

Acute rheumatic fever (ARF) is a consequence of an autoimmune response to infection with the Gram-positive bacteria Streptococcus pyogenes or group A Streptococcus (GAS) 1. ARF afflicts about 0.5 million people every year, and both environmental and genetic factors contribute to the etiology of ARF and its chronic sequelae, rheumatic heart disease (RHD) 2. ARF/RHD is more prevalent in developing countries and is mainly associated with poverty and other determinants of poor health, which facilitates rapid transmission of GAS. Although rare, ARF also persists in some developed countries especially in disadvantaged communities 3. GAS infection causes a variety of diseases ranging from pharyngitis to severe life-threatening conditions like necrotizing fasciitis and toxic shock syndrome. GAS activates both the innate and adaptive immune systems and elicits a potent inflammatory response that helps to restrict bacterial replication and dissemination. ARF develops as post-infectious sequelae to acute inflammation in genetically predisposed individuals and is manifested as a systemic inflammatory autoimmune reaction 2 – 4 weeks after the pharyngeal infection 1. Whereas the contributions of both humoral and cell-mediated immune responses in pathogenesis of ARF are well studied, the role of the innate immune system has not been explored in detail 2. Moreover, the immune and inflammatory pathways that link GAS to the progression of ARF to RHD are also poorly understood.
In this issue, Kim et al. report a dysregulated IL-1β – GM-CSF cytokine axis in ARF patients as a potential mechanism that drives development of RHD upon recurrent GAS infections 4. GAS induced persistent production of IL-1β and enhanced production of GM-CSF in peripheral blood mononuclear cells (PBMCs) from ARF patients. A strong positive correlation between the levels of IL-1β and GM-CSF suggests a role for IL-1β in promoting the expression of GM-CSF in these cells. GAS also drives expansion of GM-CSF- and IFNγ -expressing Th1 CD4+ T cells; these pathogenic CD4+ T cells upon trafficking to the heart can cause progressive heart valve dysfunction. Indeed, the level of IP-10/CXCL10, a potent Th1 chemoattractant, was found to be elevated in the systemic circulation of ARF patients 4. These findings collectively imply IL-1β - dependent expansion followed by IP-10-mediated recruitment of IFNγ− and GM-CSF-expressing Th1 CD4+ T cells as a causative factor triggering chronic autoimmune valvulitis in ARF patients. Overall, the study by Kim et al. delineates a convincing sequence of immunological events that promotes development of cardiac dysfunction and RHD. However, the mechanism by which GAS induces persistent IL-1β production in PBMCs from ARF patients, which is the primary event initiating this cascade, remains elusive.
GAS infection activates a variety of extracellular and intracellular pattern recognition receptors and induces the production of various inflammatory mediators including IL-1β 5. IL-1β is critical for host protection during GAS infection, and mice lacking IL-1R are highly susceptible to the infection 6. Moreover, patients receiving IL-1 inhibitor treatment develop GAS infections at much higher rates, further suggesting a key role for IL-1β in host defense against infection.
Because of its potent inflammatory nature and multitude of biological functions, the induction, processing and secretion of IL-1β are tightly regulated. IL-1β induced upon infection remains in an inactive zymogen form in the cytosol. The processing of pro–IL-1β into the biologically active form and its release into the extracellular milieu is regulated by cytosolic inflammasome complexes 7. These multiprotein complexes assembled via homotypic protein interactions consists of a sensor, the adaptor protein apoptosis-associated speck-like protein containing a CARD domain (ASC) and the inactive pro-form of cysteine protease caspase-1 (Figure 1). The inflammasome sensors include nucleotide-binding domain–like receptors (i.e., NLRP1, NLRP3, and NLRC4), the HIN-200 family member absent in melanoma 2 (AIM2), and pyrin, and these sensors initiate the assembly of inflammasome complexes in response to infections or other immunologic challenges 7. Once assembled, the inflammasome complex facilitates processing of caspase-1 into its active form, which in turn mediates the processing of pro–IL-1β. Gasdermin D (GSDMD), another substrate of caspase-1, forms membrane pores that disrupt cell membrane integrity, resulting in a lytic form of cell death called pyroptosis. The release of bioactive IL-1β is thought to be mediated by pyroptotic cell lysis, however, recent studies have also reported lysis-independent release of IL-1 cytokines via the GSDMD pores.
Figure 1: Possible role of inflammasomes in pathogenesis of Rheumatic Heart Disease (RHD).
