

Abstract
Nonenzymatic catalytic substrates have been engineered using toehold-mediated DNA strand displacement, and their programmable applications range from medical diagnosis to molecular computation. However, the complexity, stability, scalability, and sensitivity of those systems are plagued by network leakage. A novel way to suppress leakage is to increase its energy barrier through four-way branch migration. Presented here, we designed multi-arm junction substrates that simultaneously exploit four-way branch migration, with a high-energy barrier to minimize leakage, and three-way branch migration, with a low-energy barrier to maximize catalysis. Original feed forward, autocatalytic, and cross-catalytic systems have been designed with polynomial and exponential amplification that exhibit the modularity of linear substrates and the stability of hairpin substrates, creating a new phase space for synthetic biologist, biotechnologist, and DNA nanotechnologists to explore. A key insight is that high-performing circuits can be engineered in the absence of intensive purification and/or extensive rounds of design optimization. Without adopting established leakage suppression techniques, the ratio of the catalytic rate constant to the leakage rate constant is more than 2 orders of magnitude greater than state-of-the-art linear and hairpin substrates. Our results demonstrate that multi-arm junctions have great potential to become central building blocks in dynamic DNA nanotechnology.
Graphical Abstract
INTRODUCTION
Toehold-mediated strand displacement1 accelerates DNA invasion reactions through branch migration using short single-stranded sequences, called toeholds. Owing to the predictable Watson−Crick binding of DNA,2 this elegant concept enables the control of reaction kinetics3,4 and is fundamental to the construction of dynamic DNA systems.5 One of the key components among them is nonenzymatic catalytic substrates,6,7 which are used for biomarker sensing,8 molecular computation,9–11 and triggered self-assembly.12,13 Two common catalytic substrates are hairpin substrates12,14–16 and linear substrates,10,11,17,18 both of which exist in metastable states prior to being triggered. Once triggered by an external catalyst that reduces the energy barrier, a conformational change proceeds from a metastable state to a lower energy state.
The greatest challenge for catalytic substrates is initiation of the reaction in the absence of a catalyst, known as leakage, which limits the engineering of more complex, scalable, and sensitive tools. Total leakage occurs because of an insufficient energy barrier between the metastable state and equilibrium and is classified as either initial leakage or asymptotic leakage.19 To address total leakage, select studies have introduced: (1) highly purified DNA strands,19 (2) the concept of availability and mutual availability,20 (3) DNA clamps,7,9,10,12 (4) DNA mismatches,20,21 and (5) locked nucleic acids (LNAs).22 Although these methods are effective at reducing leakage caused by breathing23 and/or defective DNA,24 they are repair kits for leaky substrates. It is more desirable to have a substrate whose intrinsic leakage is small in the presence of breathing and defective strands. Hence, a key insight here is the design of substrates that have an intrinsically higher energy barrier for the leakage pathway and a lower energy barrier for the catalytic pathway. Considering that all strand displacement, including leakage, proceeds through branch migration, we engineered energy barriers based on the branch migration process.
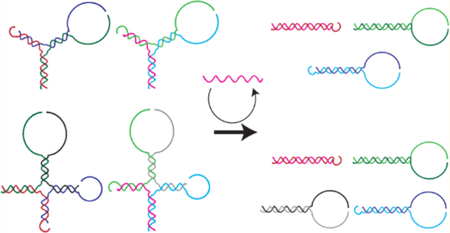
There are two classes of branch migration: three-way branch migration25 and four-way branch migration.26 Three-way branch migration occurs when a single-stranded oligonucleotide displaces a second strand in a duplex along a complementary region (Figure 1A). Four-way branch migration occurs when two duplexes exchange their hybridized strands along a complementary region (Figure 1B). An important difference between the two methods is the higher energy barrier for four-way branch migration versus three-way branch migration. This energy difference can be seen in their intuitive energy landscape (IEL)27 and results in 2−5 orders of magnitude slower reaction rates.28 The higher energy barrier of four-way branch migration will be due to the larger “sawtooth amplitude”27 of the branch migration steps, shown as the large difference between ΔGs3 (∼5 kcal/mol) and ΔGs4 (∼10 kcal/mol) (Figure 1A, B). Therefore, incorporating four-way branch migration, for leakage pathways, and three-way branch migration, for catalytic pathways, offers the potential for extremely low leakage rates and fast catalytic rates. To implement this strategy, the entropy-driven system by Zhang et al. (Figure 1C) was considered.17 This system uses a linear substrate, which is shown as a three-stranded DNA complex. Because of the condition for a single-stranded DNA to be the fuel strand, the leakage of the system occurs through three-way branch migration, resulting in a lower energy barrier for the leakage reaction. However, it is possible to make entropy-driven systems without a single-stranded DNA strand. The solution is the adoption of multi-arm junctions31 as catalytic substrates (Figure 1D). The upper panel uses three-arm junction substrates, and the lower panel uses four-arm junction substrates. In both cases, the catalytic reactions are driven by entropy gains, similar to linear substrates, while the leakage reactions proceed through four-way branch migration.
