

Abstract
Background:
Efforts to promote the cessation of harmful alcohol use are hindered by the affective and physiological components of alcohol withdrawal (AW), which can include life-threatening seizures. Although previous studies of AW and relapse have highlighted the detrimental role of stress, little is known about genetic risk factors.
Methods:
We conducted a genome-wide association study (GWAS) of AW symptom count in uniformly assessed subjects with histories of serious AW, followed by additional genotyping in independent AW subjects.
Results:
The top association signal for AW severity was in sortilin-family neurotrophin receptor gene SORCS2 on chromosome 4 (European-American meta-analysis n = 1,478, P = 4.3 × 10−9). There were no genome-wide significant findings in African-Americans (n = 1,231). Bioinformatic analyses were conducted using publicly available high-throughput transcriptomic and epigenomic datasets, showing that in humans SORCS2 is most highly expressed in the nervous system. The identified SORCS2 risk haplotype is predicted to disrupt a stress hormone-modulated regulatory element that has tissue-specific activity in human hippocampus. We used human neural lineage cells to demonstrate in vitro a causal relationship between stress hormone levels and SORCS2 expression, and show that SORCS2 levels in culture are increased upon ethanol exposure and withdrawal.
Conclusions:
Taken together, these findings indicate that the pathophysiology of withdrawal may involve the effects of stress hormones on neurotrophic factor signaling. Further investigation of these pathways could produce new approaches to managing the aversive consequences of abrupt alcohol cessation.
Keywords: alcohol withdrawal, human genetics, genome-wide association study, stress hormones
Introduction:
Heavy drinking is one of the greatest contributors to morbidity and mortality globally (Lim et al, 2012), but attempts to quit drinking can be accompanied by highly aversive physiological and affective symptoms (Becker, 2008). In the most severe cases, acute alcohol withdrawal (AW) can have life-threatening consequences, such as seizures and delirium tremens (DTs) (Schuckit, 2014). Benzodiazepines are a mainstay of AW symptom management, but benzodiazepines present potential risks, and have addictive properties (Trevisan et al, 1998). Persistent, subacute AW symptoms can promote relapse to heavy drinking (Becker, 2008; Trevisan et al, 1998), an effect that can be compounded by stress (Becker, 2012).
Prior work in humans has indicated that distinct genetic loci may influence different aspects of substance dependence, such as initiation and maintenance (Fanous & Kendler, 2005). AW especially has several features that make it a suitable target for genetic association studies. Research conducted with human twins, and long-term selective breeding strategies in rodents, have shown that AW is partly heritable, and that its genetic basis is at least partly distinct from other aspects of alcohol dependence (AD) (Crabbe & Phillips, 1993; Slutske et al, 1999). An analysis of genetic influences on DSM-IV criteria for AD in a large sample of alcohol-exposed twins showed that distinguishable genetic risk factors loaded onto withdrawal and continued use (Kendler et al, 2012). These factors were separate from genetic factors influencing tolerance and excessive drinking, or loss of control and social dysfunction, pointing to the existence of distinct risk variants for these different components of AD. Similar conclusions have been drawn from animal experiments. For example, specially bred rodents with more severe convulsions upon withdrawal from ethanol or barbiturates, and greater sensitivity to chemiconvulsants, do not exhibit abnormalities in acute behavioral tolerance to administered ethanol (Crabbe & Kosobud, 1986; Fehr et al, 2004). Furthermore, the dramatic and dangerous manifestations of AW in patients make it both readily identifiable and clinically important.
Previous studies of genetic influences on AW have focused mostly on binary phenotypes, such as the presence or absence of either simple AW or rare complications of severe AW, notably seizures and DTs (Koehnke et al, 2002; Preuss et al, 2006; Rujescu et al, 2005; Wernicke et al, 2003; Wetherill et al, 2014). Results from studies of single nucleotide polymorphisms (SNPs) in candidate genes have been inconsistent (Schmidt & Sander, 2000; van Munster et al, 2007). We uniformly assessed withdrawal symptom history in a large number of AD individuals, and conducted a genome-wide association study (GWAS) to identify risk variants associated with AW symptom count. The top findings were assessed in independent clinical samples and via bioinformatic analyses, followed by experimental manipulations in a human neural cell line. A flow chart for the present work is shown in Figure 1, which provides an overview of the human genetic studies (green), bioinformatic analyses (orange), and in vitro experiments (blue). This integration of multiple independent datasets, coupled with diverse computational and experimental approaches, facilitated interrogation of the biological influences on AW severity.
Figure 1: Flow chart outlining the present study.
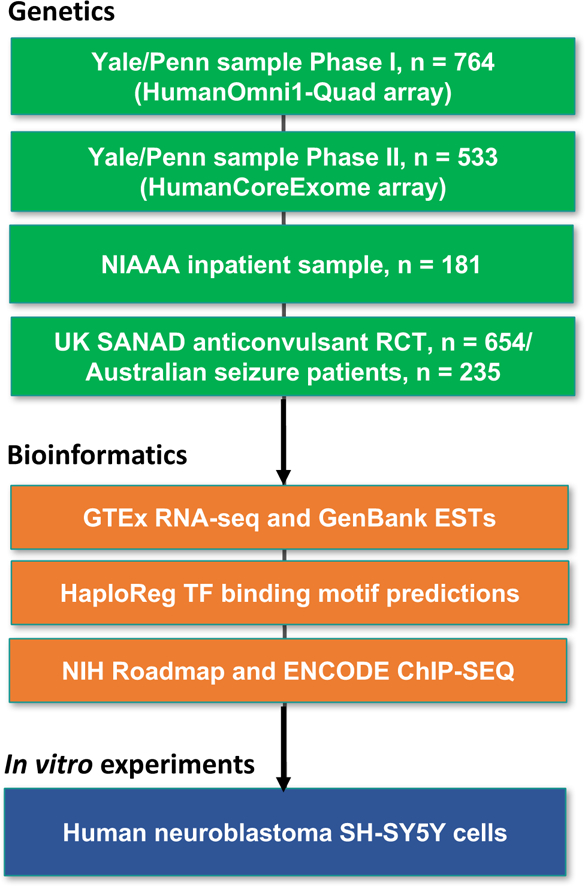
National Institute on Alcohol Abuse and Alcoholism (NIAAA), Standard and New Antiepileptic Drugs (SANAD), randomized controlled trial (RCT), expressed sequence tag (EST), transcription factor (TF).
