

Abstract
Large portions of the genome undergo alternative pre-mRNA splicing in often intricate patterns. Alternative splicing regulation requires extensive control mechanisms since errors can have deleterious consequences and may lead to developmental defects and disease. Recent work has identified a complex network of regulatory RNA elements which guide splicing decisions. In addition, the discovery that transcription and splicing are intimately coupled has opened up new directions into alternative splicing regulation. Work at the interface of chromatin and RNA biology has revealed unexpected molecular links between histone modifications, the transcription machinery, and non-coding RNAs (ncRNAs) in the determination of alternative splicing patterns.
More than 90% of human transcripts are alternatively spliced in a highly tissue-specific and developmental manner [1●●]. Even relatively modest changes in alternative splicing can have dramatic consequences, including altered cellular responses, cell death, and uncontrolled proliferation that can lead to disease [2,3]. Alternative splicing specificity is commonly thought to be achieved by the combinatorial recruitment of basal splicing factors to regulatory RNA motifs on the pre-mRNA [4,6●●]. The availability of splicing factors depends primarily on their expression levels, their nuclear localization, and post-translational modifications modulating their activity [4]. However, expression profiles of splicing factors often do not correspond to their expected abundance [5] and it remains unclear how cell type-specific, lineage-specific, and tissue-specific alternative splicing patterns are established, maintained, and altered in physiological settings such as differentiation and development.
A new dimension in the regulation of alternative splicing emerged about a decade ago with the demonstration that alternative pre-mRNA splicing is intimately linked to transcription [7,8]. Many splicing events occur co-transcriptionally and the splicing machinery is physically linked to the transcriptional apparatus via association of splicing factors with the elongating RNA polymerase II (RNAP II) [7–9]. These findings have recently been extended by numerous studies demonstrating that alternative splicing is sensitive to RNAP II elongation rate, chromatin structure, and histone modifications [10●●,11●●,12●●,13●●,14●●,15●●,16●●]. These observations make it clear that a full understanding of alternative splicing regulation requires integration of the RNA-based regulatory components with those of transcription and chromatin. Such an integrated view of alternative splicing regulation is now beginning to emerge.
An RNA splicing code
Alternatively spliced exons are defined by subsets of regulatory sequence elements which recruit trans-acting splicing factors to the pre-mRNA and act as enhancers or silencers. Computational comparison of RNA elements present in alternatively spliced, but not constitutive exons, followed by mutational analysis, together with screening of random libraries of short sequences inserted into splicing reporters has led to the characterization of a myriad of splicing motifs over the years [17,18]. However, the inability to survey large numbers of alternative splicing events comprehensively and systematically made it difficult to test the universality and predictive power of those RNA motifs.
The recent advent of genome-wide expression analysis using deep RNA sequencing methods has now allowed systematic bioinformatic analyses to probe the general validity of known regulatory sequences and to identify new ones [1●●,19]. The culmination of this approach is the analysis of the splicing patterns of over 3500 cassette-type alternative exons across 27 mouse tissues ranging from embryonic stages to adult [6●●]. This analysis compiled more than 1000 features, based on well-known RNA motifs, newly discovered motifs, and RNA secondary structures, and correlated the patterns of tissue-specific alternative splicing with the presence of those RNA features around the alternatively spliced exon. Using these motifs alone, splicing patterns could be correctly predicted with 65–90% accuracy depending on the splicing factor [6●●].
These computational studies now provide the basis for more extensive and specific analysis of the role of RNA motifs in tissue-specific regulation of alternative splicing. A key advance has been the development of methods to systematically map at the genome-wide level the binding sites of RNA-binding proteins using crosslinking immunoprecipitation coupled to high-throughput sequencing (CLIP-Seq) [20,21]. These studies reveal a much greater complexity of splicing regulation characterized by bind-ing sites far away from exons in the intronic regions and splicing factors acting as activators or repressors depending on the relative position of the binding site around the exon [20–22].
