

Abstract
Deconstructive functionalization involves C–C bond cleavage followed by bond construction on one or more of the constituent carbons. For example, ozonolysis1 and olefin metathesis2, 3 have allowed each carbon in C–C double bonds to be viewed as a functional group. Despite the significant advances in deconstructive functionalizations involving scission of C–C double bonds, there are very few methods that achieve C(sp3)–C(sp3) single bond cleavage/functionalization, especially in relatively unstrained cyclic systems. Here, we report a deconstructive strategy to transform saturated nitrogen heterocycles such as piperidines and pyrrolidines, important moities in bioactive molecules, into halogen-containing acyclic amine derivatives through sequential C(sp3)–N/C(sp3)–C(sp3) single bond cleavage followed by C(sp3)–halogen bond formation. The resulting acyclic haloamines serve as versatile intermediates that are transformed into a variety of structural motifs through substitution reactions. In this way, skeletal remodeling of cyclic amines, which constitutes a scaffold hop, can be achieved. The value of this deconstructive strategy has been demonstrated through the late-stage diversification of proline-containing peptides.
The development of technologies that enable the late-stage diversification of bioactive, heterocycle-containing molecules (Fig. 1a) should facilitate access to under-explored chemical space4. Over the past two decades, significant effort has been dedicated to the development of methods to functionalize C–H bonds at a late stage, which has enabled the fine-tuning of substituents on nitrogen heterocycles, enhancing their functional group diversity (Fig. 1b)5, 6. In the medicinal chemistry community, there is growing demand for methods that modify not only the periphery (as in C–H functionalization) but also, the core framework of molecules (i.e., achieve skeletal diversity), a concept referred to as ‘scaffold hopping’7, 8. However, few methods are known that achieve deconstructive functionalization, for example with unstrained cyclic amines9–12. One recent example generated an aldehyde intermediate that can be further transformed to install C–O, C–C, and C–N bonds13.
Figure 1. Development of a deconstructive halogenation of cyclic amines.
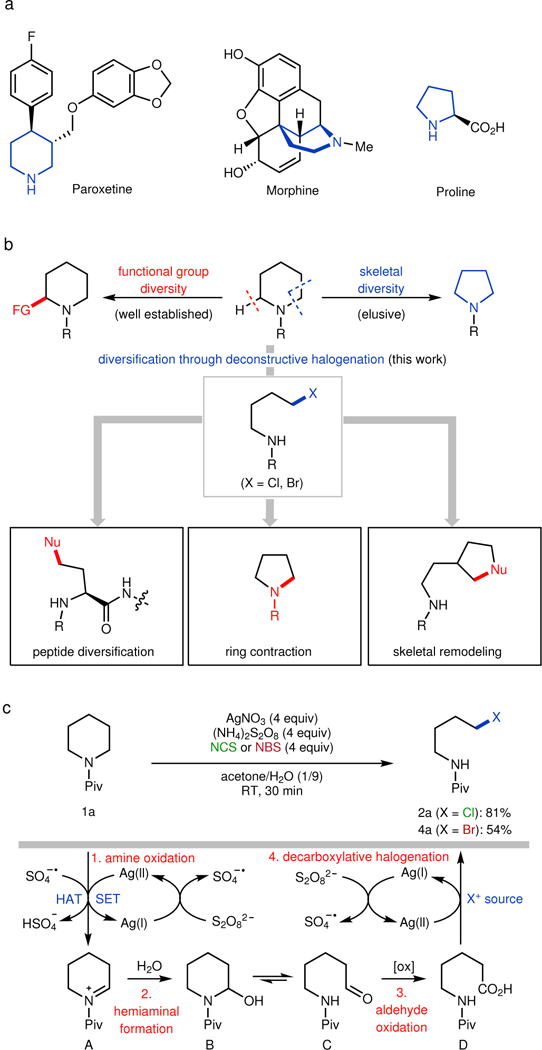
a, Representative bioactive molecules containing saturated nitrogen heterocycles. b, Deconstructive halogenation enables diversification of saturated nitrogen heterocycles. c, Proposed mechanism for silver-mediated deconstructive halogenation. FG, functional group; Nu, nucleophile; Piv, pivaloyl; NCS, N-chlorosuccinimide; NBS, N-bromosuccinimide; HAT, hydrogen-atom transfer; SET, single electron transfer.