Materials and Methods:
Recruitment and assessment of human subjects: Yale-Penn sample
Details on this sample have been published previously (Gelernter et al, 2014a; Gelernter et al, 2014b; Gelernter et al, 2014c). Briefly, adults with histories of serious substance use (alcohol, opioid, or cocaine) disorders and controls were recruited primarily via community advertisements and word of mouth as part of ongoing studies of the genetics of alcohol and drug dependence at five sites in the Eastern United States. The sample consisted of small nuclear families originally collected for linkage studies, and unrelated individuals. Exclusion criteria included a history of psychotic disorders (schizophrenia, bipolar disorder), serious head injury, or inability to read English at a sixth grade level. There was a minimum age cutoff of 18 years for subjects recruited as affecteds, and 25 for unaffecteds, so that unaffecteds had passed through the peak period of risk for developing a substance use disorder. Subjects gave written informed consent as approved by the institutional review board at each site, and certificates of confidentiality were obtained from the National Institute on Drug Abuse (NIDA) and the National Institute on Alcohol Abuse and Alcoholism (NIAAA).
In-person interviews were conducted by trained interviewers using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA), a comprehensive polydiagnostic instrument yielding reliable information on DSM-IV diagnoses and diagnostic criteria (Pierucci-Lagha et al, 2007; Pierucci-Lagha et al, 2005) (available at https://nidagenetics.org/filebrowser/download/3765). The SSADDA covers psychiatric and substance use disorders, as well as social history and demographic information. Phenotype and genotype data are being released via dbGAP (accession number phs000425.v1.p1).
Microarray genotyping and quality control: Yale-Penn sample
DNA was extracted from blood, saliva, or immortalized lymphoblastoid cell lines. Subjects were genotyped in two phases. In Phase I, a group of subjects was genotyped on the Illumina HumanOmni1-Quad v1.0. In Phase II, a second group of subjects was genotyped on the Illumina HumanCoreExome array. Extensive details on genotyping, quality control, data cleaning, and imputation for subjects genotyped on the HumanOmni1-Quadv1.0 (Phase I) have been published previously (Gelernter et al, 2014a; Gelernter et al, 2014b; Gelernter et al, 2014c). For the present study, only unrelated probands were retained for analysis, and ancestry outliers were removed based on the first 10 principal components (PCs) in self-reported European-Americans (EAs) and African-Americans (AAs) (Patterson et al, 2006; Price et al, 2006). For the HumanCoreExome genotyping platform (Phase II), genotype data processing is described in Supplemental Material.
Subject selection and AW symptom count GWAS: Yale-Penn sample
After data cleaning, we conducted a GWAS of AW symptom count. Subjects were retained for this study if they: 1) met DSM-IV criteria for lifetime AD (American Psychiatric Association, 2000), and 2) endorsed having experienced at least one of the AW symptoms referenced in Table 1 for most of two days or longer. The SSADDA elicits only symptoms lasting most of two days or longer, which differentiates them from the common symptoms of a hangover. Following the approach used in our previous studies (Gelernter et al, 2014a; Gelernter et al, 2014b; Gelernter et al, 2014c; Zhou et al, 2017), instead of using a cutoff to create a binary phenotype we sought to maximize power by analyzing quantitative phenotype data. For each individual, the total number of AW symptoms experienced for two days or longer was tallied (Figure 2) and used as the dependent variable in the GWAS. Exploratory multivariate analysis techniques validated the use of the additive symptom count sum score to capture importance variance in the AW symptom data (Supplemental Material and Methods, and Figure S1). The dataset was imputed and imputed genotype dose data were used for the GWAS, which was carried out in Plink v1.07 (Purcell et al, 2007), with minor allele frequency (MAF) and INFO cutoffs of 0.03 and 0.7, respectively. Low-frequency and rare variants with MAF below 0.03 were not examined because of the lower reliability of imputation for these variants (1000 Genomes Project Consortium, 2012). For the AW symptom count GWAS, 1,297 EAs and 1,231 AAs were included. Analyses were conducted separately in EAs and AAs. Within each population, analyses were run separately on subjects genotyped on the HumanOmni1-Quadv1.0 (Phase I) and HumanCoreExome arrays (Phase II), followed by meta-analysis using METAL (Willer et al, 2010). All analyses were adjusted for sex, age, and 10 PCs. Secondary analyses with additional covariates were conducted as described below in the Results section. The cutoff for genome-wide significance was defined using the standard value of P = 5 × 10−8.
Table 1. Alcohol withdrawal symptoms.
Questions were preceded by the following introduction, and the stem was repeated often: “People who cut down, stop, or go without drinking after drinking steadily for some time may not feel well. These feelings are more intense and can last longer than the usual hangover. When you stopped, cut down or went without drinking, did you ever experience any of the following problems for most of the day for 2 days or longer?”
| 1. Did you have the shakes (hands trembling)? |
| 2. Were you unable to sleep? |
| 3. Did you feel anxious? |
| 4. Did you feel depressed or irritable? |
| 5. Did your heart beat fast or did you sweat? |
| 6. Did you have nausea or vomiting? |
| 7. Did you feel physically weak? |
| 8. Did you have headaches? |
| 9. Did you see or hear things that weren’t there? |
| 10. Were you fidgety or restless? |
Figure 2: Alcohol withdrawal symptom counts.