Despite the high accuracy of splicing outcome predictions using this RNA motif ‘code’, RNA sequences alone are not sufficient to account for tissue-specific and develop-mental splicing regulation. Other regulatory mechanisms must provide specificity. The discovery that the majority of splicing events occur co-transcriptionally allows for the possibility that transcription and chromatin play a key role in alternative splicing regulation in vivo.
Regulation of alternative splicing by transcription
Pre-mRNA splicing and its regulation in vivo cannot be considered an isolated process since much of splicing occurs co-transcriptionally. While RNAP II still synthesizes the pre-mRNA, the splicing machinery is already recruited to the nascent transcript and splices out the emerging introns before transcription is terminated, suggesting regulatory crosstalk between the two processes [7,8]. Classic experiments demonstrated a strong link between RNAP II elongation rate and alternative splicing outcome [23,24]. Using RNAP II mutants or inserting pausing elements in reporter minigenes, it was shown that pausing or slowing down of the polymer-ase favors exon inclusion in the fibronectin and fibroblast growth factor receptor 2 (FGFR2) genes [25,26]. These results led to a ‘kinetic model’ of splicing proposing that slowing or pausing of RNAP II increases the window of time an upstream weak exon can recruit the splicing machinery before the splicing sites of a stronger down-stream exon emerge from the polymerase complex (Figure 1a).
Figure 1.
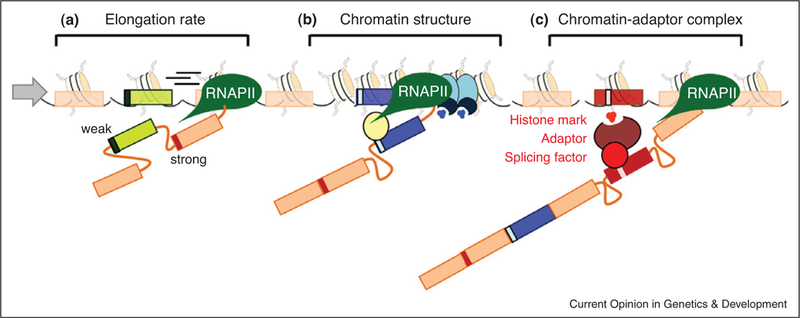
The role of chromatin in alternative splicing. (a) RNAP II elongation rate affects recruitment of the splicing machinery. Fast elongation favors inclusion of a downstream exon with strong splice sites. (b) A change in chromatin conformation, such as localized heterochromatinization (blue ovals and higher density of nucleosomes), slows down RNAP II which favors recruitment of splicing factors (yellow oval) to the weaker exon (blue rectangle), inducing exon inclusion. (c) Histone modifications (small red circles) can directly recruit splicing factors via a chromatin-adaptor system (red ovals) which consists of a chromatin-binding protein that reads the histone marks and modulates recruitment of the splicing factor to the pre-mRNA (red rectangle).
Physiological support for a role of RNAP II kinetics in alternative splicing regulation comes from the observation that ultraviolet irradiation affects co-transcriptional alternative splicing of DNA damage-responsive genes through hyperphosphorylation of the RNAP II C-terminal domain (CTD) [12]. Furthermore, inhibition of RNAP II elongation in human T-cells with DRB or camptothecin changes the pattern of alternative splicing of a subset of genes, mostly involved in RNA processing, by favoring inclusion of an exon that creates RNA instability and transcript degradation [13]. In agreement with a kinetic model, the average strength of the 5’ and 3’ splice sites of the RNAP II sensitive alternative exons is weaker than of the downstream constitutive exons [13].
Further evidence for a role for RNAP II kinetics in the coordinated regulation of gene expression is the transient accumulation of RNAP II phosphorylated on serine 5’ which is typically enriched at promoters, around the 3’ end of introns in yeast [27●●]. This RNAP II pausing is cyclic and splicing-dependent, suggesting that there are synchronized rounds of splicing that require pausing of the RNAP II right before the first exon [27●●]. The end of the pause correlates with phosphorylation of serine 2, the elongating form of RNAP II, and recruitment of several splicing factors possibly constituting a splicing-dependent transcriptional checkpoint that abrogates any further transcription until a spliceosome is assembled [27●●]. RNAP II accumulation has also been observed toward the end of yeast genes with shorter terminal exons, whereas intronless and inefficiently spliced genes lack RNAP II pausing, suggesting that another checkpoint at the end of the gene may also exist to guarantee that splicing is finished before the termination and release of the transcript from the RNAP II complex [9].
