

Recent work applying neutron and X-ray scattering to elucidate the molecular mechanisms of volatile anesthetics is reviewed.
Keywords: anesthesia, membranes, neutron scattering, synaptic plasticity, lipid rafts
Abstract
This article reviews recent work in applying neutron and X-ray scattering towards the elucidation of the molecular mechanisms of volatile anesthetics. Experimental results on domain mixing in ternary lipid mixtures, and the influence of volatile anesthetics and hydrostatic pressure are placed in the contexts of ion-channel function and receptor trafficking at the postsynaptic density.
1. Introduction
The ability of a great many gases and volatile compounds to produce general anesthesia remains a mystery. These include noble gases (xenon, krypton and argon), small hydrocarbons (n-alkanes and cyclopropane), halogenated carbon compounds and even nitrogen, which is well known for nitrogen narcosis in deep-water diving. The mystery is nearly as old as modern chemistry. The first report of inhalational anesthesia was by a young Humphry Davy in 1800 (Davy, 1800 ▸). He had synthesized nitrous oxide and after inhaling some, stumbled across the laboratory, banging his shin into a chair. In his report, he noted that the impact did not hurt and that nitrous oxide might be useful for preventing pain during surgery. Despite the enthusiasm for recreational uses of nitrous oxide, inhalational anesthesia did not come into clinical practice until after the demonstration of ether anesthesia at Massachusetts General Hospital in 1846. In subsequent years, a large variety of volatile compounds were discovered to have anesthetic properties, including chloroform and simple alkanes (Franks, 2006 ▸). Inhalational anesthetics in current use include halogenated compounds (halothane, sevoflurane and isoflurane) as well as nitrous oxide and xenon (Sanders et al., 2003 ▸). Pharmacological and other data on many anesthetics of theoretical and clinical importance, or usefulness in research, have been tabulated (Roth & Miller, 1986 ▸). Molecular mechanisms of action continue to be investigated, but understanding general anesthesia produced by gases and volatile compounds is still a challenge (Sonner & Cantor, 2013 ▸).
In about 1900, the Meyer–Overton correlation between anesthetic potency and the solubility of anesthetic compounds in oils was established, suggesting a connection between the lipids of cell membranes and anesthetic action (Meyer, 1899 ▸; Overton, 1901 ▸). An early proposal was that anesthetics decreased the viscosity of cell membranes. However, measurements indicated that clinical concentrations of anesthetics did not appreciably modify membrane viscosity. The next proposal to garner support was the idea that anesthetic partitioning into membranes would expand and thicken the membrane. This proposal was bolstered by the observation that the application of 100 bar pressure reversed anesthesia in experimental animals (Johnson & Flagler, 1950 ▸; Lever et al., 1971 ▸). This pressure reversal was thought to result from pressure returning the membrane to its normal thickness. However, experiments at the Institut Laue–Langevin in the 1980s demonstrated that hydrostatic pressure thickens fluid membranes, rather than thinning them (Braganza & Worcester, 1986 ▸). Other experimental evidence against this theory was obtained in 1979 by X-ray and neutron diffraction studies of artificial bilayer membranes (dimyristoylphosphatidylcholine with 40% molar cholesterol) to which anesthetics were applied. These studies demonstrated that doses of anesthetics well above clinical concentrations produced no significant changes in the structure of this bilayer membrane (Franks & Lieb, 1979 ▸).
At about the same time, the actions of nonvolatile anesthetics on neurotransmitter receptors and ion channels were being investigated. Substantial progress was made with the discovery that barbiturates bind specifically to γ-aminobutyric acid (GABA) receptors (Macdonald & Barker, 1978 ▸; Nicoll, 1978 ▸), producing enhanced inhibition throughout the central nervous system. Opioids were also found to bind to specific receptors that mediated their effects (Lord et al., 1977 ▸; Martin et al., 1976 ▸). This indicated that standard ligand–receptor complex formation may be important in anesthesia. However, these compounds were not anesthetics of the volatile type which give the Meyer–Overton correlation. Despite considerable effort investigating receptor binding by volatile anesthetics, no receptor has been identified as necessary and sufficient for the action of inhalational anesthetics. Receptors that bind some inhalational anesthetics only bind a small subset of these compounds. Volatile anesthetics do affect neurotransmitter receptors and ion channels directly or indirectly (Sonner & Cantor, 2013 ▸). Experiments on mice with specific receptors or ion channels eliminated through genetic knockout demonstrate that these mice require higher doses of anesthetics but still become anesthetized (Heurteaux et al., 2004 ▸; Himukashi et al., 2005 ▸; Quinlan et al., 1998 ▸). The phenomena of pressure reversal and additivity of doses of different anesthetics (Eger et al., 2008 ▸) are also difficult to explain in terms of anesthetic binding to receptors. For example, the pressures required to affect enzyme–substrate binding are at least an order of magnitude greater than those required for reversal of anesthesia (Boonyaratanakornkit et al., 2002 ▸; Girard et al., 2010 ▸).
A new approach to understanding the effects of volatile anesthetics has recently emerged. Over the past two decades, separate lines of investigation converged to give a new perspective on lipids in cell membranes. Ternary mixtures of lipids, which are mixtures of saturated and unsaturated phospholipids combined with cholesterol, were found to produce membranes with distinct liquid ordered and liquid disordered domains (Veatch & Keller, 2003 ▸). The phase boundaries (with composition and temperature) and physical properties of these mixtures have been studied in considerable detail (Feigenson, 2009 ▸). Nearly simultaneously, the extraction of cell membranes with gentle detergents revealed a relatively detergent-resistant, cholesterol-rich fraction of lipids that also contained a very significant proportion of cell signaling proteins (Lingwood & Simons, 2010 ▸). The model of a homogeneous lipid bilayer has therefore been replaced by that of a complex assembly of lipids and proteins arranged in a bilayer with functional segregation of signaling proteins into cholesterol-rich lipid domains called rafts (Lingwood & Simons, 2010 ▸). While domains of different lipid phases are not visible with light microscopy in mammalian cells at physiological temperature (with possible exceptions such as the postsynaptic density), stimulated emission depletion-fluorescence correlation spectroscopy of live mammalian cells demonstrates local trapping of lipids in domains at the nanometre scale for periods of up to 10 s (Vicidomini et al., 2015 ▸). Thus, there has been intense interest in exploring the properties of lipid-raft mixtures, ternary mixtures of phospholipids and cholesterol that exhibit phase separation into distinct liquid ordered and liquid disordered domains (Feigenson, 2009 ▸; Veatch & Keller, 2003 ▸).
2. Diffraction from multilayers of ternary mixtures
In view of these developments, we revisited the issue of inhalational anesthetic effects on lipid membranes and focused on using raft-forming lipid mixtures. Following initial X-ray studies, interest soon converged on xenon as a particularly good test of anesthetic effects. Neutron scattering studies were required for this work because the large X-ray attenuation of xenon at about 1 bar made X-ray studies extremely difficult. The minimum alveolar concentration (MAC) for xenon is 0.6–0.7 bar (Dickinson et al., 2007 ▸).