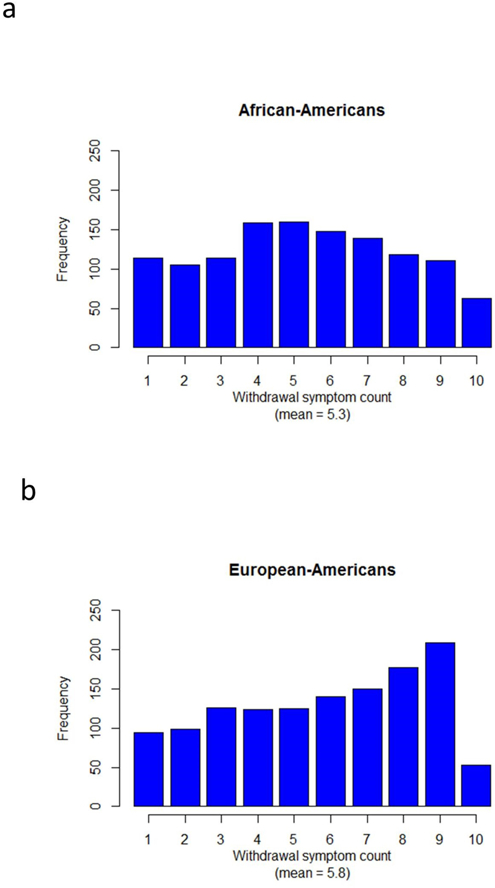
Symptom count totals are shown separately for (a) African-Americans (AAs) and (b) European-Americans (EAs).
Alcohol dependent sample from the NIAAA
Rs11731003 was the most highly significant polymorphism associated with lifetime AW symptom count in the above described GWAS of Yale-Penn EA subjects with a history of persistent AW symptoms. We genotyped rs11731003 in an independent sample of AD subjects admitted to the NIAAA Inpatient Unit, and clinically managed for AW prior to participation in other research studies. As described previously (Huang et al, 2014), all of these subjects were recruited for voluntary admission at the NIH Clinical Center, met DSM-IV criteria for AD, did not have active psychosis or cognitive impairment, and had consumed their most recent alcoholic beverage within the past 48 hours. Biospecimens from a total of n = 181 EA individuals who required treatment for AW (110 male, 71 female) were available for analysis. Informed consent was obtained as approved by the NIH IRB. Rs11731003 was genotyped by the TaqMan method using an assay-on-demand (assay ID: C___1213779_20) from Applied Biosystems (Foster City, CA). Data were processed using SDS 2.4 software (Applied Biosystems) at NIAAA. We used general linear regression to test for an association between rs11731003 and the maximum Clinical Institute Withdrawal Assessment-Alcohol revised (CIWA-Ar) (Sullivan et al, 1989) score. There were few TT genotypes observed in this small sample, so T allele carriers (CT and TT) were binned for analysis. The regression model was adjusted for age, sex, and the Alcohol Dependence Scale (ADS) (Skinner & Horn, 1984) score obtained at the time of enrollment.
Standard and New Antiepileptic Drugs (SANAD) randomized controlled trial sample and Australian seizure patient community sample
These two longitudinally tracked samples of seizure disorder patients have been described previously (Speed et al, 2014). Briefly, British patients with seizure disorders who enrolled in the SANAD trial were randomized in equal numbers to approved anticonvulsant medications (Marson et al, 2007a; b). After imputation, rs11731003 was tested in SANAD subjects (n = 654) for an association with the achievement of 12-month seizure remission, adjusting for non-genetic predictors of clinical outcome. These analyses were carried out as detailed by the authors of the original GWAS publication (Speed et al, 2014), from whom the results were obtained. Rs11731003 was also similarly imputed and evaluated in a non-randomized community sample of Australian seizure disorder patients (n = 235) treated with medications chosen by their physicians (Speed et al, 2014).
Publicly available human gene expression, histone modification, and transcription factor (TF) binding data
Human tissue transcriptome data (GTEx analysis release v4, dbGAP accession phs000424.v4.p1) were accessed and visualized via the GTEx Portal (www.gtexportal.org) (Melé et al, 2015). The “UCSC Genes” (Rosenbloom et al, 2015) and “Human ESTs” (Benson et al, 2010) annotation tracks were obtained from the UCSC Genome Browser (hg19) (Kent et al, 2002). Also obtained from the USCS Genome Browser were annotation tracks with results from chromatin immunoprecipitation followed by sequencing (ChIP-SEQ) experiments performed in human brain by NIH Roadmap consortium investigators (Roadmap Epigenomics Consortium et al, 2015) to measure Histone H3 Lysine 27 acetylation (H3K27ac), a histone mark typical of active enhancers (Shlyueva et al, 2014). Predictions of TF binding motif disruptions by SORCS2 risk haplotype SNPs were obtained from the public HaploReg (v4) resource (Ward & Kellis, 2012).
In addition, a full set of non-coding regulatory element annotations across 125 different cell and tissues types were retrieved for the risk haplotype SNPs using HaploReg (v4) (Ward & Kellis, 2012). These previously published annotations were generated by application of a Hidden Markov Model (HMM) that used 12 different histone marks to categorize regulatory sites based on a classification scheme containing 25 different ranked chromatin states (Ernst & Kellis, 2015). The UCSC Genome Browser was also used to access ENCODE consortium TF ChIP-SEQ data. The ENCODE consortium examined binding of multiple TFs in different subsets of cell lines under a variety of experimental conditions, and a total of 457 ChIP-SEQ datasets were available (ENCODE Project Consortium, 2012). To prioritize the most relevant TF binding activity at the genomic locus of interest, we filtered ChIP-SEQ peak calls using the most stringent available score cutoff (1000 on a scale of 0 to 1000) (ENCODE Project Consortium, 2012).
Treatment of human neuroblastoma SH-SY5Y cells with dexamethasone (DEX)
The SH-SY5Y cell line was cultured in DMEM F-12 containing 10% fetal bovine serum and 100 IU/ml Penicillin Streptomycin. Cells were treated with DEX for 24 hours, after which RNA was purified using the Nucleospin gel and PCR clean-up kit from Macherey-Nagel. mRNA was converted to cDNA and the 3’ end of human SORCS2 was amplified with Titanium one-step RT-PCR kit (Clontech) using the primer pair 5’-CAAGGAAGAGGAGCCTCTCGAGTGATA-3’ and 5’–GTGGTTCTGTGCCCTCCGTGGGTGAAA-3’. Human GAPDH served as a loading control and was amplified using the primer pair 5’-ACCATGGAGAAGGCTGGGGCTCATTT–3’ and 5’-ATGGCATGGACTGTGGTCATGAGTCCTT-3’. Amplified transcripts were separated on a 1.5% agarose gel and quantification of bands performed with ImageJ software. Further information on transcript generation and quantification is provided in Supplemental Materials and Methods.