Figure 1.
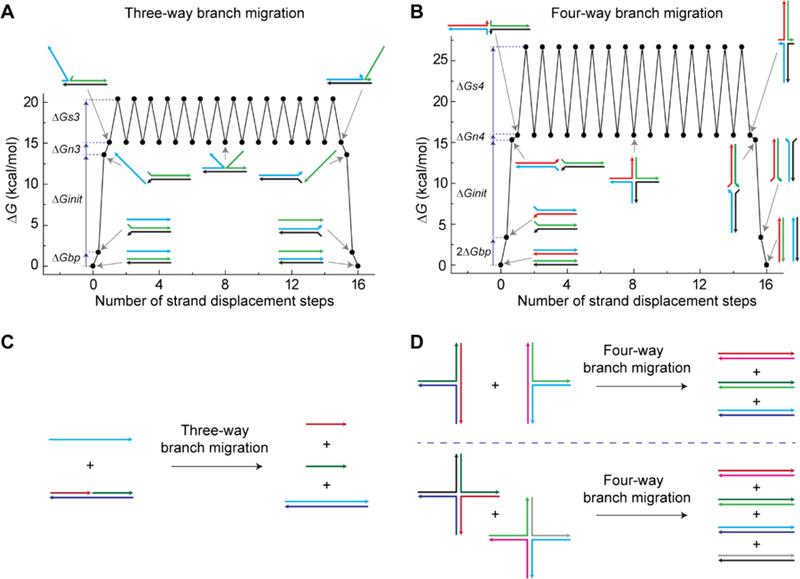
Design strategy of multi-arm junction substrates. DNA strands are shown as colored lines with arrowheads representing 3′ ends. (A) The IEL of a leakage through three-way branch migration, which was adapted from Srinivas et al.27 The first step is the free energy cost of breaking a base pair, ΔGbp = 1.7 kcal/mol. The second step is the free energy cost of an initial binding, ΔGinit = 11.9 kcal/mol. The third step is the free energy cost of a nucleation, ΔGn3 = 1.5 kcal/mol, which is the sum of a base pair gain, −ΔGbp = −1.7 kcal/mol, and the cost of introducing two single-stranded DNA overhangs at an overhang-free nick, ΔG2ov = 3.2 kcal/mol. The final step is the “sawtooth amplitude” of three-way branch migration, ΔGs3 = 5.3 kcal/mol. ΔGbp, ΔGinit, ΔG2ov, and ΔGs3 were taken from Srinivas et al.27 (B) The IEL of a leakage through four-way branch migration. The first step requires to break two base pairs (2ΔGbp), and the second step is the initial binding (ΔGinit). The third step is the free energy cost of a nucleation, ΔGn4 = 0.6 kcal/mol, which is the sum of two base pairs gain, −2ΔGbp = −3.4 kcal/mol, and the cost of introducing a four-arm junction ΔG4aj = 4 kcal/mol.29 The final step is the sawtooth amplitude of four-way branch migration, ΔGs4 = 10.8 kcal/mol, which was calculated based on the step time difference between three-way branch migration4 and four-way branch migration.30 (C) The original entropy-driven system based on a linear substrate.17 Toeholds and single-stranded DNA tails for output formation are removed for simplicity. The leakage reaction occurs through three-way branch migration. (D) The novel entropy-driven system is based on three-arm junction substrates (upper panel) and four-arm junction substrates (lower panel). Leakage reactions occur through four-way branch migration for both cases.
