

Abstract
DNA methylation is essential for gene regulation, transposon silencing, and imprinting. Although the generation of specific DNA methylation patterns is critical for these processes, how methylation is regulated at individual loci remains unclear. Here we show that a family of four putative chromatin remodeling factors, CLASSY (CLSY) 1–4, are required for both locus-specific and global regulation of DNA methylation in Arabidopsis. Mechanistically, these factors act in connection with RNA polymerase-IV (Pol-IV) to control the production of 24-nucleotide small interfering RNAs (24nt-siRNAs), which guide DNA methylation. Individually, the CLSYs regulate Pol-IV-chromatin association and 24nt-siRNA production at thousands of distinct loci, and together, they regulate essentially all 24nt-siRNAs. Depending on the CLSYs involved, this regulation relies on different repressive chromatin modifications to facilitate locus-specific control of DNA methylation. Given the conservation between methylation systems in plants and mammals, analogous pathways likely operate in a broad range of organisms.
Introduction
The use of small non-coding RNAs to silence transposons and other foreign genetic elements via the deposition of repressive chromatin modifications is a highly conserved strategy employed by eukaryotic organisms to ensure genome stability1,2. Unlike in animals and fungi, where the biogenesis of these non-coding RNAs is initiated by Pol-II, in plants they are generated by two plant-specific RNA polymerases, Pol-IV and Pol-V. These polymerases evolved from Pol-II3,4 and play central roles in the RNA-directed DNA Methylation (RdDM) pathway5,6. Briefly, Pol-IV generates short single-stranded RNAs7,8 that are copied into double-stranded RNAs by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) and cleaved into 24nt-siRNAs by DICER-LIKE PROTEIN 3 (DCL3)9. These 24nt-siRNAs are then loaded into ARGONAUTE (AGO) effector complexes, including AGO4, AGO6 and AGO910. Pol-V generates longer non-coding transcripts11 that serve as scaffolds for the recruitment of additional RdDM factors including 24nt-siRNA-loaded ARGONAUTE proteins12–14. Ultimately, these interactions lead to the recruitment of DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2)15,16 and the deposition of DNA methylation throughout the genome.
Once established, maintenance pathways take over to ensure the faithful inheritance of DNA methylation patterns5. Despite the existence of robust maintenance pathways, DNA methylation patterns are not static, and can differ between cell types17–22, tissues23–26, and even generations, depending on the organism27. The processes through which such differences in DNA methylation profiles arise, or are modulated during development, remain poorly understood. Yet, they are clearly important, as aberrant patterns of DNA methylation can result in developmental defects in plants28,29 and are associated with numerous diseases in humans, including cancer30,31.
To gain insight into the regulation of DNA methylation patterns, we investigated the functions of four SNF2-related, putative chromatin remodeling factors, CLSY1–4, in connection with the Pol-IV and SAWADEE HOMEODOMAIN HOMOLOG1 (SHH1)32–35 components of the RdDM pathway. CLSY1, the founding member of the CLSY family, was initially identified from a genetic screen for the spreading of gene silencing and was linked to Pol-IV function based on reduced 24nt-siRNA levels at several genomic loci and immunolocalization experiments36. Consistent with these observations, CLSY1 was subsequently found to co-purify with Pol-IV33,35 and SHH133, to facilitate de novo DNA methylation37, and to play a weak role in controlling DNA methylation at RdDM targets38. However, the global effects of clsy1 mutants on 24nt-siRNA levels, the functional connections between CLSY1, SHH1 and Pol-IV, and an in-depth analysis of the effects of clsy1 mutants on DNA methylation patterns and gene silencing remain to be determined. Furthermore, the roles of CLSY2, CLSY3 and CLSY4, which also co-purify with Pol-IV, remain completely unknown.
Results
The CLSY family controls 24nt-siRNA levels in a locus-specific manner
To examine the roles of the CLSY family in the RdDM pathway, T-DNA insertion mutants for each CLSY genes were obtained. Gene expression profiling in these mutants confirmed disruption of the corresponding transcripts and demonstrated that there are no obvious compensatory gene expression effects observed between family members (Supplementary Fig. 1a and Supplementary Table 1). The effects of these mutants on 24nt-siRNAs were then determined by small RNA profiling (Supplementary Table 2) and compared to a Pol-IV mutant (nrpd1, hereafter termed pol-iv) as well as three wild-type controls. After determining loci that produce small RNAs based on both unique- and multi-mapping reads (Supplementary Fig. 1b and Supplementary Table 3), a core set of 13,253 24nt-siRNA clusters were identified using ShortStack39 (Supplementary Table 3 and 4a). These core clusters were detected in all three wild-type replicates and account for more than 92% of the mapped 24nt-siRNAs in each experiment (Supplementary Fig. 1c). As expected based on previous studies40,41, the expression of these 24nt-siRNA clusters are highly dependent on Pol-IV (Supplementary Fig. 1d, e). In each clsy mutant largely non-overlapping subsets of reduced 24nt-siRNA clusters were identified using DESeq242 (fold change (FC)≥2 and false discovery rate (FDR)≤0.01; Fig. 1a, Supplementary Fig. 1f, and Supplementary Table 4). The clsy1 mutant affected the most 24nt-siRNA clusters, while clsy3 and clsy4 displayed an intermediate effect, and clsy2 only affected a small number of loci (Fig. 1a). Quantification of 24nt-siRNA levels over these reduced 24nt-siRNA clusters revealed strong decreases that are specific to each mutant and approached the levels observed in pol-iv (Fig. 1b and Supplementary Fig. 1g). Further attesting to the robustness of these phenotypes, similar results were observed using only uniquely mapping reads (Supplementary Fig. 1h) or using data from an independent, biological replicate (Supplementary Fig. 1i). In addition to depending on different CLSY family members, these four groups of 24nt-siRNA clusters also differ in their wild-type expression levels (Fig. 1b and Supplementary Fig. 1g) as well as their size (Supplementary Fig. 1j), which may contribute to their differential regulation. In total, the clsy-dependent 24nt-siRNA clusters identified here represent approximately 25% of the 24nt-siRNA producing loci genome-wide (Fig. 1a), which account for 62.7% of all the 24nt-siRNAs present in wild-type plants (Supplementary Fig. 1k). Similar differential expression analyses for 21nt- and 22nt-siRNA clusters, which include miRNAs, revealed essentially no down-regulated clusters (Supplementary Table 5). Taken together, these findings demonstrate that the CLSY proteins act as potent, locus-specific regulators of 24nt-siRNA expression.
Figure 1. The CLSY family controls 24nt-siRNA levels in a locus-specific manner.
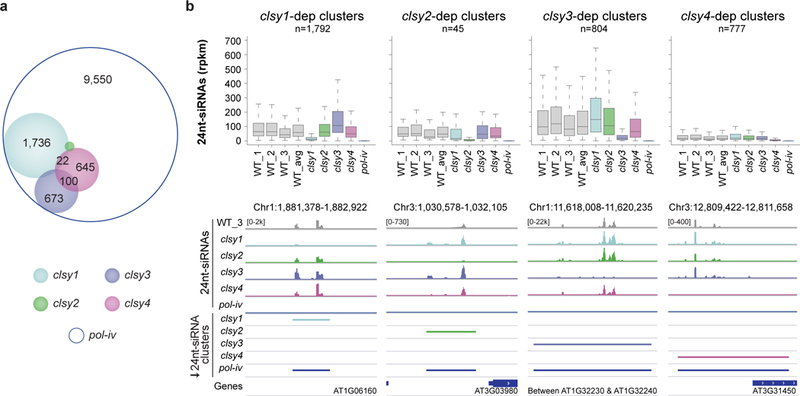
(a) Scaled Venn diagram based on the reduced 24nt-siRNA clusters provided in Supplementary Table 4 showing the relationships between loci with reduced 24nt-siRNA levels in the clsy single mutants. For readability, only overlaps >20 are labeled. A small number of overlaps between clsy2 and clsy3 are not shown due to spatial constraints, but an unscaled Venn diagram showing all the overlaps is present in Supplementary Figure 1f. (b) Boxplots showing 24nt-siRNA levels (reads per kilobase per million; rpkm) in each clsy single mutant compared to each other, wild-type (WT) controls, and pol-iv. Here, and in all subsequent figures, the boxplots show the interquartile range (IQR) with the median shown as the black line and the whiskers corresponding to 1.5 times the IQR. Above each plot, the numbers of clusters (n) are indicated and biological replicates for the WT controls are designated as WT_1, WT_2, and WT_3, with the average signal from these replicates designated as the WT_avg. These boxplots represent a single experiment, but confirmatory data from an independent biological replicate and from additional alleles are presented in Supplementary Figs. 1i and 3, respectively. Below each boxplot are genome browser screen shots showing the levels of 24nt-siRNAs (reads per 10 million; rp10m) at representative clsy-dependent 24nt-siRNA clusters. The scale for each panel is indicated in brackets, where k indicates 1000.
