

Abstract
Angiogenesis is an essential process involved in various physiological, including placentation, and pathological, including cancer and endometriosis, processes. Melatonin (MLT), a well-known natural hormone secreted primarily in the pineal gland, is involved in regulating neoangiogenesis and inhibiting the development of a variety of cancer types, including lung and breast cancer. However, the specific mechanism of its anti-angiogenesis activity has not been systematically elucidated. In the present study, the effect of MLT on viability and angiogenesis of human umbilical vein endothelial cells (HUVECs), and the production of vascular endothelial growth factor (VEGF) and reactive oxygen species (ROS), under normoxia or hypoxia was analyzed using Cell Counting kit 8, tube formation, flow cytometry, ELISA and western blot assays. It was determined that the secretion of VEGF by HUVECs was significantly increased under hypoxia, while MLT selectively obstructed VEGF release as well as the production of ROS under hypoxia. Furthermore, MLT inhibited the viability of HUVECs in a dose-dependent manner and reversed the increase in cell viability and tube formation that was induced by hypoxia/VEGF/H2O2. Additionally, treatment with an inhibitor of hypoxia inducible factor (HIF)-1α (KC7F2) and MLT synergistically reduced the release of ROS and VEGF, and inhibited cell viability and tube formation of HUVECs. These observations demonstrate that MLT may serve dual roles in the inhibition of angiogenesis, as an antioxidant and a free radical scavenging agent. MLT suppresses the viability and angiogenesis of HUVECs through the downregulation of HIF-1α/ROS/VEGF. In summary, the present data indicate that MLT may be a potential anticancer agent in solid tumors with abundant blood vessels, particularly combined with KC7F2.
Keywords: melatonin, hypoxia, vascular endothelial growth factor, reactive oxygen species, angiogenesis
Introduction
Angiogenesis, an essential process in which new blood vessels are formed, not only serves a crucial role in physiological processes, including embryonic development and wound repair, but also in pathological conditions, including chronic inflammation, cancer, heart disease and diabetic retinopathy (1,2). It exerts an important effect in promoting aggressive tumor activity, including tumor growth, metastasis and invasion (3). Progression of cancer is characterized by the stimulation of pro-angiogenic factors, including hypoxia, vascular endothelial growth factor (VEGF), cytokine interleukin-6 and metalloproteinases, in the growing endothelial cells (4). VEGF has been demonstrated to be one of the most important angiogenesis growth factors that induces permeability, proliferation, migration and tube formation (5). Numerous stimuli, including hypoxia, cytokines and oxidative stress, can increase VEGF expression (2,6). The inhibition of neoangiogenesis is considered to be an important potential strategy for efficient and effective antitumor agents that prevent cancer proliferation and metastasis (7,8).
Reactive oxygen species (ROS), including H2O2 and superoxide anion radicals, are a family of reactive molecules that serve a pivotal role in numerous cellular processes, including metabolism, differentiation, proliferation and cell death, by regulating critical signaling pathways (9). ROS are generated under various physiological and pathological conditions, including inflammation (10) and ischemia (11). Excessive levels of ROS result in an imbalance in the metabolism of these reactive intermediates as a result of oxidative stress, and is associated with various diseases, including cancer and cardiovascular and neurodegenerative diseases (12). The hypoxia inducible factor (HIF)-1α response to hypoxia is associated with hypoxia-induced production of ROS (13). Accumulating evidence indicates that VEGF and ROS exert a critical role in vascular pathophysiology (14,15). However, the interaction between VEGF and ROS remains controversial. Certain studies have demonstrated that VEGF could take advantage of ROS as messenger intermediates downstream of VEGF receptor 2, thus affecting cellular proliferation and tube formation (16,17). Other studies indicated that ROS trigger the induction of VEGF, but the underlying interaction remains ambiguous (18-20). KC7F2, as an exogenous HIF-1α translation inhibitor, is involved in cancer-associated angiogenesis, and can be evaluated in terms of its anti-proliferative actions and effects on the metabolism of cancer cells (21). Furthermore, KC7F2 can also be used as a HIF-1α inhibitor (22).
Melatonin (MLT), a well-known natural hormone secreted primarily in the pineal gland, has attracted attention due to its various effects in numerous critical physiological, including sleep, the circadian rhythm and antioxidant cell protection against free radicals, and pathological, including tumor progression, processes (23). Accumulating evidence indicates that MLT is capable of directly scavenging radicals and radical-associated reactants, as well as indirectly stimulating anti-oxidative enzymes (24,25). It has been reported that MLT can inhibit the development of a variety of cancer types (26), including liver (27-29), lung (30,31), breast (32-34), pancreatic and brain cancer (35). However, the underlying mechanism of MLT varies in different cancer types (36). Additionally, reports indicated that MLT decreases the expression of HIF-1α and VEGF caused by different factors in various cultivated cells, particularly hypoxia-induced accumulation of HIF-1α protein and expression of VEGF (37-39). Accumulating evidence indicates that the antitumor effect of MLT is associated with the inhibition of angiogenesis (24,40,41). It has been reported that ROS produced under hypoxic conditions inactivate the PHD2 enzyme and thus prevent its degradation (42). Therefore, ROS stabilizes HIF-1α, allowing it to act as a transcription factor. However, the specific mechanism of anti-angiogenesis activity of MLT has not been systematically elucidated. Therefore, the aim of the present study was to investigate the effect of MLT on HUVECs and the associated underlying mechanism.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China) and cultured in a mixture containing RPMI-1640, 10% fetal bovine serum (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA), 100 IU/ml penicillin and 100 μg/ml streptomycin (Sangon Biotech Co., Ltd., Shanghai, China). The cells were incubated at 37°C in a humidified atmosphere which was maintained at 5% CO2. All experiments were performed with HUVECs that had been cultured for ≤6 passages.
Hypoxia treatment
Control cells were incubated for 6 or 24 h under normoxic conditions (21% O2, 5% CO2 and 74% N2 at 37°C) in a humidified incubator. According to the manufacturer’s protocols, hypoxic conditions (termed hypoxia) were induced using an airtight modular incubator chamber (Billups-Rothenberg, Inc., San Diego, CA, USA). Briefly, the cells [1×104 cells/well in cell viability assay; 2×104 cells/well in tube formation assay; and 1×105 cells/well in western blotting, ELISA and flow cytometry (FCM) assays] were sealed in the modular incubator chamber with a sterile 1X PBS reserve to maintain humidity, and then purged with a reduced O2 gas mixture (1% O2, 5% CO2 and 94% N2) at 37°C for 6 h or 24 h.