Ethanol withdrawal in SH-SY5Y culture
SH-SY5Y cells were cultured until achieving 90% confluency. Cells were washed in phosphate buffered saline and incubated in serum-free DMEM F-12 (Lonza) with or without 150 mM (690 mg%) ethanol. The “Withdrawal” group was incubated with DMEM F-12 and 150 mM ethanol for the first 72 hours followed by a 24 hour withdrawal-period in pure DMEM F-12. The “EtOH” group was incubated with DMEM F-12 for the first 24 hours followed by 72 hours in DMEM F-12 and 150 mM ethanol. The control group received DMEM F-12 for all 96 hours. Cell morphology was examined at the end of the experiment, which showed no differences between the groups. SORCS2 and GAPDH expression was quantified via RT-PCR using the primer sequences described above.
Results:
GWAS of alcohol withdrawal (AW) symptom count identifies SORCS2
Descriptive information for the Yale-Penn sample included in the GWAS is provided in Table 2. After meta-analysis of the subjects genotyped in Phase I (on the Illumina HumanOmni1-Quadv1.0) and Phase II (on the Illumina HumanCoreExome array), two genome-wide significant (GWS) loci were identified in EAs (Table 3). The most significant locus was rs11731003 (MAF = 0.15, β = 0.90, standard error (SE) = 0.16, P = 7.0 × 10−9) in SORCS2 on chromosome 4 (regional association plot, Figure 3), followed by the intergenic SNP rs10052506 (β = 0.83, SE = 0.15, P = 1.22 × 10−8) on chromosome 5 (regional association plot, Figure S2; quantile-quantile (QQ) and Manhattan plots, Figure S3). There was support for these signals in both Phase I and Phase II subjects (Table 3). The QQ plots showed minimal evidence of inflation. No SNPs reached GWS in the AA sample (Figure S3).
Table 2. Overview of Yale-Penn genome-wide association study (GWAS) sample.
All subjects included in the present study met criteria for DSM-IV lifetime alcohol dependence (AD), and reported a history of alcohol withdrawal (AW) symptoms lasting at least two days. Standard deviation (SD).
| European-Americans | African-Americans | ||
|---|---|---|---|
| Sample size | 1,297 | 1,231 | |
| Male | |||
| Sample size (%) | 855 (65.9) | 770 (62.5) | |
| Mean age (SD) | 40.1 (11.2) | 42.6 (9.0) | |
| Mean number of AW symptoms (SD) | 5.7 (2.7) | 5.2 (2.6) | |
| Female | |||
| Sample size (%) | 442 (34.1) | 461 (37.5) | |
| Mean age (SD) | 39.3 (11.0) | 40.1 (8.4) | |
| Mean number of AW symptoms (SD) | 5.9 (2.7) | 5.5 (2.7) |
Table 3. Genome-wide association study (GWAS) meta-analysis results from European-Americans (EAs).
Results are shown for the two loci that reached genome-wide significance (GWS) in the GWAS of lifetime alcohol withdrawal (AW) symptom count that was conducted in subjects with a history of persistent AW symptoms. GWAS subjects (Yale-Penn sample) were interviewed using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA). Phase I subjects were genotyped on the Illumina HumanOmni1-Quadv1.0 array, and Phase II subjects were genotyped on the Illumina HumanCoreExome array. Rs11731003 was also genotyped and evaluated in an independent smaller sample of inpatient EAs being treated for acute AW (NIAAA sample), who were monitored using the Clinical Institute Withdrawal Assessment-Alcohol revised (CIWA-Ar). The Direction column shows, for each sample, the direction of effect of the minor allele. Base pair (BP), chromosome (CHR), minor allele (MA), minor allele frequency (MAF), single nucleotide polymorphism (SNP).
| SSADDA | CIWA-Ar | Direction | Meta | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phase I (n = 764) |
Phase II (n = 533) |
Meta (n = 1,297) |
n = 181 | n = 1,478 | ||||||||
| SNP | CHR | BP | MA/MAF | P | P | P | P | P | ||||
| rs11731003 | 4 | 7,390,222 | T/0.15 | 2.6E-4 | 4.6E-6 | 7.0E-9 | 0.20 | +++ | 4.3E-9 | |||
| rs10052506 | 5 | 79,598,903 | A/0.16 | 1.5E-6 | 2.5E-3 | 1.2E-8 | ++ | |||||
Figure 3: Genome-wide association study (GWAS) of alcohol withdrawal (AW) symptom count identifies SORCS2.
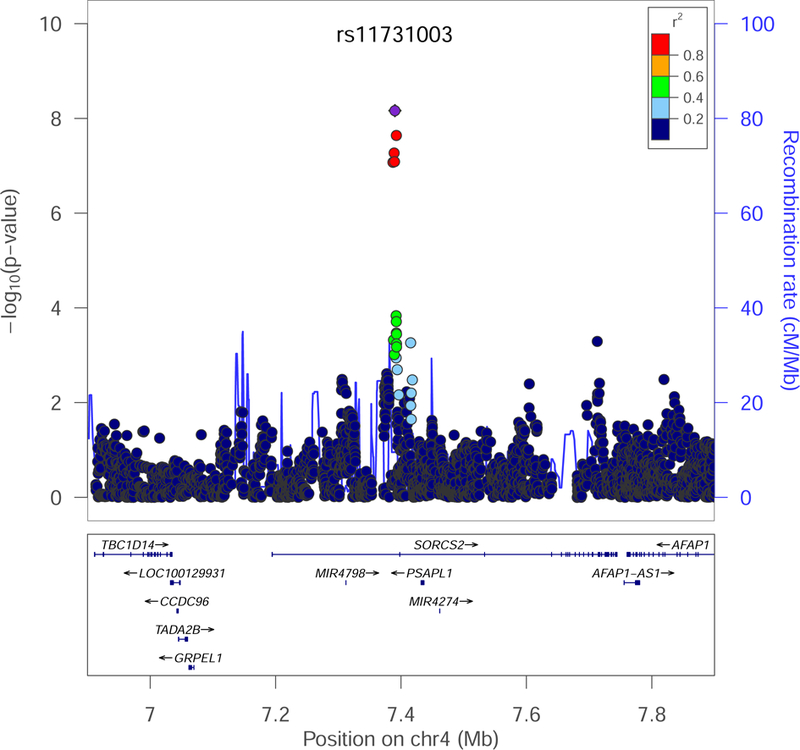
Regional association plot (genome build hg19) of single nucleotide polymorphisms (SNPs) passing quality control, showing a genome-wide significant (GWS) association in European-Americans (EAs) between lifetime AW symptom count and rs11731003 (P = 7.0 × 10−9) in SORCS2.
