

Abstract
Biofilms are surface-bound, structured microbial communities underpinning persistent bacterial infections. Biofilms often create acidic pH microenvironments, providing opportunities to leverage responsive drug delivery systems to improve antibacterial efficacy. Here, the antibacterial efficacy of novel formulations containing pH-responsive polymer nanoparticle carriers (NPCs) and farnesol, a hydrophobic antibacterial drug, were investigated. Multiple farnesol-loaded NPCs, which varied in overall molecular weight and corona-to-core molecular weight ratios (CCRs), were tested using standard and saturated drug loading conditions. NPCs loaded at saturated conditions exhibited ~300% greater drug loading capacity over standard conditions. Furthermore, saturated loading conditions sustained zero-ordered drug release over 48 hours, which was 3-fold longer than using standard farnesol loading. Anti-biofilm activity of saturated NPC loading was markedly amplified using Streptococcus mutans as a biofilm-forming model organism. Specifically, reductions of ~2–4 log colony forming unit (CFU) were obtained using microplate and saliva-coated hydroxyapatite biofilm assays. Mechanistically, the new formulation reduced total biomass by disrupting insoluble glucan formation and increased NPC-cell membrane localization. Finally, thonzonium bromide, a highly potent, FDA-approved antibacterial drug with similar alkyl chain structure to farnesol, was also loaded into NPCs and used to treat S. mutans biofilms. Similar to farnesol-loaded NPCs, thonzonium bromide-loaded NPCs increased drug loading capacity ≥ 2.5-fold, demonstrated nearly zero-order release kinetics over 96 hours, and reduced biofilm cell viability by ~6 log CFU. This work provides foundational insights that may lead to clinical translation of novel topical biofilm-targeting therapies, such as those for oral diseases.
Graphical Abstract
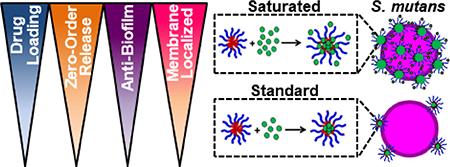
Novel polymer nanoparticle formulation improved drug loading, demonstrated zero-order release, and amplified S. mutans anti-biofilm activity via increased membrane localization.
Introduction
Biofilms underpin major public health and economic burdens, accounting for >80% of microbial-based infections and resulting in billions of U.S. dollars in healthcare costs annually (1, 2). In particular, oral biofilms that are responsible for causing dental caries, or tooth decay, contribute greatly to this burden due to their high prevalence in humans (>30% of schoolchildren and >90% of adult population worldwide) (3–5) with annual expenditures exceeding $120 billion in the U.S. alone (6–8). Biofilms are challenging to treat because bacteria produce a protective exopolysaccharide (EPS) matrix that affords antimicrobial drug tolerance (9–11). Furthermore, drug retention in the oral cavity remains a major hurdle limiting clinical relevance of topically applied anti-biofilm treatments. Bacterial pathogens, such as the cariogenic Streptococcus mutans (S. mutans), create EPS-rich and highly acidic microenvironments within the biofilm, making them difficult to reach by topical treatments often removed by salivary clearance (11–13). These microenvironments, located at or near the tooth surface, are known to reach pH values of 5.5 or lower that can dissolve the enamel, leading to dental caries (12, 14, 15). Therefore, potential treatments must bind and remain within the EPS matrix while also enduring or responding to the acidic conditions within biofilms.
To overcome these hurdles, recent efforts have focused on using nanoparticles as drug delivery vehicles for anti-biofilm therapeutics. Multiple strategies, including the use of liposomes or polymer nanoparticles as drug delivery carriers and metal nanoparticles as antibacterial agents, have shown varying degrees of promise in recent years (16–19). Among these approaches, cationic amphiphilic polymers have been shown to have selectivity for and inherent bactericidal activity against S. mutans, resulting in biofilm inhibition (20–23). Additionally, previous studies have investigated combinatorial effects between cationic polymers and antibiotics (24, 25). However, none of the carriers designed to treat oral biofilms have overcome every delivery hurdle due to limited fundamental understanding of nanoparticle design parameters and interactions between nanoparticle drug delivery carriers and bacteria and/or biofilm matrix. Therefore, an opportunity exists to use a known antibacterial drug with cationic, pH-responsive polymer nanoparticle carriers (NPCs) in new formulations against an established biofilm-forming model organism to better understand how both the drug and NPC affect antibacterial efficacy.
Previous work has shown the hydrophobic drug farnesol has antimicrobial effects against a range of pathogens, including S. mutans (26–28). Farnesol disrupts bacterial cell membranes, resulting in increased proton permeability that affects cell-membrane function (26, 29, 30). However, topical farnesol treatments require multiple treatments of high drug concentrations to inhibit oral biofilm growth (31) due to hydrophobicity-related solubility limits and poor biofilm retention (30, 32, 33). To overcome these hurdles, polymeric nanoparticles have been used to improve targeted drug delivery and increase residence time of antimicrobials within biofilms (34–36). The use of drug delivery systems, including polymeric nanoparticles, has been shown to improve farnesol delivery and effectiveness against oral biofilms (35, 37–39). We previously established that the antibacterial efficacy of farnesol is improved through delivery using pH-responsive polymer nanoparticle carriers (NPCs) (38, 39). However, while the farnesol-NPC formulation had maximum drug loading capacities between 22% and 27%, it only yielded ~1 log reduction in biofilm bacterial viability (38, 39). Interestingly, formulations using saturated hydrophobic drug conditions have been shown to increase drug loading (e.g. > 50% increase) within nanoparticles (40). Since antibacterial activity directly correlates with drug concentration (41), increased drug loading should improve antibacterial effectiveness.
Here we investigate how drug loading formulations as well as pH-responsive NPC design parameters can be exploited to enhance efficacy of farnesol and thonzonium bromide (a FDA-approved drug that is structurally similar to farnesol) against planktonic and biofilm S. mutans cells. New saturated drug-NPC formulations improve drug loading capacity, slow drug release, and leverage inherent antibacterial activity associated with cationic NPCs. NPC performance is evaluated multiple ways ranging from traditional microplate antibacterial and antibiofilm assays, to biofilm treatment models with tooth-surface mimetics, to mechanistic investigation of saturated farnesol-NPC formulation inhibition of S. mutans viability in planktonic and biofilm states. Ultimately, this work is intended to help provide foundational insights that may lead to clinical translation of novel topical antimicrobial therapies, such as those used against oral biofilms.
Experimental
Materials
Unless otherwise specified, all materials were supplied by Sigma-Aldrich. Dimethylaminoethyl methacrylate (DMAEMA) and butyl methacrylate (BMA) were purified by distillation prior to use. The chain transfer agent (CTA) used for reversible addition-fragmentation chain transfer (RAFT) polymerization, 4-cyano-4-[(ethylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (ECT), and propylacrylic acid (PAA) were synthesized as described previously (42–44). 2,2-Azobisisobutyronitrile (AIBN) was recrystallized from methanol. All water used was deionized and distilled with resistivity of 18 MΩ unless otherwise specified.
Polymer Synthesis and Characterization
Cationic corona block (Block 1) synthesis
Poly(dimethylaminoethyl methacrylate), or p(DMAEMA), was synthesized via RAFT polymerization using ECT as the CTA and AIBN as the radical initiator. Distilled DMAEMA was mixed with specific molar ratios of ECT and AIBN so the [monomer]/[CTA]/[Initiator] = 500/5/1, 2950/5/1, 4000/5/1, 1000/1/1, or 950/9.5/1 in dimethylformamide (DMF) at 40 wt% to obtain the different Block 1 molecular weights shown in Table 1. The reaction vessel was purged with nitrogen using a Schlenk line for 45 minutes and polymerized at 60 °C for 6 hours in an oil bath. The reaction was terminated by exposing the reactants to atmospheric oxygen. The product was precipitated and washed 4 times in 80:20 pentane:diethyl ether with centrifugation and dried under vacuum overnight.
Table 1.
Polymer and NPC characteristicsΔ used in this study
| Polymers |
NPC |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Corona Block |
Core Block |
Diblock Copolymers |
||||||||||||||||
| Polymer | DP | Block 1Mn1(kDa) | PDI | DP | Block 2Mn1(kDa) | %DMAEMA2 | %BMA2 | %PAA2 | OverallMn1(kDa) | PDI | CCR | Size3(nm) | ζ4(mV) | |||||
| NP12/2 | 100 | 12.4 | α | 1.05 | 40 | 2.1 | α | 17 | 57 | 26 | 14.5 | α | 1.04 | 5.9 | ν | 7.7 ± 2.9 | ρ | 18.3 ± 3.8 |
| NP12/3a | 100 | 12.4 | α | 1.05 | 65 | 3.1 | α | 11 | 38 | 51 | 15.5 | α | 1.05 | 4.0 | λ | 20.5 ± 5.7 | σ | 15.8 ± 5.0 |
| NP12/3b | 100 | 12.4 | α | 1.05 | 65 | 3.2 | α | 2 | 54 | 44 | 15.6 | α | 1.05 | 3.9 | κ | 17.1 ± 4.5 | ρ | 20.1 ± 3.5 |
| NP12/3c | 100 | 12.4 | α | 1.05 | 65 | 3.2 | α | 18 | 38 | 44 | 15.6 | α | 1.04 | 3.8 | κ | 12.9 ± 4.7 | ρ | 16.1 ± 4.3 |
| NP12/4 | 100 | 12.4 | α | 1.05 | 65 | 3.5 | α | 5 | 62 | 33 | 15.9 | α | 1.04 | 3.5 | κ | 13.8 ± 4.3 | ρ | 18.6 ± 4.1 |
| NP12/5 | 100 | 12.4 | α | 1.05 | 120 | 5.2 | α | 1 | 66 | 33 | 17.6 | α | 1.04 | 2.4 | ι | 20.6 ± 5.3 | σ | 21.3 ± 4.4 |
| NP12/12 | 100 | 12.4 | α | 1.05 | 300 | 11.6 | α | 7 | 73 | 20 | 24.0 | β | 1.06 | 1.1 | θ | 29.4 ± 7.3 | σ | 23.2 ± 4.1 |
| NP13/2 | 105 | 12.7 | α | 1.07 | 35 | 2.4 | α | 8 | 57 | 35 | 15.2 | α | 1.04 | 5.3 | ν | 9.1 ± 3.8 | ρ | 20.5 ± 4.4 |
| NP13/3 | 105 | 12.7 | α | 1.07 | 35 | 3.1 | α | 27 | 59 | 15 | 15.8 | α | 1.04 | 4.1 | λ | 15.1 ± 4.1 | ρ | 20.4 ± 3.9 |
| NP13/4 | 105 | 12.7 | α | 1.07 | 35 | 3.5 | α | 37 | 57 | 6 | 16.2 | α | 1.04 | 3.6 | κ | 18.6 ± 4.6 | ρ | 19.7 ± 4.2 |
| NP13/5 | 105 | 12.7 | α | 1.04 | 40 | 4.7 | α | 51 | 27 | 22 | 17.4 | α | 1.06 | 2.7 | ι | 11.3 ± 5.2 | ρ | 19.6 ± 4.4 |
| NP13/31 | 100 | 13.0 | α | 1.04 | 1000 | 30.8 | β | 8 | 66 | 26 | 43.8 | γ | 1.02 | 0.4 | η | 69.1 ± 20.8 | τ | 14.3 ± 3.9 |
| NP47/25 | 600 | 47.1 | γ | 1.08 | 1000 | 24.9 | β | 2 | 46 | 52 | 72.0 | δ | 1.02 | 1.9 | θ | 63.5 ± 21.3 | τ | 19.6 ± 5.1 |
| NP52/15 | 800 | 52.2 | γ | 1.13 | 350 | 14.6 | α | 33 | 32 | 35 | 66.8 | δ | 1.05 | 3.6 | κ | 30.8 ± 9.4 | σ | 18.1 ± 4.4 |
| NP99/50 | 1000 | 98.9 | ε | 1.05 | 2000 | 49.6 | γ | 3 | 58 | 39 | 148.5 | ε | 1.05 | 2.0 | θ | 106.0 ± 53.2 | υ | 20.0 ± 4.0 |
| NP99/104 | 1000 | 98.9 | ε | 1.05 | 4000 | 104 | ε | 3 | 69 | 29 | 202.9 | ε | 1.05 | 1.0 | η | 90.9 ± 40.7 | υ | 24.6 ± 3.8 |
As characterized by
gel permeation chromatography
nuclear magnetic resonance
dynamic light scattering, and
electrophoretic light scattering.