Cell viability assay
HUVECs were seeded into 96-well plates at a density of 1×104 cells/well. The conditioned medium was aspirated and 100 μl fresh Cell Counting kit 8 (CCK8) solution (Dojindo Molecular Technologies, Inc., Kunamoto, Japan) with serum-free RPMI-1640 medium was carefully added to each well. According to the manufacturer’s protocols, the plates were then incubated at 37°C for 0.5-4 h in the dark. The absorbance was periodically detected using a microplate reader at 450 nm.
Detection of ROS and VEGF by flow cytometry (FCM)
After treatment with or without MLT (1×10-5 M) under normoxia or hypoxia (according to the aforementioned conditions) at 37°C for 6 h, or pretreatment with H2O2 (50 μM; based on its cyto-toxicity, a dose of 50 μM H2O2 was selected for subsequent experiments) or VEGF (5 ng/ml) for 4 h, followed by MLT (1×10-5 M) at 37°C for 6 h, and then the release of ROS of HUVECs was detected by the Reactive Oxygen Species Assay kit (cat. no. S0033; Beyotime Institute of Biotechnology, Haimen, China). The levels of intracellular ROS generation were determined by incubating the cells in serum-free RPMI-1640 supplemented with 10 mM 2,7-dichlorofluorescein diacetate (DCFH-DA; Beyotime Institute of Biotechnology) or PBS (as the blank control) in the dark at 37°C for 30 min. DCFH-DA can be converted to the fluorescent dichlorofluorescein by ROS. Briefly, the cells were pretreated with different compounds (MLT, H2O2, VEGF, KC7F2, MLT plus VEGF, or MLT plus KC7F2; the concentrations of these compounds were the same as aforementioned) at 37°C for 6 h and/or conditions (hypoxia and normoxia, according to the aforementioned conditions). The cells were then rinsed and washed with cold PBS, followed by incubation with 1 mmol/l DCHF-DA in the dark at 37°C for 30 min. The cells were then trypsinized and washed with PBS again and resuspended in serum-free RPMI-1640 medium (1×106 cells/1 ml medium) for the FCM assay. FCM was performed on a Beckman Cyan flow cytometer (Beckman Coulter, Inc., Brea, CA, USA) using CellQuest software (version 7.1; Beckman Coulter, Inc.). At least 15,000 events were analyzed. All experiments were performed with biological triplicates and data are representative of at least three independent experiments.
Additionally, the median fluorescence intensity of VEGF was measured with a FCM assay. Cells were washed with PBS once and then trypsinized and collected by gently centrifugation at 350 × g at 4°C for 6 min. A commercial kit, eBioscience™ Intracellular Fixation/Perm Buffer (cat. no. 88-8824-00; eBio-science; Thermo Fisher Scientific, Inc., Waltham, MA, USA), was used to fix and rupture the membrane according to the manufacturer’s protocols. After centrifugation at 150 × g for 10 min at room temperature, the precipitate was resuspended in 1 ml 0.9% physiological saline and centrifuged at 150 × g for 10 min at room temperature. The precipitate was then resuspended in 150 μl 0.9% physiological saline and blocked with human AB serum (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 4°C for 30 min. Subsequently, a VEGF antibody [human VEGF allophycocyanin (APC)-conjugated antibody; 5 μl; 1:20; cat. no. IC2931A; R&D Systems, Inc., Minneapolis, MN, USA] or isotype antibody (mouse IgG2A APC-conjugated antibody; 5 μl; 1:20; cat. no. IC003A; R&D Systems, Inc.) was then incubated with the cells at room temperature in the dark for 30 min. Cells were centrifuged 350 × g at 4°C for 6 min and washed with PBS twice to remove the non-specific binding antibody. Analysis was performed on a Beckman flow cytometer with CellQuest software.
Cell treatment
To examine the effect of different reagents, HUVECs were cultivated with serum-free RPMI-1640 medium containing 1×10-5 M MLT (Sigma-Aldrich; Merck KGaA) alone at 37°C for 6 h, and/or pretreated with H2O2 (50 μM; Sigma-Aldrich; Merck KGaA), KC7F2 (20 μM; Sigma-Aldrich; Merck KGaA) and VEGF (5 ng/ml; R&D Systems, Inc.) at 37°C for 4 h. The vehicles (1% PBS for VEGF group and 1% DMSO for other groups) were used as the controls. The cell culture supernatant was collected following MLT exposure at 37°C for 6 h and stored at -80°C for further study.
Tube formation assay
Basement membrane extracellular matrix (Matrigel; BD Biosciences, San Jose, CA, USA) was thawed at 4°C overnight. Pipette tips (200 μl) and a 96-well plate were also kept at 4°C overnight, and the plate and tips were placed on ice during the entire experiment. Primary Matrigel (60 μl) was loaded in each well, and the plate was incubated at 37°C for 30 min to allow the matrix to polymerize. The pretreated HUVECs were resuspended and recounted to achieve the appropriate cell density (2×104 cells/well). The plate was kept at room temperature for 15 min and then transferred to the incubator at 37°C. After 4-6 h incubation, the capillary-like tube formation was quantified by counting numbers of junctions/enclosed circles in 5 randomly selected optical fields using an Olympus BX51+DP70 fluorescence microscope (Olympus Corporation, Tokyo, Japan; magnification, ×40 or ×100).
Assessment of the secretion levels of VEGF
The VEGF protein secreted into the conditioned medium by HUVECs was measured with a commercially available human VEGF ELISA kit (cat. no. 1117342; DAKEWE, Inc., Shenzhen, China; http://www.biocity.net/). The conditioned medium was collected and centrifuged at 350 × g at 4°C for 15 min to remove cellular debris, and then recollected and stored at -80°C until the ELISA assay was performed.