In this context, ring-opening chlorination/bromination would generate versatile intermediate en route to diverse cyclic amines by coupling to a variety of nucleophiles (Fig. 1b). Furthermore, deconstructive halogenation of proline-containing peptides would furnish versatile intermediates for the late-stage diversification of these medicinally important entities14. Although ring-opening chlorination of cyclic amines is known, the existing methods to effect this transformation are limited to 3–5-membered, N-alkyl substituted, cyclic amines because of competing N-dealkylation15. Recently, our laboratory introduced a silver-mediated deconstructive strategy to transform cyclic amine derivatives into fluorine-containing acyclic amine derivatives using Selectfluor® via homolytic ring-opening of hemiaminal intermediates16. On the basis of mechanistic insights gained from our deconstructive fluorination protocol, we questioned whether it would be possible to access acyclic chloro/bromoamines from cyclic amines using our deconstructive strategy. Upon examination of existing reports on silver-catalyzed halogenation reactions, we recognized that simple replacement of Selectfluor® with N-halo-reagents such as N-chlorosuccinimide (NCS) or N-bromosuccinimide (NBS) would be unproductive presumably due to their lower oxidation potential17. Therefore, a distinct approach would be required to oxidize Ag(I) to Ag(II) in order to achieve deconstructive bromination/chlorination.
A detailed mechanistic proposal for our envisioned, highly orchestrated, reaction sequence is depicted in Fig. 1c. We theorized that consistent with existing precedent18, in the presence of persulfate anion, Ag(I) will be oxidized to Ag(II) with concomitant disproportionation of the persulfate anion into sulfate dianion and sulfate radical anion. N-acylated cyclic amines 1would then undergo a hydrogen-atom transfer (HAT) with the resulting sulfate radical anion to give an α-amino radical19. Subsequent oxidation by Ag(II) via single electron transfer (SET) would lead to iminium ion A. An alternative pathway wherein a Ag(II) species [E° (Ag2+/Ag+) = +1.98 V vs SCE]20 oxidizes N-acylated cyclic amines (e.g., 1a: [Epa = +2.02 V vs saturated calomel electrode (SCE)]) (see Supplementary Fig. S1) to the radical cation via SET followed by HAT using the sulfate radical anion to generate the same iminium ion, A, is also possible. The resulting iminium ion A would then be trapped by H2O to give hemi-aminal B. The heterolytic cleavage of the C–N bond would then occur through an equilibrium between hemi-aminal B and aldehyde C, the latter being subsequently oxidized to carboxylic acid D21, setting the stage for a silver-catalyzed decarboxylative halogenation17, 22. This strategy would represent a general method for deconstructive diversification as the electrophile is independent of the initial redox cycle.
We commenced our investigations of the proposed deconstructive halogenation by evaluating a broad range of silver salts, halogenating reagents, and solvent combinations. After extensive screening, we identified the optimized conditions shown in Fig. 1c that employs cheap and commercially available AgNO3, (NH4)2S2O8, and NCS in a 1:9 (v/v) mixture of acetone/H2O at room temperature. Upon subjecting N-pivaloyl piperidine (1a) to the optimized conditions, we obtained 81% yield of the desired acyclic chlorinated product 2a. Likewise, a bromine-atom could be readily incorporated to afford 4a in 54% yield by switching the electrophilic halogenating reagent to NBS. It is worth noting that this method can be performed without the strict exclusion of air. Control experiments established the importance of both silver and persulfate, as no formation of the desired chlorinated product was observed in the absence of the silver salt or persulfate additive. The optimized conditions employ 4 equivalents of AgNO3, whereas lower amounts led to diminished yields presumably due to substrate/product inhibition by binding to the silver salt (see Supplementary Table 1 for details).