Abbreviations: CCR, corona-to-core molecular weight ratio; DP, target degree of polymerization; PDI, polydispersity index (Mw/Mn); ζ, zeta potential. Groups identified for data analysis: Mn, α = 1–20 kDa, β = 20–40 kDa, γ = 40–60 kDa, δ = 60–80 kDa, ε ≥ 80 kDa; CCR, η ≤ 1, θ = 1–2, ι = 2–3, κ = 3–4, λ = 4–5, ν ≥ 5; Size, ρ = 1–20 nm, σ = 20–40 nm, τ = 60–80 nm, υ ≥ 80 nm.
Hydrophobic core block (Block 2) synthesis
Poly(dimethylaminoethyl methacrylate)-b-poly(dimethylaminoethyl methacrylate-co-butyl methacrylate-co-propylacrylic acid), or p(DMAEMA)-b-p(DMAEMA-co-BMA-co-PAA), was synthesized using RAFT polymerization with p(DMAEMA) as the macroCTA, AIBN as the radical initiator, and 15–25 wt% DMAEMA, 50–55 wt% BMA, and 25–30 wt% PAA dissolved in DMF (40 wt% monomers, initiator, and macroCTA to solvent volume). The target degree of polymerization (DP), or [monomer]/[CTA], was varied according to Table 1 to control block Mn, and [ECT]/[AIBN] = 5 for all polymerizations except both blocks for NP99/50 and NP99/104 and the second blocks of NP13/31 and NP47/25 where [CTA]/[initiator] = 1 and the first block of NP13/31 where [CTA]/[initiator] = 9.5. After purging with nitrogen for 45 minutes, the reaction proceeded at 60 °C for 24 hours. The reaction was terminated by exposing the reactants to atmospheric oxygen. The product was precipitated 4 times using 80:20 pentane:diethyl ether and centrifugation before being dried overnight under vacuum.
Polymer purification and storage
Dried diblock copolymer was removed from vacuum and dissolved in ~5 mL 100% ethanol in a 50 mL conical tube. Once the raw polymer was completely dissolved, ~25 mL 1X DPBS was added to the tube. The combined solution was transferred into pre-wetted 6–8 kDa dialysis membrane tubing (Spectrum Laboratories), which was securely clipped at each end. This tubing was placed in water for ≥4 days with ~2–3 water changes each day (8–10 changes total) to remove contaminants. The dialyzed solution was frozen at −80 °C and lyophilized for ≥4 days using a Labconco FreeZone 2.5 freeze dryer. The lyophilized polymer was stored in closed containers at room temperature until use.
Polymer Characterization
Molecular weights and polydispersities (PDI, Mw/Mn) of first block and diblock copolymers were determined using gel permeation chromatography (GPC, Shimadzu Technologies) with a miniDAWN TREOS multi-angle light scattering detector (Wyatt Technology) in line with an Optilab T-rEX refractive index detector (Wyatt Technology). High Performance Liquid Chromatography (HPLC) grade DMF + 0.05 mM LiCl (0.2 μm filtered) was used as the mobile phase at a flow rate of 0.35 mL/min through a TSKgel SuperH-H guard column and TSKgel SuperHM-N column (Tosoh Biosciences) at 60 °C. ASTRA® 6.1 light scattering software (Wyatt Technology) and a previously reported dn/dc value of 0.06 (45, 46) were used to calculate molecular weight. Diblock copolymer composition was characterized using 1H NMR spectroscopy (Bruker Avance 400), as described previously (42).
Polymer Labeling
Polymers used for characterization interactions with S. mutans were labeled with Alexa Fluor® 488 cadaverine (Life Technologies, Eugene, OR, USA). Labeling was performed at 1:1:5 molar ratios of polymer : Alexa Fluor® 488 cadaverine : 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) for ~4 hours in pH 7.4 phosphate buffer using 5 mM N-hydroxysulfosuccinimide (sulfo-NHS). Labeled polymers were purified via dialysis using 6–8 kDa dialysis membrane tubing (Spectrum Laboratories) in water at room temperature for ≥1 day with 4 separate water changes. Once free Alexa Fluor® 488 cadaverine was no longer observed in the dialysis water, the polymer samples were frozen for at least 1 hour at −80 °C and lyophilized.
NPC Self-Assembly Characterization
A Zetasizer Nano ZS (Malvern Panalytical) was used to measure size and zeta potential. NPC size measurements were performed via dynamic light scattering (DLS) analysis using lyophilized polymer concentrations of ~0.2–0.3 mg/mL fully dissolved in 1X PBS and passed through a 0.45 μm PVDF aqueous syringe filter into disposable cuvettes. Zeta potential was determined using polymer concentrations of ~0.2–0.5 mg/mL in 90:10 water:1X PBS solutions and filtered using 0.45 μm PVDF aqueous syringe filters into disposable p1070 capillary cells to ensure sample conductivity values permitted analysis via General Purpose analysis in the Malvern Zetasizer software.
NPC Critical Micelle Concentration Determination
NPC critical micelle concentration (CMC) was completed using fluorescence spectral emission shifts of 6-propionyl-2-(dimethylamino)naphthalene (PRODAN, Molecular Probes, Eugene, OR, USA) caused by partitioning differences into hydrophilic versus hydrophobic NPC regions, as described previously (38, 39, 47). PRODAN was dissolved in methanol at 24 μM, and 10 μL of this PRODAN solution was aliquoted into each well of a black 96-well plate. After drying overnight, a range of NPC solutions (0.5 ng/mL to 0.1 mg/mL) were diluted using 1X DPBS, and 100 μL/well of each sample solution was loaded into the 96-well plates containing PRODAN. All samples were loaded in triplicate, and plates were sealed and incubated at 4 °C overnight. PRODAN fluorescence was measured at two wavelengths (Ex/Em1: 360 nm/435 nm for hydrophilic detection and Ex/Em2: 360 nm/520 nm for hydrophobic detection) using a Tecan plate reader. The ratio of hydrophilic to hydrophobic emission (Em1/Em2) was plotted against the log10 polymer concentration, linear regression lines were applied to each phase of the graph, and the CMC was determined as the concentration at which these regression lines intersect (see Supplemental Figure S2).
Characterization of NPC Drug Loading
NPCs were loaded with farnesol as described previously (38, 39) with a few modifications. Briefly, farnesol/1X DPBS emulsions at 1.0 mg/mL were prepared using tip sonication (Fisher Scientific Sonic Dismembrator Model 100 at 4W power setting) for ~30 seconds and immediately added to pre-weighed lyophilized diblock copolymers to achieve desired polymer NPC concentrations (e.g., 0.5 mg/mL or 2.7 mg/mL) in 20 mL glass scintillation vials. These solutions were sonicated (VWR 50T Ultrasonic Cleaner) for 15 minutes to enable drug loading. NPCs loaded with farnesol were concentrated using 3 kDa centrifugal filters (Amicon Ultra 0.5 mL, Millipore, USA). The concentrate was recovered and 1X DPBS was added to return the sample volume to 0.5 mL. This solution was diluted with an equal amount of ethanol and passed through a 0.45 μm PVDF aqueous syringe filter before the amount of farnesol loaded was measured via high performance liquid chromatography (HPLC) using a 20 μL injection volume. HPLC analysis was conducted using a gradient mobile phase consisting of HPLC-grade methanol and water (10% to 90% MeOH) and a Kromasil C18 column (50 mm × 4.6 mm, 5 μm particle size, 100 Å pore size from Supelco, Bellefonte, PA, USA) with a flow rate of at 0.5 mL/min over 20 minutes. The column effluent was monitored with a variable wavelength UV−vis detector at 210 nm (Shimadzu Technologies). Relative area-under-the-curve values for the farnesol peaks that occur between ~15–17 minutes were compared to a standard curve of known farnesol concentrations to determine the concentration of farnesol loaded. The amount of farnesol loaded was used to calculate drug loading efficiency (100% x (Wtloaded)/Wt0) and drug loading capacity (100% x (Wtloaded/WtNPC + Wtloaded)) where Wtloaded is the amount of loaded drug, Wt0 is the initial amount of drug used, and WtNPC is the amount of polymer used.