Western blot analysis for HIF-1α and VEGF
HUVECs were seeded (2×105 cells/well) in a 6-well plate and incubated at 37°C for 24 h, then treated with MLT (1×10-5 M) or left untreated under normoxic or hypoxic conditions (according to the aforementioned conditions) at 37°C for 6 h prior to protein isolation. The cells in culture were rinsed twice with ice-cold PBS and treated with radioimmunoprecipitation assay lysis buffer supplemented with 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (all from Beyotime Institute of Biotechnology) on ice for 30 min, then centrifuged at 13,400 × g at 4°C for 25 min. The supernatant was collected. Protein concentration was measured by bicinchoninic acid protein assay (Beyotime Institute of Biotechnology). Equal amounts of total protein (15 μg) were segregated by 10% SDS-PAGE (Epizyme Biotechnology, Shanghai, China) and transferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 10% non-fat milk in TBS containing 0.1% Tween-20 (TBST) for 1 h at room temperature. Subsequently, the membranes were probed overnight at 4°C with primary antibodies against HIF-1α (rabbit; 1:500 dilution; cat. no. 36169S), VEGF (rabbit; 1:1,000 dilution; cat. no. 2463S) or β-actin (rabbit; 1:3,000 dilution; cat. no. 4970S; Cell Signaling Technology, Inc., Danvers, MA, USA). This was followed by washing with 1X TBST buffer four times and incubation with the corresponding horseradish peroxidase-conjugated secondary antibody (1:5,000 dilution; cat. no. 2357; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at room temperature for 2 h. Immunoreactive bands were visualized using a Western Blotting Luminol Reagent (ECL) kit (Pierce; Thermo Fisher Scientific, Inc.).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Analysis of HIF-1α (HIF1A) and VEGF mRNA expression in HUVECs was conducted via RT-qPCR following incubation of cells with 1 mM MLT and/or hypoxic conditions (according to the aforementioned conditions) at 37°C for 6 h. Total RNA was isolated from HUVECs using TRIzol® reagent (Takara Bio, Inc., Otsu, Japan), according to the manufacturer’s protocol. The A260/A280 nm absorbance ratio was maintained at 1.8-2.0. For cDNA synthesis, RT was performed using PrimeScript RT Master mix (Takara Bio, Inc.), according to the manufacturer’s instructions. qPCR was performed with an ABI 7900HT system (Applied Biosystems; Thermo Fisher Scientific, Inc.) with SYBR® Green PCR Master mix (Takara Bio, Inc.), according to the manufacturer’s instructions. The thermocycling conditions were: 95°C for 30 min for 1 cycle; 94°C for 5 sec and 60°C for 34 sec for 40 cycles; finally, an extension step at 72°C for 10 min. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were used for normalization, and the relative expression levels of HIF1A and VEGF were calculated using the 2-ΔΔCq method (43). The primer sequences were as follows: HIF1A forward, 5’-GAA CGT CGA AAA GAA AAG TCT CG-3’ and reverse, 5’-CCT TAT CAA GAT GCG AAC TCA CA; VEGF forward, 5’-GGG CAG AAT CAT CAC GA A GT-3, and reverse, 5’-AAA TGC TTT CTC CGC TCT GA-3; and GAPDH forward, 5’-GGA GCG AGA TCC CTC CAA AAT-3 and reverse, 5’-GGC TGT TGT CAT ACT TCT CAT GG-3.
Statistical analysis
All data are presented as the mean ± standard error of the mean. Student’s t-test for two groups comparisons or one-way ANOVA with the Bonferroni’s post-hoc test for multiple comparisons was performed using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
MLT suppresses the viability and angiogenesis of HUVECs
To investigate the influence of MLT on the proliferation and angiogenesis of HUVECs in vitro, CCK8 and tube formation assays were performed. As depicted in Fig. 1A, MLT treatment suppressed the viability of HUVECs in a dose-dependent manner (P<0.001 for Ctrl vs. 10-5 M MLT or P<0.0001 for Ctrl vs. 10-3 M MLT). Additionally, MLT (1×10-5 M) resulted in a significantly reduced level of tube formation of HUVECs, compared with the control (Fig. 1B and C; P<0.001). In addition, hypoxia condition promoted the tube formation of HUVECs, and this effect was reversed by MLT (Fig. 1B and C; P<0.05 for Nor Ctrl vs. Nor MLT or P<0.001 for Hyp Ctrl vs. Hyp MLT). These data indicate that MLT restricts the viability and angiogenesis of HUVECs in vitro.
Figure 1.

MLT suppresses the viability and angiogenesis of HUVECs. (A) HUVECs were treated with MLT at various concentrations (1×10-9, 1×10-7, 1×10-5 or 1×10-3 mM) or left untreated for 24 h. The viability of HUVECs was determined using a Cell Counting kit 8 viability assay. (B) HUVECs were treated with MLT (1×10-5 M) under normoxia or hypoxia conditions, and imaged under an Olympus microscope. Magnification, ×40 or ×100. (C) The statistics histogram for. (B) Data are presented as the mean ± standard error of the mean. *P<0.05, ***P<0.001 or ****P<0.0001, compared with the Ctrl group. Ctrl, control; MLT, melatonin; HUVECs, human umbilical vein endothelial cells; Nor Ctrl, Ctrl HUVECs under normoxia condition; Nor MLT, MLT-treated HUVECs under normoxia condition; Hyp Ctrl, Ctrl HUVECs under hypoxia condition; Hyp MLT, MLT-treated HUVECs under hypoxia condition.
MLT obstructs hypoxia-induced VEGF production of HUVECs
It is widely acknowledged that VEGF is an important angiogenic factor that stimulates the formation of new blood vessels (5). To elucidate the molecular mechanisms of MLT associated with tube formation, the expression of VEGF following treatment with MLT was determined. It was observed that MLT significantly restricted the expression of VEGF at the mRNA and protein levels (Fig. 2A-C; P<0.0001, compared with the Ctrl groups). Additional treatment with recombinant human VEGF protein dose-dependently increased the viability of HUVECs (Fig. 2D; P<0.01 for Ctrl vs. 1 ng/ml VEGF, P<0.001 for Ctrl vs. 2.5 ng/ml VEGF or P<0.0001 for Ctrl vs. 5 ng/ml VEGF), while this effect was weakened by MLT (Fig. 2E; P<0.01 for Ctrl vs. MLT, P<0.001 for VEGF vs. MLT+VEGF). The same inhibition occurred in the tube formation assay, with MLT counteracting VEGF-stimulated tubular network formation (Fig. 2F; P<0.05 for Ctrl vs. MLT or P<0.001 for VEGF vs. MLT+VEGF). These data indicate that the inhibitory effect of MLT on viability and angiogenesis of HUVECs is dependent on VEGF.
Figure 2.

The inhibitory effect of MLT on the viability and angiogenesis of HUVECs is dependent on VEGF. HUVECs were treated with MLT (1×10-5 M) for 24 h, then the expression of VEGF was determined by (A) reverse transcription-quantitative polymerase chain reaction, (B) ELISA and (C) western blotting. (D) HUVECs were stimulated with or without recombinant human VEGF (1-5 ng/ml), then cell viability was detected by CCK8 assay. Cells were pretreated with VEGF (5 ng/ml) for 4 h followed by MLT for a further 2 h, then (E) cell viability and (F) tube formation were analyzed via CCK8 and tube formation assays, respectively. Data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01, ***P<0.001 or ****P<0.0001, compared with the Ctrl group. NS, no significant difference; VEGF, vascular endothelial growth factor; HUVECs, human umbilical vein endothelial cells; CCK8, Cell Counting kit 8; Ctrl, control; MLT, melatonin.