To determine whether the 24nt-siRNA clusters regulated by the clsy single mutants represent the totality of loci controlled by these factors, all 6 combinations of clsy double mutants were generated and their small RNA profiles and reduced 24nt-siRNA clusters were determined (Supplementary Table 4, Fig. 2a, b and Supplementary Fig. 2a, b). This revealed two double mutants (clsy1,2 and clsy3,4) that showed clear synergistic relationships, affecting more loci (Fig. 2a) and displaying stronger reductions in 24nt-siRNA levels relative to their respective single mutants (Fig. 2b). Notably, these findings are consistent with previous phylogenetic analyses, as CLSY1 and CLSY2 form one subgroup while CLSY3 and CLSY4 form another36. As observed for 24nt-siRNA clusters dependent on individual CLSY proteins, the reductions in 24nt-siRNAs observed at the clsy1,2- and clsy3,4-dependent clusters were largely specific to the corresponding mutants (Fig. 2b and Supplementary Fig. 2c). In total, these clsy doubles control 67% of all 24nt-siRNA clusters (Fig. 2c), which equates to 88% of all 24nt-siRNAs present in wild-type plants (Supplementary Fig. 2d), revealing a second layer of locus-specific regulation that relies on distinct pairs of CLSY proteins.
Figure 2. Specific CLSY pairs regulate 24nt-siRNAs at non-overlapping and spatially distinct genomic loci.
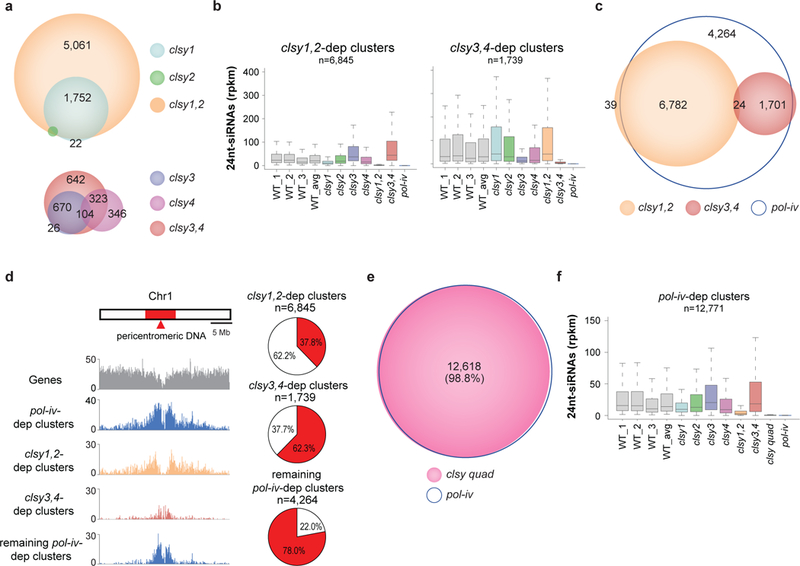
(a, c, and e) Scaled Venn diagrams showing the relationships between loci with reduced 24nt-siRNA levels in the indicated clsy single, double, and quadruple mutants. For readability, only overlaps >20 are labeled except for panel e where the % overlap between both samples is shown instead. (b and f) Boxplots showing 24nt-siRNA levels in each clsy single, double or quadruple mutant compared to each other, WT controls and pol-iv, from a single experiment. Confirmatory data using additional alleles are presented in Supplementary Fig. 3. (d) Chromosome 1 view of 24nt-siRNA clusters dependent on the genotypes indicated on the left, where the scale is the number of clusters per 100kb bin. The red region corresponds to pericentromeric DNA56. The pie charts represent the genome wide (i.e. Chr1–5) distributions. Chromosomal views for Chr2–5 are present in Supplementary Figure 2e.
To further examine the relationship between the clsy1,2- and clsy3,4-dependent 24nt-siRNA clusters, their overlap with each other and their genomic distributions were determined. Not only do these CLSY pairs regulate mutually exclusive sets of 24nt-siRNAs clusters (Fig. 2c), they also show preferential enrichment for chromosome arms (clsy1,2-dependent clusters) or pericentromeric heterochromatin (clsy3,4-dependent clusters), revealing a striking distribution of labor amongst the CLSY family (Fig. 2d and Supplementary Fig. 2e). Notably, the remaining pol-iv-dependent 24nt-siRNA clusters, which were not significantly affected in either double mutant, show an even more extreme partitioning within the genome, with 78% residing in pericentromeric heterochromatin (Fig. 2d and Supplementary Fig. 2e). These clusters are lowly expressed (Supplementary Fig. 2c, d) and, like the clsy3,4-dependent 24nt-siRNA clusters, they tend to be larger in size (Supplementary Fig. 2f). To determine whether these remaining loci are redundantly controlled by all four CLSY proteins, a clsy quadruple mutant was generated. In this mutant, greater than 98% of all pol-iv-dependent 24nt-siRNA clusters were reduced (Fig. 2e) and the levels of 24nt-siRNAs at these clusters were near zero (Fig. 2f). Finally, the effects and locus-specificities of the clsy single, double and quadruple mutants on 24nt-siRNA levels were confirmed with additional mutant alleles for all four CLSY genes (Supplementary Fig. 3) Together, these findings demonstrate that the four CLSY proteins act individually as highly locus-specific regulators of 24nt-siRNAs and together as the master regulators of essentially all Pol-IV-dependent 24nt-siRNAs.
The CLSY family controls global DNA methylation patterns
To assess the effects of the clsy-dependent 24nt-siRNA losses on DNA methylation patterns, whole genome bisulfite sequencing experiments were conducted (Supplementary Table 6). In Arabidopsis, the patterns of DNA methylation can be broadly classified into two categories43,44: Methylation at transposons and repeats, which is established via the RdDM pathway and occurs in all sequence contexts (CG, CHG, and CHH, where H=A, T, or C), and gene body methylation, which is restricted to the CG context and is established via mechanisms that remain poorly understood45. Thus, to best evaluate the roles of the clsy mutants, differentially methylated regions (DMRs) for each genotype were determined independently for the CG, CHG, and CHH contexts (FC≥40%, 20%, or 10% for CG, CHG, and CHH DMRs, respectively, relative to three wild-type controls with an FDR≤0.01; Fig. 3a and Supplementary Table 7). Consistent with roles for the CLSY family in RdDM, this analysis revealed a high degree of overlap between hypo DMRs and reduced 24nt-siRNA clusters, especially for non-CG DMRs in the clsy double and quadruple mutants (Fig. 3a). Furthermore, even at DMRs that failed to overlap with reduced 24nt-siRNA clusters, 24nt-siRNA levels were still decreased (Supplementary Fig. 4). Thus, at non-CG DMRs, reduced DNA methylation is highly correlated with 24nt-siRNA losses. In contrast, a similar analysis at CG DMRs showed minimal overlap with reduced 24nt-siRNA clusters in the clsy mutants (Fig. 3a) and revealed that the vast majority of these regions have little to no 24nt-siRNAs (Supplementary Fig. 4), suggesting they likely represent natural variation in methylation at body-methylated genes rather than defects in targeting methylation at RdDM loci. Nonetheless, the small subset of CG DMRs that do overlap with reduced 24nt-siRNA clusters (Supplementary Fig. 4a) showed a clear reduction in 24nt-siRNAs, nearly phenocopying pol-iv mutants. Together, these comparisons reveal the subset of loci where reductions in 24nt-siRNA levels result in the most significant changes in DNA methylation for each sequence context.
Figure 3. 24nt-siRNA losses in clsy mutants result in reduced DNA methylation.