With the optimized conditions in hand, we proceeded to investigate the scope of the deconstructive halogenation process (Fig. 2). An N-substituted piperidine derivative bearing a tert-butoxycarbonyl group (Boc, 1b), gave the desired chlorinated products in a combined 52% yield of 2b, along with formimide product 3b, which results from homolytic C–C bond cleavage of hemi-aminal B16. Unlike the bulky pivaloyl group which favors linear aldehyde C over hemiaminal B in the equilibration of the two species, the less sterically congested Boc group presumably favors B (see Fig. 1c). Bromination using NBS led to a mixture of mono and dibrominated products 5b and 6b in 65% combined yield. Upon switching the group on nitrogen to benzoyl (Bz, 1c), secondary amide products 2c and 4c were obtained as the major products along with formimide products 3c and 5c. In all cases, the secondary amide product and corresponding formimide are easily separated. Saturated heterocycles with various ring sizes (1d−1f) underwent deconstructive halogenation in moderate to good yields (55%−77% combined yield), whereas the deconstructive bromination of 1d led to 5,6-dihydro-4H-1,3-oxazine through autocyclization of desired alkyl bromide 4d (see the Supplementary Information for details)23. Substituents at the 2- and 4-position on piperidines are also well tolerated (1g−1i, 53%−80%). Polycyclic compounds such as 1j are also readily functionalized, paving the way for late-stage derivatization in more complex polycyclic frameworks. Halogenated amino acid derivatives (2k, 2l and 4k) are accessed in 3 steps from L-proline and L-pipecolic acid, which may serve as versatile intermediates to other unnatural amino acids.
Figure 2. Deconstructive halogenation: cyclic amine scope.

Only isolated yields are shown. Reaction conditions: 1 (0.1 mmol), NXS (4 equiv), (NH4)2S2O8 (4 equiv), acetone: H2O (1:9), room temperature, 0.5 h. Boc, tert-butoxycarbonyl; Bz, benzoyl; BRSM, based on recovered starting material. *5,6-dihydro-4H-1,3-oxazine was obtained (See the Supplementary Information for details).
Next, the skeletal remodeling of piperidine scaffolds bearing other reactive groups was examined (Fig. 3a). Oxidative ring-opening of 7 and engaging the pendant 2-nitrobenzenesulfonamide (NsNH) nucleophile with the incipient aldehyde group in 8 ultimately yielded corresponding lactam 9. The choice of halogenating reagent led to divergence in the products that were formed. For example, when carboxylic acid 10 was subjected to the deconstructive chlorination conditions, dichloro compound 11 was obtained through decarboxylative17 and deconstructive chlorination, and was directly transformed to azetidine 12 via double nucleophilic displacement with NsNH2. Alternatively, when NBS was used as the halogenating agent, in situ generated alkyl bromide 13 was engaged by the carboxylic acid group to form the corresponding lactone 14 in 44% yield.
Figure 3. Applications of deconstructive halogenation.

a, Skeletal remodeling of cyclic amines. b, Dehomologation of cyclic amines. *Yields in bracket represent the average yield per step. Ns, 2-nitrobenzenesulfonamide; DBU, 1,8-diazabicyclo(5.4.0)undec-7-ene; DMF, N,N-dimethylformamide.
Given the aforementioned importance of scaffold hopping in cyclic systems7, 8, we have also pursued the ring contraction of piperidines (Fig. 3b). Few reports exist that detail the ring contraction of piperidines to pyrrolidines24–26. Deconstructive bromination of N-benzoyl piperidine (1c) with dibromohydantoin followed by cyclization of the resulting bromoamine with NaOtBu furnished N-benzoyl pyrrolidine (15) in 89% (94% average yield per step) in just two steps with only one chromatographic purification step. Notably, this process can also be conducted in one-pot, albeit in lower yield (unoptimized) due to the competing displacement of the newly installed halogen group by the imide byproduct from the halogenating reagent. This ring contraction process also proceeds for a series of simple cyclic amines such as 2- and 4-methyl substituted piperidines and azepane (16, 18 and 20, 35%−60% yield over 2 steps). These results demonstrate a powerfully direct approach to achieving deep-seated structural modifications.