MLT suppresses the expression of VEGF induced by hypoxia
Hypoxia stimulates the formation of new capillary vessels to counteract low oxygen tension. HUVECs were cultured under normoxic or hypoxic conditions. As depicted in Fig. 3A, hypoxia significantly enhanced HIF1A mRNA levels (P<0.05, compared with the Ctrl group). By contrast, MLT exerted a significant inhibitory effect on the transcription of HIF1A (Fig. 3B; P<0.01, compared with the Ctrl group). Subsequently, it was determined that the mRNA and protein level of VEGF were significantly upregulated in HUVECs under hypoxia condition (Fig. 3C and D; P<0.01 for normoxia group vs. hypoxia group or P<0.001 for normoxia group vs. hypoxia group). Additionally, the results of western blotting demonstrated that MLT notably suppressed the expression of HIF-1α and VEGF that was upregulated due to hypoxia (Fig. 3E-G). These results indicate that MLT can inhibit hypoxia-induced VEGF expression and further obstruct VEGF-induced angiogenesis of HUVECs.
Figure 3.

MLT obstructs the expression of VEGF induced by hypoxia. HUVECs were (A) cultured under normoxia or hypoxia conditions, (B) or treated with or without MLT (1×10-5 M) for 24 h. The mRNA expression of HIF1A was detected by RT-qPCR. (C) HUVECs were cultured under normoxia or hypoxia conditions for 24 h, and the mRNA expression of VEGF was detected by RT-qPCR. (D) The secretion of VEGF was detected by ELISA. (E) HUVECs were treated with or without MLT (1×10-5 M) under the condition of normoxia or hypoxia for 24 h, and then the expression of HIF-1α and VEGF was measured by western blotting. (F) The statistics histogram of HIF-1α. (G) The statistics histogram of VEGF. Data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01 or ***P<0.001. VEGF, vascular endothelial growth factor; HUVECs, human umbilical vein endothelial cells; Ctrl, control; MLT, melatonin; HIF-1α, hypoxia inducible factor-1α; HIF1A, HIF-1α; RT-qPCR, reverse transcription-quantitative polymerase chain reaction.
MLT suppresses the release of ROS, particularly under the condition of hypoxia
ROS can act as signaling molecules in a variety of cellular processes, including regulating gene transcription, cell growth, differentiation, apoptosis and metabolism (12,44). Furthermore, emerging evidence indicated that ROS acts as a key element in stabilizing HIF-1α. To gain an insight into the association between hypoxia and ROS, HUVECs were treated with or without MLT under the condition of normoxia or hypoxia. MLT markedly suppressed the release of ROS by HUVECs (Fig. 4A and B; P<0.001, compared with the Ctrl group). ROS release was increased under the condition of hypoxia, and this effect was reversed by MLT (Fig. 4A and B; P<0.001 for hypoxia vs. hypoxia+MLT).
Figure 4.

MLT suppresses the release of ROS, particularly under the condition of hypoxia. (A and B) The release of ROS was detected by flow cytometry following treatment with or without MLT (1×10-5 M) under normoxia or hypoxia for 6 h. Data are presented as the mean ± standard error of the mean, ***P<0.001. MFI, mean fluorescence intensity; Ctrl, control; MLT, melatonin; ROS, reactive oxygen species; NS, no significant difference.
Positive feedback between ROS and VEGF in HUVECs
To identify the association between ROS and VEGF, HUVECs were stimulated with different concentrations of recombinant human VEGF. Exogenous VEGF promoted the release of ROS in a dose-dependent manner (Fig. 5A; P<0.05 for Ctrl vs. 2.5 ng/ml VEGF or P<0.01 for Ctrl vs. 5 ng/ml VEGF). H2O2 is frequently applied as the representative ROS in modeling and inducing oxidative stress, thus, H2O2 was used as an inducer of ROS (13,14). Treatment with H2O2 enhanced the production of ROS to a certain extent (Fig. 5B; P<0.05 for Ctrl vs. 100 μM H2O2 or Ctrl vs. 200 μM H2O2; or P<0.001 for Ctrl vs. 500 μM H2O2 or Ctrl vs.1,000 μM H2O2). Based on its cytotoxicity, a dose of 50 μM H2O2 was selected for subsequent experiments. As indicated in Fig. 5C, the expression of VEGF was significantly upregulated by H2O2 (P<0.01 compared to ctrl group).
Figure 5.

Positive feedback between ROS and VEGF in HUVECs. (A and B) HUVECs were treated with (A) recombinant human VEGF (0-5 ng/ml) or (B) H2O2 (0-1,000 μM) for 4 h, and then ROS release of cells was detected by FCM. (C) HUVECs were treated with H2O2 (50 μM) for 4 h, and then the expression of VEGF in HUVECs was detected by FCM. Data are presented as the mean ± standard error of the mean, *P<0.05, **P<0.01 or ***P<0.001, compared with the Ctrl. MFI, mean fluorescence intensity; ROS, reactive oxygen species; Ctrl, control; FCM, flow cytometry; VEGF, vascular endothelial growth factor; HUVECs, human umbilical vein endothelial cells.
MLT inhibits the viability and angiogenesis of HUVECs by disrupting the crosstalk between ROS and VEGF
As indicated by the aforementioned results, exogenous H2O2 stimulates the expression of ROS and VEGF in HUVECs. HUVECs were pretreated with H2O2 (50 μM) or VEGF (5 ng/ml) for 4 h, and then cultured with or without MLT for 2 h. It was determined that the release of ROS and the induction of VEGF upregulated by H2O2 were inhibited by MLT (Fig. 6A-D; P<0.01 for Ctrl vs. MLT or P<0.001 for H2O2 vs. MLT+H2O2). Furthermore, MLT reversed the stimulatory effect of H2O2 on VEGF expression in HUVECs (Fig. 6E; P<0.001 for H2O2 vs. MLT+H2O2). To examine the effects of H2O2 treatment on HUVECs, cell viability in response to H2O2 stimulation was detected. H2O2 was observed to promote the viability of HUVECs (Fig. 6F; P<0.001 for Ctrl vs. H2O2). However, treatment with additional MLT significantly weakened this effect (Fig. 6F; P<0.01 for H2O2 vs. MLT+H2O2). Additionally, MLT suppressed the tube formation of HUVECs that was induced by H2O2 or VEGF (Fig. 6G and H; P<0.05 for H2O2 vs. MLT+H2O2 or P<0.001 for VEGF vs. MLT+VEGF).
Figure 6.