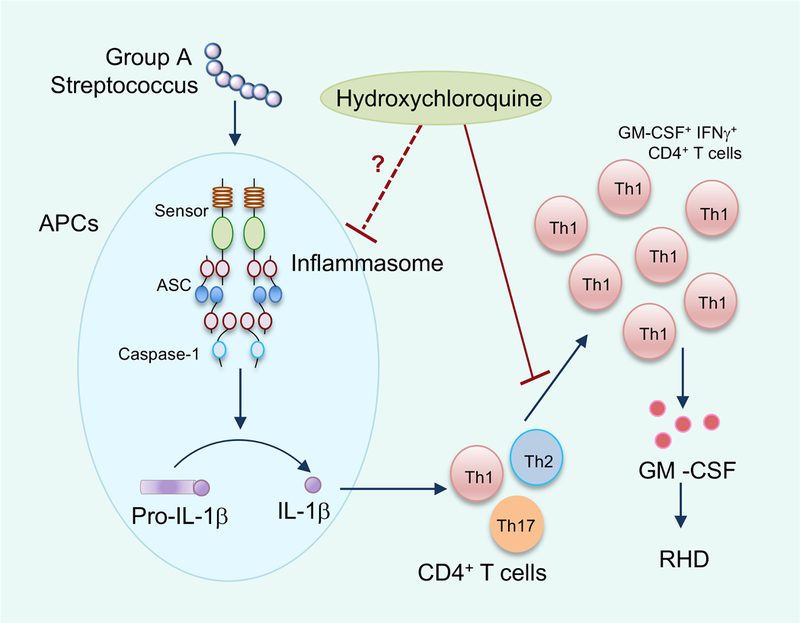
Infection by Group A Streptococcus (GAS) activates the inflammasome leading to processing and release of IL-1β. IL-1β facilitates the expansion of GM-CSF-expressing Th1 CD4+ T cells. These cells traffic to the heart valve and the inflammatory cytokines secreted from these cells induce valvulitis, cardiac dysfunction and subsequent RHD in high-risk patients with acute rheumatic fever (ARF). Hydroxychloroquine (HCQ) limits the expansion of Th1 CD4+ T cells. HCQ could also have a possible function in limiting inflammasome activation.
GAS infection triggers activation of the inflammasome, and the inflammasome activators include the secreted toxins pore-forming streptolysin O (SLO) and ADP-ribosyltransferase SpyA, as well as the GAS virulence factor M1 protein 6, 8 (Figure 1). The persistent release of IL-1β in PBMCs from ARF patients suggests a hyperactive inflammasome due to host factors, since GAS infection did not induce a similar response in cells from healthy donors4. Previous studies have reported polymorphisms in genes encoding proteins in both innate and adaptive immune pathways that could confer genetic susceptibility to ARF 1. It is not known whether polymorphisms in inflammasome components are also associated with genetic predisposition to ARF. Another intriguing possibility is the epigenetic remodeling of inflammasome-associated genes due to recurrent GAS infections that leads to altered gene expression upon reinfection 9. Of note, inflammasomes have been shown to have a key role in innate memory or ‘trained immunity’ (long-term epigenetic reprogramming of innate immune cells) and therefore, the dysregulated cytokine secretion could also be due to immunological imprinting in these genes 10. Interestingly, levels of MYD88 and CASPASE1 are elevated in PBMCs from ARF patients during the peak of disease. Additional studies are warranted to further investigate whether priming (transcriptional induction) and/or activation of the inflammasome is differentially regulated in PBMCs from ARF patients. In addition to IL-1β, processing and release of IL-18 is also regulated by inflammasomes 7. IL-18 is a potent IFNγ-inducing factor, and IL-18 could also be contributing to the functionality of inflammatory Th1 CD4+ T cells. Therefore, the inflammasome could possibly be impacting multiple aspects of the inflammatory milieu that drive the progression of RHD.
An exciting clinical implication of the present study is the identification of the antimalarial drug hydroxychloroquine (HCQ) as a potential therapy to reduce the risk of progression to RHD in ARF patients 4 (Figure 1). The authors demonstrate that HCQ limits IL-1β -mediated expansion of GM-CSF-expressing CD4+ T cells, however the mechanism by which HCQ exerts this effect is not known. Although HCQ has been used to treat autoimmune diseases for decades, its mode of action is still incompletely understood. HCQ has been shown to modulate lysosomal acidification and thereby affect antigen processing and TLR signaling 11. Moreover, HCQ was also shown to act as an ion channel inhibitor that restricts NLRP3 inflammasome activation and IL-1β release by inhibiting Ca++- activated K+ channels 12. HCQ also limits NLRP3 inflammasome activation by preventing the release of lysosomal cathepsins 13. Inflammasome activation induced by GAS is also linked to endocytic pathway and ionic balance and the GAS M1 protein requires both clathrin-mediated endocytosis as well as potassium efflux for inflammasome activation 8. The pore-forming toxin streptolysin O (SLO) could also be affecting the cellular potassium levels that lead to subsequent NLRP3 inflammasome activation 7.