In both phases of the Yale-Penn study, rs11731003 in SORCS2 was well imputed (r2 of 0.93 and 0.87), and the significant association was observed whether the data were fit with linear (above) or ordinal models (P = 1.5 × 10−8). All models were adjusted for the first 10 PCs, sex, and age; further exploratory adjustment for the number of years spent drinking regularly to intoxication did not materially alter the observed significance of rs11731003 (P = 9.0 × 10−9). Another measure of drinking quantity, maximum number of standard drinks ever consumed in 24 hours (MAXDRINKS), is influenced by the efficiency of alcohol metabolizing enzymes (Bierut et al, 2012). Additional adjustment for MAXDRINKS also did not change the GWS of the relationship between rs11731003 and AW symptom count (P = 2.9 × 10−8). We genotyped and evaluated rs11731003 in a small independent sample of EAs receiving inpatient treatment for acute AW (NIAAA sample), testing for association of rs11731003 with CIWA-Ar score. Meta-analysis of the results (n =181, P = 0.2) with our GWAS of lifetime AW symptom count slightly strengthened the initial association (meta-analysis P = 4.3 × 10−9, Table 3).
The most serious cases of AW can include seizures, and anticonvulsants have been studied as treatments for AW symptoms (Minozzi et al, 2010). However, consistent with prior observations (Schuckit, 2014) the majority of subjects in our GWAS (92%) did not report any history of AW-related seizures, limiting our ability to investigate the relevance of the SORCS2 risk SNP to the course of seizures during AW. We therefore evaluated rs11731003 in the SANAD trial (Marson et al, 2007a; b), a unique study in which seizure disorder patients were prospectively randomized between treatments, thereby allowing for the identification of prognostic factors. In the SANAD trial (Speed et al, 2014), the risk allele (T) of rs11731003 was associated with greater likelihood of failing to achieve the pre-specified endpoint of seizure remission at 12 months (n = 654, P = 3.2 × 10−3). There was no such association in a smaller non-randomized community sample of seizure disorder patients from Australia (Speed et al, 2014), in which anticonvulsants were selected and managed by the treating physician (n = 235, P = 0.82).
SORCS2 risk haplotype disrupts TF binding motifs within a stress hormone-responsive enhancer active in human hippocampus
In humans, SORCS2 is most highly expressed in the nervous system (Figure S4) (Melé et al, 2015). Lead SNP rs11731003 (Figure 4, upper panel, purple) tags a haplotype that includes seven SNPs in very high (r2 > 0.95) linkage disequilibrium (LD) with rs11731003 in the 1000 Genomes CEU reference population (Figure 4, upper panel, red) (1000 Genomes Project Consortium, 2012), and which maps to an intron in SORCS2 (Figure 4, upper panel, black). The UCSC Genome Browser gene prediction annotation track also contained a predicted transcription start site (TSS) (Figure 4, upper panel, light blue) at the intronic location of the risk haplotype. Intronic TSSs for non-coding transcripts have been shown to mark the site of active regulatory elements (Kowalczyk et al, 2012). Examination of the GenBank (Benson et al, 2010) catalogue of human expressed sequence tags (EST) identified an EST captured from human hippocampus (Figure 4, upper panel, magenta) in an experimental analysis of oligo-cap cDNA libraries from 164 different human tissues types and cell lines, suggesting activation of an intronic regulatory element specifically in human hippocampus (Kimura et al, 2006).
Figure 4: SORCS2 risk haplotype disrupts transcription factor (TF) binding motifs within a stress hormone-regulated enhancer element active in human hippocampus.
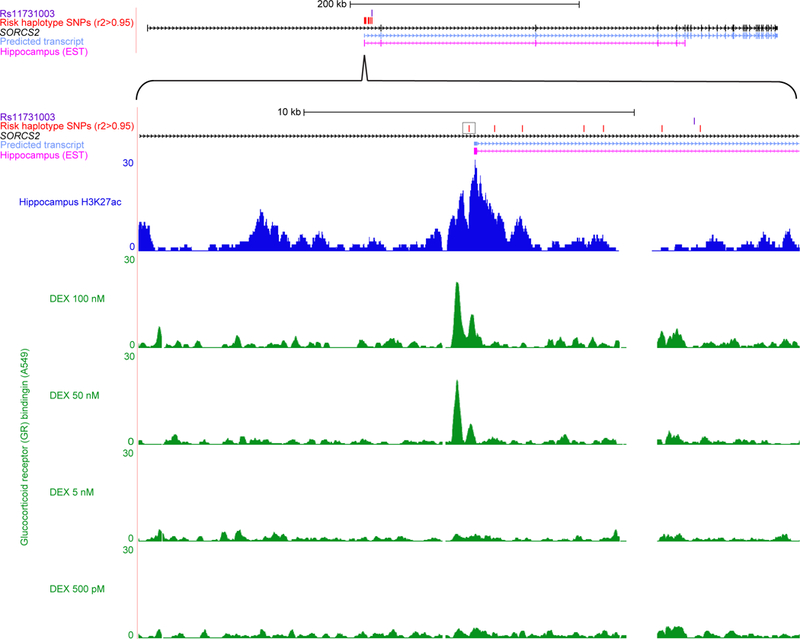
Genomic region of interest are shown at two degrees of zoom. Upper panel: SORCS2 risk single nucleotide polymorphism (SNP) rs11731003 (purple); 1000 Genomes (1000 Genomes Project Consortium, 2012) SNPs (red) in r2 > 0.95 with lead SNP rs11731103; SORCS2 (black); bioinformatically predicted transcript (light blue) (Rosenbloom et al, 2015) suggesting the presence of an enhancer at the intronic transcription start site (TSS) (Kowalczyk et al, 2012); expressed sequence tag (EST) from hippocampus (magenta) consistent with enhancer activation, captured after generation of oligo-cap cDNA libraries from 164 different human tissues and cell lines by Kimura et al (Kimura et al, 2006). Lower panel: rs4367173 (black box) is predicted to disrupt multiple TF binding motifs (Table S1) (Ward & Kellis, 2012), and evaluation of 12 chromatin marks across 125 human tissue and cell types showed that regulatory activity at this genomic position was concentrated in brain (Table S2) (Ernst & Kellis, 2015); NIH Roadmap consortium (Roadmap Epigenomics Consortium et al, 2015) chromatin immunoprecipitation followed by sequencing (ChIP-SEQ) data from human hippocampus for the H3K27ac (dark blue) chromatin modification typical of active enhancers (Shlyueva et al, 2014); examination of the highest confidence TF ChIP-SEQ peak calls from ENCODE Consortium (ENCODE Project Consortium, 2012) cell-line data identified glucocorticoid receptor (GR) binding (green) in A549 immortal epithelial cells characterized by Reddy et al (Reddy et al, 2012; Reddy et al, 2009), and showed that GR binding is completely dependent on treatment with synthetic stress hormone dexamethasone (DEX).