MLT inhibits the viability and angiogenesis of HUVECs by disrupting the crosstalk between ROS and VEGF. (A) HUVECs were pretreated with H2O2 (50 μM) for 4 h, followed by MLT (1×10-5 M) for 6 h, and then the release of ROS was detected by flow cytometry. (B) The statistics histogram for (A). (C) HUVECs were pretreated with VEGF (5 ng/ml) for 4 h, followed by MLT (1×10-5 M) for 6 h, and then the release of ROS was detected by flow cytometry. (D) The statistics histogram for. (C) Following treatment with H2O2 for 4 h and MLT for 6 h, (E) the expression of VEGF and (F) viability of HUVECs was detected by flow cytometry and a Cell Counting kit 8 assay, respectively. (G) Following stimulation with MLT, H2O2 or VEGF alone, or combined treatment with MLT. Tube formation of HUVECs was measured with a tube formation assay. (H) The statistics histogram for (G). Data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01 or ***P<0.001. Blank, blank control (PBS treatment); MFI, mean fluorescence intensity; ROS, reactive oxygen species; Ctrl, control; VEGF, vascular endothelial growth factor; HUVECs, human umbilical vein endothelial cells; Ctrl, control; MLT, melatonin.
MLT combined with KC7F2 severely disrupts the growth and angiogenesis of HUVECs by targeting the hypoxia/ROS/VEGF axis
KC7F2 has been reported to have an inhibitory effect on the expression of HIF-1α. KC7F2 decreased cell viability in a dose-dependent manner (Fig. 7A; P<0.01 for Ctrl vs. 20 μM KC7F2 or P<0.001 for Ctrl vs. 200 μM KC7F2, compared with the Ctrl group). It was also identified that a dose of 20 μM KC7F2 suppressed the release of ROS (Fig. 7B; P<0.001 for Ctrl vs. KC7F2), production of VEGF (Fig. 7C; P<0.001 for Ctrl vs. KC7F2), cell viability (Fig. 7D; P<0.01 for Ctrl vs. KC7F2) and tube formation (Fig. 7E and F; P<0.01 for Ctrl vs. KC7F2) in HUVECs. Combined treatment with KC7F2 and MLT resulted in reduced ROS release, VEGF production, cell viability and angiogenesis of HUVECs, compared with KC7F2 alone (Fig. 7B-F; P<0.05 for KC7F2 vs. MLT+KC7F2 or P<0.01 for KC7F2 vs. MLT+KC7F2). These results indicate that MLT combined with KC7F2 suppresses the growth and angiogenesis of HUVECs by targeting the hypoxia/ROS/VEGF axis.
Figure 7.

MLT combined with KC7F2 severely disrupts the growth and angiogenesis of HUVECs by targeting the hypoxia/ROS/VEGF axis. (A) HUVECs were treated with KC7F2 (0, 0.2, 2, 20 or 200 μM) for 4 h, and then cell viability was analyzed with a CCK8 assay. HUVECs were pretreated with KC7F2 (20 μM) or VEGF (5 ng/ml) for 4 h, then treated with MLT (1×10-5 M) for 6 h. (B) The release of ROS, (C) the expression of VEGF, (D) cell viability and (E) tube formation were analyzed by (B and C) flow cytometry, (D) CCK8 and (E) tube formation assays (E). (F) The statistics histogram of tube formation. Data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01 or ***P<0.001. Blank, blank control (PBS treatment); MFI, mean fluorescence intensity; ROS, reactive oxygen species; CCK8, Cell Counting kit 8; Ctrl, control; VEGF, vascular endothelial growth factor; HUVECs, human umbilical vein endothelial cells; MLT, melatonin.
Discussion
Numerous studies in recent years indicated that MLT markedly inhibits the proliferation and metastasis of cancer cells (26,27,30,32,36), and therefore could suppress the progression and development of cancer, reduce the risk of mortality and adverse events, and even improve prognosis and quality of life. Tumor growth, development and metastasis are associated with angiogenesis, particularly for solid tumor types (2,6,8,36,45,46). Endothelial cells in the tumor microenvironment serve a pivotal role in the development and progression of cancer through modulating angiogenesis (8,45). Previous studies demonstrated that pharmacological concentrations of MLT have a direct anti-angiogenic effect though the suppression of proliferation in vascular endothelial cells, as well as an indirect effect via inhibition of pro-angiogenesis cytokines, including VEGF, epidermal growth factor and insulin-like growth factor (37,41,47). VEGF is one of the most potent pro-angiogenic cytokines, which specifically triggers the proliferation of endothelial cells and increases permeability (48). However, the underlying mechanisms of this remain poorly elucidated. Previous studies in this area have only been preliminary and the effect of MLT on VEGF expression and the underlying mechanisms remain ambiguous (37,41,47). In the present study, MLT markedly restrained viability and disrupted tube formation in HUVECs, and this effect was dependent on VEGF. Additionally, 1×10-5 M MLT decreased the cell viability. However, 1×10-3 M MLT significantly suppressed cell viability to 30%, and these HUVECs would be dead. Therefore, in the present study 1×10-5 M was selected in the subsequent trials, and the effect of MLT on the apoptosis and death of HUVECs should be studied further.
Tumor angiogenesis is a key process for delivering oxygen and nutrients to growing tumors, and therefore is considered an essential characteristic of the microenvironment in solid tumor types (49). The production of certain angiogenic cytokines and growth factors is regulated by hypoxia, while tumor angiogenesis and subsequent rapid tumor growth also further accelerate tumor hypoxia (37,38). This feedback mechanism is a critical factor resulting in pathogenic features of cancer, including poor treatment prognosis, and progression to malignancies and metastatic disease (48). In the present study, it was determined that MLT could inhibit hypoxia-induced HIF-1α expression in HUVECs at mRNA level and protein level. However, the role of MLT on HIF1A at transcriptional level in nuclear extracts and the detailed mechanism require further study. Additionally, it was observed that hypoxia stimulated VEGF production. Inhibitor of HIF-1α significantly downregulated VEGF expression. However, whether the regulation of hypoxia on VEGF depends on the specific transcriptional activation of HIF1A on VEGF remains to be further studied.