Highly oriented multi-lamellar stacks of lipid bilayers at 1–2 mg cm−2 were formed on thin microscope cover glass substrates as described previously (Weinrich et al., 2012 ▸) by slow evaporation of solvent from solutions in ethanol or 80% ethanol/20% water at 37–40°C in air, followed by 15 min in vacuum. We constructed a sealed chamber of aluminium, pressure-tested to 8 bar, to enclose the lipid samples. The chamber temperature was controlled by circulating a thermocontrolled liquid through its base and top. Temperature was monitored with sensors in the bath and attached to different points on the chamber. Humidity was maintained at 98% with saturated salt solution (Worcester et al., 1996 ▸). Xenon gas was slowly introduced into the chamber through a bubbler filled with distilled water to aid constant hydration of the lipid multilayers. Particular care was taken when changing pressure in the chamber to avoid rapid pressure changes and maintain constant temperature and humidity. Glass substrates with deposited multilayers were rotated in the incident cold-neutron beam (wavelength λ = 5.0 Å, Δλ/λ = 0.01) through angles θ relative to the incident beam (Dura et al., 2006 ▸). Diffracted neutrons were detected at angles 2θ (referred to as θ–2θ scans). Thus, the neutron momentum transfer (q = 4πsinθ/λ) normal to the bilayer plane probes the structure of the membrane along the bilayer normal. A high-efficiency pencil detector registered two series of lamellar diffraction peaks which corresponded to stacked Lo and Ld domains with different Bragg spacings.
The effect of the noble gas xenon on the first-order peaks of dipalmitoylphosphatidylcholine (DPPC)/dioleoylphosphatidylcholine (DOPC)/cholesterol and DOPC/sphingomyelin (SPM)/cholesterol samples is illustrated in Fig. 1 ▸ at 28 and 27°C, respectively, and 98% humidity (Weinrich & Worcester, 2013 ▸). Diffraction from the Lo phase predominates in air or helium. Xenon produced a marked increase in the Ld phase in both the first- and second-order peak intensities. At 3.2 MAC, the ratio of Lo to Ld intensities in the DPPC sample decreased by 21% and at 6.4 MAC it decreased by 35%. The sphingomyelin sample behaved similarly, with a 39% change in the ratio of Lo to Ld intensities in response to xenon at 4.6 MAC. The complex structure of the aligned multilayer stacks, with the connecting elements between Lo and Ld domains not oriented to the bilayer normal (Mills et al., 2008 ▸), is partially responsible for the increase in the Ld phase. Reducing the pressure of xenon to atmospheric pressure resulted in a prompt reversal of the intensity ratio, giving an increase in Lo and a decrease in Ld, with the ratio for 1 bar xenon for both mixtures remaining about 14% below that observed in air.
Figure 1.

(a) Neutron diffraction. First-order diffraction peaks for a multilayered sample of deuterated d62-DPPC/DOPC (1:1) with 20% cholesterol. The first peak is from the Lo phase and the second is from the Ld phase. The black trace is for helium at atmospheric pressure, the red trace is for xenon at two times atmospheric pressure (3.2 MAC) and the blue trace is for xenon at four times atmospheric pressure (6.4 MAC). Experiments were performed at 28°C and 98% relative humidity. Traces are Gaussian fits to data. q = 4πsinθ/λ is the neutron momentum transfer. Bars indicate standard errors (1 atm = 101 kPa). (b) Neutron diffraction. First-order diffraction peaks for a multilayered sample of deuterated d31-palmitoyl sphingomyelin/DOPC (1:1) with 20% cholesterol. The first peak is from the Lo phase and the second is from the Ld phase. The black trace is for air and the red trace is for xenon at three times atmospheric pressure (4.6 MAC). Experiments were performed at 27°C and 98% relative humidity. Traces are Gaussian fits to data. Bars indicate standard errors (1 atm = 101 kPa). Reprinted with permission from Weinrich & Worcester (2013 ▸). Copyright 2013 American Chemical Society.
As illustrated in Fig. 2 ▸, the changes in the ratios of first-order Lo and Ld diffraction-peak areas increase linearly with anesthetic concentrations for both xenon and nitrous oxide (MAC = 1 bar) and both of the lipid-raft mixtures. Such changes are also observed in response to all other volatile anesthetics that we have tested, including halogenated anesthetics (halothane, isoflurane and chloroform), as well as hexane, and are of comparable magnitude (Table 1 ▸). These studies were mainly performed by X-ray diffraction. Stable vapor concentrations of volatile liquid anesthetics were obtained using solutions of anesthetics in hexadecane. Such solutions are close to ideal and the vapor pressures follow Raoult’s law, so the solution provides a reservoir of anesthetic at essentially constant chemical potential (King et al., 1985 ▸; White et al., 1981 ▸). Vapor concentrations were sampled with gas syringes and measured with an Agilent 6850 chromatograph to be sure of the concentrations. X-ray diffraction was performed with a Rigaku Ultima-III diffractometer fitted with a sealed chamber, the base of which had Peltier temperature control. Samples were prepared as for neutron diffraction. Introducing anesthetic/hexadecane solutions into the sealed chamber with a syringe did not change the temperature by more than 0.1°C and the humidity remained constant to within 0.5%.
Figure 2.

Change in the ratio of first-order peak areas versus concentration as MAC. Solid squares are for an SPM/DOPC/cholesterol mixture with xenon, solid triangles are for an SPM/DOPC/cholesterol mixture with nitrous oxide, open squares are for a DPPC/DOPC/cholesterol mixture with xenon and open triangles are for a DPPC/DOPC/cholesterol mixture with nitrous oxide. The MAC used for xenon is 0.63 bar and that for nitrous oxide is 1.04 bar. The dashed line represents the least-squares linear fit; the adjusted r 2 is 0.89. Bars represent standard errors. Reprinted with permission from Weinrich & Worcester (2013 ▸). Copyright 2013 American Chemical Society.
Table 1. X-ray diffraction data for lipid-raft mixtures in the presence of halogenated anesthetics.