Chromatin as a regulator of alternative splicing
If the elongation rate of RNAP II contributes to alternative splicing, how is elongation rate controlled? One obvious way is by chromatin structure. It is generally assumed that transcription through nucleosome-dense genome regions, such as compacted chromatin, is slower than through nucleosome-poor regions. Although direct in vivo evidence of changes in elongation rate within a given gene are lacking [28●●], nucleosomes have been shown to behave in vitro as fluctuating barriers that locally increase pause density, slow pause recovery, and reduce the apparent pause-free velocity of RNAP II [29]. Moreover sequence-dependent nucleosome positioning in the body of a transcription unit impairs RNAP II progression, provokes RNAP II accumulation upstream of the positioned nucleosome, and reduces transcription of a reporter minigene [30], suggesting that nucleosome density and chromatin structure may regulate RNAP II kinetics.
In agreement with a role for nucleosome positioning in alternative splicing regulation, genome-wide mapping of microccocal nuclease-digested chromatin in plants, Caenorhabditis elegans and human T-cells and gametes has revealed enrichment of nucleosomes at exons [31●●,32●●,33●●,34●●,35●●,36●●,37●●]. This accumulation is independent of gene expression levels [32●●,35●●] and seems to be determined by the DNA sequences defining introns [38]. Moreover, while isolated exons flanked by long introns are highly enriched in nucleosomes [34●●], alternatively spliced exons present slightly less enrichment than constitutive exons [31●●], suggesting a link between nucleosome positioning and alternative splicing regulation. It can be envisioned that the specific nucleosome enrichment at exons, particularly at weak exons, acts as an RNAP II pausing barrier to assure the coordinated recruitment and assembly of the spliceosome (Figure 1b). In support, genome-wide mapping of RNAP II also reveals a non-random enrichment at exons [31●●,36●●,39●●].
In addition to nucleosomes, histone modifications and DNA methylation are also non-randomly distributed over exons and may therefore contribute to splicing regulation [31●●,32●●,34●●,36●●,37●●]. High-throughput mapping of 42 histone marks in human T-cells showed preferential distribution of several histone marks at exons [31●●,32,34●●]. Most notably, H3K36me3 marks highly transcribed exons and H3K27me2 marks lowly transcribed exons [34●●]. Interestingly, H3K36me3 is more enriched in constitutive exons than in alternatively spliced ones, possibly defining them as such [37●●]. In support, distinctive subsets of histone modifications have been correlated with particular splicing patterns in several genes. In the neuron-specific gene NCAM, membrane depolarization locally increases H3K9 acetylation and H3K36 methylation around the alternatively spliced exon reducing its inclusion [11]. In the case of the fibronectin gene, RNAi-mediated heterochromatinization of exon EDI increases H3K9 and H3K27 methylation favoring inclusion [10●●]. Similarly, CD44, PKN2, and TAF4B are marked by higher levels of H3K9me3 and the chromatin-binding protein HP1g along the alternatively spliced exons favoring its inclusion [16]. Finally, in several poly-pyrimidine-tract binding protein (PTB)-dependent genes, exclusion of the PTB-dependent exon correlates with H3K36me3 levels, whereas inclusion correlates with high H3K27me3 and H3K4me3 [14●●]. Importantly, modulation of these and other splicing-specific chromatin signatures by inhibitors, overexpression of histone modifiers, or knockdown experiments induces changes in alternative splicing outcome in a predictable way, suggesting that some histone modifications play a direct role in regulating alternative splicing [10●●,11,14●●,16].