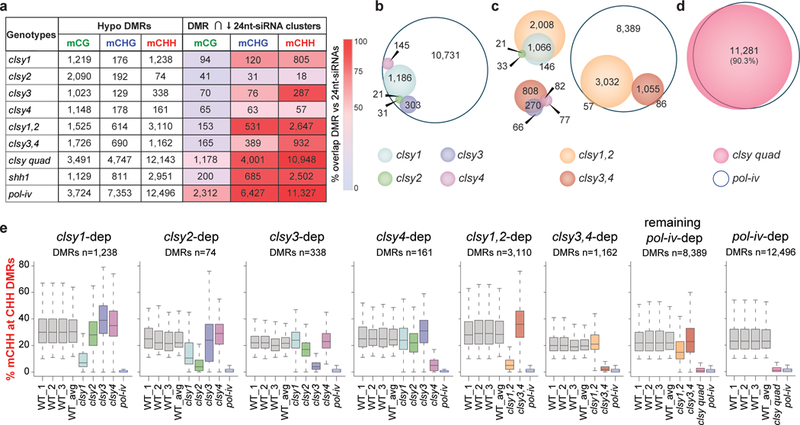
(a) Table showing the numbers of hypo DMRs in the genotypes and methylation contexts indicated, where H=A, T, or C. The number of these DMRs that overlap (∩) with reduced 24nt-siRNA clusters (“DMR ∩ ↓ 24nt-siRNA clusters”) is also indicated and shaded from light blue to red based on the percentage of total DMRs represented. (b-d) Scaled Venn diagrams of hypo CHH DMRs showing the relationships between loci regulated by the clsy single, double, and quadruple mutants, respectively. For readability, only overlaps >20 are labeled except for panel d where the % overlap is shown instead. For panel b, a small number of overlaps are not shown due to spatial constraints, but an unscaled Venn diagram showing all the overlaps is present in Supplementary Figure 5a. (e) Boxplots showing the levels of CHH methylation at the hypo CHH DMRs identified in each clsy single, double or quadruple mutant as compared to each other, WT controls, and pol-iv. These boxplots represent a single experiment including three independent WT controls.
As expected based on the presence of pathways controlling the maintenance of DNA methylation in the CG and CHG contexts5, the largest effects on DNA methylation observed in the RdDM mutants were in the CHH context. Consistent with their 24nt-siRNA phenotypes, each clsy single mutant affected DNA methylation at largely distinct sets of DMRs. Once again, clsy1 was the strongest with 1,238 CHH DMRs, clsy3 and clsy4 had 338 and 161, respectively, and clsy2 was the weakest with just 74 (Fig. 3a, b and Supplementary Fig. 5a). Further paralleling the effects observed for 24nt-siRNAs, the clsy double mutants showed additive effects at mutually exclusive sets of CHH DMRs (Fig. 3a, c) and the quadruple mutant showed the strongest effect, overlapping with >90% of the CHH DMRs identified in pol-iv (Fig. 3a, d). Quantification of DNA methylation levels at all the non-CG DMRs (Fig. 3e and Supplementary Fig. 5b), as well as the CG DMRs overlapping with reduced 24nt-siRNA clusters (Supplementary Fig. 5c), revealed the strongest reductions in DNA methylation levels in the corresponding mutant backgrounds. In addition, quantification of DNA methylation levels at all the reduced 24nt-siRNA clusters, not just those corresponding to DMRs, revealed similar trends: CG methylation levels were minimally affected, while stronger reductions were observed in the non-CG contexts in a genotype-specific manner (Supplementary Fig. 5d). Together, these findings demonstrate that the locus-specific reductions in 24nt-siRNA levels observed in the clsy single, double and quadruple mutants result in locus-specific decreases in DNA methylation.
Figure 5. The CLSY proteins are required for Pol-IV chromatin association at 24nt-siRNA producing loci.
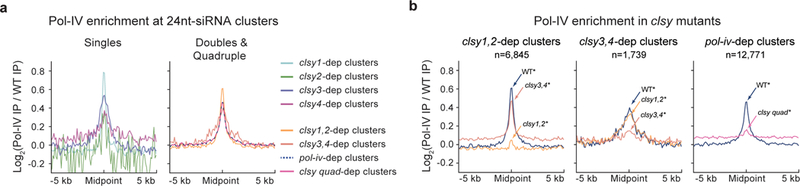
(a and b) Profile plots showing Pol-IV enrichment at all the different classes of clsy-dependent 24nt-siRNA clusters in a WT background (the pNRPD1::NRPD1–3xFLAG line) or the indicated clsy mutant backgrounds, respectively, from two sets of ChIP-seq data (see Supplementary Table 10). The asterisk (*) indicates that these lines are also homozygous for both the NRPD1–3xFLAG transgene and the nrpd1 mutant.
The CLSY family is required for DNA methylation-mediated silencing
Given the known roles of DNA methylation in gene silencing, transcriptome profiling experiments were conducted to identify RdDM targets up-regulated in pol-iv and clsy mutants (Supplementary Table 1, 8 and 9). These analyses revealed a total of 177 genes, repeats, and unannotated transcripts up-regulated at least 2-fold in pol-iv mutants. Although the clsy single mutants displayed weak expression phenotypes, at least one locus regulated predominantly by each mutant was identified (Fig. 4a, Supplementary Fig. 6a, and Supplementary Table 9). Of these single mutants, clsy4 was by far the strongest. However, the vast majority of pol-iv loci were redundantly controlled by all four CLSY proteins, as the clsy quadruple mutant regulated approximately 50% of all pol-iv up-regulated loci and nearly 80% of those were at least 5-fold up-regulated (Fig. 4a and Supplementary Table 9). To determine the extent to which the observed changes in gene expression correlate with altered 24nt-siRNA and DNA methylation profiles, these features were plotted side-by-side for all 177 loci (+/− 2kb) in the pol-iv and clsy quadruple mutants (Fig. 4b). On aggregate, these loci showed lower levels of 24nt-siRNAs and DNA methylation. For approximately half of the genes, and the majority of unannotated transcripts and repeats, discrete regions with more strongly reduced 24nt-siRNAs and DNA methylation levels were apparent either within the transcript itself or in the flanking 2kb regions (Fig. 4b). Indeed, further characterization of these loci revealed a high degree of overlap (80–100%) with the previously identified reduced 24nt-siRNA clusters and hypo DMRs (Supplementary Fig. 6b, c and Supplementary Tables 4 and 7). In contrast, similar reductions were not observed in the clsy2 single mutant, which is the weakest mutant overall and thus served as a negative control (Supplementary Fig. 6d). Nonetheless, like the pol-iv and clsy quadruple mutants, two of the three loci up-regulated in the clsy2 mutant were associated with reduced 24nt-siRNA clusters and hypo DMRs (Supplementary Fig. 6e). Together, these findings support the conclusion that these up-regulated loci in the clsy mutants are normally silenced by DNA methylation that is controlled by the RdDM pathway.
Figure 4. The CLSY family controls the expression of RdDM targets.
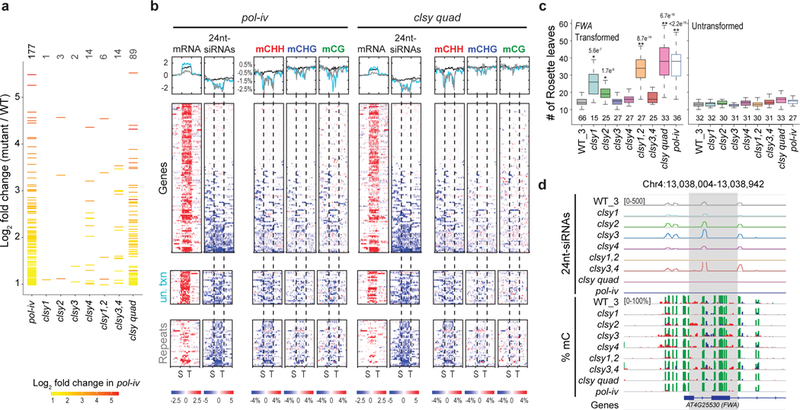
(a) Plot showing the expression level of pol-iv-up-regulated loci (represented as horizontal slashes) in the clsy single, double and quadruple mutants. The slashes in all genotypes are colored based on the expression level of up-regulated loci in pol-iv and the number of up-regulated loci in each mutant is indicated above. (b) Heatmaps and profile plots showing the expression levels of the up-regulated TAIR10 genes (n=115), unannotated transcripts (un. txn; n=26), and TAIR10 repeats (n=36) shown in a as well as the corresponding 24nt-siRNA and DNA methylation levels at these same loci. For the mRNA and 24nt-siRNA analyses, the Log2 fold change in expression is plotted and for the DNA methylation analysis, the percent difference in methylation is plotted. Color bars indicating the scales are shown below. The heatmaps include 2kb flanking the transcription start site (S) and the transcription termination site (T) and were ranked based on the 24nt-siRNA and mCHH values in both mutants (pol-iv and the clsy quad). The profiles of the genes, un. txn, and repeats are in black, light blue, and grey, respectively. (c) Boxplots showing the number of leaves produced before flowering in FWA transformed T0 plants (Left) or untransformed plants (Right). The number of independent transformants (or untransformed plants) used for each genotype is shown below the boxplots. p-values ≤1e−4 calculated using Wilcoxon sum tests relative to the WT_3 control are shown above. (d) Genome browser screen shot showing the levels of 24nt-siRNAs (rp10m) and DNA methylation at the endogenous FWA gene in the indicated genotypes. For each set of data, the scale is indicated in brackets, with CG, CHG, and CHH methylation shown in green, blue, and red, respectively. The region showing the most prominent reduction in CHH methylation is highlighted in grey. The expression data presented in panels a, b, and d corresponds to two biological replicates of each genotype.