Risk haplotype SNP rs4367173 (Figure 4, lower panel, black box) is computationally predicted to disrupt binding motifs for multiple TFs (Table S1) (Ward & Kellis, 2012), including TFs such as Egr1 and Sp4 that have well-known roles in hippocampal physiology (Penke et al, 2014; Zhou et al, 2005). Signal intensity data from ChIP-SEQ experiments performed on human hippocampus by the NIH Roadmap consortium (Roadmap Epigenomics Consortium et al, 2015) using antibodies for the H3K27ac histone modification, which marks active enhancers (Shlyueva et al, 2014), provided further empirical evidence for the presence of an active enhancer element in human hippocampus (Figure 4, lower panel, dark blue). In human hippocampus the genomic position of rs4367173 was designated as “Active Enhancer 1” by a recently developed classifier algorithm that categorizes regulatory sites based on histone modification data (Ernst & Kellis, 2015). Although several other tissues and cell types showed some evidence of regulatory potential at this genomic position, the designation of “Active Enhancer 1” was restricted exclusively to tissue samples taken from the adult brain (Table S2) (Ward & Kellis, 2012). The GTEx project (Melé et al, 2015) provided suggestive evidence from outside the context of AW that the risk alleles may be relevant to SORCS2 expression at baseline (Figure S5).
To gain insight into the specific cell signaling mechanisms that control this regulatory element, we examined TF binding data generated by the ENCODE consortium from human cell lines (ENCODE Project Consortium, 2012). Restricting our analysis of the SORCS2 enhancer element to only those TFs with ChIP-SEQ peak calls receiving the highest possible confidence scores, we identified two bound TFs, both in immortal epithelial cell lines: STAT3 in MCF10A-Er-Src cells (Fleming et al, 2015), and the glucocorticoid receptor (GR) in A549 cells (Reddy et al, 2012; Reddy et al, 2009). The GR binding assays were performed at increasing doses of the cortisol analog dexamethasone (DEX) to mimic the physiological effects of stress (Reddy et al, 2012; Reddy et al, 2009). GR binding (Figure 4, lower panel, green) was present only after treatment with higher doses of DEX. Thus, expression of SORCS2 and full enhancer activation are both preferentially found in neural tissue, and in vitro assays in cell culture show that GR binds the enhancer element in a stress hormone-responsive manner.
Stress hormone administration and ethanol withdrawal both induce SORCS2 upregulation
Having found evidence that a genetic risk allele for severe AW may impede a stress-initiated SORCS2 upregulatory program in human brain, we sought to demonstrate the existence of a direct relationship between stress hormones and SORCS2 expression. Because 50 nM was the concentration of DEX needed to induce GR binding at the enhancer element (Figure 4, lower panel, green), we treated SH-SY5Y human neuroblastoma cells with this same concentration of DEX, which resulted in a 38.4% increase in SORCS2 expression (P = 0.006, Figure 5). Next, we evaluated whether SORCS2 upregulation is observed in an experimental model of AW. SH-SY5Y cells were exposed to ethanol, and SORCS2 expression was then quantified prior to and after ethanol removal. We found that SORCS2 expression was increased by 40.2% following initial ethanol exposure (P = 0.015), and by 60.1% 24 hours after withdrawal of ethanol (P = 0.002) (Figure 6).
Figure 5: Cortisol analog dexamethasone (DEX) upregulates SORCS2 expression in human neuroblastoma SH-SY5Y cells.
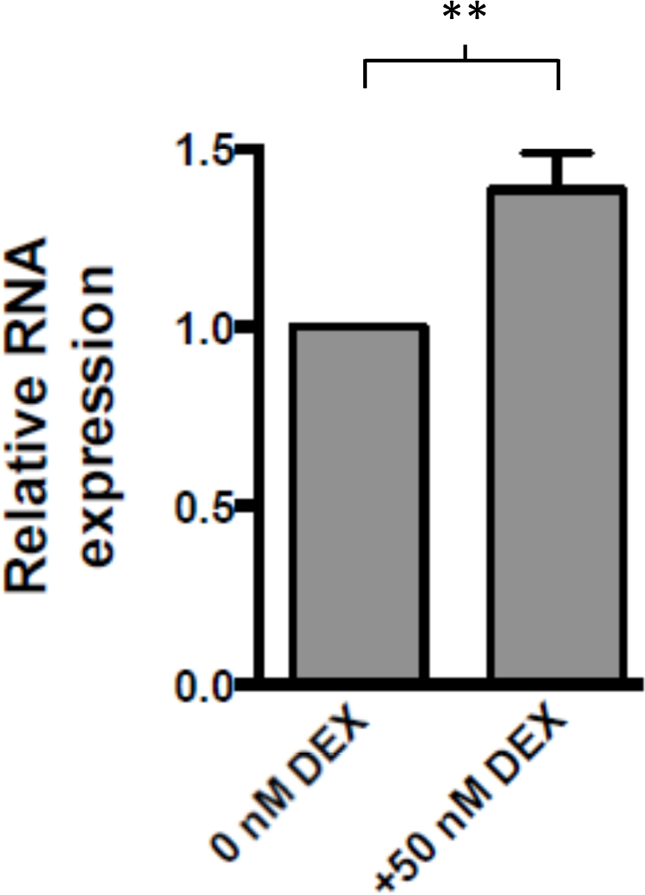
Treatment of SH-SY5Y cells with 50 nM DEX induced a 38.4% increase in SORCS2 transcription, as quantified by RT-PCR (P =0.006, two-tailed unpaired t-test).