One function of histone marks over alternatively spliced genome regions may be to establish and maintain nucleosome density in the region which in turn might regulate RNAP II elongation rate [24] (Figure 1b). However, it remains to be seen whether changes in chromatin structure are capable to elicit alterations in RNAP II elongation rate of the magnitude required to cause alternative splicing patterns to change. In addition, it is unclear whether kinetic regulation alone can account for competition between splice sites of equal strength and situations where more than two splice sites are involved.
Regulation of alternative splicing by chromatin-splicing adaptor complexes
Apart from modulating RNAP II kinetics, histone modifications have recently been shown to also act by a more direct mechanism. Observations on splicing events which are dependent on PTB suggest that histone modifications can direct the recruitment of splicing factors to the nascent RNA via formation of a chromatin-splicing adaptor complex (Figure 1c) [14●●]. This model is based on the analysis of several PTB-dependent genes, including the well characterized alternative splicing model FGFR2 [14●●,26]. In these experiments, exclusion of the PTB-dependent exon correlated with increased H3K36me3 levels and a reduction in H3K36me3 levels favored inclusion of the regulated exon [14●●]. Modulation of these and other key histone modifications resulted in the alteration of splicing patterns, indicating that these modifications can drive splicing outcome. The molecular link between chromatin and the splicing machinery is the chromatin-binding adaptor protein MRG15, which reads the histone mark and recruits the splicing factor PTB to the pre-mRNA [14●●] (Figure 1c).
The function of such chromatin-splicing adaptor complexes is likely to facilitate the recruitment of splicing regulators to weak splice sites. Co-transcriptionally spliced mRNA, but not splicing factors, can be isolated from chromatin fractions, suggesting that the affinity of splicing factors for nascent RNA is intrinsically weak [9]. Histone marks may increase the affinity and stabilize the binding of splicing factors to their RNA motifs. In sup-port, genome-wide analysis indicates that the PTB-de-pendent exons which utilize this mechanism are characterized by relatively weak PTB-recognition sites, suggesting that chromatin-splicing adaptor complexes may facilitate recruitment of PTB to them, whereas exons with strong PTB sites do not require such aid [14●●].
In addition to the H3K36me3/MRG15/PTB complex, other adaptor complexes have been described, although their function in splicing control has not been established yet [24]. The H3K4me3-binding protein CHD1 is important for the recruitment of U2snRNP to the first exon of active genes [40] and the H3K9me3-binding protein HP1 co-immunoprecipitates with ASF/SF2 and several hnRNPs splicing repressors [41,42], suggesting that the combinatorial enrichment of histone marks may act cooperatively to modulate alternative splicing out-come.
Histone modifications may affect the recruitment of splicing factors linearly, with relative levels of the different histone marks determining the abundance of the chromatin-binding proteins that would compete for the recruitment of the corresponding splicing factor to the target RNA (Figure 2a). Alternatively, histone modifications may act in a combinatorial manner with histone marks cross-talking to each other and either favoring or repressing the recruitment of a key chromatin-binding adaptor protein and the interacting splicing factor (Figure 2b and c).
Figure 2.
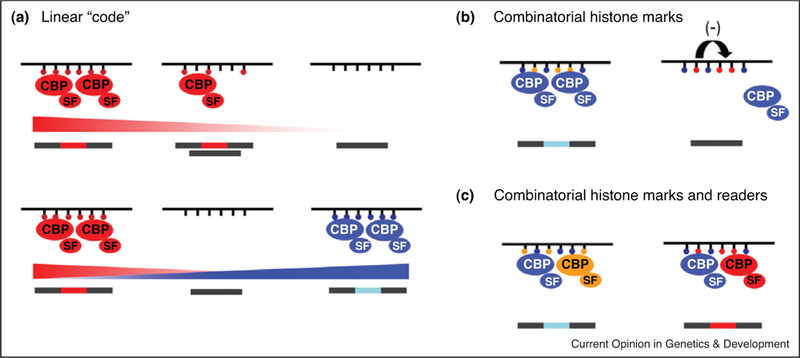
A ‘histone code’ in alternative splicing. Histone modifications may transmit their information in several modes. (a) Histone marks may act linearly with increasing levels of a single histone mark recruiting increasing levels of a chromatin-adaptor protein complex leading to increased usage of a given site. Competing levels of different histone marks modulate the recruitment of competing chromatin-adaptor complexes determining the final splicing outcome. (b) Histone modifications may act in combination by favoring (left) or inhibiting (right) the recruitment of a single chromatin-splicing complex. (c) Multiple histone marks may recruit in combination multiple chromatin-adaptor complexes that will favor or inhibit exon inclusion. CBP: chromatin-binding protein; SF: splicing factor.