%Δ1H indicates the percentage change in the ratio of first-order diffraction peaks Lo/Ld upon adding anesthetic. %MAC indicates the concentration used as a percentage of the minimum alveolar concentration necessary for humans. At least two experiments on separate samples were performed for each anesthetic. Mean values are tabulated with standard errors. Reprinted with permission from Weinrich & Worcester (2013 ▸). Copyright 2013 American Chemical Society.
| Mixture | Anesthetic | %Δ1H | %MAC |
|---|---|---|---|
| DOPC:DPPC/cholesterol 1:1/0.2 | |||
| Halothane | 31 ± 3 | 270 ± 60 | |
| Isoflurane | 17 ± 2 | 121 ± 22 | |
| Chloroform | 22 ± 2 | 167 ± 17 | |
| DOPC:SPM/cholesterol 1:1/0.2 | |||
| Halothane | 21 ± 5 | 540 ± 20 | |
| Isoflurane | 20 ± 3 | 300 ± 60 | |
| Chloroform | 25 ± 8 | 262 ± 26 |
Fig. 3 ▸ illustrates the time course of the mixing/demixing transition of DPPC/DOPC/cholesterol in response to hexane introduced as a vapor from a mixture with hexadecane. As the concentration of hexane rises in the chamber with evaporation from the hexadecane solution, the ratio of ordered to disordered peak heights falls, reaching a stable and persistent minimum that was maintained for many hours. When the hexane/hexadecane solution was withdrawn and replaced with pure hexadecane more than 12 h later, the ratio recovered to the starting value as the hexane was absorbed into the hexadecane.
Figure 3.
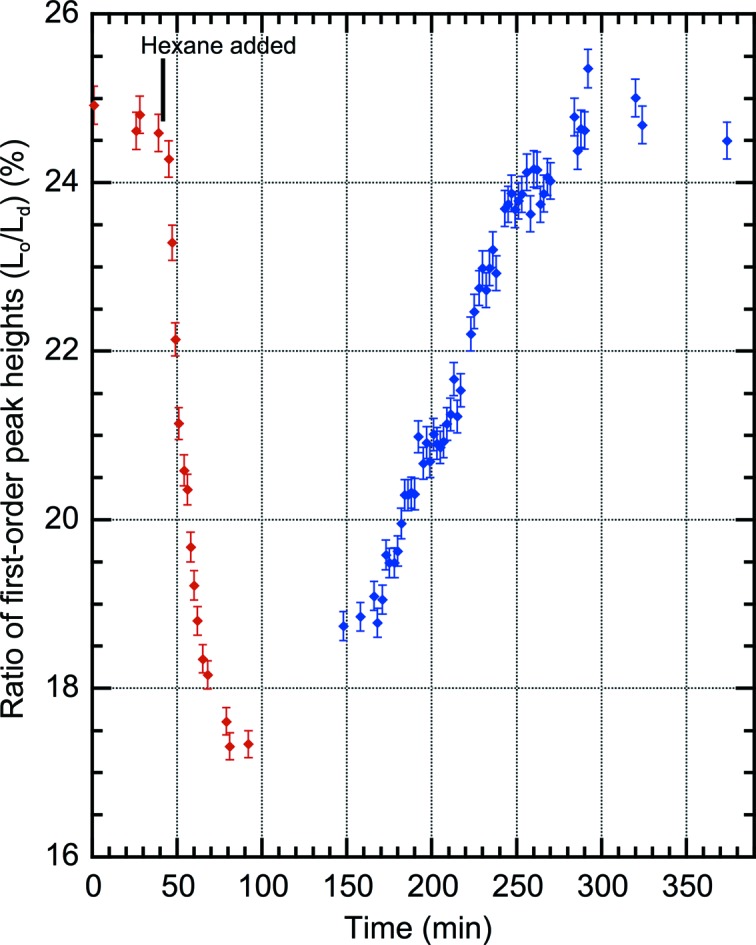
Time course of first-order X-ray diffraction for a multilayered sample of 1:1 DPPC/DOPC with 20% cholesterol hydrated at 98% relative humidity at 27°C. Data are plotted as the ratio of first-order peak heights for the two domains: Lo/Ld. At 40 min, n-hexane was introduced into the sample chamber as a solution in n-hexadecane (1/10 by volume) to maintain an n-hexane partial pressure of 0.042 bar, which is the partial pressure required for narcosis in mice (White et al., 1981 ▸). The decrease in Lo/Ld results from mixing of Lo lipids into the Ld phase. At about 100 min equilibrium is reached and the sample chamber was kept undisturbed for 12 h to test stability. Subsequent data (blue) have 720 min subtracted from the time in order to include the results upon removal of n-hexane by replacing the hexane solution with n-hexadecane on the same plot. Anesthetic doses for n-hexane (MAC) in vivo are uncertain because one metabolic product is a neurotoxin. Error bars indicate standard error.
Using the fully deuterated form of DPPC and in-plane neutron diffraction, we were able to demonstrate that the shifts observed are independent of the stacking of domains in multilayers and are truly dependent on the movement of lipid between phases (Weinrich et al., 2012 ▸). Fig. 4 ▸ illustrates in-plane neutron diffraction by oriented d62-DPPC/dilauroylphosphatidylcholine (DLPC) multilayer membranes in the region of the chain diffraction. Scattering in the q range from 1.4 to 1.5 Å−1 depends strongly on the deuterium content of the fatty-acid chains. The negative neutron scattering length of hydrogen makes the net scattering length of the CH2 group small and negative, whereas that for CD2 is large and positive. Consequently, only the chains of d62-phospholipids in noncrystalline phospholipids produce observable in-plane neutron scattering. For a mixture of H and D chains, such as in a d62-DPPC/DLPC mixture, the intensity of the chain diffraction depends on the size and the number of domains consisting of primarily d62 chains and on the order of the d62 chains within these domains. Separation of d62-DPPC into two-dimensional domains increases the intensity, while mixing of the H and D lipids decreases the intensity. This in-plane neutron diffraction method does not require vertical alignment of the lipid domains into three-dimensional domains as required by lamellar diffraction, and is especially effective for observing changes in the composition of lateral domains (Stamm, 1982 ▸). The mixing transition for d62-DPPC/DLPC is broad, beginning at about 22°C and extending to 31°C. Other techniques have found similarly broad mixing transitions in other lipid mixtures (Trudell et al., 1975 ▸). However, the breadth of these transitions is not apparent in the published phase diagrams for these mixtures (Feigenson, 2009 ▸). Halothane at 1.5 mol% (about 2 MAC) produced a marked shift of about 5°C in the mixing transition towards lower temperatures (an order of magnitude larger than the shift in the main melting transition of pure DPPC induced by anesthetic concentrations of octanol; Heimburg & Jackson, 2007 ▸), while 7.5 mol% F6 (1,2-dichlorohexafluorocyclobutane, a non-immobilizer control) produced a shift of about half this magnitude.
Figure 4.