Chronic hypoxia is the primary cause of high concentration ROS formation within tumor cells (50). ROS, which are produced by numerous physiological oxidative activities and stress reactions in the body, are also associated with numerous physiological, including cell autophagy and immunoregulation, and pathophysiological, including tumor progression, processes (51-54). At low levels, ROS exhibit beneficial effects, serving as signaling molecules by regulating intracellular signals and maintaining homeostasis, including in the processes of proliferation, differentiation, adhesion, migration, invasion and apoptosis; however, at high levels, ROS are deleterious to cells, resulting in damage to proteins, lipids and DNA, and eventually resulting in autophagy and programmed cell death (55,56). An increased level of ROS has been demonstrated to promote cell proliferation, cell migration, cell survival and epithelial-mesenchymal transition through activating the mitogen-activated protein kinase and Ras-extracellular signal-regulated kinases pathway (57,58), finally resulting in oxidative stress, which initiates carcinogenesis (59). Previous evidence indicates that ROS may act as second messengers in response to hypoxia, and serve an important role in stabilizing HIF-1α protein and inducing the production of angiogenic factors. Furthermore, chemical antioxidants suppress HIF-1α accumulation and inhibit the transcription of VEGF via a mechanism that involves ROS (55).
In the present study, it was determined that hypoxia enhances the release of ROS and VEGF expression. Additionally, there is a positive feedback mechanism between ROS and VEGF, which contributes to the high viability and angiogenesis of HUVECs. Notably, MLT could effectively suppress these effects, particularly in combination with KC7F2 (a novel small molecule HIF-1α translation inhibitor). Collectively, these results indicate that MLT inhibits the viability and angiogenesis of HUVECs by targeting the HIF-1α/VEGF/ROS axis. Wang et al (60) demonstrated that MLT downregulates the MLT nuclear receptor RZR/RAR related orphan receptor (ROR)γ expression causing growth-inhibitory and anti-angiogenesis activity in human gastric cancer cells in vitro and in vivo. However, whether RZR/RORγ is involved in the effect of MLT on the HIF-1α/VEGF/ROS axis requires further investigation. Additionally, ROS can regulate the B-cell lymphoma 2 family via direct and indirect mechanisms (61,62), indicating that tumor growth is also inversely associated with the level of ROS. Therefore, the effect of KC7F2 and ROS on the apoptosis of HUVECs should be studied further.
In conclusion, as depicted in Fig. 8, the present observations demonstrate that hypoxia upregulates the level of ROS and VEGF in a direct manner and HIF-1α-dependent manner. ROS production results in an increase in the viability and angiogenesis of HUVECs. As an important angiogenesis growth factor, VEGF can directly promote angiogenesis of HUVECs. Furthermore, VEGF increases ROS production and amplifies the stimulatory effect of ROS on the viability and angiogenesis of HUVECs. MLT serves a dual role in the inhibition of angiogenesis, acting directly as a growth-associated inhibitor, or indirectly as an antioxidant and free radical scavenging agent. MLT suppresses the viability and angiogenesis of HUVECs through downregulating the hypoxia/HIF-1α/ROS/VEGF pathway in vitro. As an inhibitor of HIF-1α, KC7F2 can exert an anti-angiogenesis effect synergistically with MLT in vitro. In summary, the present data indicate that MLT may be a potential anticancer agent in solid tumors with abundant blood vessels, particularly in combination with KC7F2. However, further in vivo studies and trials are required.
Figure 8.
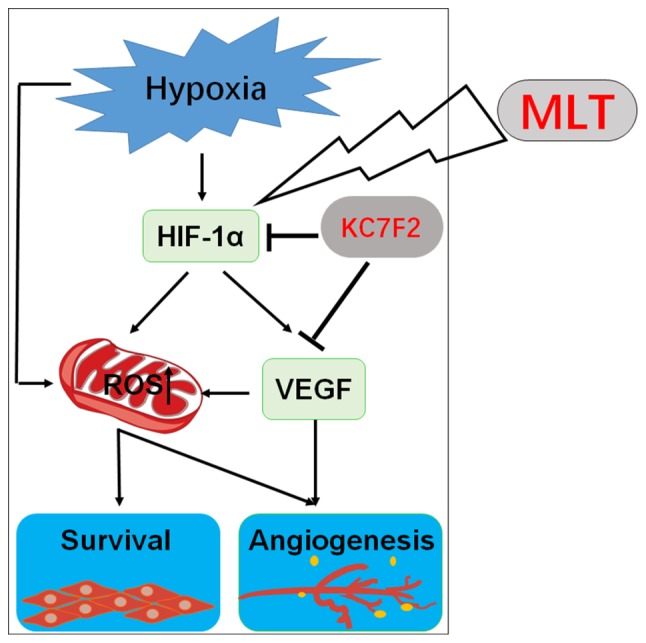
Schematic presentation of the anti-angiogenesis role of MLT via targeting of the HIF-1α/ROS/VEGF axis. Under the condition of hypoxia, the release of ROS and expression of VEGF are enhanced, which appears to be dependent on HIF-1α due to KC7F2 (a novel small molecule HIF-1α translation inhibitor) inhibiting these effects. There is a positive feedback mechanism between ROS and VEGF, which contributes to the high viability and angiogenesis of HUVECs. MLT could effectively suppress hypoxia/HIF-1α/ROS/VEGF-induced growth and angiogenesis of HUVECs, particularly in combination with KC7F2. HIF-1α, hypoxia-inducible factor-1α; HUVECs, human umbilical vein endothelial cells; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor; MLT, melatonin.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Major Research Program of National Natural Science Foundation of China (grant nos. 91542108, 81471513, 31671200, 81571509 and 81401273), the Shanghai Rising-Star Program (grant no. 16QA1400800), the Development Fund of Shanghai Talents (grant no. 201557), the Oriented Project of Science and Technology Innovation from the Key Laboratory of Reproduction Regulation of NPFPC (grant no. CX2017-2) and the Program for Zhuoxue of Fudan University, the Program of Jiangsu Key Youth Medical Talents (grant no. QNRC2016244) and the Introduction Project of Suzhou Clinical Medicine Expert Team (grant no. SZYJTD201708).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
JC conducted all experiments and prepared the figures and the manuscript. HLY, CJG, and YKL assisted with flow cytometry analysis. JS, RZ, YYH and XYZ assisted in the study design and critically revised the manuscript for important intellectual content. MQL designed the study, supervised the project and edited the manuscript. All the authors were involved in writing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling-Role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/B:MCBI.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 2.Gacche RN, Meshram RJ. Targeting tumor micro-environment for design and development of novel anti-angiogenic agents arresting tumor growth. Prog Biophys Mol Biol. 2013;113:333–354. doi: 10.1016/j.pbiomolbio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Park JH, Yoon J, Park B. Pomolic acid suppresses HIF1 α/VEGF-mediated angiogenesis by targeting p38-MAPK and mTOR signaling cascades. Phytomedicine. 