These emerging data on the role of histone modifications in alternative splicing regulation draw strong parallels between the role of epigenetic modifications in transcription and in splicing. In the same way characteristic subsets of histone marks define promoter subtypes and gene activity, subsets of histone marks may also define and represent different splicing signatures [24]. Such a model would explain the long-standing conundrum of how cells establish and maintain their alternative splicing patterns and how they change them during differentiation and development. Just as epigenetic modifications are known to define cell type-specific transcription programs that are maintained over cell divisions, epigenetic modifications may also establish and maintain cell type-specific alternative splicing programs during cell differentiation by using them as a molecular memory to bookmark splicing patterns.
The emerging role of ncRNAs in alternative splicing
ncRNAs have recently emerged as novel regulators of alternative splicing. One mode of control by ncRNAs is the regulation of the expression of key splicing factors by short microRNAs during development and differentiation (Figure 3a). An example is the neuron-specific microRNA miR-124 that directly regulates expression of the splicing repressor PTB [43]. A reduction in PTB levels triggers a neuronal-specific splicing program essential for the correct differentiation of progenitor cells into mature neurons during development [43]. During mouse myoblast differentiation, there is upregulation of the muscle-restricted miR-133 which downregulates the splicing factor nPTB triggering a new muscle-specific splicing program [44]. Finally, the miRNA miR-23a/b inhibits the protein levels of the splicing regulator CUGBP and ETR-3like factor (CELF) that regulates nearly half of developmentally regulated splicing transitions in the heart [45] (Figure 3a).
Figure 3.
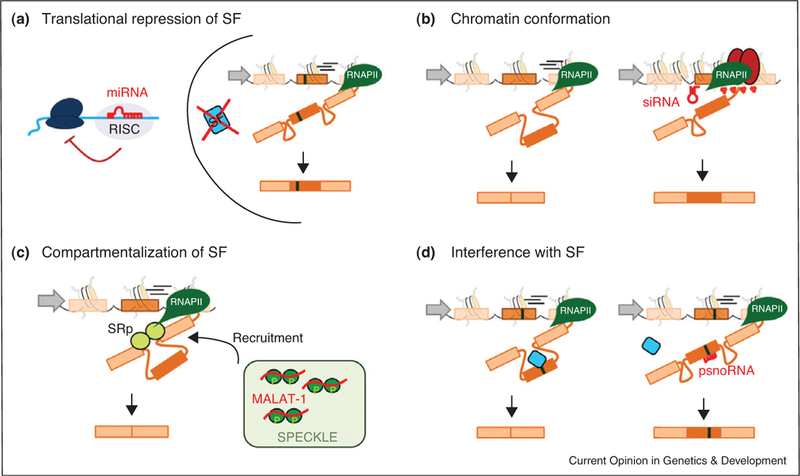
Mechanisms of alternative splicing regulation by ncRNAs. (a) MicroRNAs (red hairpin) regulate the protein levels of key developmental splicing factors (SF, blue rectangle). (b) siRNA-mediated heterochromatinization (red ovals) of a weak exon favors its inclusion. (c) The long intergenic ncRNA MALAT-1 (red line) maintains a pool of inactive SR proteins (dark green spheres) stored in splicing factor compartments (speckle). Splicing factors are released from speckles when needed. (d) The binding of a psnoRNA (red line) by sequence complementarity to an RNA silencer in the exon interferes with the recruitment of a splicing factor (blue rectangle) and subsequent exon inclusion.