RESULTS AND DISCUSSION
Single-Layer Catalytic System with Three-Arm Junction Substrates.
We first tested a single-layer catalytic system using a three-arm junction substrate (Figure 2A). In this system, the substrate S1 reacts with the catalyst C1 to produce the intermediate I1. Then, I1 reacts with the second substrate S2 to release the catalyst C1. Since both of the reactions proceed through three-way branch migration, the catalytic reaction was expected to be fast. In comparison, the leakage reaction proceeds through four-way branch migration, which was expected to be extremely slow (see Figure S1 for the overview, and Figure S2 for details of the catalytic and the leakage pathways). The product P2 reacts with the reporter complex R and displaces the dye strand D from the quencher, which monitors the reaction kinetics via fluorescence emission (Figure 2B). Kinetic experiments with 10 nM of substrate showed no detectable leakage in the absence of the catalyst C1 (0 pM), and addition of 500 pM or 1 nM catalyst showed quick catalytic reactions (Figure 2C). The catalytic rate constant (kcat) was measured from the 500 pM catalytic reaction and was 2.84 × 1013 M−2s−1 (Figure S3A), which is similar to a linear substrate (kcat = 2.86 × 1013 M−2s−1)20 and faster than a catalytic hairpin assembly (CHA) system (kcat = 4.72 × 1011 M−2s−1).19 The 50 pM catalytic reaction showed that the reaction nearly stalled at 10 h, yielding catalytic turnover of ∼30. This value is less than a linear substrate, which showed catalytic turnover between 80 and 100 at 24 h.32 Catalytic turnover is known to be very sensitive to the quality of DNA strands19,32 and thus can be readily improved by using better purified DNA. We also observed the saturation of the catalytic speed for higher catalyst concentrations (Figure S3B) due to the involved unimolecular four-way branch migration where the intermediate I2 is converted into products P2 and P3 (Figure S2C). The influence of this unimolecular reaction becomes stronger as the catalyst concentration becomes higher, and hence the 1 nM catalytic reaction showed worse fitting than the 500 pM catalytic reaction (Figure S3C). The numerical integration steps in Section 3 of the Supporting Information reproduced the saturation behavior at high concentrations of catalyst as well as lower concentrations of catalyst such as 1 nM and 500 pM (Figure S7A).
Figure 2.
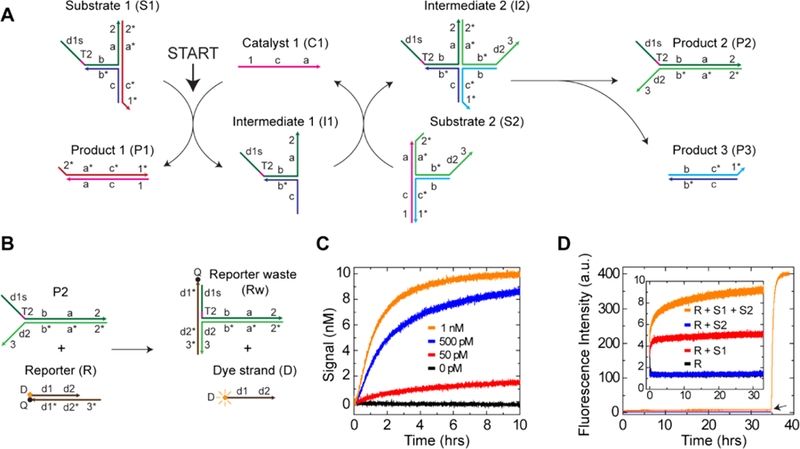
Single-layer catalytic system with three-arm junction substrates. (A) A schematic of the catalytic pathway. Functionalities of DNA sequences are represented by domains, which are unique segments of continuous oligonucleotides. Asterisks represent complementary domains, domains with toeholds are represented by numbers, and specificity domains are letters. Domain T2 of S1 represents 2 nt thymidine. (B) Reporting reaction. The reporter complex R has both the dye (D) and the quencher (Q), resulting in quenched fluorescence. The reaction between P2 and R releases the dye strand D and increases fluorescence intensity. (C) Kinetic traces with different concentrations of the catalyst C1. Fluorescence intensity was normalized so that 10 nM corresponds to the maximum fluorescence intensity and 0 nM corresponds to the initial intensity. [S1] = [S2] = 10 nM, [R] = 20 nM. (D) Kinetic traces of the leakage and background reactions. [S1] = [S2] = 500 nM, [R] = 700 nM. The leakage trace is R + S1 + S2, and other traces were performed to measure the background signals. Fluorescence intensity was not normalized. The black arrow shows the addition of catalyst C1 to obtain the maximum fluorescence intensity.