HPLC was also used to quantify thonzonium bromide loading. In 20 mL glass scintillation vials, pre-calculated volumes of 1X DPBS were combined with pre-weighed lyophilized diblock copolymers and known volumes of thonzonium bromide fully dissolved in DMSO to achieve desired polymer NPC and drug concentrations. These solutions were bath sonicated, centrifuged, diluted with PBS, filtered, and analyzed using HPLC. The mobile phase consisted of HPLC-grade acetonitrile and 0.1% formic acid with the following flow conditions: 0.5 mL/min flow rate; gradient elution 0–3.5 min 20% acetonitrile, 3.5–10 min 80% acetonitrile.
Characterization of NPC Drug Release
Drug release from NPCs was characterized using dialysis as described previously (38, 39, 48). After loading, NPC/PBS solution was transferred into pre-wetted 6–8 kDa dialysis membrane tubing (Spectrum Laboratories), which was securely clipped at each end. The filled dialysis membranes were placed in phosphate buffer at pH 4.5 or 7.2 and dialyzed at 37 °C with buffer changes occurring after each sample collection time point to ensure sink conditions. For farnesol release studies, samples were collected at 0, 2, 4, 6, 12, 24, 48, and 72 hours. For thonzonium bromide release studies, samples were collected at 0, 2, 4, 6, 24, 48, 72, and 96 hours. Samples were immediately frozen and stored at −80 °C until drug concentration of each sample was assessed via HPLC as described above for drug loading characterization. First-order release fits of the data were performed using GraphPad Prism software (v.6.07), and release rate constants (kobs) and release half-times (t1/2) were calculated according to the first order release equation (% release = 100 × (1 − e−k*t)), where % release is the percent of drug release at time t, and k is the observed kinetic constant of drug release (kobs). The following relationship between kobs and t1/2 was used for value conversions: t1/2 = ln(2)/kobs. First derivatives of the release fit equations were calculated by GraphPad Prism to yield graphical depictions of the change in release rates over time.
Polymer Shipping
For each sample shipment from Rochester, NY to Philadelphia, PA, two frozen gel packs were removed from −80 °C at least 3 hours prior to the planned shipment drop-off time and stored in a closed standard laboratory foam shipping container held at room temperature. This step enabled sufficient pre-packout temperature conditioning for the container and warmed the gel packs prior to sample packing to prevent sample freezing. Samples consisting of lyophilized polymer and/or aqueous solutions containing NPC and/or drugs in 20 mL scintillation vials were either wrapped in protective dunnage (e.g., packing peanuts, paper towels, and/or aluminum foil) or loaded in cardboard boxes. These protected sample containers were placed in the foam shipping container between the two frozen gel packs and covered in additional protective dunnage to maintain interior container temperatures between 0 and 8 °C for at least 24 hours during overnight shipment. This level of temperature control maintained sample stability (e.g., no detectable physical changes; data not shown). Test shipments between labs confirmed reproducible temperature control throughout the duration of a representative overnight shipment (Supplemental Figure S9). Upon shipment receipt, aqueous samples were stored at 2–8 °C and lyophilized polymer samples were stored at room temperature until use.
Minimum Inhibitory Concentration Testing
The minimum inhibitory concentration (MIC) of farnesol-loaded nanoparticles was tested using planktonic Streptococcus mutans cells using a microdilution (microplate) assay as described previously (49). S. mutans UA159 serotype c (a biofilm-forming and cariogenic model organism) cells were grown in ultra-filtered (10 kDa cutoff; Millipore) tryptone-yeast extract (UFTYE) broth containing 1% glucose (w/v) at 37 °C and 5% CO2 to mid-exponential phase. A range of different concentrations (0.125 to 64 μg/ml) of farnesol-loaded nanoparticles were prepared in UFTYE broth at pH 7.0 and 5.0, and dispensed into a 96-well plate (90 μl per well). Ten μl of S. mutans suspension (106 colony forming units (CFU)/ml) was then added into each well containing UFTYE and test agents. After 24 h incubation at 37 °C with 5% CO2, the MIC was determined as the lowest concentration of the test agent that inhibited bacterial growth as described previously (49). The MICs were determined in triplicates in three different experiments. Supplemental Figure S3 summarizes the MIC testing process and the CFU growth assay used to evaluate the antibacterial effectiveness against planktonic S. mutans.
Minimum Concentration for Biofilm Killing (MCBK) Assay
The minimum concentration for biofilm killing (MCBK) by farnesol-loaded nanoparticles was determined by microdilution (microplate) assay. Each well was inoculated with ~2 × 105 CFU of S. mutans per ml in UFTYE containing 1% (w/v) sucrose at 37 °C with 5% CO2 for 16 h. After overnight incubation, planktonic cells were carefully removed and biofilms were washed 3 times with PBS. The preformed biofilms were used to assess the antibiofilm activity of test formulations. Serial dilutions of farnesol-loaded nanoparticles in UFTYE at pH 5.0 or pH 7.0 as well as controls (PBS and NPC or farnesol alone) were prepared and added to the preformed biofilms. Following 16 h incubation, the treatment solution was carefully removed. The treated biofilms were then washed 3 times with PBS, resuspended in PBS, and homogenized by sonication (30 s pulse at an output of 7 W; Branson Sonifier 150; Branson Ultrasonics, Danbury, CT, USA). The homogenized biofilm suspension was serial diluted and plated onto blood agar for CFU counting. Untreated, preformed biofilms were also included to determine the baseline CFU. The MCBK is defined as the lowest concentration that resulted in 99.99% killing of untreated baseline biofilm cells (50). The MCBKs were determined in triplicates in three independent experiments. Supplemental Figure S5 summarizes the MCBK testing process used to evaluate antibacterial effectiveness against 16-hour S. mutans biofilms.
Saliva-coated Hydroxyapatite Biofilm Assay
Antibiofilm effect of farnesol-loaded nanoparticles on S. mutans biofilm formation was assessed following a modified topical treatment regimen (38). Briefly, four treatment solutions were used to treat biofilms: PBS, free nanoparticles (CCR ~4, 0.5 mg/ml in PBS), farnesol-loaded nanoparticles (0.5 mg/ml loaded with 1.0 mg/ml farnesol in PBS), and free farnesol (1.0 mg/ml in PBS). S. mutans biofilms were formed on saliva-coated hydroxyapatite (sHA) discs (surface area, 2.7 ± 0.2 cm2, Clarkson Chromatography Products Inc., South Williamsport, PA, USA) vertically suspended in 24-well plates using a custom-made wire disc holder, mimicking the smooth surfaces of the pellicle-coated tooth (10, 38, 51). Each sHA disc was inoculated with ~2 × 105 CFU of S. mutans per ml in UFTYE containing 1% (w/v) sucrose at 37 °C with 5% CO2. The sHA discs and biofilms were treated with the above-described solutions for 10 min, and transferred to culture media. The first treatment was applied directly after salivary pellicle formation (sHA) and the treated discs were transferred to culture media. Biofilms were allowed to form on the discs without interruption for 6 h at which point a second treatment was applied. The next day, biofilms were treated three times (at 20, 26, and 32 h) and the culture media was changed twice (at 20 h and 30 h). After 44 h, biofilms were removed and homogenized by sonication in 0.89% NaCl (30 s pulse; 7 W). CFU and dry-weight of biofilm were determined as described elsewhere (10, 38).
Inhibition of Insoluble Glucan Formation Assays
Insoluble glucans were produced by purified S. mutans-derived exoenzyme glucosyltransferase B (GtfB) immobilized on poly-L-lysine coated MatTek dish, and labelled with 1 μM Alexa Fluor 647-dextran conjugate (647/668 nm; Molecular Probes) as described previously (52). Briefly, MatTak dishes were pre-conditioned with poly-L-lysine solution (0.1 w/v in H2O; Sigma-Aldrich, St. Louis, MO, USA) overnight to facilitate GtfB immobilization. 30 units of GtfB were adsorbed to the poly-L-lysine coated dish for 1 h at room temperature. Then, the dish was gently washed with PBS to remove unbounded enzyme. Subsequently, 1 ml of treatment solution was mixed with sucrose substrate (200 mmol/L sucrose, 40 μmol/L dextran 9000, 0.02% sodium azide in adsorption buffer, pH 6.5) and incubated at 37 °C for 2 h. Four treatment solutions were used for this assay: PBS, free nanoparticles (CCR4, 0.5 mg/ml in PBS), farnesol-loaded nanoparticles (0.5 mg/ml loaded with 1.0 mg/ml farnesol in PBS), and free farnesol (1.0 mg/ml in PBS). After incubation, the total amount of glucans formed was determined by the phenol-sulfuric acid method (10). In addition, time-lapsed confocal imaging was performed to visualize inhibition of EPS production using a 20X (numerical aperture, 1.0) water immersion objective. Images were reconstructed and quantified by Image J.
Confocal Imaging of Farnesol-loaded Nanoparticles Binding/Penetration and Bacterial Killing
Time-lapse high-resolution confocal fluorescence imaging was performed to assess the binding/penetration of farnesol-loaded nanoparticles into bacterial cell and its killing efficacy. S. mutans UA159 was grown in UFYTE broth at 37 °C and 5% CO2 to mid-exponential phase. Standard or saturated CCR ~4 nanoparticles were added to actively growing S. mutans (108 CFU/ml) at a concentration of (128 μg/ml) at pH 4.5. We used nanoparticles conjugated with Alexa Fluor® 488 (490/525 nm; Molecular Probes) to visualize nanoparticle binding and localization on the cell surface, and Syto 82 (541/560 nm; Molecular Probes) was used for labeling bacterial cells. For live/dead imaging, all bacteria and dead cells were labeled with Syto 9 (485/498 nm; Molecular Probes) and propidium iodide (PI, 535/617 nm; Molecular Probes). Confocal images were acquired in the same field of view at 0, 20, 40, and 60 min using Zeiss LSM 800 upright single-photon laser scanning microscope Airyscan pinhole-detection technology with a 40X (numerical aperture, 1.2) water immersion objective for super-resolution imaging. Images were reconstructed and analyzed by ImageJ.
Statistical Analysis
Statistical analyses were performed using two-tailed Student t-tests with Welch’s correction, One-way ANOVA with Tukey’s or Dunnett’s correction for multiple comparisons, or Two-way ANOVA with Tukey’s multiple comparisons at p-values of p < 0.05 as indicated in the figure captions.
Results and Discussion
Based on recent studies indicating saturated conditions can enhance drug loading (40, 53), this approach was investigated using farnesol-loaded p(DMAEMA)-b-p(DMAEMA-co-BMA-co-PAA) NPCs. Polymer composition and physical characteristics, including molecular weight, corona-to-core molecular weight ratio (CCR), hydrophobic core composition, and size (39, 48) were examined using a panel of sixteen NPCs in conjunction with saturated loading to enhance overall NPC-farnesol anti-biofilm efficacy (Table 1; Supplemental Figure S1).