Neutron diffraction of 1:1 d62-DPPC/DLPC oriented multilayers on glass with q (1.4–1.5 Å−1) directed parallel to the plane of the membrane. Neutron diffraction in this plane is generated by the d62-lipid chains and is greatly reduced by mixing of d62-lipid chains with h62-lipid chains so that d62-lipid chains are no longer adjacent. The midpoint of the phase-mixing transition for the native lipid mixture is about 29°C (squares) and corresponds well to the phase diagrams established by calorimetry (van Dijck et al., 1977 ▸). The addition of 1.5 mol% halothane (measured at 27°C) decreases the transition temperature to 25°C (circles), but 7.5 mol% F6 only decreases the transition to 27°C (triangles). Inset: neutron counts collected in θ/2θ scans across the chain diffraction peak for the 1:1 d62 DPPC/DLPC oriented multilayers at three temperatures: at the beginning, midpoint and end of the temperature scan (top, middle and bottom traces, respectively) in the absence of halothane. Peaks were integrated in the range q = 1.46–1.52 Å−1 and background counts outside this region were subtracted to obtain the plotted chain diffraction intensity. Error bars represent one standard deviation and are from counting statistics. Reprinted with permission from Weinrich et al. (2012 ▸). Copyright 2012 American Chemical Society.
3. Small-angle neutron scattering
The use of lipid multilayers has been a very convenient technique and most of our measurements have been made in this way, with very extensive use of X-rays. However, there are some disadvantages.
(i) Selective stacking requires less than full hydration (usually 98% humidity is used) and domains that are larger than nanoscale.
(ii) The Bragg spacings of the Lo and Ld phases must be different by at least about 2% to resolve the two series of peaks in the lower orders.
(iii) Including proteins to study their influence on the transitions produces difficulties associated with stack formation and lack of full hydration.
All of these problems have been encountered in recent studies that have investigated extending our work to include proteins and the use of more physiological lipid mixtures. Small-angle neutron scattering (SANS) provides a method that avoids all of these problems (Pencer et al., 2007 ▸).
We used the SANS technique to measure the effect of hydrostatic pressure on mixing/demixing transitions in small unilamellar vesicles (SUVs) of DPPC/DOPC/cholesterol (Worcester & Weinrich, 2015 ▸). Both fatty-acid chains in the DPPC of the lipid mixture were fully deuterated. The D2O/H2O solvent mixture was prepared to have the same neutron scattering length density as the lipid mixture when fully mixed at 33°C, thus minimizing the scattering intensity at this temperature. As demixing occurs at lower temperatures, the contrast between the solvent and both types of domains increases, resulting in increased scattering. Small unilamellar vesicles (SUVs) were made from the same mixture (2:2:1) of DOPC/DPPC/cholesterol by sonication (Masui et al., 2008 ▸) of the dried lipid mixture resuspended in 45% D2O/H2O, taking particular care to maintain the lipid mixture above the mixing temperature at all times (Pencer et al., 2007 ▸). The clear suspension of vesicles was measured by dynamic light scattering to confirm a uniform distribution of vesicles of 30 nm diameter. The lipid concentration for neutron scattering was 5 mg ml−1. Scattering was performed on the 30 m SANS instrument at NIST (Glinka et al., 1998 ▸). The detector was placed at 5 m and the neutron wavelength was 6.0 Å (10% full-width at half-maximum). A pressure cell with sapphire windows was temperature-controlled with a circulating bath and a thermocouple was used to monitor the cell temperature. Temperature steps of 2°C were used and the cell was allowed to equilibrate for 1 h before measurements. Hydrostatic pressure was applied using a hand pump. Data were analyzed using the NIST SANS IGOR macros (Kline, 2006 ▸).
Fig. 5 ▸(a) displays the raw intensity data obtained from vesicles at 25°C. As the pressure increased from atmospheric pressure to 31 MPa, the scattering increased correspondingly, indicating the growth of two distinct domains with different scattering length densities. Quantification of the domain separation is best accomplished using the scattering invariant Q (Pencer et al., 2005 ▸). 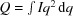 and is a function of the relative proportions of the two domains (Pencer et al., 2007 ▸). Fig. 5 ▸(b) demonstrates the relationship between temperature, pressure and the scattering invariant Q.
and is a function of the relative proportions of the two domains (Pencer et al., 2007 ▸). Fig. 5 ▸(b) demonstrates the relationship between temperature, pressure and the scattering invariant Q.
Figure 5.

(a) Small-angle neutron scattering intensity versus
q for d62-DPPC/DOPC/cholesterol (2:2:1 molar ratio) unilamellar vesicles in D2O/H2O (45% D2O) at 25°C matched to the scattering length for vesicles at atmospheric pressure at 33°C: pink inverted triangles, 0 MPa; green triangles, 10 MPa; cyan circles, 21 MPa; purple squares, 31 MPa. The detector is at 5 m and the neutron wavelength is 6 Å. (b) Scattering invariant Q (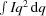 ) for the SANS data as a function of pressure and temperature. The dotted line is drawn to guide the eye through the points at 0 MPa. The arrow indicates ΔT, while ΔP is the MPa value at 25°C and the ratio of these values gives dT/dP, assuming linearity. The dT/dP values thus obtained are 0.26, 0.24 and 0.20°C MPa−1 from top to bottom. Error bars indicate standard error. Reprinted with permission from Worcester & Weinrich (2015 ▸). Copyright 2015 American Chemical Society.
) for the SANS data as a function of pressure and temperature. The dotted line is drawn to guide the eye through the points at 0 MPa. The arrow indicates ΔT, while ΔP is the MPa value at 25°C and the ratio of these values gives dT/dP, assuming linearity. The dT/dP values thus obtained are 0.26, 0.24 and 0.20°C MPa−1 from top to bottom. Error bars indicate standard error. Reprinted with permission from Worcester & Weinrich (2015 ▸). Copyright 2015 American Chemical Society.
The scattering invariant Q increases nonlinearly up to 10 MPa. Beginning at 21 and 31 MPa, there appears to be a roughly constant relationship between temperature and pressure: −1°C ≃ 4 MPa, i.e. an increase of pressure by 4 MPa reduces the invariant scattering equivalently to a 1°C reduction in temperature. Thus, measurements on the mixing transition gave values of dT/dP in the range −0.2 to −0.3°C MPa−1.
Previous work using differential scanning calorimetry and electron spin resonance probes on saturated phospholipids (Albon & Sturtevant, 1978 ▸; Ipsen & Mouritsen, 1988 ▸) and binary mixtures of saturated phospholipids with cholesterol (Shimshick & McConnell, 1973 ▸) also demonstrated the antagonistic effects of temperature and pressure on membrane fluidity, for which dT/dP in a general sense is the change in temperature required to offset the effects of a change in pressure. These studies yielded dT/dP values in the range −0.13 to −0.21°C MPa−1 (also consistent with studies on synaptic and myelin fractions of goldfish brain; Chong et al., 1983 ▸). Pressure and anesthetics produce antagonistic effects on the transitions between gel and liquid disordered states in these model systems (Kamaya et al., 1979 ▸; Mountcastle et al., 1978 ▸; Trudell et al., 1973 ▸, 1975 ▸). Our work extends these findings to more physiologically relevant ternary lipid mixtures, demonstrating that hydrostatic pressure at physiologically relevant magnitudes antagonizes domain mixing in a ternary lipid mixture and is consistent with the physiological significance of anesthetic effects on domain mixing. As mentioned above, most mammalian cells do not exhibit microscopically visible domains. The influence of the cytoskeleton (Arumugam et al., 2015 ▸) or other complex membrane-protein interactions may be responsible for inhibiting domain formation. However, blebs derived from mammalian cell plasma membranes demonstrate domains and anesthetics promote domain mixing in this system (Gray et al., 2013 ▸). which is quantitatively similar to what we have observed in model mixtures.