2016;23:1716–1726. doi: 10.1016/j.phymed.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frezzetti D, Gallo M, Maiello MR, D’Alessio A, Esposito C, Chicchinelli N, Normanno N, De Luca A. VEGF as a potential target in lung cancer. Expert Opin Ther Targets. 2017;21:959–966. doi: 10.1080/14728222.2017.1371137. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Dabrosin C, Yin X, Fuster MM, Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B, Ribatti D, et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. 2015;35(Suppl):S224–S243. doi: 10.1016/j.semcancer.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SH, Jeong D, Han YS, Baek MJ. Pivotal role of vascular endothelial growth factor pathway in tumor angiogenesis. Ann Surg Treat Res. 2015;89:1–8. doi: 10.4174/astr.2015.89.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delli Carpini J, Karam AK, Montgomery L. Vascular endothelial growth factor and its relationship to the prognosis and treatment of breast, ovarian, and cervical cancer. Angiogenesis. 2010;13:43–58. doi: 10.1007/s10456-010-9163-3. [DOI] [PubMed] [Google Scholar]
- 9.Park WH. Effects of antioxidants and MAPK inhibitors on cell death and reactive oxygen species levels in H2O2-treated human pulmonary fibroblasts. Oncol Lett. 2013;5:1633–1638. doi: 10.3892/ol.2013.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–2992. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 12.Tapeinos C, Larranaga A, Sarasua JR, Pandit A. Functionalised collagen spheres reduce H2O2 mediated apoptosis by scavenging overexpressed ROS. Nanomedicine. 2018;14:2397–2405. doi: 10.1016/j.nano.2017.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol. 2002;22:566–573. doi: 10.1161/01.ATV.0000012262.76205.6A. [DOI] [PubMed] [Google Scholar]
- 15.Maraldi T, Prata C, Caliceti C, Vieceli Dalla, Sega F, Zambonin L, Fiorentini D, Hakim G. VEGF-induced ROS generation from NAD(P)H oxidases protects human leukemic cells from apoptosis. Int J Oncol. 2010;36:1581–1589. doi: 10.3892/ijo_00000645. [DOI] [PubMed] [Google Scholar]
- 16.Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, Galeotti T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem. 2002;277:3101–3108. doi: 10.1074/jbc.M107711200. [DOI] [PubMed] [Google Scholar]
- 17.Oshikawa J, Kim SJ, Furuta E, Caliceti C, Chen GF, McKinney RD, Kuhr F, Levitan I, Fukai T, Ushio-Fukai M. Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2012;302:H724–H732. doi: 10.1152/ajpheart.00739.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 19.Chen S, Zhou Y, Zhou L, Guan Y, Zhang Y, Han X. Anti-neovascularization effects of DMBT in age-related macular degeneration by inhibition of VEGF secretion through ROS-dependent signaling pathway. Mol Cell Biochem. 2018;448:225–235. doi: 10.1007/s11010-018-3328-6. [DOI] [PubMed] [Google Scholar]
- 20.Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125:10–19. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, Van Meir EG. Identification of a novel small molecule HIF-1alpha translation inhibitor. Clin Cancer Res. 2009;15:6128–6136. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan G, Zhang Y, Lu Y, Liu L, Shi D, Wen Y, Yang L, Ma Q, Liu T, Zhu X, et al. The HIF-1α/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells. Cancer Lett. 2015;357:254–264. doi: 10.1016/j.canlet.2014.11.034. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, Chen J, Lu X, Shen C, Zeng J, Chen L, Pei Z. Melatonin protects against rotenone-induced cell injury via inhibition of Omi and Bax-mediated autophagy in Hela cells. J Pineal Res. 2012;52:120–127. doi: 10.1111/j.1600-079X.2011.00926.x. [DOI] [PubMed] [Google Scholar]
- 24.Goradel NH, Asghari MH, Moloudizargari M, Negahdari B, Haghi-Aminjan H, Abdollahi M. Melatonin as an angiogenesis inhibitor to combat cancer: Mechanistic evidence. Toxicol Appl Pharmacol. 2017;335:56–63. doi: 10.1016/j.taap.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 25.Su SC, Hsieh MJ, Yang WE, Chung WH, Reiter RJ, Yang SF. Cancer metastasis: Mechanisms of inhibition by melatonin. J Pineal Res. 2017;62:e12370. doi: 10.1111/jpi.12370. [DOI] [PubMed] [Google Scholar]
- 26.Reiter RJ, Rosales-Corral SA, Tan DX, Acuna-Castroviejo D, Qin L, Yang SF, Xu K. Melatonin, a full service anti-cancer agent: Inhibition of initiation, progression and metastasis. Int J Mol Sci. 2017;18:E843. doi: 10.3390/ijms18040843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carbajo-Pescador S, Ordonez R, Benet M, Jover R, García-Palomo A, Mauriz JL, González-Gallego J. Inhibition of VEGF expression through blockade of Hif1alpha and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer. 2013;109:83–91. doi: 10.1038/bjc.2013.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang TH, Wu CH, Yeh CT, Su SC, Hsia SM, Liang KH, Chen CC, Hsueh C, Chen CY. Melatonin suppresses hepato-cellular carcinoma progression via lncRNA-CPS1-IT-mediated HIF-1 α inactivation. Oncotarget. 2017;8:82280–82293. doi: 10.18632/oncotarget.19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Pi H, Reiter S, Guo RJ, Zhang P, Li L, Li Y, Cao M, Tian Z, Xie LJ, et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11:1037–1051. doi: 10.1080/15548627.2015.1052208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Z, Yang Y, Fan C, Han J, Wang D, Di S, Hu W, Liu D, Li X, Reiter RJ, Yan X. Melatonin as a potential anticarcinogen for non-small-cell lung cancer. Oncotarget. 2016;7:46768–46784. doi: 10.18632/oncotarget.8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plaimee P, Weerapreeyakul N, Barusrux S, Johns NP. Melatonin potentiates cisplatin-induced apoptosis and cell cycle arrest in human lung adenocarcinoma cells. Cell Prolif. 2015;48:67–77. doi: 10.1111/cpr.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Proietti S, Cucina A, Reiter RJ, Bizzarri M. Molecular mechanisms of melatonin’s inhibitory actions on breast cancers. Cell Mol Life Sci. 2013;70:2139–2157. doi: 10.1007/s00018-012-1161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Proietti S, Catizone A, Masiello MG, Dinicola S, Fabrizi G, Minini M, Ricci G, Verna R, Reiter RJ, Cucina A, Bizzarri M. Increase in motility and invasiveness of MCF7 cancer cells induced by nicotine is abolished by melatonin through inhibition of ERK phosphorylation. J Pineal Res. 2018;64:e12467. doi: 10.1111/jpi.12467. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez-Gonzalez A, Gonzalez A, Alonso-Gonzalez C, Menéndez-Menéndez J, Martínez-Campa C, Cos S. Complementary actions of melatonin on angiogenic factors, the angiopoietin/Tie2 axis and VEGF, in co-cultures of human endothelial and breast cancer cells. Oncol Rep. 2018;39:433–441. doi: 10.3892/or.2017.6070. [DOI] [PubMed] [Google Scholar]