These diblock co-polymers that self-assemble into NPCs (42, 47, 54) were synthesized via reversible addition fragmentation chain transfer (RAFT) polymerizations (Figure 1A). RAFT was used because it is easily adaptable for a variety of functional monomers and can be used to precisely control molecular weight and polydispersity index of synthesized polymers (Mw/Mn, PDI < 1.3) (38, 39, 42). Since degree of polymerization (DP) controls the actual number of repeating monomers in a polymer chain, DP of block 1 (100–1000) and block 2 (35–4000) were varied, as shown in Table 1. Moreover, DP impacts the block 1 and block 2 molecular weights, which spanned 12.4–98.9 kDa and 2.1–104 kDa, respectively. Furthermore, as shown in Table 1, NPC sizes were 7.7–106.0 nm, while NPC zeta potentials were 15.8–24.6 mV. Molecular weight and the related DP are known to directly affect NPC size (55–58) while CCR is independent of overall molecular weight and, thus, is not expected to impact NPC size (Table 1).
Figure 1. Saturated farnesol-loaded NPC formulation improved drug loading capacity.

A) Scheme showing NPC polymer composition and self-assembly in aqueous conditions. B) Chemical structure of farnesol and cartoon showing standard approach of drug loading in hydrophobic cores. C) Cartoon showing saturated drug loading formulation where drug is both loaded into and surrounding NPC. D) Dynamic Light Scattering results showing increases in NPC size for standard loaded and saturated loaded NP12/3a and NP12/12 compared to NPC alone (and compared to theoretical values based on volumetric diameter calculations). Data shown as average and standard deviation from n≥3 independent measurements. # p ≤ 0.0001 versus 0 mg/mL Far/2.7 mg/mL NPC CCR 4 measured group; $ p ≤ 0.01 versus 1.0 mg/mL Far/2.7 mg/mL NPC CCR 4 measured group from two-way ANOVA with Tukey’s multiple comparison’s test. E) Drug loading capacity and F) drug loading efficiency graphs comparing NP12/3a (CCR 4) and NP12/12 (CCR 1) with standard farnesol loading to saturated farnesol loading approach. Data shown as average and standard deviation from n=2–4 independent experiments. & = no significant difference, *p ≤ 0.05, ****p ≤ 0.0001 versus standard loading from unpaired Student t-test. Abbreviations: AIBN, 2,2′-azobis(2-methylpropionitrile); BMA, butyl methacrylate; CTA, chain transfer agent; DMAEMA, dimethylaminoethyl methacrylate; DMF, dimethylformamide; DP, degree of polymerization; ECT, 4-cyano-4-[(ethylsulfanylthiocarbonyl)sulfanyl]pentanoic acid; PAA, propylacrylic acid.
A critical limitation to micelle-based drug delivery systems is drug loading capacity (59). Previous studies demonstrated the anti-biofilm potential of farnesol-loaded p(DMAEMA)-b-p(DMAEMA-co-BMA-co-PAA) NPC, which exhibited up to 4-fold higher efficacy against S. mutans biofilms in vitro than free farnesol alone (38, 39). However, a modest ≤ 1-log reduction in bacterial viability (colony forming units) was observed in these studies (38, 39). Therefore, further development of this therapeutic approach is crucial for clinical relevance. Recently, formulations using saturated hydrophobic drug conditions in aqueous solution showed significantly increased drug loading (e.g. > 50% increase) into polymeric nanoparticles (40). Moreover, the drug to polymer ratio proportionally increased drug loading and encapsulation efficiency (53). With these findings in mind, the saturated drug formulation was employed for two NPCs (NP12/12 and NP12/3a, black dashed boxes in Table 1) that closely resembled NPCs with modest antibacterial effectiveness using standard farnesol loading formulations (38, 39). Specifically, NPCs had first to second block molecular weight ratios, or CCRs, of 1 (NP12/12, CCR 1) (38) or 4 (NP12/3a, CCR 4) (39). Similar NPCs showed high biofilm retention property, albeit with low bacterial killing activity potentially due to standard drug loading formulations, which resulted in ~22–27wt% of farnesol (38, 39). Therefore, the effects of standard and saturated farnesol-NPC formulations (Figures 1B & 1C, respectively) on drug loading were characterized.
Saturated loading increases NP12/12 (CCR 1) size from ~29 nm to ~59 nm while standard loading conditions only increase NPC size to ~45 nm (Figure 1D). Similarly, NP12/3a (CCR 4) increases in size from ~14 nm to ~34 nm and ~81 nm for standard and saturated loading conditions, respectively (Figure 1D). NPC size increases for standard farnesol loading conditions have been directly attributed to farnesol partitioning into the micelle core, which is consistent with our observations (38). Results here show ~62% loading capacity using CCR 1 and CCR 4 NPCs with the saturated condition versus standard loading (~20%), an improvement of ~300% (Figure 1E). These results likely stem from the saturated drug concentration realizing greater farnesol partitioning into the hydrophobic core to reduce the free energy of the system (40, 60). Indeed, critical micelle concentration (CMC) testing confirmed that the saturated farnesol-loaded NPC formulation improved NPC stability regardless of CCR (Supplemental Figure S2). Therefore, more farnesol is incorporated into the hydrophobic core of the NPC and stabilizes the NPC for saturated formulation conditions than the standard formulation conditions. Importantly, this increased drug loading capacity had little effect on drug loading efficiency for the NPCs tested (Figure 1F).
Based on improved drug loading, saturated formulation release kinetics were investigated for NP12/3a and NP12/12 at neutral and acidic pH conditions. Using sink conditions to mimic salivary clearance in the oral environment, drug release profiles showed distinct behaviors for free drug alone, the standard formulation, and the saturated formulation over time for each of the NPCs tested (Figure 2A & B). As expected, rapid drug availability was observed for free drug alone with release rate constants (kobs) greater than 0.6 hr−1 and release half-times (t1/2) of ~1 hour at both pH 4.5 and pH 7.2. Using standard loading conditions at pH 4.5, NP12/3a and NP12/12 had kobs values of ~0.11 hr−1 and t1/2 ~6 hours, which aligned with previous kobs values (0.09–0.31 hr−1) and t1/2 values (2–7 hours) for similar NPCs in acidic conditions (38, 39). At pH 7.5, NP12/3a and NP12/12 with standard loading conditions had kobs values of 0.1 and 0.08 hr−1 and t1/2 values of 7.3 hours and 9.0 hours, respectively, compared to previous kobs values (0.05–0.15 hr−1) and t1/2 values (4–15 hours) for similar NPCs in neutral conditions (38, 39). However, the saturated formulation release profiles differed significantly from the standard formulation release profiles. Using saturated loading conditions at pH 4.5, the kobs values for both NP12/3a and NP12/12 were approximately one-third (0.03 and 0.04 hr−1, respectively) and the release half-lives were more than three times greater (22.9 and 19.0 hours, respectively) versus standard formulation controls (Figure 2C). Moreover, the kobs and t1/2 values for NP12/3a (0.02 hr−1 and 36.6 hours) and NP12/12 (0.03 hr−1 and 22.4 hours) at pH 7.2 differed by 2–5 fold versus standard formulation loaded controls (Figure 2C). Both formulations also exhibited nearly zero-order release over the first 12 hours with slightly more pH-responsiveness and prolonged zero-order release up to 72 hours for the saturated farnesol formulation using NP12/3a (Figure 2B). Interestingly, others have also seen nearly zero-order release up to 48 hours when using methyoxyPEG-(mPEG-)-PLA diblock copolymers with saturated drug loading conditions (61). Similarly, reduction in overall polymer:drug ratios has been used to improve control over drug release (62) while increased drug encapsulation efficiency and tunable release kinetics have been observed when adding a co-surfactant to polymer NPC formulations (63). Therefore, the reduced NPC concentration coupled with farnesol partitioning into the hydrophobic core likely contributed to zero-order release kinetics observed for the saturated formulation.
Figure 2. Saturated farnesol-loaded NPC formulation lengthened release times and slowed release rates over 48 hours.

A & B) Farnesol release kinetics, including release rates (insets), for farnesol and standard and saturated loading formulations using A) NP12/12 (CCR 1) and B) NP12/3a (CCR 4) at pH 4.5 and pH 7.2. Data shown as average and standard error from n=3 independent experiments. C) Half-time of release (t1/2) and release rate constants (k) for standard and saturated loading formulations using NP12/12 (CCR 1) and NP12/3a (CCR 4) at pH 4.5 and pH 7.2. Abbreviations: CCR, Corona-to-Core molecular weight Ratio; Sat, saturated farnesol formulation; Std, standard farnesol formulation.
Given the improved drug loading and prolonged, zero- order farnesol release kinetics observed, the effect of the saturated formulation as a biofilm treatment was investigated. Using a microplate S. mutans biofilm assay where PBS, farnesol only, NP12/3a only, and NP12/12 only were included as controls, the saturated farnesol-NPC formulations resulted in a 2–4 log colony forming unit (CFU) reduction in bacteria viability for both NP12/12 and NP12/3a (Figures 3A-B). This result is likely due to overall higher drug dosing, as the formulation change did not dramatically improve other aspects of NPC-mediated delivery (e.g., pH-triggered release and binding). The saturated formulation conditions yield drug loading efficiencies of ~80–85% and drug loading capacities of ~62% (Figure 1E-F), which are ~45-fold greater than the published minimum inhibitory concentration (MIC, ~0.014 mg/mL) of farnesol for S. mutans (49). In comparison, the use of standard formulation conditions correlates to drug loading capacities of ~17–25%, which are ~18-fold greater farnesol concentrations than the MIC for S. mutans and are comparable to previously observed drug loading capacity results (~20–27%) (38, 39).
Figure 3. Saturated farnesol-loaded NPC formulation improved anti-biofilm efficacy.

16-hour biofilm CFU/mL data comparing standard loaded and saturated loaded NP12/12 (i.e. CCR 1, A) and NP12/3a (i.e. CCR 4, B) to controls (i.e. NPCs, farnesol, and PBS alone) showing improved anti-biofilm efficacy (e.g., lower CFU/mL) for the saturated formulations at pH 7 and pH 5.* p ≤ 0.05 and ns = no significant difference from Student t-test with Welch’s correction. Data shown as average and standard deviation from n=2–6 independent experiments. Abbreviations: CCR, Corona-to-Core molecular weight ratio; CFU, colony forming unit; Far, Farnesol; Sat, Saturated formulation; Std, Standard Formulation.
