung und abfangreaktionen. Chem. Ber 127, 1137–1142 (1994). [Google Scholar]
- 26.Drinkuth S, Groetsch S, Peters E, Peters K & Christl M 1-Methyl-1-azacyclohexa-2,3-diene(N–B)borane—generation and interception of an unsymmetrical isodihydropyridine. Eur. J. Org. Chem 14, 2665–2670 (2001). [Google Scholar]
- 27.Elliott RL et al. Cycloadditions of cephalosporins. A comprehensive study of the reaction of cephalosporin triflates with olefins, acetylenes, and dienes to form [2+2] and [4+ 2] adducts. J. Org. Chem 62, 4998–5016 (1997). [Google Scholar]
- 28.Elliott RL, Takle AK, Tyler JW & White J Cycloadditions of cephalosporins. A general synthesis of novel 2,3-fused cyclobutane and cyclobutene cephems. J. Org. Chem 58, 6954–6955 (1993). [Google Scholar]
- 29.Elliott RL et al. Cycloadditions of cephalosporins. The formation of [4+ 2] adduct with 5-membered heterocycles. J. Org. Chem 59, 1606–1607 (1994). [Google Scholar]
- 30.Musch PW, Scheidel D & Engles B Comprehensive model for the electronic structures of 1,2,4-cyclohexatriene and related compounds. J. Phys. Chem. A 107, 11223–11230 (2003). [Google Scholar]
- 31.Emanuel CJ & Shelvin PB Mechanism of the reaction of atomic carbon with pyrrole. Evidence for the intermediacy of a novel dehydropyridinium ylide. J. Am. Chem. Soc 116, 5991–5992 (1994). [Google Scholar]
- 32.Pan W & Shelvin PB The chemistry of the N-methyl-3-dehydropyridinium ylid. J. Am. Chem. Soc 119, 5091–5094 (1997). [Google Scholar]
- 33.Pan W, Balci M & Shelvin PB Thiacyclohexatriene–thiopheneylcarbene rearrangement. A sufur analog of the cycloheptatetraene–phenylcarbene rearrangement. J. Am. Chem. Soc 119, 5035–5036 (1997). [Google Scholar]
- 34.Wang J & Sheridan RS A singlet aryl-CF3 carbene: 2-benzothienyl(trifluoromethyl)carbene and interconversion with a strained cyclic allene. Org. Lett 9, 3177–3180 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Schöneboom JC, Groetsch S, Christl M & Engles B Computational assessment of the electronic structure of 1-azacyclohexa-2,3,5-triene(3δ2-1H-pyridine) and its benzo derivative (3δ2-1H-quinoline) as well as generation and interception of 1-methyl-3δ2-1H-quinoline. Chem. Eur. J 9, 4641–4649 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Christl M & Drinkuth S 3δ2-Chromene (2,3-didehydro-2H-1-benzopyran): generation and interception. Eur. J. Org. Chem 2, 237–241 (1998). [Google Scholar]
- 37.Vitaku E, Smith DT & Njardarson JT Analysis of structural diversity, substitution pattern, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 57, 10257–10274 (2014). [DOI] [PubMed] [Google Scholar]
- 38.McMahon TC et al. Generation and regioselective trapping of a 3,4-piperidyne for the synthesis of functionalized heterocycles. J. Am. Chem. Soc 137, 4082–4085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinck T & Linder M On the method-dependence of transition state asynchronicity in Diels–Alder reactions. Phys. Chem. Chem. Phys 15, 5108–5114 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Houk KN & Bickelhaupt FM Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem. Int. Ed 56, 10070–10086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christl M et al. The stereochemical course of the generation and interception of a six-membered cyclic allene: 3δ2-1H-napthalene (2,3-didehydro-1,2-dihydronaphthalene). Eur. J. Org. Chem 2006, 5045–5058 (2006). [Google Scholar]
- 42.Carry J-C, Brohan E, Perron S & Bardouillet P-E Chiral supercritical fluid chromatography in the preparation of enantiomerically pure (S)-(+)-tert-butyl-3-hydroxyazepane-1-carboxylate. Org. Process Res. Dev 17, 1568–1571 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.