Next, kinetic experiments were performed with higher concentration of substrates to quantify the leakage and other background reactions (Figure 2D). Other than the leakage (R + S1 + S2), the reaction (R + S1) also showed an observable fluorescence increase. In addition, we observed that the maximum fluorescence intensity with 500 nM of substrate was ∼30% less than the value expected from 10 nM of substrate, which would be due to a weak reaction between the reporter waste Rw and the released dye strand D (Figure S4A, B). Therefore, the leakage rate constant, kleak, was calculated after subtraction of the background (R + S1), following fluorescence intensity normalization in order to consider the nonlinearity between the fluorescence intensity and the concentration (Figure S4C). The leakage rate constant was 2.20 × 10−2 M−1S−1, which is in good agreement with the rate constant for four-way branch migration with a zero-toehold (3.4 × 10−2 M−1 s−1)28 and is more than 2 orders of magnitude smaller than a linear substrate (8.12 M−1 s−1).20 As a metric to compare the performance of different catalytic substrates, the ratio kcat/kleak was summarized in Table 1. The data show that the three-arm junction substrate has more than 2 orders of magnitude larger kcat/kleak than other high-performing substrates. Note that the hairpin system compared in Table 1 was optimized and operated at higher temperature and lower salinity than the other systems, which will affect the reaction rates. We did not compare other factors, such as maximum catalytic turnover and the initial leakage, because these factors are highly influenced by the quality of DNA strands.19 In addition, even in the absence of a leakage between S1 and S2, single-stranded tails on those substrates (domains d1s-T2 of S1 and domains d2−3 of S2) may cooperatively displace the D strand from R and produce a fluorescence signal. In order to estimate this background reaction, R was mixed with two single-stranded DNA strands, one with domains d1s-T2 and the other with domains d2–3, and we observed a significant fluorescence signal (Figure S4D, E). Therefore, the actual leakage rate is likely smaller than the calculated value above. We also designed the same system with specificity domains shortened from 22 nt to 16 nt, and it showed similar performance (Figure S8).
Table 1.
Ratio of the Catalytic to Leakage Rates Constants (kcat/kleak) for Different Catalytic Substrates
Two-Layer Feed Forward Catalytic System with Three-Arm Junction Substrates.
Signal cascaded systems for higher signal amplification or molecular computation require smooth connection between multiple layers using outputs of one layer as inputs into other layers. An associative toehold,33 which is applied to our design, was originally used for hairpin substrates, and its reaction speed was 2 orders of magnitude slower than a single-stranded invader with a 8 nt toehold. Although a longer toehold was demonstrated to speed up the reaction, it is not suitable for a toehold exchange reaction4 because a long toehold inhibits its spontaneous dissociation. In contrast with a hairpin substrate, after the investigation of the design parameters (Figure S9A–D), a three-arm junction substrate only showed approximately three times the slow down of a 6 nt toehold (Figure S9E, F).