Based on the release kinetics shown in Figure 2, approximately 60–100 μg/mL of farnesol would be delivered to the biofilm over the 16-hour treatment period using saturated conditions. However, standard loaded NPC release profiles deliver ~2–3-fold greater amounts of farnesol (150–180 μg/mL) versus the saturated loading condition yet bacterial inhibition is far greater for the saturated condition. This paradox may be explained by considering the interactions of the cationic and hydrophobic components of the NPCs with EPS matrix and bacterial cell membranes. Recent studies have shown that cationic and hydrophobic particles penetrate and distribute more homogeneously throughout a biofilm compared to cationic-only particles (64, 65) that are capable of strongly interacting with the EPS matrix (38) at the solvent-biofilm interface. Hence, NPC-associated farnesol in the saturated loading condition may improve biofilm penetration as it effectively increases the hydrophobicity of the NPC, rather than the farnesol sequestered in the core, as in the standard loading condition. As it is well established that farnesol interacts non-specifically with bacterial membranes (29, 30, 33, 66), excess NPC-associated farnesol may also improve NPC-bacteria interactions, thereby enhancing antibacterial activity. In contrast, standard loaded NPCs may have limited antibacterial activity and greater diffusional hindrances within the biofilm due to high affinity EPS matrix and bacterial membrane binding (38). Taken together, these insights suggest that the saturated farnesol formulation may improve antibacterial effectiveness due to more uniform drug concentrations (i.e., higher farnesol availability in situ) and longer durations of drug release within biofilms compared to the standard formulation. Regardless of the mechanism, to the best of our knowledge, this report is the first to test a saturated drug loaded NPC formulation for anti-biofilm treatment applications. It should also be noted that similar to our results, saturated loading of the antipsychotic drug haloperidol in mPEG-PLA diblock copolymers showed greater efficacy (i.e., attenuated ketamine-induced psychosis) versus standard loading (40, 61).
The saturated farnesol formulation was further tested using a panel of NPCs that span a range of values for several key polymer design parameters to determine if these parameters, in conjunction with saturated loading, can be tuned to enhance antibacterial effectiveness. The parameters investigated were overall molecular weight, hydrophilic corona block (i.e., Block 1) molecular weight, hydrophobic core block (i.e., Block 2) molecular weight, corona-to-core molecular weight ratio (CCR), and NPC size (Table 1, Figure 4A). The polymers were grouped based on these characteristics for data analysis (Table 1, Figure 4A). Minimum inhibitory concentration (MIC) and minimum concentration for biofilm killing (MCBK) tests using planktonic and biofilm cells of S. mutans, respectively, were performed for each NPC loaded with saturated farnesol conditions as well as unloaded NPCs at pH 5 and pH 7. See Supporting Information (Table S1 and Figures S3-S6) for details of the MIC and MCBK results in addition to unloaded NPC data. Samples from each of the MIC and MCBK tests were plated on agar using serial dilutions to quantify CFUs, and the farnesol-loaded NPC results are shown in Figure 4B and Figure 4C, respectively. As shown in Figure 4B, five separate NPC formulations were found to have pH-responsive CFU/mL reductions of approximately 2 logs (e.g., NP12/3a, NP12/3b, NP12/3c, NP12/12, and NP13/5). Another six NPC formulations showed pH-dependent CFU/mL reductions of approximately 1 log (e.g. NPCs NP52/15, NP99/50, NP13/4, NP13/3, NP13/2, NP12/2), and the remaining NPC formulations showed less than 1 log CFU/mL reduction between pH 7 and pH 5 (Figure 4B). In contrast, the MCBK data revealed CFU/mL reductions of approximately 4 log versus untreated or PBS controls and pH-responsive CFU/mL reductions of approximately 2 log from for nearly all 16 NPCs tested. However, no clear differences in pH-responsive CFU/mL reductions were observed among the NPCs tested.
Figure 4. MIC and MCBK testing of saturated farnesol-loaded NPC formulation against planktonic and biofilm S. mutans, respectively, showed pH-responsive CFU/mL reductions of ~2 log.

A) Cartoon showing the five NPC design parameters investigated in this study. B) Effectiveness of saturated farnesol-loaded NPCs (16 μg/mL farnesol, 8 μg/mL NPC) against planktonic S. mutans at pH 5 and pH 7 in CFU/mL. C) Effectiveness of saturated farnesol-loaded NPCs (128 μg/mL farnesol, 64 μg/mL NPC) against 16-hour S. mutans biofilms at pH 5 and pH 7 in CFU/mL. Black dotted line and grey dashed line represent average CFU/mL values for NPC formulations at pH 7 (~1.7×107 CFU/mL) and pH 5 (~3.0×105 CFU/mL), respectively. D-F) S. mutans CFU/mL fold change between pH 7 and pH 5 based on (D) overall diblock (squares), Block 1 (triangles), and Block 2 (circles) molecular weight, (E) NPC diameter (i.e., size), and (F) CCR. Solid shapes indicate results from planktonic testing, and open shapes indicate results from 16-hour biofilm testing. Data shown as average and standard deviation from n=2 independent MIC and MCBK tests with at least 3 replicates per test. Abbreviations: CCR, Corona-to-Core molecular weight Ratio; Mn, molecular weight.
From the MIC and MCBK data, NPCs were grouped by design parameters (see Table 1, Figure 4A) and the CFU/mL fold-change between pH 7 to pH 5 was calculated and averaged. The results of this analysis are shown in Figure 4D-F, where higher fold-change values (i.e. y-axis) represent greater pH-responsive antibacterial effectiveness. In Figure 4D, an inverse relationship between pH-responsive antibacterial effectiveness against planktonic S. mutans (solid shapes) and molecular weight was observed; lower Block 1 Mn, Block 2 Mn, and Total Mn values (≤ 20 kDa) had ~2 log reductions in CFU/mL. However, the microplate S. mutans biofilm results (open shapes) in Figure 4D showed approximately 2 log reductions in CFU/mL regardless of molecular weight. Although previous studies have shown that high levels (> 300 μg/mL) of farnesol are necessary to demonstrate antibacterial effectiveness against S. mutans biofilms (28, 31, 67), this result suggested that the saturated formulation conditions improve anti-biofilm effectiveness since a sub-inhibitory concentration of farnesol (128 μg/mL) was used as part of the bacteriostatic (rather than bactericidal) nature of this assay.
NPC size exhibited binary effects, where NPCs with small diameters (< 40 nm) had greater antibacterial effects (~2 log) against planktonic S. mutans (solid shapes) compared to NPCs with large diameters (> 60 nm) that showed nearly no change (Figure 4E). However, the S. mutans biofilm results (open shapes) in Figure 4E again showed 2 log CFU/mL reductions regardless of NPC size. This finding was surprising given that the average S. mutans biofilm pore size has been estimated to be 20–50 nm (68) and the saturated loading NPC diameter is 50–100% larger than the standard loading condition (Figure 1). However, due to NPC treatment, disrupted biofilm development is likely to yield greater pore sizes for NPC diffusion and further anti-biofilm efficacy. While S. mutans biofilm results (open shapes) in Figure 4F also showed 2 log CFU/mL reductions across the CCR range tested, CCR significantly impacted pH-responsive antibacterial effectiveness against planktonic S. mutans (solid shapes). Specifically, antibacterial efficacy increased more than 50-fold as CCR increased from ≤ 1 to 4 and decreased by 12-fold for CCR values greater than 5 (Figure 4F). These results aligned well with previously published findings (38, 39), which used standard farnesol loading conditions and determined that NPCs with Block 1 Mn values ≤ 20 kDa, small sizes (≤ 20 nm), and CCR ~4 demonstrated pH-responsive antibacterial efficacy (≤ 2.4 log CFU/mL reductions). Due to the similarities to previous anti-biofilm effectiveness data obtained using standard physicochemical characterizations (39), these biological screening assays (i.e., MIC and MCBK) are beneficial for screening NPC design parameters and may be useful complementary analyses when investigating optimal NPC design features. However, the MIC and MCBK data also highlight striking dichotomies in antibacterial efficacy of saturated farnesol-loaded NPCs between planktonic and biofilm S. mutans. Additionally, the drug concentration conditions employed for these assays were different. Clear trends in antibacterial efficacy were observed using a farnesol concentration (16 μg/mL) similar to the published farnesol MIC for planktonic S. mutans (14–28 μg/mL) (49). However, the farnesol concentration used for MCBK testing (128 μg/mL) was below farnesol levels known to inhibit S. mutans biofilms in vitro (~300 μg/mL) (28, 49). This finding suggests that the saturated formulation conditions improved pH-responsive anti-biofilm activity even though the differences among the NPC design parameters tested did not significantly impact anti-biofilm effectiveness.
Although planktonic and microplate biofilm screening approaches to test antibacterial treatments may be useful for initial evaluations, testing a more clinically relevant in vitro biofilm model is necessary to fully substantiate therapeutic efficacy. In this model, saliva-coated hydroxyapatite (as pellicle-coated tooth mimetics) and a topical treatment regimen (twice-daily followed by dip-wash to remove excess) were used to simulate clinical conditions (Figure 5A). Since no significant differences were observed across the various NPC design parameters in the MCBK test results, NP12/12 and NP12/3a were again selected from the NPC panel and tested with the saturated farnesol loading formulation using a saliva-coated hydroxyapatite disk biofilm assay (26). In contrast to previously published results with standard loading in which NPCs with high CCR values (~4) were most effective against S. mutans biofilms, the saturated farnesol formulation approach substantially improved anti-biofilm effectiveness against S. mutans regardless of CCR (Figure 5). As shown in Figure 5B, saturated farnesol formulations used with NP12/12 (CCR 1) or NP12/3a (CCR 4) reduced biofilm viability in terms of CFU/mL by approximately 2 logs. However, the standard farnesol-loaded NPC formulations for both of these NPCs showed modest effects on biofilm cell viability (Figure 5B). Furthermore, the saturated farnesol-loaded NPC formulation for NP12/12 and NP12/3a reduced S. mutans biofilm dry weight by 90% and 94% of the PBS control, respectively, while the standard farnesol loaded NPC formulation had limited effects (Figure 5C). Therefore, the saturated farnesol-loaded NPC formulation demonstrated significantly enhanced anti-biofilm effectiveness in vitro against S. mutans versus the standard farnesol-loaded NPC formulation approach. It should also be noted that the lack of a difference in biofilm viability and dry weight between NP12/12 and NP12/3a (Figure 5) further supports the microplate biofilm results shown in both Figure 3 and Figure 4C where no change in pH-responsive anti-biofilm efficacy was observed regardless of the CCRs tested.