4. Discussion and implications
Lipid modulation of protein function can occur through several mechanisms (Dart, 2010 ▸). Many membrane signaling proteins are localized to lipid rafts (Dart, 2010 ▸). Ion channels from many different families are sensitive to the lipid environment surrounding them (Poveda et al., 2014 ▸). Nicotinic acetylcholine receptors (nAchRs) require an anionic lipid and cholesterol for full functioning (Morales et al., 2006 ▸). Anionic lipids bind to KcsA, the bacterial potassium channel, modulating the conformation of the channel (Molina et al., 2015 ▸). Lipids are integral to the structure of the Kv1.2 potassium channel (Long et al., 2007 ▸). Metabotropic glutamate receptors are modulated by cholesterol (Kumari et al., 2013 ▸), and many ion channels are affected by phosphatidylinositol 4,5-biphosphate (PIP2; Poveda et al., 2014 ▸). Phosphatidylinositol 3,4,5-triphosphate (PIP3) is localized to membrane rafts (Hansen, 2015 ▸) and maintains α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor clustering (Arendt et al., 2010 ▸). Beyond the modulation of individual channels, lipids also affect channel–channel interactions and thus the clustering of channels. In the case of KcsA (Molina et al., 2015 ▸) and nAchR (Barrantes, 2014 ▸), clustering appears to have very significant effects on channel function.
Mixing/demixing transitions in membranes are second-order phase transitions, entailing compositional changes in the separate domains throughout the transition. Thus, such phase changes could affect ion channels which are sensitive to lipid composition. We recently demonstrated that gramicidin, a model ion channel, has a much shorter lifetime in the ordered phase of a lipid-raft mixture, and that phase mixing shifts the distribution of lifetimes (Weinrich et al., 2017 ▸). Fig. 6 ▸ illustrates the presence of two separate populations of gramicidin channels with different characteristic lifetimes in a planar bilayer of DOPC/sphingomyelin/cholesterol. These separate populations exist well above the mixing temperature for this lipid mixture as determined by fluorescence microscopy, thus demonstrating the effects of nanoscopic lipid domains on ion-channel function.
Figure 6.

(a) Gramicidin channels in 5:2 DOPC/cholesterol (CHL) at 30°C, 1 M KCl, 100 mV. The horizontal bar is 2 s in length. The vertical bar is 3 pA. (b) Logarithmically binned histogram of gramicidin lifetimes in 5:2 DOPC/CHL at 30°C and single-exponential fit to log probability. (c) Gramicidin channels in 1:1:1 DOPC/SPM/CHL at 32°C (7°C above the miscibility transition for this mixture), 1 M KCl, 100 mV. The horizontal bar is 2 s in length. The vertical bar is 3 pA. (d) Logarithmically binned histogram of gramicidin lifetimes in 1:1:1 DOPC/SPM/CHL at 32°C with single-exponential (green trace) and double-exponential fits (fuchsia trace). Reprinted with permission from Weinrich et al. (2017 ▸). Copyright 2017 Royal Society of Chemistry.
Additionally, membrane tension modulates N-methyl-d-aspartate (NMDA) receptors (Kloda et al., 2007 ▸), as well as potassium channels (Schmidt & MacKinnon, 2008 ▸) and sodium channels (Morris & Juranka, 2007 ▸). Anesthetic-induced changes along the phase-transition boundary can produce changes in the lateral pressure profile. Such changes in the lateral pressure profile could significantly modulate the function of transmembrane proteins (Cantor, 1997 ▸).
Glutamate is the most important excitatory neurotransmitter in the mammalian brain and AMPA receptors are the most prominent excitatory receptors. These receptors undergo extensive trafficking along the plasma membrane and undergo endocytosis, recycling and transport back to the postsynaptic density (Anggono & Huganir, 2012 ▸). This changes the synaptic strength by changing the numbers of AMPA receptors at the synapse, including synapses without AMPA receptors, which are named ‘silent synapses’. These aspects of synaptic strength have been of great interest to research on memory formation for many years, but their possible connections to the actions of volatile anesthetics have not previously been investigated.
AMPA receptors are tightly clustered in postsynaptic domains of about 70 nm in diameter comprised of about 20 channels. The clusters are highly dynamic and can form or disperse in minutes (Nair et al., 2013 ▸). Binding and crowding by scaffolding proteins within the postsynaptic density are responsible for limiting the diffusion of AMPA receptors at the synapse (MacGillavry et al., 2011 ▸, 2013 ▸). Movements of AMPA receptors inside synapses are fast enough to impact synaptic transmission on a millisecond timescale and the regulation of AMPA receptor mobility impacts the fidelity of synaptic transmission (Choquet & Triller, 2013 ▸). Czöndör et al. (2012 ▸) developed a quantitative model of AMPA receptor trafficking at synapses, and found that decreases in the binding affinity between receptors and scaffold proteins resulted in decreased numbers of AMPA receptors at the synapse within seconds, while changes in the balance between endocytosis and exocytosis of receptors could result in significant changes in receptor levels within a minute. The important concept for anesthesia is that movement of AMPA receptors away from the postsynaptic density leaves them unresponsive to released neurotransmitter. Such movement may result from anesthetic action on lipid mixing/demixing or on receptor interactions with scaffold and other postsynaptic proteins.
The function of AMPA receptors is dependent on their interaction with transmembrane AMPA receptor regulatory proteins (TARPs), a family of membrane-associated proteins (Kott et al., 2007 ▸; Twomey et al., 2016 ▸). Most notably, stargazin binds to the membrane domains of the AMPA receptor (Ben-Yaacov et al., 2017 ▸). This TARP protein binds both to the membrane as well as to postsynaptic density protein 95 (PSD-95), another TARP protein. Decreased binding of stargazin to the membrane causes increased binding to PSD-95, resulting in relative immobilization of AMPA receptors in domains and an increased size of miniature excitatory postsynaptic currents (Hafner et al., 2015 ▸). Other glutamate receptor families also share this functional dependence on membrane-associated proteins (Sheng et al., 2015 ▸; Fang et al., 2003 ▸). Inhalational anesthetics disrupt protein interactions between the PDZ domains of several such proteins, including PSD-95 and potassium channel Kv1.4, as well as the the glutamate receptor subunit GluA2 (Tao et al., 2015 ▸) and the NMDA receptor (Fang et al., 2003 ▸). Also of interest in this regard is the demonstration that metabotropic glutamate receptors oscillate between active and resting states (Olofsson et al., 2014 ▸), suggesting the possibility that perturbations of the surrounding environment could shift their equilibrium points. It is apparent that there are multiple points involving protein–protein interactions necessary for AMPA receptor functioning and trafficking where perturbations of the lipid environment could be disruptive.