As an additional test of the CLSY specificities, their roles in the establishment of DNA methylation were assessed using a well-vetted de novo methylation assay involving the transformation of an unmethylated FWA transgene into each mutant background46. In this assay, failure to methylate and silence the incoming transgene results in an increase in the number of leaves produced prior to flowering. Compared to the untransformed controls, several of the FWA-transformed clsy mutants showed delayed flowering (Fig. 4c). In addition to clsy1, which was previously shown to display a late-flowering phenotype in FWA assays37, clsy2 mutants also showed a slight delay, while clsy3 and clsy4 flowered at or near the normal number of leaves. This phenotype was enhanced in the clsy1,2 double, which flowered nearly as late as the clsy quadruple and pol-iv mutants. Notably, the specificities observed for this de novo assay match those observed at the endogenous FWA gene, where 24nt-siRNA production depends on CLSY1 and CLSY2 (Fig. 4d). These findings represent the first examples wherein bone fide components of the RdDM pathway (i.e. CLSY3 and CLSY4) are not required to establish methylation in the FWA de novo assay and demonstrate that the locus specificity observed for the CLSY family extends to the establishment phase of the RdDM pathway.
The CLSY family is required for Pol-IV chromatin association
To gain mechanistic insights into the roles of the CLSY proteins, enrichment of Pol-IV at 24nt-siRNA-producing loci was determined by chromatin immunoprecipitation and sequencing (ChIP-seq) experiments using a previously characterized tagged Pol-IV line (pNRPD1::NRPD1–3xFLAG34) crossed into various clsy mutant backgrounds (Supplementary Table 10). In a wild-type background, Pol-IV was enriched at all classes of clsy-dependent 24nt-siRNA clusters and, consistent with previous Pol-IV ChIP-seq experiments34, Pol-IV was most enriched at highly expressed 24nt-siRNA clusters (e.g. clsy1-dependent loci) and less enriched at lowly expressed clusters (e.g. clsy4-dependent loci) (Fig. 5a). In the clsy1,2 or clsy3,4 mutant backgrounds, Pol-IV enrichment was specifically reduced at the loci regulated by these factors, and in the clsy quadruple mutant Pol-IV enrichment was depleted at all 24nt-siRNA loci (Fig. 5b and Supplementary Fig. 7a, b). In the clsy single mutants, reductions in Pol-IV enrichment were most clearly observed at clsy1- and clsy3-dependent loci (Supplementary Fig. 7c). For the clsy2 mutant, where only a few reduced 24nt-siRNA clusters were identified (n=45), or the clsy4 mutant, where the reduced 24nt-siRNA clusters are lowly expressed even in wild-type plants (Fig. 1b), global reductions were difficult to observe. However, individual examples of Pol-IV reduction in these mutants were identified (Supplementary Fig. 7d), and in both cases these weaker mutants (clsy2 and clsy4) enhanced their stronger mutant counterparts (clsy1 and clsy3, respectively; Supplementary Fig. 7a). Taken together, these findings demonstrate that the CLSY proteins are required for the locus-specific association of Pol-IV at chromatin.
The CLSY proteins rely on different chromatin modifications
In addition to the CLSY family, one other Pol-IV-associated factor, the methyl-H3K9 reader SHH1, is known to regulate 24nt-siRNA expression and function at the level of Pol-IV chromatin association32–35. Consistent with previous results33,34, 24nt-siRNA profiling revealed that ~50% of the core 24nt-siRNA clusters were at least 2-fold reduced in shh1 mutants (Fig. 6a). Comparison of shh1-dependent 24nt-siRNA clusters and hypo CHH DMRs with those identified in the clsy1,2 or clsy3,4 double mutants show a nearly complete, and highly specific overlap between shh1 and clsy1,2 (Fig. 6a-d), revealing a genetic connection between these mutants. Further supporting this relationship, analysis of 24nt-siRNA levels over all pol-iv-dependent clusters demonstrated that shh1 and either the shh1,clsy1 double or the shh1,clsy1,2 triple mutants have similarly reduced 24nt-siRNA levels, while the shh1,clsy3,4 triple mutant phenocopies the clsy quadruple and pol-iv mutants (Fig. 6e). Based on these findings, the hypothesis that CLSY1 and CLSY2 are required for the association of SHH1 with Pol-IV in vivo was tested by a series of co-immunoprecipitation experiments. Indeed, this interaction was specifically disrupted in clsy1,2 mutants, with less than ~12.5% of the wild-type level of NRPD1, the largest subunit of Pol-IV, co-purifying with SHH1 (Fig. 6f and Supplementary Fig. 8). Given the known connections between SHH1 and H3K9 methylation, the dependence of 24nt-siRNA production at CLSY1- and CLSY2-regulated loci on H3K9 methylation was also determined. In the suvh4,5,6 triple mutant, where H3K9 methylation levels are globally reduced, but not eliminated47, 24nt-siRNA levels at clsy1,2-dependent, but not clsy3,4-dependent loci, were significantly reduced (Supplementary Table 2, Fig. 6g and Supplementary Fig. 9). As the reductions in 24nt-siRNA levels in the suvh4,5,6 mutant were not as strong as those observed in the clsy1,2 and shh1 mutants, publicly available data48 was used to further investigate the relationship between the residual H3K9 di-methylation and 24nt-siRNA abundances in this mutant. At clsy1,2-dependent loci, regions that retain more H3K9 di-methylation in the suvh4,5,6 mutant also retain more 24nt-siRNAs (Supplementary Fig. 9a, b), further supporting the notion that 24nt-siRNAs at these loci are regulated in an H3K9me-dependent manner. Finally, consistent with previous observations that H3K9 methylation depends on CG methylation47,49,50, 24nt-siRNA levels at clsy1,2-dependent loci were also reduced in the met1 and ddm1 mutants (Fig. 6g). Although some roles for CG methylation independent of H3K9 methylation cannot be excluded, these findings support a model in which CLSY1 and CLSY2 mediate the interaction between SHH1 and Pol-IV to control 24nt-siRNA production at clsy1,2-dependent loci in a highly H3K9 methylation-dependent manner.
Figure 6. The CLSY1/2 and CLSY3/4 proteins regulate Pol-IV in connection with repressive chromatin marks.
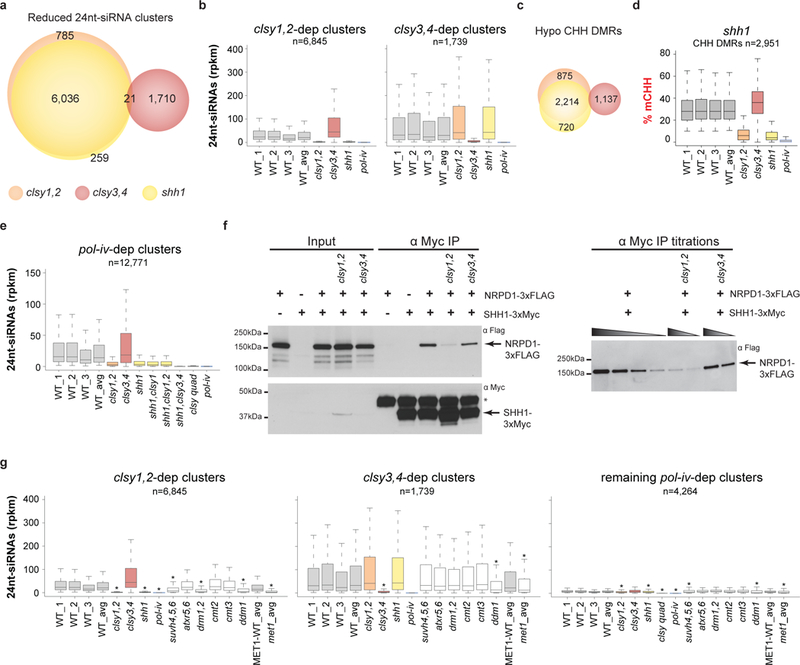
(a and c) Scaled Venn diagrams of reduced 24nt-siRNA clusters and hypo CHH DMRs, respectively, showing the relationships between loci regulated by the shh1 single and clsy1,2 or clsy3,4 double mutants. For readability, only overlaps >20 are labeled. (b, e and g) Boxplots showing the levels of 24nt-siRNAs at the reduced 24nt-siRNA clusters identified in the clsy double mutants, b and g, or pol-iv single mutant, e, in the genotypes indicated below. In g, the asterisks (*) indicate a p-value <2.2e−16 calculated using a Wilcoxon sum test relative to the WT_avg control for all samples except for met1, which was calculated relative to the MET1-WT control. The p-values for all other samples are >0.05. These boxplots represent a single experiment including three independent WT controls. (d) Boxplot showing the levels of CHH methylation at the hypo CHH DMRs identified in the shh1 single mutant as compared to the clsy double mutants and pol-iv. This boxplot represents a single experiment including three independent WT controls. (f) Cropped Western blots showing the levels of NRPD1–3xFLAG or SHH1–3xMyc from co-immunoprecipitation (co-IP) experiments in the genetic backgrounds indicated above each lane. For each blot the antibody (α) used is indicated in the upper right corner and the sizes of the protein markers are indicated on the left. An asterisk (*) marks a background band present in the α-Myc IP and the bands corresponding to the NRPD1–3xFLAG and SHH1–3xMyc proteins are marked with arrows. For the IP titrations, the gradient triangles represent a series of 2-fold dilutions starting from undiluted IP samples. Uncropped images are shown in Supplementary Fig. 8.