Another type of short ncRNA, the siRNAs, have also been shown to impact alternative splicing. Experimental targeting of a siRNA complementary to the downstream intronic region of the alternatively spliced exon EDI of the fibronectin gene induces a localized hetero chromatinization and increases inclusion [10●●]. This observation raises the intriguing, albeit untested, possibility that RNAi-mediated transcriptional gene silencing may also be involved in alternative splicing regulation by modulating chromatin conformation near alternatively sliced gene regions (Figure 3b).
A different mode of action for a ncRNA in splicing regulation is exemplified by the long ncRNA MALAT-1. This ncRNA has been suggested to act as a buffer to control the pool of available splicing-competent serine-arginine rich (SR) proteins in the nucleoplasm [46]. In this scenario, inactive SR-proteins are sequestered in nuclear splicing speckles by binding to MALAT-1 until needed. Down-regulation of MALAT-1 increases the levels of active SR proteins in the nucleoplasm and leads to enhanced exon inclusion in a number of genes [46] (Figure 3c).
Although these mechanisms are indirect, there is the intriguing possibility of a more direct regulation of alternative splicing by complementary ncRNAs which bind directly to target pre-mRNAs. In support of such mechanisms, the brain-specific small nucleolar RNA (snoRNA) HB-52II is processed into smaller variants, the psnoRNAs, which are exported from the nucleolus to the nucleoplasm for binding by sequence complementarity to a silencing element in exon Vb of the serotonin receptor 5-HT2C pre-mRNA [47,48]. The binding of the psoRNAs to the exonic splice silencer probably interferes with the binding of a splicing repressor, favoring inclusion of the alternatively spliced exon [47,48] (Figure 3d). It will be interesting to see how many other alternative splicing events rely on such direct ncRNA regulation.
Conclusion
Regulation of alternative splicing is a multifactorial process. Although contributions from RNA motifs and the splicing machinery have long been appreciated, a significant role for chromatin, epigenetic modifications, and the transcription machinery are rapidly emerging. How precisely these factors contribute to regulation and how they interface with the splicing machinery remain to be deter-mined. Regardless, these findings open up new possibilities for the understanding of how a complex organism with thousands of highly tissue-specific and developmentally regulated alternative splicing programs can establish and maintain patterns of alternative splicing with a limited number of splicing regulators. Although we are beginning to uncover the effect of chromatin on RNA splicing, remarkably little is known about the impact of splicing on transcription and chromatin structure. Initial observations suggest that active recruitment of splicing factors to co-transcriptionally spliced genes is required for proper RNAP II elongation [49,50]; however, the real impact of splicing regulation upon chromatin structure, histone modification distribution, and transcription factors recruitment is still poorly understood.
It seems likely that what little we know about the link between chromatin and splicing is just the tip of the iceberg. A better understanding of the impact of histone modifications in the regulation, establishment and maintenance of cell-specific splicing programs will be essential for a full appreciation of the complexity of alternative splicing regulation. Importantly, these considerations also have practical implications since they may allow the development of novel and more efficient therapies against splicing-specific diseases.
Footnotes
Papers of particular interest, published within the period of review, have been highlighted as:
●of special interest
●●of outstanding interest
References and recommended reading
- 1.●●.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB: Alternative isoform regulation in human tissue transcriptomes. Nature 2008.One of the first genome-wide studies based on RNA deep-sequencing which revealed that large portions of the human genome are alternatively spliced in a highly tissue-specific fashion.
- 2.Wang GS, Cooper TA: Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 2007, 8:749–761. [DOI] [PubMed] [Google Scholar]
- 3.Venables JP: Aberrant and alternative splicing in cancer. Cancer Res 2004, 64:7647–7654. [DOI] [PubMed] [Google Scholar]
- 4.Grosso AR, Gomes AQ, Barbosa-Morais NL, Caldeira S, Thorne NP, Grech G, von Lindern M, Carmo-Fonseca M: Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res 2008, 36:4823–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graveley BR: Alternative splicing: regulation without regulators. Nat Struct Mol Biol 2009, 16:13–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.●●.Barash Y, Calarco JA, Gao WJ, Pan Q, Wang XC, Shai O, Blencowe BJ, Frey BJ: Deciphering the splicing code. Nature 2010, 465:53–59.This systematic computational analysis defines a ‘‘splicing code’’ based on the combinatorial use of RNA motifs.