Based on these results, we constructed a two-layer feed forward catalytic system (Figure 3A). Kinetic data showed extremely slow leakage, resulting in the large signal-to-noise ratio for the catalyst detection (Figure 3B). It will be possible to make more sensitive feed forward systems by using higher quality DNA strands to gain better catalytic turnover19,32 or by connecting more layers. In addition, a cross-catalytic system was also constructed by feeding back the signal from the second to the first layer (Figure S12). The leakage source was investigated for both catalytic systems by conducting a background check, and the results indicated that the initial leakage was the primary source of leakage (Figure S13). We also constructed a feed forward system and an autocatalytic system based on 16 nt specificity domains (Figure S14), although they showed larger leakage than the 22 nt designs presented here.
Figure 3.
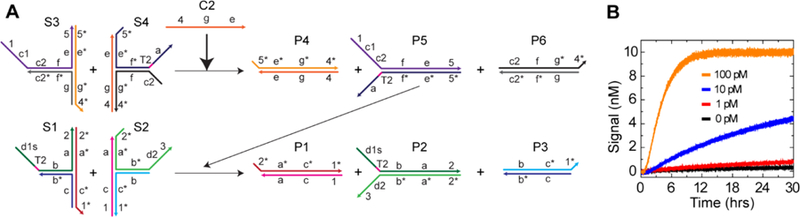
Two-layer feed forward catalytic system with three-arm junction substrates. (A) A simplified schematic of the catalytic pathway (see Figure S10 for details). The substrates S3 and S4 in the first layer produce the product P5, whose single-stranded sequence acts as the input catalyst for the second layer. The substrates in the second layer (S1, S2) are the same as the single-layer catalytic system (Figure 2) with the same reporting system. Domain c = domains c1 + c2. (B) Kinetic traces with different concentrations of the catalyst C2. [S1] = [S2] = [S3] = [S4] = 10 nM, and [R] = 20 nM.
Autocatalytic System with Four-Arm Junction Substrates.
The design principle of a three-arm junction substrate can be generalized and extended to other multi-arm junctions. Here, we used a four-arm junction. The reaction converts two substrates S5 and S6 into four products P1, P2, P8, and P9, and the new combinations of domains on each product can trigger downstream reactions (Figure S15). In order to characterize the catalytic and leakage reaction, a single-layer catalytic system was initially designed (Figure S16). The result showed a very slow catalytic reaction when all of the toeholds were 6 nt long (Figure S16B). Therefore, a suitable toehold design was examined (Figure S16C–F), together with the connection between multiple layers (Figure S9G, H). Based on the results, an autocatalytic system was constructed with an extended toehold for both toehold domains 1 and 2 (Figure 4A). The reaction kinetics for the leakage showed good stability without the initiation of clear leakage up to 1.5 h (Figure 4B), resulting in a better detection limit. Owing to the limited catalytic turnover of catalytic systems that are based on DNA strand displacement,32 autocatalytic or other exponential amplification systems, whose amplification is not limited by maximum catalytic turnover, can be a reasonable candidate for biomarker sensor applications. Because of the very small asymptotic leakage and the quick catalytic reaction (Table 1, Figure S17A–), the reduction of the initial leakage is critical for the improvement of the sensitivity. Note that, although three-arm and four-arm junction structures have appeared in dynamic DNA systems, using hairpin substrates12 or associative toeholds34 for instance, those multi-arm junction structures exist as products without further structural changes. On the contrary, our work demonstrates that the energy stored in multi-arm junction structures can be catalytically released when coupled with complementary multi-arm junctions. Beyond their application in structural DNA nanotechnology,35 this research provides a highly novel perspective of multi-arm junctions as a vital tool for dynamic DNA nanotechnology.
Figure 4.
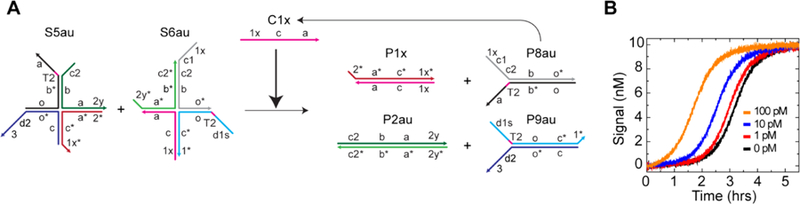
Autocatalytic system with four-arm junction substrates. (A) A simplified schematic of the autocatalytic system (see Figure S19 for details). P8au has catalytic domains to perform exponential amplification. Two nt were added at the 5′ end of domain 1 and the 3′ end of domain 2 to generate domains 1x and 2y, respectively. (B) Kinetic traces with different concentrations of the catalyst C1x. [S5au] = [S6au] = 10 nM, [R] = 20 nM.