Alternatively, the genetic interactions between shh1, suvh4,5,6, and the clsy mutants clearly demonstrate that CLSY3 and CLSY4 facilitate Pol-IV function independent of both SHH1 and H3K9 methylation (Fig. 6 and Supplementary Fig. 9c, d). Thus, we sought to determine whether CLSY3 and CLSY4 rely on any other epigenetic features to facilitate Pol-IV localization. To this end, 24nt-siRNA levels at clsy3,4-dependent loci were profiled in mutants controlling DNA methylation in all three contexts (drm1,2, cmt2, and cmt3), as well as mutants controlling the deposition of several known repressive histone modifications (suvh4,5,6 and atxr5,6; Supplementary Table 2). Of these mutants, only those controlling methylation in the CG context, ddm1 and met1, showed significantly reduced 24nt-siRNA levels (Fig. 6g), demonstrating that 24nt-siRNA production at loci controlled by CLSY3 and CLSY4 depends on CG methylation. However, it remains unknown whether these CLSYs rely directly on CG methylation or if they instead depend on other chromatin modifications or heterochromatin features that, like H3K9 methylation, rely on CG methylation.
Discussion
A major unanswered question in the field of epigenetics is how specific patterns of DNA methylation are generated and modulated—a critical step in deciphering epigenetic processes in both normal development and disease. As Pol-IV “kicks off” the RdDM pathway by initiating the biogenesis of 24nt-siRNAs, which ultimately guide DNA methylation in a sequence specific manner, understanding the regulation of this polymerase is essential to determining how specific DNA methylation patterns are generated. Previously, we identified the CLSY proteins as components of the Pol-IV complex(es)35 and here we show they act as locus-specific regulators of both 24nt-siRNA production and DNA methylation. This locus-specific behavior differs from previously characterized RdDM factors, as none rival the degree or comprehensive nature of the specificities displayed by the CLSY family. Overall, these findings not only shed light on the regulation of Pol-IV, but also uncover a long-sought layer of complexity within the RdDM pathway that enables the locus-specific control of DNA methylation patterns.
Investigation into the locus-specific behavior of the CLSYs revealed that different chromatin modifications are required for the production of 24nt-siRNAs depending on the CLSY proteins involved. For loci regulated by CLSY3 and CLSY4, CG methylation is required, but the connections (direct or indirect) between CG methylation and CLSY3 and CLSY4 remain to be elucidated. Perhaps further characterization of factors like HISTONE DEACETYLASE 6, which participate in both the CG methylation and RdDM pathways51,52, will shed light on these connections. For loci regulated by CLSY1 and CLSY2, our analyses provide a direct link to H3K9 methylation, as these two CLSY proteins are required for the association between the H3K9me2 reader, SHH1, and the Pol-IV complex. Finally, for the remaining loci that are redundantly controlled by all four CLSYs, it remains unclear whether different modes of regulation are employed as these 24nt-siRNA clusters are expressed at low levels in all mutants tested (Fig. 6g). Together, these results reveal that specific chromatin features, including, but not limited to, CG and H3K9 methylation, can be leveraged to generate locus-specific control over DNA methylation. Indeed, such mechanisms appear to be conserved between plants and animals, as a similar, though less locus-specific, mechanism was recently identified in Drosophila wherein the core transcriptional machinery was shown to be linked to repressive histone marks in connection with the H3K9me3 reader, Rhino53. Furthermore, given the widespread conservation of SNF2 chromatin remodeling factors in general, and the specific conservation of the CLSY family in crops including rice54,55 and maize54, we anticipate that our findings will be informative for understanding the mechanisms governing the establishment of specific DNA methylation patterns in diverse organisms.
Online methods
Plant Materials
All plant materials used in this study were in the Columbia-0 (Col-0) ecotype and unless otherwise specified, plants were grown in Salk greenhouses with long-day conditions. Newly characterized CLSY T-DNA insertion mutant lines include: clsy1–10 (SALK_204860C)57, clsy3–2 (SALK_204501C)57, clsy4–2 (WiscDsLox472B9)58, clsy2–1 (GABI-Kat line 554E02)59, clsy2–2 (SAIL_484_F03)60, clsy3–1 (SALK_040366) and clsy4–1 (SALK_003876)57. Unless otherwise specified, the clsy1–7, clsy2–2, clsy3–1, and clsy4–1 alleles were utilized. Previously published mutant lines include: clsy1–7 (SALK_018319)61, nrpd1–4 (SALK_083051)62, shh1–1 (SALK_074540C)35, drm1–2,drm2–2 (drm1,2; SALK_031705 and SALK_150863, respectively)63, cmt2–7 (WiscDsLox7E02)47, cmt3–11 (SALK_148381)63, met1–3 (CS16387)64, ddm1–2 (EMS allele)65, atxr5,atxr6 (atxr5,6; SALK_130607 and SAIL_240_H01, respectively)66, and suvh4,suvh5,suvh6 (suvh4,5,6; SALK_41474, GABI-Kat 263C05, Garlic_1244_F04.b.1a, respectively)67. The pNRPD1::NRPD1–3xFLAG and pSHH1::SHH1–3xMyc transgenic lines were previously characterized in Law et al.35.
Small RNA isolation, library preparation, sequencing and data processing
Small RNA isolation:
4 un-opened flower buds (stage 12 and younger) from individual mutant plants as well as 3 individual wild-type (WT) controls were collected, frozen in liquid nitrogen and kept at −80°C until use. The total RNA extraction and small RNA enrichment were performed as previously described68 with the following minor modifications: (1) for the small RNA enrichment step an equal volume of 20% polyethylene glycol 8000/2M NaCl was added to each total RNA sample and (2) the ZR-small RNA ladder (Zymo Research, Cat# R1090) was used to determine the gel region corresponding to the 17–29 nucleotide (nt) size range. The resulting small RNAs were then used for library preparation with the NEBNext Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs, Cat# E7300) following the user’s manual. The final library products were further purified using an 8% polyacrylamide gel to excise 130–160nt products relative to the pBR322 DNA-MspI Digest ladder (New England Biolabs, Cat# E7323AA). The libraries were pooled and sequenced (single end 50bp, SE50) on a HiSeq 2500 machine (Illumina).
Small RNA data processing and mapping:
The adapter sequences in the de-multiplexed small RNA (smRNA) sequencing reads were trimmed using cutadapt (v1.9.1) and reads longer than 15nt were kept for further analyses69. The trimmed smRNA reads were then mapped to the Arabidopsis genome (TAIR10) using ShortStack (v3.8.1)39, allowing 1 mismatch (--mismatches 1) and employing either the multi-mapping mode (--mmap f) or the no multi-mapping, none mode (--mmap n). Subsequently, a custom JSON filter (JSON_findPerfectMatches_and_TerminalMisMatches_v3) was employed to keep only perfectly matching reads and reads with a single mismatch at their 3’ terminus, as such mismatches were recently identified as a feature of Pol-IV-dependent RNAs7. The smRNA reads passing this custom filter were then used to call small RNA clusters using ShortStack with the —mincov 20, pad 100, --dicermin 21 and —dicermax 24 options. The number of 21–24nt smRNA clusters identified were extracted using a custom perl script (splitpancakesbysize_shortStack_v3.8.1.pl) and are presented in Supplementary Table 3. To facilitate further analysis, the smRNA reads passing the JSON filter (bam file format) were used to generate a “Tag Directory” using the makeTagDirectory script from the HOMER (Hypergeometric Optimization of Motif EnRichment) package70. The Tag Directory was then split into sub-TagDirectories by smRNA size (20–25nt) using a custom perl script (splitTagDirectoryByLength.dev2.pl).