Regarding the side effects of anesthetics, it is clear that some off-target effects, for example cardiac arrhythmias and myocardial depression, stem from effects on non-neural ion channels (Zanghi & Jevtovic-Todorovic, 2017 ▸). Anesthetic disturbances in the development and maturation of the central nervous system may be owing to effects on glial cells as well as the effects of decreased synaptic activity on neurons (Zanghi & Jevtovic-Todorovic, 2017 ▸), some of which are potentially reversible through the application of receptor agonists (Huang et al., 2016 ▸). Regarding the most-feared issue of apoptosis in the developing brain (Ikonomidou et al., 2001 ▸; Zanghi & Jevtovic-Todorovic, 2017 ▸), the issue is less clear. Mitochondria are crucial components of the apoptotic pathway, releasing cytochrome c and other factors that direct the cell towards cell death (Boulbrima et al., 2016 ▸). VDAC, the voltage-dependent anion channel, is the most prevalent protein in the outer mitochondrial membrane and regulates the flow of ATP and metabolites (Colombini, 2012 ▸), maintaining cell viability. VDAC forms oligomeric clusters in lipid micelles (Ujwal et al., 2009 ▸) and native membranes (Gonçalves et al., 2007 ▸), and oligomeric clustering of VDAC in mitochondria is coupled to the induction of apoptosis (Boulbrima et al., 2016 ▸; Keinan et al., 2010 ▸). Lipid composition affects mitochondrial function (Mårtensson et al., 2017 ▸) and specifically the function of VDAC (Rostovtseva et al., 2006 ▸), and there are some suggestions of direct anesthetic interactions with VDAC (Weiser et al., 2014 ▸). It is not known whether the outer mitochondrial membrane exhibits phase separation at the nanoscopic level under normal conditions; however, there appears to be a complex relationship between the lipids in the mitochondrial outer membrane and proteins mediating the permeabilization of the outer membrane that leads to apoptosis (Patwardhan et al., 2016 ▸).
Further research is needed to elucidate the role of lipid domains and lipid mixing/demixing in the modulation of ion-channel function. This work may identify new strategies for the development and use of safer, more effective volatile anesthetics, of which xenon is currently of much interest. We emphasize that to produce anesthesia, the effects of volatile anesthetics on lipid mixing must ultimately disrupt the normal function of neurotransmitter receptors, and these effects in no way contravene their binding to specific sites on ion channels. Rather, anesthetic effects on lipid mixing/demixing are another, possibly very important, component of a complex physiological phenomenon.
Note added in proof. Pavel et al. (2018 ▸) recently demonstrated that anesthetics mobilize phospholipase D2 from lipid rafts to bind to TREK-1 channels, potentiating their activity through the production of phosphatidic acid. The increased TREK-1 activity hyperpolarizes the membrane, inhibiting the transmission of neural signaling. This observation provides a mechanism for anesthetic disruption of lipid rafts to affect ion-channel function. Anesthetic disruption of lipid rafts may mobilize other palmitoylated membrane proteins, thus affecting the functions of other ion channels and cell processes.
Acknowledgments
The authors declare no competing financial interests. The authors declare that they have no conflicts of interest with the contents of this article. The identification of any commercial product or trade name does not imply any endorsement or recommendation by the National Institute of Standards and Technology.
Funding Statement
This work was funded by National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development grant .
References
- Albon, N. & Sturtevant, J. M. (1978). Proc. Natl Acad. Sci. USA, 75, 2258–2260. [DOI] [PMC free article] [PubMed]
- Anggono, V. & Huganir, R. L. (2012). Curr. Opin. Neurobiol. 22, 461–469. [DOI] [PMC free article] [PubMed]
- Arendt, K. L., Royo, M., Fernández-Monreal, M., Knafo, S., Petrok, C. N., Martens, J. R. & Esteban, J. A. (2010). Nature Neurosci. 13, 36–44. [DOI] [PMC free article] [PubMed]
- Arumugam, S., Petrov, E. P. & Schwille, P. (2015). Biophys. J. 108, 1104–1113. [DOI] [PMC free article] [PubMed]
- Barrantes, F. J. (2014). Front. Synaptic Neurosci. 6, 25. [DOI] [PMC free article] [PubMed]
- Ben-Yaacov, A., Gillor, M., Haham, T., Parsai, A., Qneibi, M. & Stern-Bach, Y. (2017). Neuron, 93, 1126–1137. [DOI] [PubMed]
- Boonyaratanakornkit, B. B., Park, C. B. & Clark, D. S. (2002). Biochim. Biophys. Acta, 1595, 235–249. [DOI] [PubMed]
- Boulbrima, A., Temple, D. & Psakis, G. (2016). Biochem. Soc. Trans. 44, 1531–1540. [DOI] [PubMed]
- Braganza, L. F. & Worcester, D. L. (1986). Biochemistry, 25, 7484–7488. [DOI] [PubMed]
- Cantor, R. S. (1997). Biochemistry, 36, 2339–2344. [DOI] [PubMed]
- Chong, P. L., Cossins, A. R. & Weber, G. (1983). Biochemistry, 22, 409–415. [DOI] [PubMed]
- Choquet, D. & Triller, A. (2013). Neuron, 80, 691–703. [DOI] [PubMed]
- Colombini, M. (2012). Biochim. Biophys. Acta, 1818, 1457–1465. [DOI] [PMC free article] [PubMed]
- Czöndör, K., Mondin, M., Garcia, M., Heine, M., Frischknecht, R., Choquet, D., Sibarita, J. B. & Thoumine, O. R. (2012). Proc. Natl Acad. Sci. USA, 109, 3522–3527. [DOI] [PMC free article] [PubMed]
- Dart, C. (2010). J. Physiol. 588, 3169–3178. [DOI] [PMC free article] [PubMed]
- Davy, H. (1800). Researches, Chemical and Philosophical Chiefly Concerning Nitrous Oxide, or Diphlogisticated Nitrous Air, and its Respiration. Bristol: Biggs & Cottle.