CONCLUSION
DNA strand displacement systems are metastable reactions that are triggered by the addition of a specific, single-stranded sequence. Catalytic substrates exploit strand displacement for programmable applications that range from medical diagnosis to molecular computation. However, the complexity, stability, scalability, and sensitivity of said systems are plagued by network leakage the Achilles’ heel of dynamic DNA nanotechnology. A novel way to suppress leakage is to increase its energy barrier through four-way branch migration. Multi-arm junction substrates were designed here to simultaneously exploit four-way branch migration (with a high-energy barrier to minimize leakage) and three-way branch migration (with a very low energy barrier to maximize catalysis). Original feed forward, autocatalytic, and cross-catalytic systems were built with polynomial and exponential amplification that exhibit the modularity of linear substrates and the stability of hairpin substrates. As the stability of hairpin substrates are desired for transcription and used for in vitro37 or in vivo applications,38 and the modularity of linear substrates are desired for circuits,39 the combined performance of multi-arm-junction substrates creates a new phase space for synthetic biologist, biotechnologists, and DNA nanotechnologist to explore. When compared to state-of-the-art hairpin and linear substrates, our multi-arm junction substrates showed very low leakages without intensive purification of DNA strands,19 the application of availability and mutual availability,20 clamps,7,9,10,12 mismatches,20,21 or LNAs.22 They also showed quick catalytic reactions, resulting in more than 2 orders of magnitude larger ratio of the catalytic to leakage reaction rates. It is expected that combinations of our design with other leakage reduction techniques mentioned above, or a proposed novel domain design,36 will further improve the performance of our systems. Additionally, the output design of the multi-arm junction substrates showed the modularity to construct a variety of signal cascades. Considering these features, multi-arm junction substrates have great potential to further explore dynamic DNA nanotechnology5 for the realization of more complex, stable, scalable, and sensitive systems. Future work will focus on optimizing the performance of multi-arm junctions, exploring novel applications for them, and testing our five-arm multi-arm junction substrate (Figure S21).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by (1) The W.M. Keck Foundation, (2) NIH grant no. K25GM093233 from the National Institute of General Medical Sciences, (3) NIH grant no. P20GM103408 from the National Institute of General Medical Sciences, Institutional Development Award (IDeA), and (4) The Micron Foundation. We also thank the Nanoscale Materials and Device Research Group at Boise State University for their support. We specifically thank B. Yurke and B. Cannon for their editorial comments and X. Olson for her experimental insight.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Yurke B; Turberfield AJ; Mills AP; Simmel FC; Neumann JL Nature 2000, 406, 605–608. [DOI] [PubMed] [Google Scholar]
- (2).SantaLucia J; Hicks D Annu. Rev. Biophys. Biomol. Struct 2004, 33, 415–440. [DOI] [PubMed] [Google Scholar]
- (3).Yurke B; Mills A Jr. Genet. Program. Evolvable Mach 2003, 4, 111–122. [Google Scholar]
- (4).Zhang DY; Winfree EJ Am. Chem. Soc 2009, 131, 17303–17314. [DOI] [PubMed] [Google Scholar]
- (5).Zhang DY; Seelig G Nat. Chem 2011, 3, 103–113. [DOI] [PubMed] [Google Scholar]
- (6).Turberfield AJ; Mitchell JC; Yurke B; Mills AP; Blakey MI; Simmel FC Phys. Rev. Lett 2003, 90, 118102. [DOI] [PubMed] [Google Scholar]
- (7).Seelig G; Yurke B; Winfree EJ Am. Chem. Soc 2006, 128, 12211–12220. [DOI] [PubMed] [Google Scholar]