Differential expression analysis:
To identify a core set of 24nt-siRNA clusters in WT plants, common clusters from three WT replicates (WT_1, WT_2, and WT_3) were determined and the overlapping regions of each cluster were kept and merged using the mergePeaks.pl script from HOMER. All differential expression analyses were conducted based on these core clusters using DESeq242 as follows: First, the raw read counts (24nt) for each cluster in each genotype, including all the corresponding WT controls, were calculated using annotatePeaks.pl script (-raw -len 1) from HOMER. These read counts were then normalized using DESeq2 with modifications to the size factor estimation in order to relate counts to total mapped reads (i.e. smRNA reads of all sizes passing the JSON filter) rather than reads associated with specific features (e.g. 24nt reads) as follows: First, size factors were calculated for all the WT replicates using the DESeq2 default method. Then, these values were compared against the corresponding number of total mapped reads in order to derive an average number of mapped reads per size factor unit. With this average value, the number of mapped reads per sample was used to calculate the size factors for the individual mutants. The derived size factors and the matrix of raw read counts for each cluster in all the mutants, as well as the WT replicates, were then used as the input for DESeq2 to call mutant-dependent differential expression of 24nt-siRNA clusters (fold change (FC) ≥2, false discovery rate (FDR) ≤0.01). For 21nt- and 22nt-smRNAs, core clusters for each size class were determined as describe above and reduced clusters were identified using DESeq2 (FC≥2 and FDR≤0.01).
Visualization and analysis of 24nt-siRNA levels:
Downstream analyses were performed using HOMER and other tools as described below. Genome browser tracks of 24nt-siRNAs were generated using the HOMER makeUCSCfile script (-fragLength 24 -norm 10000000). For each boxplot, normalized smRNA read counts for the specified 24nt-siRNA clusters were calculated using the HOMER annotatePeaks.pl script (-rpkm -len 1) and the boxplot was drawn in R using RStudio (v1.0.136). For each heatmap, the HOMER annotatePeaks.pl script (-size 10000, -hist 600, -ghist and -len 24) was used to calculate the values for each set of 24nt-siRNA clusters. A pseudocount of 1 was then added to all the data, which was then log2 transformed and visualized using the Morpheus online tool. To generate the Venn diagrams, the unique identifiers of each mutant-dependent 24nt-siRNA cluster were imported and visualized using online tools for unscaled (VENNY2.1) or scaled (VennMaster71) Venn diagrams. For the chromosome-wide views of reduced 24nt-siRNA clusters, the pericentromeric heterochromatin genomic features were marked in the IGV genome browser based on previously published regions56 and the distribution of mutant-dependent reduced 24nt-siRNA clusters were determined by bedmap72 (--count --bp-ovr 1) in 100kb bins.
DNA isolation, MethylC-seq library construction, sequencing and data processing
DNA isolation:
0.1g of un-opened flower buds (stage 12 and younger) were collected from the same individual plants as used for the smRNA-seq analyses and genomic DNA was isolated using the DNeasy Plant Mini Kit (Qiagen, Cat# 69104). 2.0μg of purified genomic DNA was then used to generate MethylC-seq libraries as described in Li et al.73. The resulting libraries were pooled and sequenced (single end 50bp, SE50) on a HiSeq 2500 machine (Illumina).
MethylC-seq data processing:
MethylC-seq reads were trimmed and analyzed using BS-Seeker2 (v2.0.9). Briefly, reads were mapped against the C-to-T converted TAIR10 reference genome using the bs_seeker2-align.py script with the bowtie aligner, allowing 2 mismatches (-m 2). Clonal reads were removed using the MarkDuplicates function within picard tools (http://broadinstitute.github.io/picard). The mapped reads were then used to calculate the methylation level at each cytosine using the bs_seeker2-call_methylation.py script, requiring a minimum coverage of 4 reads (-r 4). From these analyses, the mapability, coverage, global percent CG, CHG, and CHH methylation levels, and non-conversion rates for each library were determined (See Supplementary Table 6). In addition, wiggle (wig) files showing the percent CG, CHG and CHH methylation for each genotype at single nucleotide resolution were generated using a custom perl script (Bsseeker2_2_wiggleV2.pl) based on the BS-Seeker2 Cgmap output files.
DMR calling:
To call differentially methylated regions (DMRs) several custom perl scripts were used (Bsseeker2_methylCall2Cytosine.pl, CytosineTo100bpBin.pl, GetOnlyCommonBins.pl, DMRFtestFDR.R, SplittingDMRs.pl, and SplittingDMR2Bed.pl). These scripts identified DMRs in the CG, CHG or CHH contexts based on pair-wise comparisons between each mutant and three independent WT data sets in 100bp non-overlapping bins using the following criteria: (1) only bins with ≥4 cytosines in the specified context were included, (2) only bins in which there was sufficient coverage in both genotypes being compared were included (i.e. ≥4 reads over the required 4 cytosines in the specific context), and (3) only bins with a fold change of 40%, 20% or 10% methylation in the CG, CHG, and CHH contexts, respectively, with an adjusted p-value of ≤0.01 relative to all three WT controls were called as DMRs.
Visualization and analysis of DNA methylation levels:
The overlaps between the clsy DMRs and reduced 24nt-siRNA clusters were determined using bedops72 (--element-of 1) and the heatmap indicating the percent overlap in Figure 3a was generated using the Morpheus online tool. The overlaps between DMRs called in different genotypes were determined using bedops (--element-of 1) and visualized as Venn diagrams generated as described for the smRNA analyses. The DNA methylation levels over reduced 24nt-siRNA clusters or DMRs were determined using the HOMER tool suite. For these analyses, Tag Directories were made from each of the methyl CG, CHG, and CHH wig files in two steps. First, the wig files were converted into the tag format recognized by HOMER using a custom script (parseWig_noChr.v2.pl) and then the Tag Directories were generated using the HOMER makeTagDirectory script (-precision 3 -t). Using these Tag Directories, the percent methylation over the desired genomic regions (e.g. reduced 24nt-siRNA clusters or DMRs) were determined using the HOMER annotatePeaks.pl script (-ratio -len 1). These methylation levels were then used to generate boxplots in R using RStudio.
RNA isolation, real-time PCR, mRNAseq library construction, sequencing and data processing
RNA isolation:
4 un-opened flower buds (stage 12 and younger) were collected from the same individual plants as used for the smRNA-seq and MethylC-seq analyses and total RNA was isolated using the Quick-RNA MiniPrep kit (Zymo Research, Cat# R1055). For the Reverse Transcriptase quantitative PCR (RT-qPCR) assays, 1.0μg of DNase I-treated total RNA reverse transcribed using High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied biosystems, Cat#4374967). The RT-qPCR assays were conducted using the iTaq Universal SYBR Green Mix (Bio-Rad, Cat#172–5124) with CFX384 Real-Time System (Bio-Rad). The cDNA levels of target genes were normalized to ACTIN2 and the error bars represent the standard error between three technical replicates. The primer pairs for the CLSY genes are listed in Supplementary Table 11. For the RNA-seq libraries, 2.0μg of total RNA from each genotype was used to generate mRNA-seq libraries using the NEBNext Ultra RNA Library Prep Kit (New England Biolabs, Cat# E7530). All size selection and clean-up steps were preformed using Sera-Mag Magnetic SpeedBeads (Thermo Scientific, Cat# 65152105050250). The resulting libraries were pooled and sequenced (single end 50bp, SE50) on a HiSeq 2500 machine (Illumina).
mRNA-seq data processing:
mRNA-seq reads were mapped to the TAIR10 reference genome using STAR (v2.5.0c)74 allowing 2 mismatches (--outFilterMismatchNmax 2) and including only uniquely mapped reads (--outFilterMultimapNmax 1). The sorted bam files were then used to generate Tag Directories using the HOMER makeTagDirectory script and the TAIR10 annotation was used to obtain the raw read counts for each gene (or repeat) using the HOMER analyzeRepeats.pl script with different options for genes (rna tair10 -raw -condenseGenes -len 1) or repeats (repeats tair10 -raw -len 1). Differentially expressed genes and repeats were then determined by DESeq2 using the default parameters and employing a FC threshold of ≥2 with an FDR ≤0.05 compared to all WT controls. To identify previously un-annotated transcripts regulated by the RdDM pathway, the mRNA-seq data was re-analyzed using TopHat2 (v2.1.1)75. Briefly, the mRNA-seq reads were mapped to TAIR10 genome by TopHat2 and the output bam files were used to identify transcript assemblies using Cufflinks (v2.2.1) without using the TAIR10 annotation. The resulting transcript assemblies were merged using Cuffmerge to get the de novo transcript units (in GTF form), which were further converted into bed format using the gtfToGenePred and genePredToBed scripts. The converted bed file was then used to obtain raw read counts for each transcript using the HOMER annotatePeaks.pl script (-raw -len 1). The differentially expressed transcripts were then determined by DESeq2 as described above for genes and repeats. These differentially expressed transcripts were then compared with TAIR10 genes and repeats, and non-overlapped transcripts were designated as un-annotated transcripts.