- Dickinson, R., Peterson, B. K., Banks, P., Simillis, C., Martin, J. C., Valenzuela, C. A., Maze, M. & Franks, N. P. (2007). Anesthesiology, 107, 756–767. [DOI] [PubMed]
- Dura, J. A., Pierce, D. J., Majkrzak, C. F., Maliszewskyj, N. C., McGillivray, D. J., Lösche, M., O’Donovan, K. V., Mihailescu, M., Perez-Salas, U., Worcester, D. L. & White, S. H. (2006). Rev. Sci. Instrum. 77, 074301. [DOI] [PMC free article] [PubMed]
- Eger, E. I., Raines, D. E., Shafer, S. L., Hemmings, H. C. Jr & Sonner, J. M. (2008). Anesth. Analg. 107, 832–848. [DOI] [PMC free article] [PubMed]
- Fang, M., Tao, Y.-X., He, F., Zhang, M., Levine, C. F., Mao, P., Tao, F., Chou, C.-L., Sadegh-Nasseri, S. & Johns, R. A. (2003). J. Biol. Chem. 278, 36669–36675. [DOI] [PubMed]
- Feigenson, G. W. (2009). Biochim. Biophys. Acta, 1788, 47–52. [DOI] [PMC free article] [PubMed]
- Franks, N. P. (2006). Br. J. Pharmacol. 147, S72–S81. [DOI] [PMC free article] [PubMed]
- Franks, N. P. & Lieb, W. R. (1979). J. Mol. Biol. 133, 469–500. [DOI] [PubMed]
- Girard, E. et al. (2010). Biophys. J. 98, 2365–2373. [DOI] [PMC free article] [PubMed]
- Glinka, C. J., Barker, J. G., Hammouda, B., Krueger, S., Moyer, J. J. & Orts, W. J. (1998). J. Appl. Cryst. 31, 430–445.
- Gonçalves, R. P., Buzhynskyy, N., Prima, V., Sturgis, J. N. & Scheuring, S. (2007). J. Mol. Biol. 369, 413–418. [DOI] [PubMed]
- Gray, E., Karslake, J., Machta, B. B. & Veatch, S. L. (2013). Biophys. J. 105, 2751–2759. [DOI] [PMC free article] [PubMed]
- Hafner, A. S., Penn, A. C., Grillo-Bosch, D., Retailleau, N., Poujol, C., Philippat, A., Coussen, F., Sainlos, M., Opazo, P. & Choquet, D. (2015). Neuron, 86, 475–489. [DOI] [PubMed]
- Hansen, S. B. (2015). Biochim. Biophys. Acta, 1851, 620–628. [DOI] [PMC free article] [PubMed]
- Heimburg, T. & Jackson, A. D. (2007). Biophys. J. 92, 3159–3165. [DOI] [PMC free article] [PubMed]
- Heurteaux, C., Guy, N., Laigle, C., Blondeau, N., Duprat, F., Mazzuca, M., Lang-Lazdunski, L., Widmann, C., Zanzouri, M., Romey, G. & Lazdunski, M. (2004). EMBO J. 23, 2684–2695. [DOI] [PMC free article] [PubMed]
- Himukashi, S., Miyazaki, Y., Takeshima, H., Koyanagi, S., Mukaida, K., Shichino, T., Uga, H. & Fukuda, K. (2005). Acta Anaesthesiol. Scand. 49, 771–773. [DOI] [PubMed]
- Huang, L., Cichon, J., Ninan, I. & Yang, G. (2016). Sci. Transl. Med. 8, 344ra85. [DOI] [PMC free article] [PubMed]
- Ikonomidou, C., Bittigau, P., Koch, C., Genz, K., Hoerster, F., Felderhoff-Mueser, U., Tenkova, T., Dikranian, K. & Olney, J. W. (2001). Biochem. Pharmacol. 62, 401–405. [DOI] [PubMed]
- Ipsen, J. H. & Mouritsen, O. G. (1988). Biochim. Biophys. Acta, 944, 121–134. [DOI] [PubMed]
- Johnson, F. H. & Flagler, E. A. (1950). Science, 112, 91–92. [DOI] [PubMed]
- Kamaya, H., Ueda, I., Moore, P. S. & Eyring, H. (1979). Biochim. Biophys. Acta, 550, 131–137. [DOI] [PubMed]
- Keinan, N., Tyomkin, D. & Shoshan-Barmatz, V. (2010). Mol. Cell. Biol. 30, 5698–5709. [DOI] [PMC free article] [PubMed]
- King, G. I., Jacobs, R. E. & White, S. H. (1985). Biochemistry, 24, 4637–4645. [DOI] [PubMed]
- Kline, S. R. (2006). J. Appl. Cryst. 39, 895–900.
- Kloda, A., Lua, L., Hall, R., Adams, D. J. & Martinac, B. (2007). Proc. Natl Acad. Sci. USA, 104, 1540–1545. [DOI] [PMC free article] [PubMed]
- Kott, S., Werner, M., Körber, C. & Hollmann, M. (2007). J. Neurosci. 27, 3780–3789. [DOI] [PMC free article] [PubMed]
- Kumari, R., Castillo, C. & Francesconi, A. (2013). J. Biol. Chem. 288, 32004–32019. [DOI] [PMC free article] [PubMed]
- Lever, M. J., Miller, K. W., Paton, W. D. & Smith, E. B. (1971). Nature (London), 231, 368–371. [DOI] [PubMed]
- Lingwood, D. & Simons, K. (2010). Science, 327, 46–50. [DOI] [PubMed]
- Long, S. B., Tao, X., Campbell, E. B. & MacKinnon, R. (2007). Nature (London), 450, 376–382. [DOI] [PubMed]
- Lord, J. A., Waterfield, A. A., Hughes, J. & Kosterlitz, H. W. (1977). Nature (London), 267, 495–499. [DOI] [PubMed]
- Macdonald, R. L. & Barker, J. L. (1978). Science, 200, 775–777. [DOI] [PubMed]
- MacGillavry, H. D., Kerr, J. M. & Blanpied, T. A. (2011). Mol. Cell. Neurosci. 48, 321–331. [DOI] [PMC free article] [PubMed]
- MacGillavry, H. D., Song, Y., Raghavachari, S. & Blanpied, T. A. (2013). Neuron, 78, 615–622. [DOI] [PMC free article] [PubMed]
- Mårtensson, C. U., Doan, K. N. & Becker, T. (2017). Biochim. Biophys. Acta, 1862, 102–113. [DOI] [PubMed]
- Martin, W. R., Eades, C. G., Thompson, J. A., Huppler, R. E. & Gilbert, P. E. (1976). J. Pharmacol. Exp. Ther. 197, 517–532. [PubMed]
- Masui, T., Urakami, N. & Imai, M. (2008). Eur. Phys. J. E Soft Matter, 27, 379–389. [DOI] [PubMed]
- Meyer, H. (1899). Arch. Exp. Pathol. Pharmakol. 42, 109–118.