The virtue of this methodology is evident in the deconstructive functionalization/diversification of peptides27. As shown in Fig. 4a, L-proline-containing tripeptide 21 underwent ring-opening chlorination in 41% yield along with 15% recovered starting material (RSM). Importantly, chlorinated peptide 22 is easily transformed into a variety of products. For example, treatment of 22 with sodium methylthiolate afforded 23 in 91% yield, constituting the conversion of a proline residue into the corresponding methionine residue in only two steps. Alternatively, C–N bond formation can be achieved by treatment of 22 with sodium azide and in this way convert a proline residue or polypeptides bearing a cyclic amine (e.g., L-pipecolic acid) into a site for azide-based biorthogonal click chemistry28. In a demonstration of this tactic, 22 was azidated and then subjected to copper-catalyzed azide-alkyne cycloaddition to afford triazole 24 in 72% yield over the two steps. In addition, C–O bond formation is also easily achieved by displacement of the halogen group with benzoic acid. Treatment of 22 with NaCN in DMF led to nitrile 26 as the major product along with 5,6-dihydro-4H-1,3-oxazine 27 in 36% yield, demonstrating the feasibility of C–C bond formation. Cyclized product 27 is obtained as the sole product when 22 is treated with DBU.
Figure 4. Deconstructive chlorination of L-proline-containing peptides.

a, Deconstructive diversification of tripeptide 21. b, The tolerance for oxidizable amino acid residues. c, Deconstructive chlorination of L-phenylalanine-containing tripeptide 30. d, Deconstructive fluorination of tripeptide 21. RSM, recovered starting material; Tf, trifluoromethanesulfonyl.
Additionally, we evaluated the functional group tolerance of the deconstructive chlorination process. As shown in Fig. 4b, a variety of dipeptides bearing potentially oxidizable amino acid residues participate in this deconstructive protocol (29a−29f, 19%−44%). It is worth noting that the proline residue can be preferentially oxidized over the benzylic position (29a and 29b) and C–H bonds of the activated aliphatic side-chains bearing oxygen heteroatoms (29e and 29f). A dipeptide bearing a methionine residue 29g underwent deconstructive chlorination with oxidation of the thioether to the corresponding sulfone. Therefore, like many other oxidative processes29, 30, deconstructive halogenation leads to competing reaction with the sulfur group of methionine. Additionally, deconstructive chlorination of the challenging tripeptide substrate 30 proceeded to furnish 16% yield of the ring-opened product 31 along with 62% of recovered starting material (Fig. 4c). Given the mechanistic change in the current methodology to incorporate a heterolytic C–N cleavage (BC, Fig. 1c), over-oxidation of the hemiaminal intermediate B is generally avoided as evidenced by the ring opening fluorination of 21 to give fluorinated tripeptide 32 using the newly developed strategy (Fig. 4d). Despite the lower yields obtained in the presence of these reactive residues, the deconstructive protocol provides an expedient approach to a novel range of peptides without the need for their de novo synthesis.
Saturated heterocycles remain a prevalent structural motif that is found in a large percentage of bioactive organic molecules such as pharmaceuticals. We anticipate that deconstructive functionalization strategies will provide access to wide-ranging structural diversity at a late stage in the preparation of bioactive molecules.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS RO1 086374). J.B.R. thanks the NIH for a graduate diversity supplement fellowship ((NIGMS RO1 086374). Y.K. thanks the Japan Society for the Promotion of Science (JSPS) for an Overseas Research Fellowship. L.T.G. thanks LMU PROSA and DAAD for financial support. We thank Jeffrey Derrick for assistance with electrochemical measurements.
Footnotes
Author Information
Reprints and permissions information is available at www.nature.com/reprints.
Competing Interests: J.B.R., Y.K., L.T.G., and R. S. are listed as inventors on an initial patent application describing the Ag-mediated deconstructive halogenation of cyclic amines and subsequent transformations (052103–515P01US).
Data availability
All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.