Visualization and analysis of pol-iv-dependent up-regulated loci:
To visualize loci up-regulated in pol-iv mutants (including genes, repeats and unannotated transcripts) that are also up-regulated in the clsy mutants, a profile plot of pol-iv-dependent up-regulated loci was generated as follows: (1) From the DESeq2 output files, the up-regulated loci in pol-iv were determined (FC ≥2 and FDR ≤0.05). (2) Then FC and FDR values for this set of loci in each mutant were extracted from the DESeq2 output files and filtered with the threshold (FC ≥2 and FDR ≤0.05). The FC values passing the filter were kept and all other values were replaced with “NA”. (3) The resulting data matrix was organized by tidyr (gather), color-coded tidyr based on FC value in pol-iv and visualized by ggplot2 (geom_point) in RStudio.
To determine the correlation between 24nt-siRNAs, DNA methylation and gene expression, the set of 177 up-regulated loci in pol-iv was used to generate heatmaps and profile plots using deepTools (v2.4.0)76. For the mRNA-seq data, the sorted bam files derived from the STAR mapping were first compared to WT controls using the bamCompare tool (--ratio=log2 --scaleFactorsMethod SES -bs 10). For 24nt-siRNA data, the 24nt-siRNA bedGraph files generated by HOMER were converted into bigwig format using the bedGraphToBigWig script with default options and then compared to WT controls using the bigwigCompare tool (--ratio=log2 -bs 10). For DNA methylation data, the wig files were first converted into bigwig format using the wigToBigWig tool and then the difference between the mutants and WT controls were calculated using the bigwigCompare tool (--ratio=subtract). The resulting bigwig files were then used to calculate a matrix using computeMatrix tool (scale-regions -a 2000 --regionBodyLength 2000 -b 2000 -bs=100). Finally, the data was plotted using the plotHeatmap tool. To determine the overlaps between the up-regulated loci indicated in Fig. 4a, and reduced 24nt-siRNA clusters and hypo DMRs identified in the pol-iv, clsy quadruple, and clsy2 mutants, the bedops --element-of 1 function was used, and to determine the number of DMRs overlapping with each locus, the bedmap counts function was used (--count --bp-ovr 1).
FWA transformation assay
A previously described FWA plasmid35 was used for floral dipping77 into the following genotypes: Col-0, clsy1–7, clsy2–1, clsy3–1, clsy4–1, clsy1–7,2–1, clsy3–1,4–1, clsy1–7,2–1,3–1,4–1 and nrpd1–4. The resulting T0 seeds were selected on Linsmaier and Skoog (LS) media with 0.6% agar and Basta (25mg/L) for one week and the resistant plants were transferred to soil and grown in a growth chamber at 22°C, on a 16h light and 8h dark cycle, with 70% humidity. The number of rosette leaves produced prior to bolting were determined and plotted using R in RStudio and the p-values were calculated using Wilcoxon rank sum tests.
ChIP, ChIP-seq library preparation, sequencing and data processing
ChIP:
A FLAG-tagged Pol-IV line, pNRPD1::NRPD1–3xFLAG in an nrpd1–4 mutant background35 was crossed into the following mutants: clsy1–7, clsy2–2, clsy3–1, clsy4–1, clsy1–7,2–2, clsy3–1,4–1, clsy1–7,2–2,3–1,4–1 and shh1–1. The progeny of these crosses were screened by drug-resistance to select for lines homozygous for the tagged Pol-IV transgene and genotyped by PCR to isolate lines homozygous for each mutant background, including the nprd1–4 allele. The ChIP was performed as previously described in Law et al.34. For each genotype, 2.0g of un-opened flower buds (stage 12 and younger) were collected, ground to a fine powder in liquid nitrogen, and crosslinked with 1% formaldehyde (Sigma, Cat# F8775) for 20min at room temperature with slow rotation. The chromatin was then fragmented to ~500bp by sonication and the lysate was incubated with anti-FLAG M2 Magnetic beads (Sigma, Cat# M8823) at 4°C for 2h. The beads were washed 5 times, for 5min at 4°C and eluted twice using 150μL of 3xFLAG peptide [0.1 mg/mL] (Sigma, Cat# F4799) at room temperature, rotating for 15min each time. The crosslinking was reversed by incubation at 65°C overnight, and the DNA was purified using a Phenol:Chloroform:Isoamyl Alcohol kit (Thermo Scientific, Cat# 17908). ChIP libraries were prepared from the resulting DNA using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, Cat# 7645) and sequenced (single end 50bp, SE50) on a HiSeq 2500 machine (Illumina).
ChIP-seq data analysis:
Pol-IV ChIP sequencing data were aligned to TAIR10 reference genome using bowtie (v1.1.0)78 allowing 2 mismatches (-v 2) and including multi-mapping reads (--all --best --strata). Pol-IV ChIP enrichment relative to WT controls at the identified 24nt-siRNA clusters were visualized using deepTools (v2.4.0)76. Briefly the sorted bam files derived from bowtie mapping were compared to WT controls using the bamCompare tools (reference-point --referencePoint center --ratio=log2 –scaleFactorsMethod SES -bs=10) and the resulting bigwig files were used to generate a data matrix using the computeMatrix tool (reference-point --referencePoint center -a 5000 -b 5000 -bs=10). Finally, the data was plotted using the plotHeatmap or plotProfile tools. The H3 and H3K9me2 ChIP sequencing data sets were downloaded from the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession numbers GSM2837360 and GSM283735948, and were mapped and analyzed as described for the Pol-IV ChIP, including visualization using deepTools.
Co-IP and Western blotting
For these experiments, the plant lines described above in which the pNRPD1::NRPD1–3xFLAG construct was crossed into the clsy1,2 or clsy3,4 mutants were super-transformed with a previously described Myc-tagged SHH1 plasmid, pSHH1::SHH1–3xMyc35, using the floral dip method77. The resulting T0 seeds were selected on LS media with 0.6% agar and hygromycin (25mg/L) for one week and the resistant plants were then transferred to soil and grown under long-day conditions at 22°C. The two tagged control lines, pSHH1::NRPD1–3xMyc and pNRPD1::NRPD1–3xFLAG were also grown under the same conditions. Approximately 0.5g of flower buds were collected from each genotype and ground into a fine powder in liquid nitrogen with 1mL Lysis buffer (50mM Tris, pH 7.6; 150mM NaCl; 5mM MgCl2; 10% Glycerol; 0.1% NP40) containing protease inhibitors. The lysate was cleared by centrifugation at 13,000rpm for 10min at 4°C. The supernatants were incubated with 2.0μL anti-c-Myc 4A6 antibody (Millipore, Cat# 05–724) and 30μL protein G Dynabeads (Invitrogen, Cat# 10004D) at 4°C for 2h rotating slowly. The beads were then washed 5 times, for 5min, with 1mL of Lysis buffer and resuspended in 50μL SDS-PAGE loading buffer. 16μL of input and bead eluate were resolved on a 7.5% TGX Precast Protein Gel (Bio-Rad, Cat# 3450005). The proteins were then detected by Western blotting using either the anti-FLAG M2 Monoclonal Antibody-Peroxidase Conjugated antibody (Sigma, Cat# A8592) at a dilution of 1:5,000 or the anti-c-Myc 4A6 antibody at a dilution of 1:2,000. Goat anti-mouse IgG horseradish peroxidase (Bio-Rad, Cat# 170–6516) was used at a dilution of 1:10,000 as the secondary antibody. All Western blots were developed using the ECL2 Western Blotting Substrate (Pierce, Cat# 80196).