Figure 6: SORCS2 expression is increased by administration and withdrawal of ethanol (EtOH).
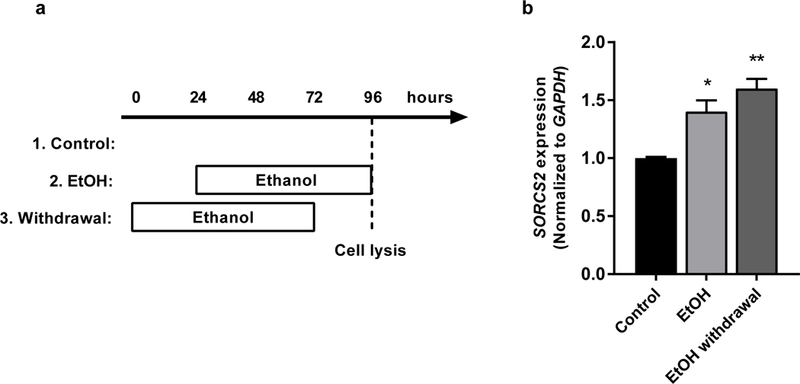
(a) The experimental timeline of EtOH exposure and withdrawal in SH-SY5Y cell cultures. (b) SORCS2 expression was increased by 40.2% following EtOH exposure (P = 0.015), and by 60.1% following EtOH withdrawal, as quantified by RT-PCR (P = 0.0022, multiple t-test adjusted p-values). In each group n = 3, and error bars represent standard error of the mean (SEM).
Discussion:
We investigated the genetic basis of lifetime AW symptom count in AD subjects via a GWAS, finding GWS risk SNPs in SORCS2 on chromosome 4, and in an intergenic region on chromosome 5. Because of the difficulty of studying intergenic SNPs, only the SORCS2 signal was evaluated further in the present study. SorCS2 is a member of the VPS10-domain-containing receptor family, which also includes sortilin and three other members (SorL1, SorCS1, and SorCS3), all of which are highly expressed in the central nervous system (Willnow et al, 2008). Our results are contextualized by multiple previous studies relating SorCS2 to a wide variety of neuropsychiatric illnesses and traits, including volume of the temporal lobe (the location of the hippocampus) (Kohannim et al, 2012), schizophrenia and bipolar disorder (Baum et al, 2008; Christoforou et al, 2011; Ollila et al, 2009), attention and ADHD (Alemany et al, 2015; Glerup et al, 2014), and Parkinson’s and Alzheimer’s diseases (Reitz et al, 2013; Visanji et al, 2015).
The SORCS2 association was present in a meta-analysis of the Yale-Penn sample a small independent sample assessed while in acute withdrawal. The same SORCS2 risk SNP also predicted poorer seizure control in patients from the SANAD trial of anticonvulsant effectiveness, although not in a smaller non-randomized community sample from Australia. Recruitment of new and larger samples will be needed for confirmation and further characterization of SNP effects across different clinical settings. The results from our GWAS and the SANAD trial are consistent with the expectation that overlapping biological pathways would be relevant to illnesses with overlapping pharmacotherapies (Henriksen, 1998; Minozzi et al, 2010; VonDran et al, 2014). As is often the case in complex trait genetics, we observed population-specific associations with common variants (CONVERGE consortium, 2015). Differing genetic backgrounds across populations and different linkage disequilibrium patterns could influence which specific polymorphisms are seen to have an effect in a particular group of patients (Polimanti et al, 2015). It is unclear whether population-specific genetic architectures, and genetic risks, underlie the prior clinical observation that EAs are more likely than AAs to experience severe AW (Chan et al, 2009), illustrating the pressing need that future studies recruit diverse populations.
Our findings validate an approach that has been supported by the literature: disaggregating AD, so that genetic influences on its distinctive components can be investigated separately, which might increase power even in the context of a modest sample size. As the overall sample size continues to grow, this same strategy may also become applicable to DTs and other less prevalent extreme phenotypes. A limitation of our study - as for most GWAS studies - is that the precise nature of the impact of risk variants on biological function remains unclear, as does the timing of their influence (i.e., do the relevant effects of the risk variants occur before, during, or after heavy alcohol consumption). In the case of intergenic SNPs such as the GWS association identified on chromosome 5, it remains unknown which genes in the region are relevant to the trait. One candidate in this chromosome 5 region is SERINC5, which like SORCS2 encodes a neural membrane protein (Inuzuka et al, 2005).
Importantly, at the intergenic SORCS2 risk locus on chromosome 4, results of ENCODE GR binding assays indicate that the risk variant may have its most pronounced effects in the context of stress, and several independent lines of evidence pointed toward a regulatory function for the risk locus in human brain. First, SORCS2 is most highly expressed in the human nervous system, and transcription at the intronic regulatory site suggested enhancer activation specifically in hippocampus. Second, bioinformatic analyses predicted disruption of binding motifs for multiple TFs, including TFs that are well-known regulators of hippocampal function. Third, histone modifications marking active enhancers were present in hippocampus and exhibited tissue-specificity.