- (8).Jung C; Ellington AD Acc. Chem. Res 2014, 47, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Seelig G; Soloveichik D; Zhang DY; Winfree E Science 2006, 314, 1585–1588. [DOI] [PubMed] [Google Scholar]
- (10).Qian L; Winfree E Science 2011, 332, 1196–1201. [DOI] [PubMed] [Google Scholar]
- (11).Qian L; Winfree E; Bruck J Nature 2011, 475, 368–372. [DOI] [PubMed] [Google Scholar]
- (12).Yin P; Choi HMT; Calvert CR; Pierce NA Nature 2008, 451, 318–322. [DOI] [PubMed] [Google Scholar]
- (13).Zhang DY; Hariadi RF; Choi HMT; Winfree E Nat. Commun 2013, 4, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dirks RM; Pierce NA Proc. Natl. Acad. Sci. U. S. A 2004, 101, 15275–15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bois JS; Venkataraman S; Choi HM; Spakowitz AJ; Wang ZG; Pierce NA Nucleic Acids Res 2005, 33, 4090–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li B; Ellington AD; Chen X Nucleic Acids Res 2011, 39, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang DY; Turberfield AJ; Yurke B; Winfree E Science 2007, 318, 1121–1125. [DOI] [PubMed] [Google Scholar]
- (18).Chen Y-J; Dalchau N; Srinivas N; Phillips A; Cardelli L; Soloveichik D; Seelig G Nat. Nanotechnol 2013, 8, 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen X; Briggs N; McLain JR; Ellington AD Proc. Natl. Acad. Sci. U. S. A 2013, 110, 5386–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Olson X; Kotani S; Padilla JE; Hallstrom N; Goltry S; Lee J; Yurke B; Hughes WL; Graugnard E ACS Synth. Biol 2017, 6, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jiang YS; Bhadra S; Li B; Ellington AD Angew. Chem., Int. Ed 2014, 53, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Olson X; Kotani S; Yurke B; Graugnard E; Hughes WL J. Phys. Chem. B 2017, 121, 2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jose D; Datta K; Johnson NP; von Hippel PH Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4231–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Temsamani J; Kubert M; Agrawal S Nucleic Acids Res 1995, 23, 1841–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Green C; Tibbetts C Nucleic Acids Res 1981, 9, 1905–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Panyutin IG; Hsieh P Proc. Natl. Acad. Sci. U. S. A 1994, 91, 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Srinivas N; Ouldridge TE; Šulc P; Schaeffer JM; Yurke B; Louis AA; Doye JPK; Winfree E Nucleic Acids Res 2013, 41, 10641–10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dabby NL Synthetic molecular machines for active self-assembly: prototype algorithms, designs, and experimental study Ph.D. Thesis, California Institute of Technology: Pasadena, CA, 2013. [Google Scholar]
- (29).Wang JS; Zhang DY Nat. Chem 2015, 7, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).McKinney SA; Freeman ADJ; Lilley DMJ; Ha T Proc. Natl. Acad. Sci. U. S. A 2005, 102, 5715–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Seeman NC; Kallenbach NR Annu. Rev. Biophys. Biomol. Struct 1994, 23, 53–86. [DOI] [PubMed] [Google Scholar]
- (32).Zhang DY; Winfree E Nucleic Acids Res 2010, 38, 4182–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chen XJ Am. Chem. Soc 2012, 134, 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhu J; Zhang L; Dong S; Wang E ACS Nano 2013, 7, 10211–10217. [DOI] [PubMed] [Google Scholar]
- (35).Seeman NC Annu. Rev. Biochem 2010, 79, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Thachuk C; Winfree E; Soloveichik D Proceedings of the 21st International Conference on DNA Computing and Molecular Programming 2015, 9211, 133–153. [Google Scholar]
- (37).Bhadra S; Ellington AD Nucleic Acids Res 2014, 42, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Green AA; Silver PA; Collins JJ; Yin P Cell 2014, 159, 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Phillips A; Cardelli LJR Soc., Interface 2009, 6, S419–S436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.