- Mills, T. T., Tristram-Nagle, S., Heberle, F. A., Morales, N. F., Zhao, J., Wu, J., Toombes, G. E., Nagle, J. F. & Feigenson, G. W. (2008). Biophys. J. 95, 682–690. [DOI] [PMC free article] [PubMed]
- Molina, M. L., Giudici, A. M., Poveda, J. A., Fernández-Ballester, G., Montoya, E., Renart, M. L., Fernández, A. M., Encinar, J. A., Riquelme, G., Morales, A. & González-Ros, J. M. (2015). J. Biol. Chem. 290, 25745–25755. [DOI] [PMC free article] [PubMed]
- Morales, A., de Juan, E., Fernández-Carvajal, A. M., Martinez-Pinna, J., Poveda, J. A., Encinar, J. A., Ivorra, I. & González-Ros, J. M. (2006). J. Mol. Neurosci. 30, 5–6. [DOI] [PubMed]
- Morris, C. E. & Juranka, P. F. (2007). Biophys. J. 93, 822–833. [DOI] [PMC free article] [PubMed]
- Mountcastle, D. B., Biltonen, R. L. & Halsey, M. J. (1978). Proc. Natl Acad. Sci. USA, 75, 4906–4910. [DOI] [PMC free article] [PubMed]
- Nair, D., Hosy, E., Petersen, J. D., Constals, A., Giannone, G., Choquet, D. & Sibarita, J. B. (2013). J. Neurosci. 33, 13204–13224. [DOI] [PMC free article] [PubMed]
- Nicoll, R. A. (1978). Science, 199, 451–452. [DOI] [PubMed]
- Olofsson, L., Felekyan, S., Doumazane, E., Scholler, P., Fabre, L., Zwier, J. M., Rondard, P., Seidel, C. A., Pin, J.-P. & Margeat, E. (2014). Nature Commun. 5, 5206. [DOI] [PubMed]
- Overton, C. E. (1901). Studies of Narcosis. London: Chapman & Hall.
- Patwardhan, G. A., Beverly, L. J. & Siskind, L. J. (2016). J. Bioenerg. Biomembr. 48, 153–168. [DOI] [PMC free article] [PubMed]
- Pavel, M. A., Petersen, E. N., Lerner, R. A. & Hansen, S. B. (2018). bioRxiv, 313973. http://dx.doi.org/10.1101/313973.
- Pencer, J., Mills, T., Anghel, V., Krueger, S., Epand, R. M. & Katsaras, J. (2005). Eur. Phys. J. E Soft Matter, 18, 447–458. [DOI] [PubMed]
- Pencer, J., Mills, T. T., Kucerka, N., Nieh, M.-P. & Katsaras, J. (2007). Methods Mol. Biol. 398, 231–244. [DOI] [PubMed]
- Poveda, J. A., Giudici, A. M., Renart, M. L., Molina, M. L., Montoya, E., Fernández-Carvajal, A., Fernández-Ballester, G., Encinar, J. A. & González-Ros, J. M. (2014). Biochim. Biophys. Acta, 1838, 1560–1567. [DOI] [PubMed]
- Quinlan, J. J., Homanics, G. E. & Firestone, L. L. (1998). Anesthesiology, 88, 775–780. [DOI] [PubMed]
- Rostovtseva, T. K., Kazemi, N., Weinrich, M. & Bezrukov, S. M. (2006). J. Biol. Chem. 281, 37496–37506. [DOI] [PubMed]
- Roth, S. & Miller, K. (1986). Molecular and Cellular Mechansims of Anesthetics, pp. 455–470. New York: Plenum.
- Sanders, R. D., Franks, N. P. & Maze, M. (2003). Br. J. Anaesth. 91, 709–717. [DOI] [PubMed]
- Schmidt, D. & MacKinnon, R. (2008). Proc. Natl Acad. Sci. USA, 105, 19276–19281. [DOI] [PMC free article] [PubMed]
- Sheng, N., Shi, Y. S., Lomash, R. M., Roche, K. W. & Nicoll, R. A. (2015). Elife, 4, e11682. [DOI] [PMC free article] [PubMed]
- Shimshick, E. J. & McConnell, H. M. (1973). Biochem. Biophys. Res. Commun. 53, 446–451. [DOI] [PubMed]
- Sonner, J. M. & Cantor, R. S. (2013). Annu. Rev. Biophys. 42, 143–167. [DOI] [PubMed]
- Stamm, M. (1982). J. Polym. Sci. Polym. Phys. Ed. 20, 235–244.
- Tao, F., Chen, Q., Sato, Y., Skinner, J., Tang, P. & Johns, R. A. (2015). Anesthesiology, 122, 776–786. [DOI] [PMC free article] [PubMed]
- Trudell, J. R., Hubbell, W. L., Cohen, E. N. & Kendig, J. J. (1973). Anesthesiology, 38, 207–211. [DOI] [PubMed]
- Trudell, J. R., Payan, D. G., Chin, J. H. & Cohen, E. N. (1975). Proc. Natl Acad. Sci. USA, 72, 210–213. [DOI] [PMC free article] [PubMed]
- Twomey, E. C., Yelshanskaya, M. V., Grassucci, R. A., Frank, J. & Sobolevsky, A. I. (2016). Science, 353, 83–86. [DOI] [PMC free article] [PubMed]
- Ujwal, R., Cascio, D., Chaptal, V., Ping, P. & Abramson, J. (2009). Channels, 3, 167–170. [DOI] [PMC free article] [PubMed]
- Van Dijck, P. W. M., Kaper, A. J., Oonk, H. A. & De Gier, J. (1977). Biochim. Biophys. Acta, 470, 58–69. [DOI] [PubMed]
- Veatch, S. L. & Keller, S. L. (2003). Biophys. J. 85, 3074–3083. [DOI] [PMC free article] [PubMed]
- Vicidomini, G., Ta, H., Honigmann, A., Mueller, V., Clausen, M. P., Waithe, D., Galiani, S., Sezgin, E., Diaspro, A., Hell, S. W. & Eggeling, C. (2015). Nano Lett. 15, 5912–5918. [DOI] [PMC free article] [PubMed]
- Weinrich, M., Nanda, H., Worcester, D. L., Majkrzak, C. F., Maranville, B. B. & Bezrukov, S. M. (2012). Langmuir, 28, 4723–4728. [DOI] [PMC free article] [PubMed]
- Weinrich, M. & Worcester, D. L. (2013). J. Phys. Chem. B, 117, 16141–16147. [DOI] [PMC free article] [PubMed]
- Weinrich, M., Worcester, D. L. & Bezrukov, S. M. (2017). Nanoscale, 9, 13291–13297. [DOI] [PMC free article] [PubMed]
- Weiser, B. P., Bu, W., Wong, D. & Eckenhoff, R. G. (2014). FEBS Lett. 588, 4398–4403. [DOI] [PMC free article] [PubMed]
- White, S. H., King, G. I. & Cain, J. E. (1981). Nature (London), 290, 161–163.
- Worcester, D. L., Hamacher, K., Kaiser, H., Kulasekere, R. & Torbet, J. (1996). Basic Life Sci. 64, 215–226. [DOI] [PubMed]
- Worcester, D. L. & Weinrich, M. (2015). J. Phys. Chem. Lett. 6, 4417–4421. [DOI] [PubMed]
- Zanghi, C. N. & Jevtovic-Todorovic, V. (2017). Neurotoxicol. Teratol. 60, 24–32. [DOI] [PMC free article] [PubMed]