Figure 5. Saturated farnesol-loaded NPC formulation improved biofilm inhibition using in vitro saliva-coated hydroxyapatite disk assay.

A) Scheme showing treatment regimen used with S. mutans biofilms grown in vitro on saliva-coated hydroxyapatite (sHA) disks. B) CFU/mL values obtained for biofilms formed on sHA disks for each treatment group tested. C) Biofilm dry weight values obtained for biofilms formed on sHA disks for each treatment group tested. Data shown as mean and standard deviation from n=6–10 independent assays. % p ≤ 0.001 versus PBS, & p ≤ 0.001 versus Farnesol, ∞ p ≤ 0.001 versus CCR 1, ● p ≤ 0.0001 versus CCR 4, ! p ≤ 0.0001 versus CCR 1 + Std Far, * p ≤ 0.05 versus CCR 4 + Std Far, # p ≤ 0.05 versus CCR 1 + Sat Far, and $ p ≤ 0.05 versus CCR 4 + Sat Far from One-way ANOVA with Tukey’s multiple comparisons test. Abbreviations: CFU, Colony Forming Unit; CCR, Corona-to-Core molecular weight Ratio; Far, farnesol; NPC, nanoparticle carrier.
Due to the observation that saturated farnesol-loaded NPC formulations exhibited improved anti-biofilm effectiveness against S. mutans regardless of CCR, mechanisms underpinning this finding were investigated. Since a previous study (39), MICs (Figure 4), and biofilm results (Figure 5) showed that NPCs with CCR values of approximately 4 exhibit higher antibacterial effectiveness, NP12/3a and NP13/4 (i.e., CCRs 4.0 and 3.6, respectively) were selected for further investigation. First, temporal glucans production by S. mutans-derived glucosyltransferases was evaluated qualitatively using time-lapse fluorescence microscopy and quantitatively using imaging and biochemical analyses, as depicted in Figure 6. The saturated farnesol-loaded NPC formulation using NP12/3a reduced total EPS amounts, as indicated by reduced signal of labeled glucans compared to the controls in Figure 6A. This signal was lower than the controls by ~30–50 fold at 20 minutes, ~4–7 fold at 40 minutes, and ~2–3 fold at 60 minutes (Figure 6B). Similarly, biochemical analyses show ~2-fold decrease in insoluble glucans mass for the saturated farnesol-loaded NPC formulation using NP13/4 compared to controls of PBS, farnesol alone, and NPC alone after 60 minutes incubation (Figure 6C), consistent with fluorescence imaging analysis. Farnesol can inhibit insoluble glucans formation by S. mutans within biofilms (28, 49). Since the EPS matrix produced by this bacterium is largely comprised of insoluble glucans (10), reduction of glucans formation could improve drug access and antibacterial effectiveness, as achieved here. Altogether, these data reveal an important antibiofilm mechanism by which saturated farnesol-loaded NPC formulations effectively inhibit glucan synthesis by bacterial glucosyltransferases.
Figure 6. Saturated farnesol-loaded NPC formulation improved biofilm inhibition due to reduced total biomass caused by prevention of insoluble glucan formation.

A) Fluorescence microscopy images showing glucan formation by GtfB using Alexa 647 sucrose over 60 minutes for PBS (control), saturated farnesol-loaded NPC, NPC alone, and farnesol alone treatments. B) Quantification of relative intensity of glucans formed in (A) using ImageJ. Data shown as average and standard deviation from n=4 independent experiments. * p ≤ 0.05 PBS versus NPC + Sat Far, # p ≤ 0.0001 Far versus NPC + Sat Far, $ p ≤ 0.001 NPC versus NPC + Sat Far, ns = no significant difference from Two-way ANOVA with Tukey’s multiple comparisons test. C) GtfB activity for PBS, farnesol alone, NPC alone, and saturated farnesol-loaded NPC groups using a colormetric assay for insoluble glucans. Data shown as average and standard deviation from n=3 independent experiments. **** p < 0.0001 from One-way ANOVA with Dunnett’s multiple comparisons test. Abbreviations: Far, farnesol; NPC, nanoparticle carrier; Sat, saturated farnesol formulation.
Additional mechanistic investigations of farnesol-loaded NPC interactions with S. mutans were performed at single-cell level using super-resolution confocal microscopy. By fluorescently labeling bacteria and NPCs (i.e., NP13/4), NPC localization on the surface of individual bacterial cells was visualized and fluorescence intensity measured. Based on the results shown in Figure 7A, the saturated formulation conditions display a thicker (halo-like) layer of NPCs completely surrounding the bacterial cells. Relative quantification of this NPC layer indicated a 7-fold increase in NPCs surrounding the bacterial cells for the saturated versus standard formulation (Figure 7B). These results indicate the NPCs in the saturated formulation can interact and partition with the cell membrane, which leads to enhanced NPC uptake and intracellular NPC and drug concentrations (Figure 7C). Interestingly, zeta potential analyses revealed no significant difference in NPC surface charge between the two formulations (Supplemental Figure S7). In addition, live/dead staining of S. mutans treated with either the standard or saturated farnesol-loaded NPC formulation reveals increased cell killing for the saturated condition (Figure 7D). Taken together, these results suggest that increased localization of farnesol-loaded NPCs surrounding the bacterial cells when using saturated formulation conditions likely contributes to enhanced NPC killing efficacy compared to standard formulation conditions.
Figure 7. Saturated farnesol-loaded NPC formulation enhanced NPC localization around planktonic S. mutans cell membranes leading to improved bacterial cell death.

A) Confocal microscopy results showing the saturated farnesol-loaded NPC formulation enhanced NPC localization around planktonic S. mutans cell membranes. Bacteria were labeled with SYTO 82 (pseudo-colored magenta) and the NPCs (i.e., NP13/4) were labeled with Alexa Fluor® 488 (green). B) Quantification of results from (A) showing relative fluorescence intensity per bacteria. Data shown as average and standard deviation using representative images from n=10 independent measurements. **** p ≤ 0.0001 as measured by an unpaired Student t-test with Welch’s correction. C) Representative confocal images showing enhanced NPC penetration into the cell membrane with the saturated formulation compared to the standard formulation. Top panel shows the projection images of the entire cell, and the bottom panel depicts a cross-section of the middle plane of a single bacterial cell indicating the NPCs localize intracellularly. The dotted lines are fiducial markers representing the outer membrane of the bacterial cell. D) Live-dead staining of S. mutans treated with standard and saturated farnesol-loaded NPCs (i.e., NP13/4) using SYTO 9 (green) to label viable bacteria and propidium iodide (red) to label dead cells.
This class of cationic nanoparticles have established affinity to biofilms (e.g., EPS) (35, 69, 70) and bacterial cells (34, 71). Previous investigations have characterized nanoparticle interactions with bacterial membranes (18, 70, 72, 73), showing that cationic polymers, such as p(DMAEMA), selectively bind Gram positive bacteria, including S. mutans, while demonstrating some antibacterial effectiveness (20, 21). Interestingly, farnesol has been identified to be a potential therapeutic adjuvant due to its ability to disrupt bacterial cell membranes and facilitate entry of other agents, such as antibiotics and possibly nanoparticles (66, 74, 75). These findings were further expanded here using single cell imaging to evaluate the spatial localization and concentration of NPCs around cells. Higher localized farnesol due to greater NPC accumulation and proximity in situ, which ultimately leads to higher uptake and internalization, improved antibacterial effectiveness. Additional studies focused on the exact biophysical mechanisms facilitating NPC-bacterial cell membrane interactions and membrane effects caused by increased, localized farnesol concentrations are warranted. Such studies should also consider evaluating other pathogens, including Gram negative bacteria and fungi.
Although farnesol has proven useful as a cost effective, model hydrophobic drug for testing NPC delivery, its clinical relevance is limited due to the high concentrations necessary to control biofilms (31). Therefore, an antibacterial agent identified via high throughput drug screening, namely thonzonium bromide, was investigated for NPC loading and antibiofilm efficacy (see Supplemental Table 1 in Ref 76). Thonzonium bromide, an FDA-approved drug used to treat outer ear infections (e.g., swimmer’s ear), was investigated due to its structurally similar alkyl chain to farnesol (Figure 8A), which is important for core loading via hydrophobic interactions with BMA, and its higher potency compared to chlorhexidine against another Gram positive bacteria, S. aureus (see Supplemental Table 1 in Ref 76). The standard thonzonium bromide-NPC formulation and saturated thonzonium bromide-NPC formulation were compared using CCR 4 NPCs. Saturated thonzonium bromide loading conditions improved drug loading capacity greater than 2.5-fold with minimal impact on drug loading efficiency (Figure 8B and Supplemental Figure S8). This result highlighted the versatility of both the NPC and the saturated drug loading formulation approach. Additionally, thonzonium bromide release kinetics were investigated using CCR 4 NPCs at neutral and acidic pH conditions. The thonzonium bromide release profiles showed less pronounced differences (vs. farnesol release profile) with respect to pH or loading conditions (Figure 8C). This observation may be due to the dominant effect of drug-NPC interactions on release versus physical changes in NPCs in response to pH (48). In particular, release limiting drug-NPC interactions may be specific to DMAEMA, BMA, or PAA effects on thonzonium bromide in the hydrophobic core since others have seen successful pH-responsive drug release using a similar drug (N-phenacylthialzolium bromide) with standard loading conditions in PLGA/chitosan NPCs (77). In addition, the thonzonium bromide formulations also showed nearly zero-order release over 96 hours (Figure 8C).
Figure 8. Saturated thonzonium bromide-loaded NPC formulation also improved drug loading capacity, lengthened release times, and improved anti-biofilm efficacy.