- 35.Lee H, Lee HJ, Jung JH, Shin EA, Kim SH. Melatonin disturbs SUMOylation-mediated crosstalk between c-Myc and nestin via MT1 activation and promotes the sensitivity of paclitaxel in brain cancer stem cells. J Pineal Res. 2018;65:e12496. doi: 10.1111/jpi.12496. [DOI] [PubMed] [Google Scholar]
- 36.Hill SM, Frasch T, Xiang S, Yuan L, Duplessis T, Mao L. Molecular mechanisms of melatonin anticancer effects. Integr Cancer Ther. 2009;8:337–346. doi: 10.1177/1534735409353332. [DOI] [PubMed] [Google Scholar]
- 37.Lai YH, Hu DN, Rosen R, Sassoon J, Chuang LY, Wu KY, Wu WC. Hypoxia-induced vascular endothelial growth factor secretion by retinal pigment epithelial cells is inhibited by melatonin via decreased accumulation of hypoxia-inducible factors-1α protein. Clin Exp Ophthalmol. 2017;45:182–191. doi: 10.1111/ceo.12802. [DOI] [PubMed] [Google Scholar]
- 38.Park SY, Jang WJ, Yi EY, Jang JY, Jung Y, Jeong JW, Kim YJ. Melatonin suppresses tumor angiogenesis by inhibiting HIF-1 alpha stabilization under hypoxia. J Pineal Res. 2010;48:178–184. doi: 10.1111/j.1600-079X.2009.00742.x. [DOI] [PubMed] [Google Scholar]
- 39.Park JW, Hwang MS, Suh SI, Baek WK. Melatonin downregulates HIF-1 alpha expression through inhibition of protein translation in prostate cancer cells. J Pineal Res. 2009;46:415–421. doi: 10.1111/j.1600-079X.2009.00678.x. [DOI] [PubMed] [Google Scholar]
- 40.Cui P, Luo Z, Zhang H, Su Y, Li A, Li H, Zhang J, Yang Z, Xiu R. Effect and mechanism of melatonin’s action on the proliferation of human umbilical vein endothelial cells. J Pineal Res. 2006;41:358–362. doi: 10.1111/j.1600-079X.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 41.González A, González-González A, Alonso-González C, Menéndez-Menéndez J, Martínez-Campa C, Cos S. Melatonin inhibits angiogenesis in SH-SY5Y human neuroblastoma cells by downregulation of VEGF. Oncol Rep. 2017;37:2433–2440. doi: 10.3892/or.2017.5446. [DOI] [PubMed] [Google Scholar]
- 42.Vriend J, Reiter RJ. Melatonin and the von Hippel-Lindau/HIF-1 oxygen sensing mechanism: A review. Biochim Biophys Acta. 2016;1865:176–183. doi: 10.1016/j.bbcan.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Pan B, Zhong W, Deng Z, Lai C, Chu J, Jiao G, Liu J, Zhou Q. Inhibition of prostate cancer growth by solanine requires the suppression of cell cycle proteins and the activation of ROS/P38 signaling pathway. Cancer Med. 2016;5:3214–3222. doi: 10.1002/cam4.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvarez-García V, González A, Alonso-González C, Martínez-Campa C, Cos S. Antiangiogenic effects of melatonin in endothelial cell cultures. Microvasc Res. 2013;87:25–33. doi: 10.1016/j.mvr.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Vijayalaxmi, Thomas CR, Jr, Reiter RJ, Herman TS. Melatonin: From basic research to cancer treatment clinics. J Clin Oncol. 2002;20:2575–2601. doi: 10.1200/JCO.2002.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Dai M, Cui P, Yu M, Han J, Li H, Xiu R. Melatonin modulates the expression of VEGF and HIF-1 alpha induced by CoCl2 in cultured cancer cells. J Pineal Res. 2008;44:121–126. doi: 10.1111/j.1600-079X.2007.00498.x. [DOI] [PubMed] [Google Scholar]
- 48.Colombo J, Maciel JM, Ferreira LC, Da Silva RF, Zuccari DA. Effects of melatonin on HIF-1 α and VEGF expression and on the invasive properties of hepatocarcinoma cells. Oncol Lett. 2016;12:231–237. doi: 10.3892/ol.2016.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Liu Q, Wang F, Ling EA, Liu S, Wang L, Yang Y, Yao L, Chen X, Wang F, et al. Melatonin antagonizes hypoxia-mediated glioblastoma cell migration and invasion via inhibition of HIF-1 α. J Pineal Res. 2013;55:21–130. doi: 10.1111/jpi.12052. [DOI] [PubMed] [Google Scholar]
- 50.Guzy RD, Humacker PT. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 51.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 52.D’Autréaux B, Toledano MB. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013;23:898–914. doi: 10.1038/cr.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diebold L, Chandel NS. Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med. 2016;100:86–93. doi: 10.1016/j.freeradbiomed.2016.04.198. [DOI] [PubMed] [Google Scholar]
- 55.Xia C, Meng Q, Lin LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–10830. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 56.Li Z, Xu X, Leng X, He M, Wang J, Cheng S, Wu H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch Virol. 2017;162:603–610. doi: 10.1007/s00705-016-3130-2. [DOI] [PubMed] [Google Scholar]
- 57.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J, Chang F, Li F, Fu H, Wang J, Zhang S, Zhao J, Yin D. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun. 2015;463:262–267. doi: 10.1016/j.bbrc.2015.05.042. [DOI] [PubMed] [Google Scholar]
- 59.Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017;387:95–105. doi: 10.1016/j.canlet.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 60.Wang RX, Liu H, Xu L, Zhang H, Zhou RX. Melatonin downregulates nuclear receptor RZR/RORγ expression causing growth-inhibitory and anti-angiogenesis activity in human gastric cancer cells in vitro an in vivo. Oncol Lett. 2016;12:897–903. doi: 10.3892/ol.2016.4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.You BR, Shin HR, Park WH. PX-12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS-dependent apoptosis. Int J Oncol. 2014;44:301–308. doi: 10.3892/ijo.2013.2152. [DOI] [PubMed] [Google Scholar]
- 62.Bauer G. Central signaling elements of intercellular reactive oxygen/nitrogen species-dependent induction of apoptosis in malignant cells. Anticancer Res. 2017;37:499–513. doi: 10.21873/anticanres.11342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.