Because of the diverse effects of HCQ on TLR signaling, cellular ionic balance and lysosomal fusion, it is tempting to speculate that the inflammasome itself could be one of the targets for HCQ during GAS infection. An easy experiment would be to test whether HCQ limits the persistent IL-1β secretion in PBMCs from ARF patients. The findings from this study suggest the potential use of HCQ along with antibiotic prophylaxis to restrict inflammatory responses in high-risk ARF patients. Another potential approach that is worthy of testing is targeting the processing and release of IL-1β and IL-18 using either selective inflammasome blockers or caspase-1 inhibitors 14, 15. Inhibitors of the NLRP3 inflammasome and caspase-1 have been tested in preclinical animal studies for various diseases and were found to be efficacious for reducing inflammation and pathology. As GSDMD is critical for release of IL-1 cytokines, agents that impair GSDMD function would also have the potential to limit the hyperinflammatory response. In addition to describing a fascinating new mechanism involved in RHD and identifying an easily accessible prophylactic agent, the current study opens up several exciting avenues for future research, including the role of the inflammasome and its potential as a therapeutic target to tackle RHD.
Acknowledgments
Sources of funding:
Dr. Kanneganti is supported by grants from the National Institutes of Health (AI101935, AI124346, AR056296, and CA163507) and American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Disclosures:
The authors declare that they have no conflict of interest.
References:
- 1.Karthikeyan G and Guilherme L. Acute rheumatic fever. Lancet. 2018;392:161–174. [DOI] [PubMed] [Google Scholar]
- 2.Carapetis JR, Beaton A, Cunningham MW, Guilherme L, Karthikeyan G, Mayosi BM, Sable C, Steer A, Wilson N, Wyber R and Zuhlke L. Acute rheumatic fever and rheumatic heart disease. Nat Rev Dis Primers. 2016;2:15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuhlke LJ, Beaton A, Engel ME, Hugo-Hamman CT, Karthikeyan G, Katzenellenbogen JM, Ntusi N, Ralph AP, Saxena A, Smeesters PR, Watkins D, Zilla P and Carapetis J. Group A Streptococcus, Acute Rheumatic Fever and Rheumatic Heart Disease: Epidemiology and Clinical Considerations. Curr Treat Options Cardiovasc Med. 2017;19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim MLM WJ; Minigo G; Keeble JL; Garnham AL; Pacini G; Smyth GK; Speed TP; Carapetis J; Wicks IP A dysregulated IL-1β -GM-CSF axis in acute rheumatic fever that is limited by hydroxychloroquine. Circulation. 2018. [DOI] [PubMed] [Google Scholar]
- 5.Fieber C and Kovarik P. Responses of innate immune cells to group A Streptococcus. Front Cell Infect Microbiol. 2014;4:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LaRock CN and Nizet V. Inflammasome/IL-1beta Responses to Streptococcal Pathogens. Front Immunol. 2015;6:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma D and Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valderrama JA, Riestra AM, Gao NJ, LaRock CN, Gupta N, Ali SR, Hoffman HM, Ghosh P and Nizet V. Group A streptococcal M protein activates the NLRP3 inflammasome. Nat Microbiol. 2017;2:1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arts RJW, Joosten LAB and Netea MG. The Potential Role of Trained Immunity in Autoimmune and Autoinflammatory Disorders. Front Immunol. 2018;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Bassler K, Klee K, Schulte-Schrepping J, Ulas T, Moorlag S, Kumar V, Park MH, Joosten LAB, Groh LA, Riksen NP, Espevik T, Schlitzer A, Li Y, Fitzgerald ML, Netea MG, Schultze JL and Latz E. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell. 2018;172:162–175 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An J, Minie M, Sasaki T, Woodward JJ and Elkon KB. Antimalarial Drugs as Immune Modulators: New Mechanisms for Old Drugs. Annu Rev Med. 2017;68:317–330. [DOI] [PubMed] [Google Scholar]
- 12.Eugenia Schroeder M, Russo S, Costa C, Hori J, Tiscornia I, Bollati-Fogolin M, Zamboni DS, Ferreira G, Cairoli E and Hill M. Pro-inflammatory Ca(++)-activated K(+) channels are inhibited by hydroxychloroquine. Sci Rep. 2017;7:1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang TT, Lv LL, Pan MM, Wen Y, Wang B, Li ZL, Wu M, Wang FM, Crowley SD and Liu BC. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018;9:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw PJ, McDermott MF and Kanneganti TD. Inflammasomes and autoimmunity. Trends Mol Med. 2011;17:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD and Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606. [DOI] [PubMed] [Google Scholar]