Supplementary Material
Acknowledgements
We thank lab members and colleagues for helpful comments and discussions, the Salk NGS core for sequencing, the bioinformatics core for technical support, and L. and C. Greenfield for charitable contributions. This work was supported by the NIH (GM112966) and Hearst Foundation to J. Law. M. Zhou was funded by the Pioneer Fund Postdoctoral Award. M. Zhou and AM. Palanca were funded by postdoctoral fellowships from the Glenn Center for Aging Research at the Salk Institute. This work was also supported by the NGS Core Facility and the Integrative Genomics and Bioinformatics Core Facility at the Salk Institute with funding from NIH-NCI CCSG: P30 014195, the Glenn Center for Aging Research at the Salk Institute, the Chapman Foundation and the Helmsley Charitable Trust.
Footnotes
Competing Financial Interests Statement
The authors declare no competing interests.
Code availability
All custom codes are provided as Supplementary Data Set 1.
Data availability
Illumina sequencing data (smRNA-seq, MethylC-seq, mRNA-seq, and ChIP-seq) has been deposited in the NCBI Gene Expression Omnibus (GEO) and are accessible through the GEO series accession number GSE99694.
URLs:
The Morpheus online tool: https://software.broadinstitute.org/morpheus/
VENNY2.1: http://bioinfogp.cnb.csic.es/tools/venny/
gtfToGenePred: http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/gtfToGenePred
genePredToBed: http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/genePredToBed
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
References
- 1.Castel SE & Martienssen RA RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14, 100–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holoch D & Moazed D RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16, 71–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haag JR & Pikaard CS Multisubunit RNA polymerases IV and V: purveyors of non-coding RNA for plant gene silencing. Nat Rev Mol Cell Biol 12, 483–92 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Zhou M & Law JA RNA Pol IV and V in gene silencing: Rebel polymerases evolving away from Pol II’s rules. Curr Opin Plant Biol 27, 154–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Law JA & Jacobsen SE Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11, 204–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matzke MA & Mosher RA RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nature reviews. Genetics 15, 394–408 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Zhai J et al. A One Precursor One siRNA Model for Pol IV-Dependent siRNA Biogenesis. Cell 163, 445–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blevins T et al. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. Elife 4(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Z et al. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2, E104 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mallory A & Vaucheret H Form, function, and regulation of ARGONAUTE proteins. Plant Cell 22, 3879–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wierzbicki AT, Haag JR & Pikaard CS Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135, 635–48 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Shami M et al. Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE-binding platforms in RNAi-related components. Genes Dev 21, 2539–44 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li CF et al. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 126, 93–106 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Wierzbicki AT, Ream TS, Haag JR & Pikaard CS RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat Genet 41, 630–4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong X et al. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 157, 1050–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bohmdorfer G et al. RNA-directed DNA methylation requires stepwise binding of silencing factors to long non-coding RNA. Plant J 79, 181–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calarco JP et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151, 194–205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibarra CA et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337, 1360–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawakatsu T et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat Plants 2, 16058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slotkin RK et al. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136, 461–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang WW, Kobayashi T, Irie N, Dietmann S & Surani MA Specification and epigenetic programming of the human germ line. Nat Rev Genet 17, 585–600 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Seisenberger S et al. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc Lond B Biol Sci 368, 20110330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh TF et al. Genome-wide demethylation of Arabidopsis endosperm. Science 324, 1451–4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widman N, Feng S, Jacobsen SE & Pellegrini M Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 9, 236–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz MD et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stricker SH, Koferle A & Beck S From profiles to function in epigenomics. Nat Rev Genet 18, 51–66 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Heard E & Martienssen RA Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pikaard CS & Mittelsten Scheid O Epigenetic regulation in plants. Cold Spring Harb Perspect Biol 6, a019315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Springer NM & Schmitz RJ Exploiting induced and natural epigenetic variation for crop improvement. Nat Rev Genet 18, 563–575 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Smith ZD & Meissner A DNA methylation: roles in mammalian development. Nat Rev Genet 14, 204–20 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Klutstein M, Nejman D, Greenfield R & Cedar H DNA Methylation in Cancer and Aging. Cancer Res 76, 3446–50 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Liu J et al. An atypical component of RNA-directed DNA methylation machinery has both DNA methylation-dependent and -independent roles in locus-specific transcriptional gene silencing. Cell Res 21, 1691–700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H et al. DTF1 is a core component of RNA-directed DNA methylation and may assist in the recruitment of Pol IV. Proc Natl Acad Sci U S A 110, 8290–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law JA et al. Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature 498, 385–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law JA, Vashisht AA, Wohlschlegel JA & Jacobsen SE SHH1, a homeodomain protein required for DNA methylation, as well as RDR2, RDM4, and chromatin remodeling factors, associate with RNA polymerase IV. PLoS Genet 7, e1002195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith LM et al. An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis. Plant Cell 19, 1507–21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg MV et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics 6, 344–54 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stroud H, Greenberg MV, Feng S, Bernatavichute YV & Jacobsen SE Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson NR, Yeoh JM, Coruh C & Axtell MJ Improved Placement of Multi-mapping Small RNAs. G3 (Bethesda) 6, 2103–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mosher RA, Schwach F, Studholme D & Baulcombe DC PolIVb influences RNA-directed DNA methylation independently of its role in siRNA biogenesis. Proc Natl Acad Sci U S A 105, 3145–50 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Henderson IR, Lu C, Green PJ & Jacobsen SE Role of RNA polymerase IV in plant small RNA metabolism. Proc Natl Acad Sci U S A 104, 4536–41 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cokus SJ et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lister R et al. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 133, 523–536 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bewick AJ & Schmitz RJ Gene body DNA methylation in plants. Curr Opin Plant Biol 36, 103–110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan SW et al. RNA silencing genes control de novo DNA methylation. Science 303, 1336 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Stroud H et al. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nature structural & molecular biology 21, 64–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inagaki S et al. Gene-body chromatin modification dynamics mediate epigenome differentiation in Arabidopsis. EMBO J 36, 970–980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soppe WJ et al. DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO J 21, 6549–59 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson L, Cao X & Jacobsen S Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12, 1360–7 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Blevins T et al. A two-step process for epigenetic inheritance in Arabidopsis. Mol Cell 54, 30–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim JM, To TK & Seki M An epigenetic integrator: new insights into genome regulation, environmental stress responses and developmental controls by histone deacetylase 6. Plant Cell Physiol 53, 794–800 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Andersen PR, Tirian L, Vunjak M & Brennecke J A heterochromatin-dependent transcription machinery drives piRNA expression. Nature advance online publication (2017). [DOI] [PMC free article] [PubMed]
- 54.Hale CJ, Stonaker JL, Gross SM & Hollick JB A novel Snf2 protein maintains trans-generational regulatory states established by paramutation in maize. PLoS Biol 5, e275 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Y et al. Analysis of rice Snf2 family proteins and their potential roles in epigenetic regulation. Plant Physiol Biochem 70, 33–42 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Yelina NE et al. Epigenetic remodeling of meiotic crossover frequency in Arabidopsis thaliana DNA methyltransferase mutants. PLoS Genet 8, e1002844 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alonso JM et al. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301, 653–7 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Woody ST, Austin-Phillips S, Amasino RM & Krysan PJ The WiscDsLox T-DNA collection: an arabidopsis community resource generated by using an improved high-throughput T-DNA sequencing pipeline. J Plant Res 120, 157–65 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Kleinboelting N, Huep G, Kloetgen A, Viehoever P & Weisshaar B GABI-Kat SimpleSearch: new features of the Arabidopsis thaliana T-DNA mutant database. Nucleic Acids Res 40, D1211–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sessions A et al. A high-throughput Arabidopsis reverse genetics system. Plant Cell 14, 2985–94 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dunoyer P et al. An endogenous, systemic RNAi pathway in plants. EMBO J 29, 1699–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Herr AJ, Jensen MB, Dalmay T & Baulcombe DC RNA polymerase IV directs silencing of endogenous DNA. Science 308, 118–120 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Chan SW et al. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in arabidopsis. PLoS Genet 2, e83 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saze H, Mittelsten Scheid O & Paszkowski J Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34, 65–9 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Vongs A, Kakutani T, Martienssen RA & Richards EJ Arabidopsis thaliana DNA methylation mutants. Science 260, 1926–8 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Jacob Y et al. ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16, 763–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ebbs ML & Bender J Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 18, 1166–76 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu C, Meyers BC & Green PJ Construction of small RNA cDNA libraries for deep sequencing. Methods 43, 110–7 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 3 (2011). [Google Scholar]
- 70.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kestler HA et al. VennMaster: area-proportional Euler diagrams for functional GO analysis of microarrays. BMC Bioinformatics 9, 67 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neph S et al. BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li D et al. The MBD7 complex promotes expression of methylated transgenes without significantly altering their methylation status. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clough SJ & Bent AF Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16, 735–43 (1998). [DOI] [PubMed] [Google Scholar]
- 78.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.