- 7.Perales R, Bentley D: ‘‘Cotranscriptionality’’: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell 2009, 36:178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore MJ, Proudfoot NJ: Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009, 136:688–700. [DOI] [PubMed] [Google Scholar]
- 9.Carrillo Oesterreich F, Preibisch S, Neugebauer KM: Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell 2010, 40:571–581. [DOI] [PubMed] [Google Scholar]
- 10.●●.Allo M, Buggiano V, Fededa JP, Petrillo E, Schor I, de la Mata M, Agirre E, Plass M, Eyras E, Abou Elela S et al. : Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol 2009, 16:717–U743.RNAi-induced localized heterochromatinization changes alternative spli-cing patterns. The finding suggests that changes in chromatin structure can have a direct role in splicing outcome.
- 11.Schor IE, Rascovan N, Pelisch F, Allo M, Kornblihtt AR: Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci U S A 2009, 106:4325–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.●●.Munoz MJ, Santangelo MSP, Paronetto MP, De la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben Dov C, Blaustein M, Lozano JJ et al. :DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 2009, 137:708–720.This study reports physiological evidence for alteration of RNAP II kinetics by DNA damage that changes the alternative splicing patterns of genes involved in cell stress.
- 13.Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, Blencowe B: Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 2011. [DOI] [PMC free article] [PubMed]
- 14.●●.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T: Regulation of alternative splicing by histone modifications. Science 2010, 327:996–1000.This study provides evidence for histone modifications as modulators of pre-mRNA splicing and characterizes chromatin-adaptor complexes as novel regulators of alternative splicing.
- 15.Batsche E, Yaniv M, Muchardt C: The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol 2006, 13:22–29. [DOI] [PubMed] [Google Scholar]
- 16.Saint-Andre V, Batsche E, Rachez C, Muchardt C: Histone H3 lysine 9 trimethylation and HP1gamma favor inclusion of alternative exons. Nat Struct Mol Biol 2011, 18:337–344. [DOI] [PubMed] [Google Scholar]
- 17.Fu XD: Towards a splicing code. Cell 2004, 119:736–738. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB: Systematic identification and analysis of exonic splicing silencers. Cell 2004, 119:831–845. [DOI] [PubMed] [Google Scholar]
- 19.Blencowe BJ: Alternative splicing: new insights from global analyses. Cell 2006, 126:37–47. [DOI] [PubMed] [Google Scholar]
- 20.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH: An RNA code for the FOX2 splicing regulator revealed by mapping RNA–protein interactions in stem cells. Nat Struct Mol Biol 2009, 16:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB: An RNA map predicting Nova-dependent splicing regulation. Nature 2006, 444:580–586. [DOI] [PubMed] [Google Scholar]
- 22.Llorian M, Schwartz S, Clark TA, Hollander D, Tan LY, Spellman R, Gordon A, Schweitzer AC, de la Grange P, Ast G et al. : Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol 2010, 17:1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kornblihtt AR, De la Mata M, Fededa JP, Munoz MJ, Nogues G: Multiple links between transcription and splicing. RNA 2004, 10:1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T: Epigenetics in alternative pre-mRNA splicing. Cell 2011, 144:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR: A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003, 12:525–532. [DOI] [PubMed] [Google Scholar]
- 26.Robson-Dixon ND, Garcia-Blanco MA: MAZ elements alter transcription elongation and silencing of the fibroblast growth factor receptor 2 exon IIIb. J Biol Chem 2004, 279:29075–29084. [DOI] [PubMed] [Google Scholar]
- 27.●●.Alexander RD, Innocente SA, Barrass JD, Beggs JD: Splicing-dependent RNA polymerase pausing in yeast. Mol Cell 2010, 40:582–593.This is the first evidence for the existence of a splicing check point coordinating transcription and splicing in yeast.
- 28.●●.Singh J, Padgett RA: Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol 2009, 16:1128–1133.This study describes the in vivo kinetics of transcription and splicing and confirms coupling of the two processes.