A) Chemical structure of thonzonium bromide. B) Drug loading capacity (left axis) and drug loading efficiency (right axis) graphs for CCR 4 NPCs comparing standard TB loading to saturated TB loading approach. & = no significant difference, *p ≤ 0.05, versus standard loading from unpaired Student t-test with Welch’s correction. Data shown as average and standard deviation using representative images from n=2–4 independent experiments. C) TB release kinetics for standard and saturated loading formulations using CCR 4 NPCs. D) CFU/mL values obtained for biofilms for each TB treatment group tested at pH 5 and pH 7. % p ≤ 0.01 versus PBS, 221E p ≤ 0.01 versus CCR 4, * p ≤ 0.05 versus CCR 4 + Std TB, $ p ≤ 0.01 versus CCR4 + Sat TB from unpaired Student t-test with Welch’s correction. Data shown as average and standard deviation from n=4 independent experiments. Abbreviations: CCR, corona-to-core molecular weight ratio; NPC, nanoparticle carrier; Sat, saturated formulation; Std, standard formulation; TB, thonzonium bromide.
The efficacy of the saturated drug-NPC formulation was further tested using thonzonium bromide-loaded NPCs in MIC and MCBK tests using planktonic and microplate biofilm S. mutans, respectively. Specifically, NPCs with CCR 1 and CCR 4 (NP12/12 and NP12/3c) loaded with saturated thonzonium bromide conditions at pH 5 and pH 7 were tested. The MIC and MCBK results from this testing are shown in Supplemental Table S2. Similar thonzonium bromide MICs were observed between pH 5 and pH 7 regardless of NPC concentration. However, the saturated thonzonium bromide formulation used with NP12/3c (CCR 4) reduced biofilm viability in terms of CFU/mL by ~6 logs while the standard formulation showed ~5 log reduction on biofilm viability (Figure 8D). Furthermore, both formulations reduced S. mutans biofilm dry weight to below the detectable limit compared to controls (data not shown). Therefore, although both thonzonium bromide-loaded NPC formulations proved effective, the saturated formulation demonstrated enhanced anti-biofilm efficacy in vitro against S. mutans compared to the standard formulation approach.
Conclusions
In this work, a novel formulation approach where a pH-responsive polymer nanoparticle carrier (NPC) combined with a saturated drug solution was investigated for use against S. mutans biofilms. This formulation approach improved farnesol drug loading capacity by ~300% with little to no effect on drug loading efficiency while sustaining zero-ordered drug release over 48 hours, which was 3-fold longer versus standard farnesol loading. Moreover, the saturated drug-NPC formulation markedly enhanced antibacterial effectiveness against planktonic and biofilm S. mutans, resulting in a 2–4 log CFU/mL reduction in bacteria viability using both microplate and sHA biofilm assays. Reduced total biomass caused by disruption of insoluble glucan synthesis and enhanced NPC localization around S. mutans cell membranes specific to the saturated drug-NPC formulation were determined to contribute to this improved biofilm inhibition. Thonzonium bromide was also tested using the saturated drug-NPC formulation and increased drug loading capacity ≥ 2.5-fold, demonstrated nearly zero-order release over 96 hours, and reduced bacterial viability by ~6-log CFU/mL as determined using the sHA biofilm assay. This work is intended to help provide fundamental insights that may lead to clinical translation of novel topical antimicrobial therapies, such as those used against oral biofilms.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the National Institutes of Health (R01 DE018023 to HK and DB, and F31 DE026944 to KS) and the National Science Foundation (DMR 1206219 to DB) for funding that supported this work. Research reported in this publication was supported by the National Institute of Dental & Craniofacial Research of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would also like to thank Paul Dunman for general advice regarding efficient MIC experimental execution and James L. McGrath for access to the Malvern Zetasizer.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Research on microbial biofilms In: National Heart L, and Blood Institute., editor. National Heart, Lung, and Blood Institute, Bethesda, MD: 2002. [Google Scholar]
- 2.Romling U, Kjelleberg S, Normark S, Nyman L, Uhlin BE, Akerlund B. Microbial biofilm formation: a need to act. J Intern Med. 2014;276(2):98–110. [DOI] [PubMed] [Google Scholar]
- 3.Petersen PE, Bourgeois D, Ogawa H, Estupinan-Day S, Ndiaye C. The global burden of oral diseases and risks to oral health. Bulletin of the World Health Organization. 2005;83(9):661–9. [PMC free article] [PubMed] [Google Scholar]
- 4.Tonetti MS, Jepsen S, Jin L, Otomo-Corgel J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. Journal of clinical periodontology. 2017;44(5):456–62. [DOI] [PubMed] [Google Scholar]
- 5.Jin LJ, Lamster IB, Greenspan JS, Pitts NB, Scully C, Warnakulasuriya S. Global burden of oral diseases: emerging concepts, management and interplay with systemic health. Oral diseases. 2016;22(7):609–19. [DOI] [PubMed] [Google Scholar]
- 6.Gift HC, Reisine ST, Larach DC. The social impact of dental problems and visits. American journal of public health. 1992;82(12):1663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng L, Zhang K, Weir MD, Melo MA, Zhou X, Xu HH. Nanotechnology strategies for antibacterial and remineralizing composites and adhesives to tackle dental caries. Nanomedicine (Lond). 2015;10(4):627–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kassebaum NJ, Bernabe E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of untreated caries: a systematic review and metaregression. Journal of dental research. 2015;94(5):650–8. [DOI] [PubMed] [Google Scholar]
- 9.Klein MI, Hwang G, Santos PH, Campanella OH, Koo H. Streptococcus mutans-derived extracellular matrix in cariogenic oral biofilms. Frontiers in cellular and infection microbiology. 2015;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koo H, Xiao J, Klein MI, Jeon JG. Exopolysaccharides produced by Streptococcus mutans glucosyltransferases modulate the establishment of microcolonies within multispecies biofilms. Journal of bacteriology. 2010;192(12):3024–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krzysciak W, Jurczak A, Koscielniak D, Bystrowska B, Skalniak A. The virulence of Streptococcus mutans and the ability to form biofilms. European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology. 2014;33(4):499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsui R, Cvitkovitch D. Acid tolerance mechanisms utilized by Streptococcus mutans. Future Microbiol. 2010;5(3):403–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falsetta ML, Klein MI, Lemos JA, Silva BB, Agidi S, Scott-Anne KK, et al. Novel antibiofilm chemotherapy targets exopolysaccharide synthesis and stress tolerance in Streptococcus mutans to modulate virulence expression in vivo. Antimicrob Agents Chemother. 2012;56(12):6201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowen WH. The Stephan Curve revisited. Odontology. 2013;101(1):2–8. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi N, Nyvad B. Caries ecology revisited: microbial dynamics and the caries process. Caries research. 2008;42(6):409–18. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Hu C, Shao L. The antimicrobial activity of nanoparticles: present situation and prospects for the future. International journal of nanomedicine. 2017;12:1227–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zazo H, Colino CI, Lanao JM. Current applications of nanoparticles in infectious diseases. Journal of controlled release : official journal of the Controlled Release Society. 2016;224:86–102. [DOI] [PubMed] [Google Scholar]
- 18.Gupta A, Landis RF, Rotello VM. Nanoparticle-Based Antimicrobials: Surface Functionality is Critical. F1000Research. 2016;5:F1000 Faculty Rev-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huh AJ, Kwon YJ. “Nanoantibiotics”: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. Journal of controlled release : official journal of the Controlled Release Society. 2011;156(2):128–45. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Nadres ET, Kuroda K. Cationic Amphiphilic Polymers with Antimicrobial Activity for Oral Care Applications: Eradication of S. mutans Biofilm. Biomacromolecules. 2017;18(1):257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magennis EP, Francini N, Mastrotto F, Catania R, Redhead M, Fernandez-Trillo F, et al. Polymers for binding of the gram-positive oral pathogen Streptococcus mutans. PloS one. 2017;12(7):e0180087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palermo EF, Lee DK, Ramamoorthy A, Kuroda K. Role of cationic group structure in membrane binding and disruption by amphiphilic copolymers. The journal of physical chemistry B. 2011;115(2):366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuroda K, DeGrado WF. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J Am Chem Soc. 2005;127(12):4128–9. [DOI] [PubMed] [Google Scholar]
- 24.Khalil H, Chen T, Riffon R, Wang R, Wang Z. Synergy between polyethylenimine and different families of antibiotics against a resistant clinical isolate of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2008;52(5):1635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siala W, Van Bambeke F, Taresco V, Piozzi A, Francolini I. Synergistic activity between an antimicrobial polyacrylamide and daptomycin versus Staphylococcus aureus biofilm. Pathogens and disease. 2016;74(5). [DOI] [PubMed] [Google Scholar]
- 26.Jeon JG, Pandit S, Xiao J, Gregoire S, Falsetta ML, Klein MI, et al. Influences of trans-trans farnesol, a membrane-targeting sesquiterpenoid, on Streptococcus mutans physiology and survival within mixed-species oral biofilms. International journal of oral science. 2011;3(2):98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koo H, Schobel B, Scott-Anne K, Watson G, Bowen WH, Cury JA, et al. Apigenin and tt-farnesol with fluoride effects on S. mutans biofilms and dental caries. Journal of dental research. 2005;84(11):1016–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koo H, Hayacibara MF, Schobel BD, Cury JA, Rosalen PL, Park YK, et al. Inhibition of Streptococcus mutans biofilm accumulation and polysaccharide production by apigenin and tt-farnesol. Journal of Antimicrobial Chemotherapy. 2003;52(5):782–9. [DOI] [PubMed] [Google Scholar]
- 29.Rowat AC, Keller D, Ipsen JH. Effects of farnesol on the physical properties of DMPC membranes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2005;1713(1):29–39. [DOI] [PubMed] [Google Scholar]
- 30.Inoue Y, Shiraishi A, Hada T, Hirose K, Hamashima H, Shimada J. The antibacterial effects of terpene alcohols on Staphylococcus aureus and their mode of action. FEMS Microbiol Lett. 2004;237(2):325–31. [DOI] [PubMed] [Google Scholar]