References
- 1.Bailey PS The reactions of ozone with organic compounds. Chem. Rev. 58, 925–1010 (1958). [Google Scholar]
- 2.Hoveyda AM & Zhugralin AR The remarkable metal-catalysed olefin metathesis reaction. Nature 450, 243–251 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Vougioukalakis GC & Grubbs RH Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 110, 1746–1787 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Galloway WRJD, Isidro-Llobet A & Spring DR Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 1, 80 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Chen W, Ma L, Paul A & Seidel D Direct α-C–H bond functionalization of unprotected cyclic amines. Nat. Chem. 10, 165–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cernak T, Dykstra KD, Tyagarajan S, Vachal P & Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Sun H, Tawa G & Wallqvist A Classification of scaffold-hopping approaches. Drug Discov. Today 17, 310–324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Stumpfe D & Bajorath J Recent advances in scaffold hopping. J. Med. Chem. 60, 1238–1246 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Shawcross AP & Stanforth SP Reaction of N-nitroaryl-1,2,3,4-tetrahydroisoquinoline derivatives with oxygen. J. Heterocyclic Chem. 27, 367–369 (1990). [Google Scholar]
- 10.Han G, McIntosh MC & Weinreb SM A convenient synthetic method for amide oxidation. Tetrahedron Lett. 35, 5813–5816 (1994). [Google Scholar]
- 11.Ito R, Umezawa N & Higuchi T Unique oxidation reaction of amides with pyridine-N-oxide catalyzed by ruthenium porphyrin: Direct oxidative conversion of N-acyl-L-proline to N-acyl-L-glutamate. J. Am. Chem. Soc. 127, 834–835 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Kaname M, Yoshifuji S & Sashida H Ruthenium tetroxide oxidation of cyclic N-acylamines by a single layer method: formation of ω-amino acids. Tetrahedron Lett. 49, 2786–2788 (2008). [Google Scholar]
- 13.Osberger TJ, Rogness DC, Kohrt JT, Stepan AF & White MC Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 537, 214–219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henninot A, Collins JC & Nuss JM The current state of peptide drug discovery: Back to the future? J. Med. Chem. 61, 1382–1414 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Yu C et al. Selective ring-opening of N-alkyl pyrrolidines with chloroformates to 4-chlorobutyl carbamates. J. Org. Chem. 82, 6615–6620 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Roque JB, Kuroda Y, Göttemann LT & Sarpong R Deconstructive fluorination of cyclic amines by carbon-carbon cleavage. Science 361, 171–174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z et al. Silver-catalyzed decarboxylative chlorination of aliphatic carboxylic acids. J. Am. Chem. Soc. 134, 4258–4263 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Anderson JM & Kochi JK Silver(I)-catalyzed oxidative decarboxylation of acids by peroxydisulfate. Role of silver(II). J. Am. Chem. Soc. 92, 1651–1659 (1970). [Google Scholar]
- 19.Dai C, Meschini F, Narayanam JMR & Stephenson CRJ Friedel–Crafts amidoalkylation via thermolysis and oxidative photocatalysis. J. Org. Chem. 77, 4425–4431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Po HN Heterocyclic and macrocyclic amine complexes of silver(II) and silver(III). Coord. Chem. Rev. 20, 171–195 (1976). [Google Scholar]
- 21.Gallopo AR & Edwards JO Kinetics and mechanisms of the spontaneous and metal-modified oxidations of ethanol by peroxydisulfate ion. J. Org. Chem. 36, 4089–4096 (1971). [Google Scholar]
- 22.Tan X et al. Silver-catalyzed decarboxylative bromination of aliphatic carboxylic acids. Org. Lett. 19, 1634–1637 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Reddy DN & Prabhakaran EN Synthesis and isolation of 5,6-dihydro-4H-1,3-oxazine hydrobromides by autocyclization of N-(3-bromopropyl)amides. J. Org. Chem. 76, 680–683 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Bandarage UK & Davies RJ A new synthesis of spiropyrrolidine–tetralones via an unexpected formal ring-contraction of 4-disubstituted piperidine to 3-disubstituted pyrrolidine. Tetrahedron Lett. 51, 6415–6417 (2010). [Google Scholar]
- 25.Aldmairi AH, Griffiths-Jones C, Dupauw A, Henderson L & Knight DW Piperidines from acid-catalysed cyclisations: Pitfalls, solutions and a new ring contraction to pyrrolidines. Tetrahedron Lett. 58, 3690–3694 (2017). [Google Scholar]
- 26.Sengupta S & Mehta G Late stage modification of peptides via C–H activation reactions. Tetrahedron Lett. 58, 1357–1372 (2017). [Google Scholar]
- 27.Wang F, He Y, Tian M, Zhang X & Fan Z Synthesis of α‑formylated N‑heterocycles and their 1,1-diacetates from inactivated cyclic amines involving an oxidative ring contraction. Org. Lett. 20, 864–867 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Kolb HC Finn MG & Sharpless KB. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Shechter Y, Burstein Y & Patchornik A Selective oxidation of methionine residues in proteins. Biochemistry 14, 4497–4503 (1975). [DOI] [PubMed] [Google Scholar]
- 30.Lin S et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.