- 29.Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C: Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science 2009, 325:626–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subtil-Rodriguez A, Reyes JC: BRG1 helps RNA polymerase II to overcome a nucleosomal barrier during elongation, in vivo. EMBO Rep 2010, 11:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.●●.Schwartz S, Meshorer E, Ast G: Chromatin organization marks exon–intron structure. Nat Struct Mol Biol 2009, 16:990–995.This study reports that nucleosomes and histone marks are non-randomly distributed along genes with specific enrichment at exons that is conserved throughout evolution.
- 32.Andersson R, Enroth S, Rada-Iglesias A, Wadelius C, Komorowski J: Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res 2009, 19:1732–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nahkuri S, Taft RJ, Mattick JS: Nucleosomes are preferentially positioned at exons in somatic and sperm cells. Cell Cycle 2009, 8:3420–3424. [DOI] [PubMed] [Google Scholar]
- 34.●●.Spies N, Nielsen CB, Padgett RA, Burge CB: Biased chromatin signatures around polyadenylation sites and exons. Mol Cell 2009, 36:245–254.Using genome-wide analysis, this extensive study identifies several histone modifications which differentially mark exons.
- 35.●●.Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcarcel J, Guigo R: Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol 2009, 16:996–1001.This study comprehensively maps nucleosomes along the genome and demonstrates that they are non-randomly distributed along genes with higher enrichment at exons defined by weaker splice sites and irrespective of the expression levels.
- 36.●●.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ et al. : Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466:388–392.This is one of the first genome-wide studies showing differential enrich-ment of DNA methylation and RNAP II specifically marking exons and correlating with nucleosome distribution along genes.
- 37.●●.Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J:Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet 2009, 41:376–381.This study provided the first evidence of selective enrichment of histone marks over exons and for differential marking of alternatively spliced exons.
- 38.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J et al. : The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brodsky AS, Meyer CA, Swinburne IA, Giles H, Keenan BJ, Liu XLS, Fox EA, Silver PA: Genomic mapping of RNA polymerase II reveals sites of co-transcriptional regulation in human cells. Genome Biol 2005:6. [DOI] [PMC free article] [PubMed]
- 40.Sims RJ, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D: Recognition of trimethylated histone h3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell 2007, 28:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loomis RJ, Naoe Y, Parker JB, Savic V, Bozovsky MR, Macfarlan T, Manley JL, Chakravarti D: Chromatin binding of SRp20 and ASF/SF2 and dissociation from mitotic chromosomes is modulated by histone H3 serine 10 phosphorylation. Mol Cell 2009, 33:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piacentini L, Fanti L, Negri R, Del Vescovo V, Fatica A, Altieri F, Pimpinelli S: Heterochromatin protein 1 (HP1a) positively regulates euchromatic gene expression through RNA transcript association and interaction with hnRNPs in Drosophila. Plos Genet 2009:5. [DOI] [PMC free article] [PubMed]
- 43.Makeyev EV, Zhang J, Carrasco MA, Maniatis T: The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 2007, 27:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boutz PL, Chawla G, Stoilov P, Black DL: MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 2007, 21:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalsotra A, Wang K, Li PF, Cooper TA: MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes Dev 2010, 24:653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA et al. : The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 2010, 39:925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kishore S, Khanna A, Zhang Z, Hui J, Balwierz PJ, Stefan M, Beach C, Nicholls RD, Zavolan M, Stamm S: The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum Mol Genet 2010, 19:1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kishore S, Stamm S: The snoRNA HBII-52 regulates alternative splicing oftheserotonin receptor 2C. Science 2006, 311:230–232. [DOI] [PubMed] [Google Scholar]
- 49.Lin SR, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD: The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol 2008, 15:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Damgaard CK, Kahns S, Lykke-Andersen S, Nielsen AL, Jensen TH, Kjems J: A 50 splice site enhances the recruitment of basal transcription initiation factors in vivo. Mol Cell 2008, 29:271–278. [DOI] [PubMed] [Google Scholar]