- 31.Rocha GR, Florez Salamanca EJ, de Barros AL, Lobo CIV, Klein MI. Effect of tt-farnesol and myricetin on in vitro biofilm formed by Streptococcus mutans and Candida albicans. BMC complementary and alternative medicine. 2018;18(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kubo I, Muroi H, Masaki H, Kubo A. Antibacterial activity of long-chain alcohols: the role of hydrophobic alkyl groups. Bioorganic & medicinal chemistry letters. 1993;3(6):1305–8. [Google Scholar]
- 33.Togashi N, Shiraishi A, Nishizaka M, Matsuoka K, Endo K, Hamashima H, et al. Antibacterial Activity of Long-Chain Fatty Alcohols against Staphylococcus aureus. Molecules. 2007;12(2):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Busscher HJ, Zhao B, Li Y, Zhang Z, van der Mei HC, et al. Surface-Adaptive, Antimicrobially Loaded, Micellar Nanocarriers with Enhanced Penetration and Killing Efficiency in Staphylococcal Biofilms. ACS nano. 2016;10(4):4779–89. [DOI] [PubMed] [Google Scholar]
- 35.Mogen AB, Chen F, Ahn SJ, Burne RA, Wang D, Rice KC. Pluronics-Formulated Farnesol Promotes Efficient Killing and Demonstrates Novel Interactions with Streptococcus mutans Biofilms. PloS one. 2015;10(7):e0133886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radovic-Moreno AF, Lu TK, Puscasu VA, Yoon CJ, Langer R, Farokhzad OC. Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics. ACS nano. 2012;6(5):4279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen F, Liu XM, Rice KC, Li X, Yu F, Reinhardt RA, et al. Tooth-binding micelles for dental caries prevention. Antimicrob Agents Chemother. 2009;53(11):4898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horev B, Klein MI, Hwang G, Li Y, Kim D, Koo H, et al. pH-activated nanoparticles for controlled topical delivery of farnesol to disrupt oral biofilm virulence. ACS nano. 2015;9(3):2390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou J, Horev B, Hwang G, Klein MI, Koo H, Benoit DSW. Characterization and optimization of pH-responsive polymer nanoparticles for drug delivery to oral biofilms. Journal of Materials Chemistry B. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hans M, Shimoni K, Danino D, Siegel SJ, Lowman A. Synthesis and Characterization of mPEG−PLA Prodrug Micelles. Biomacromolecules. 2005;6(5):2708–17. [DOI] [PubMed] [Google Scholar]
- 41.Bernier S, Surette M. Concentration-dependent activity of antibiotics in natural environments. Frontiers in microbiology. 2013;4(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. Journal of controlled release : official journal of the Controlled Release Society. 2009;133(3):221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moad G, Chong YK, Postma A, Rizzardo E, Thang SH. Advances in RAFT polymerization: the synthesis of polymers with defined end-groups. Polymer. 2005;46(19):8458–68. [Google Scholar]
- 44.Murthy N, Robichaud JR, Tirrell DA, Stayton PS, Hoffman AS. The design and synthesis of polymers for eukaryotic membrane disruption. Journal of Controlled Release. 1999;61(1–2):137–43. [DOI] [PubMed] [Google Scholar]
- 45.Gallow KC, Jhon YK, Genzer J, Loo Y-L. Influence of gradient strength and composition profile on the onset of the cloud point transition in hydroxyethyl methacrylate/dimethylaminoethyl methacrylate gradient copolymers. Polymer. 2012;53(5):1131–7. [Google Scholar]
- 46.Vesterinen A, Lipponen S, Rich J, Seppälä J. Effect of block composition on thermal properties and melt viscosity of poly(2-(dimethylamino)ethyl methacrylate), poly(ethylenen oxide) and poly(propylene oxide) block co-polymers. EXPRESS POLYMER LETTERS. 2011;5(9):754–65. [Google Scholar]
- 47.Malcolm DW, Freeberg MAT, Wang Y, Sims KR Jr., Awad HA, Benoit DSW. Diblock Copolymer Hydrophobicity Facilitates Efficient Gene Silencing and Cytocompatible Nanoparticle-Mediated siRNA Delivery to Musculoskeletal Cell Types. Biomacromolecules. 2017;18(11):3753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baranello MP, Bauer L, Benoit DS. Poly(styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromolecules. 2014;15(7):2629–41. [DOI] [PubMed] [Google Scholar]
- 49.Koo H, Rosalen PL, Cury JA, Park YK, Bowen WH. Effects of Compounds Found in Propolis on Streptococcus mutans Growth and on Glucosyltransferase Activity. Antimicrobial Agents and Chemotherapy. 2002;46(5):1302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao J, Klein MI, Falsetta ML, Lu B, Delahunty CM, Yates JR III, et al. The Exopolysaccharide Matrix Modulates the Interaction between 3D Architecture and Virulence of a Mixed-Species Oral Biofilm. PLOS Pathogens. 2012;8(4):e1002623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hwang G, Koltisko B, Jin X, Koo H. Nonleachable Imidazolium-Incorporated Composite for Disruption of Bacterial Clustering, Exopolysaccharide-Matrix Assembly, and Enhanced Biofilm Removal. ACS Appl Mater Interfaces. 2017;9(44):38270–80. [DOI] [PubMed] [Google Scholar]
- 53.Dora CP, Singh SK, Kumar S, Datusalia AK, Deep A. Development and characterization of nanoparticles of glibenclamide by solvent displacement method. Acta poloniae pharmaceutica. 2010;67(3):283–90. [PubMed] [Google Scholar]
- 54.Malcolm DW, Varghese JJ, Sorrells JE, Ovitt CE, Benoit DSW. The Effects of Biological Fluids on Colloidal Stability and siRNA Delivery of a pH-Responsive Micellar Nanoparticle Delivery System. ACS nano. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perrier S 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules. 2017;50(19):7433–47. [Google Scholar]
- 56.FitzGerald PA, McDonald DM, Warr GG. The effect of degree of polymerization on intra- and interchain micellization of a tail-type cationic polysoap. Soft Matter. 2013;9(9):2711–6. [Google Scholar]
- 57.Shinoda R, Saito T, Okita Y, Isogai A. Relationship between Length and Degree of Polymerization of TEMPO-Oxidized Cellulose Nanofibrils. Biomacromolecules. 2012;13(3):842–9. [DOI] [PubMed] [Google Scholar]
- 58.Kleinebudde P, Jumaa M, El Saleh F. Influence of the degree of polymerization on the behavior of cellulose during homogenization and extrusion/spheronization. AAPS pharmSci. 2000;2(3):E21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Ren T, Gou J, Zhang L, Tao X, Tian B, et al. Strategies for improving the payload of small molecular drugs in polymeric micelles. Journal of controlled release : official journal of the Controlled Release Society. 2017. [DOI] [PubMed] [Google Scholar]
- 60.Hans ML. Synthesis, characterization, and application of biodegradable polymeric prodrug micelles for long-term drug delivery Drexel University; 2005.
- 61.L. HM, Christina M, S. ER, Kayla M, Yuling L, J. SS, et al. Evaluation of in vitro release and in vivo efficacy of mPEG‐PLA‐haloperidol conjugate micelle‐like structures. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2007;83B(2):422–30. [DOI] [PubMed] [Google Scholar]
- 62.Fu X, Ping Q, Gao Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. Journal of microencapsulation. 2005;22(7):705–14. [DOI] [PubMed] [Google Scholar]
- 63.Sharma A, Soliman GM, Al-Hajaj N, Sharma R, Maysinger D, Kakkar A. Design and Evaluation of Multifunctional Nanocarriers for Selective Delivery of Coenzyme Q10 to Mitochondria. Biomacromolecules. 2012;13(1):239–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Witten J, Ribbeck K. The particle in the spider’s web: transport through biological hydrogels. Nanoscale. 2017;9(24):8080–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X, Yeh Y-C, Giri K, Mout R, Landis RF, Prakash YS, et al. Control of nanoparticle penetration into biofilms through surface design. Chemical Communications. 2015;51(2):282–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jabra-Rizk MA, Meiller TF, James CE, Shirtliff ME. Effect of Farnesol on Staphylococcus aureus Biofilm Formation and Antimicrobial Susceptibility. Antimicrobial Agents and Chemotherapy. 2006;50(4):1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koo H, Pearson SK, Scott-Anne K, Abranches J, Cury JA, Rosalen PL, et al. Effects of apigenin and tt-farnesol on glucosyltransferase activity, biofilm viability and caries development in rats. Oral microbiology and immunology. 2002;17(6):337–43. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, Nadezhina E, Wilkinson KJ. Quantifying Diffusion in a Biofilm of Streptococcus mutans. Antimicrobial Agents and Chemotherapy. 2011;55(3):1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ong TH, Chitra E, Ramamurthy S, Siddalingam RP, Yuen KH, Ambu SP, et al. Chitosan-propolis nanoparticle formulation demonstrates anti-bacterial activity against Enterococcus faecalis biofilms. PloS one. 2017;12(3):e0174888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Q, Kang F, Gao Y, Mao X, Hu X. Sequestration of nanoparticles by an EPS matrix reduces the particle-specific bactericidal activity. Scientific Reports. 2016;6:21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huo S, Jiang Y, Gupta A, Jiang Z, Landis RF, Hou S, et al. Fully Zwitterionic Nanoparticle Antimicrobial Agents through Tuning of Core Size and Ligand Structure. ACS nano. 2016;10(9):8732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hayden SC, Zhao G, Saha K, Phillips RL, Li X, Miranda OR, et al. Aggregation and Interaction of Cationic Nanoparticles on Bacterial Surfaces. Journal of the American Chemical Society. 2012;134(16):6920–3. [DOI] [PubMed] [Google Scholar]
- 73.Feng ZV, Gunsolus IL, Qiu TA, Hurley KR, Nyberg LH, Frew H, et al. Impacts of gold nanoparticle charge and ligand type on surface binding and toxicity to Gram-negative and Gram-positive bacteria. Chemical Science. 2015;6(9):5186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gomes F, Cerca N, Azeredo J, Oliveira R, Teixeira P, Leite B. Farnesol as Antibiotics Adjuvant in Staphylococcus epidermidis Control In Vitro. The American Journal of the Medical Sciences. 2011;341(3):191–5. [DOI] [PubMed] [Google Scholar]
- 75.Bandara HMHN, Herpin MJ, Kolacny D, Harb A, Romanovicz D, Smyth HDC. Incorporation of Farnesol Significantly Increases the Efficacy of Liposomal Ciprofloxacin against Pseudomonas aeruginosa Biofilms in Vitro. Molecular pharmaceutics. 2016;13(8):2760–70. [DOI] [PubMed] [Google Scholar]
- 76.Jacobs AC, Didone L, Jobson J, Sofia MK, Krysan D, Dunman PM. Adenylate kinase release as a high-throughput-screening-compatible reporter of bacterial lysis for identification of antibacterial agents. Antimicrob Agents Chemother. 2013;57(1):26–36. (see Supplemental Table 1 in this reference). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin JH, Feng F, Yu MC, Wang CH, Chang PC. Modulation of periodontitis progression using pH-responsive nanosphere encapsulating metronidazole or N-phenacylthialzolium bromide. Journal of periodontal research. 2018;53(1):22–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.