Based on experimental results, ethanol and glucocorticoids each appear to be able to influence SORCS2 expression independently. Regarding the mechanism of glucocorticoid-dependent SORCS2 upregulation, we first observed that rising stress hormone levels lead to a dose-dependent increase in GR binding at the location of the SORCS2 enhancer. Subsequently, in human SH-SY5Y neuroblastoma cells, we confirmed the existence of a causal relationship between increased stress-hormone levels and increased SORCS2 expression. Regarding the mechanism of SORCS2 upregulation following ethanol exposure and withdrawal, it may involve one or more of the many TFs that bind at the enhancer (Table S1), however the exact pathways remain to be clarified.
Given the stress of experiencing serious AW, we surmise that at the organismal level there are neuroprotective effects of SORCS2 upregulation during AW, which are reinforced by glucocorticoid-dependent SORCS2 upregulation. Our results are consistent with prior work that has uncovered SORCS2 upregulation in animal models of status epilepticus (VonDran et al, 2014), which, like AW, also involves neuronal hyperexcitation. Risk alleles that disrupt enhancers and impair SORCS2 upregulation during AW could interfere with salutary neurotrophin signaling cascades that rely on SorCS2 (Glerup et al, 2016; Nykjaer & Willnow, 2012). Future studies could use CRISPR-Cas9 to introduce AW-severity risk alleles into experimentally tractable iPSC-derived hippocampal neurons, or even organoid systems. Such approaches could lead to a fuller understanding of how human-specific regulatory elements help orchestrate a response to stressful and potentially dangerous clinical situations.
In summary, we identified novel risk variants in SORCS2 that increase the severity of AW in chronic alcoholism. The SORCS2 risk haplotype is predicted to disrupt a stress hormone-regulated enhancer that is active in human hippocampus, a brain region that is well known for its role in AW. In vitro experiments in human neural lineage cells confirm a novel role for stress hormones as a net upregulator of SORCS2, and show that SORCS2 upregulation also occurs following the initiation and cessation of ethanol exposure. Based on our results, which span multiple levels of analysis, further investigation is warranted into the role of SORCS2 and its downstream signaling partners in responding to AW. Better management of AW symptoms has treatment implications not only for acute AW, which can become life threatening, but also AD itself, where the lasting dysphoric component of withdrawal can affect the likelihood of relapse (Heilig et al, 2010).
Supplementary Material
Acknowledgments:
Recruitment and assessment were overseen at Yale School of Medicine and the APT Foundation by James Poling, Ph.D. and Aryeh Herman, Ph.D., at McLean Hospital by Roger Weiss, M.D., at the Medical University of South Carolina by Kathleen Brady, M.D., Ph.D., at the University of Connecticut Health Center by Henry R. Kranzler, M.D., and at the University of Pennsylvania, initially by David Oslin, M.D. and then by Henry R. Kranzler, M.D. Genotyping services for our GWAS study were provided by the Center for Inherited Disease Research (CIDR) and the Yale University Center for Genome Analysis. We are grateful to Ann Marie Lacobelle, Christa Robinson, Benedicte Vestergaard and Anja Aagaard Danneskjold for their technical assistance, to the SSADDA interviewers, led by Yari Nuñez and Michelle Slivinsky, to Richard Sherva and Ryan Koesterer for their assistance with data processing and quality control, and to John Farrell and Alexan Mardigan for database management assistance. We gratefully acknowledge Dr. Doug Speed for his assistance with data from the seizure treatment datasets. We also gratefully acknowledge Melanie Schwandt, Colin Hodgkinson and Mary-Anne Enoch for assistance with genotyping and statistical analyses at the NIAAA.
This work was funded by the National Institutes of Health [RC2 DA028909, R01 DA12690, R01 DA12849, R01 DA18432, R01 AA11330, R01 AA017535, D43TW009087-01, MSTP 5T32GM007205-38, CTSA 8UL1TR000142 TL1, F30 DA037665]; the US Veterans Affairs Connecticut and Veterans Affairs Philadelphia Mental Illness Research, Education and Clinical Centers; a US Department of Veterans Affairs VISN1 Career Development Award, and the Lundbeck Foundation. Genotyping services at CIDR at The Johns Hopkins University were supported by the National Institutes of Health [contract N01-HG-65403]. Some analyses were conducted on the Yale Biomedical Supercomputer, funded by the National Center for Research Resources, National Institutes of Health [S10 RR19895-01]. Genotype and phenotype data are being deposited into dbGAP (phs000425.v1.p1).
Although unrelated to the current study, Dr. Kranzler has been a consultant or advisory board member for Indivior and Lundbeck. He is also a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by AbbVie, Alkermes, Amygdala Neurosciences, Arbor Pharmaceuticals, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, and Pfizer. Drs. Kranzler, Gelernter, and Smith are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. Although also unrelated to the current study, Dr. Nykjær is founder and CSO of InsuSense Therapeutics ApS, and he has also consulted for Lundbeck.
Footnotes
Conflict of Interest:
No other authors declare possible conflicts.
References:
- 1000 Genomes Project Consortium (2012) An integrated map of genetic variation from 1,092 human genomes. Nature, 491(7422), 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemany S, Ribasés M, Vilor-Tejedor N, Bustamante M, Sánchez-Mora C, Bosch R, Richarte V, Cormand B, Casas M, Ramos-Quiroga JA & Sunyer J (2015) New suggestive genetic loci and biological pathways for attention function in adult attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet [DOI] [PubMed]
- American Psychiatric Association (2000) Diagnostic and Statistical Manual of Mental Disorders, 4th edition.
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, Schulze TG, Cichon S, Rietschel M, Nöthen MM, Georgi A, Schumacher J, Schwarz M, Abou Jamra R, Höfels S, Propping P, Satagopan J, Detera-Wadleigh SD, Hardy J & McMahon FJ (2008) A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry, 13(2), 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC (2008) Alcohol dependence, withdrawal, and relapse. Alcohol Res Health, 31(4), 348–61. [PMC free article] [PubMed] [Google Scholar]
- Becker HC (2012) Effects of alcohol dependence and withdrawal on stress responsiveness and alcohol consumption. Alcohol Res, 34(4), 448–58. [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J & Sayers EW (2010) GenBank. Nucleic Acids Research, 39(Supplement 1), D32–D37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Goate AM, Breslau N, Johnson EO, Bertelsen S, Fox L, Agrawal A, Bucholz KK, Grucza R, Hesselbrock V, Kramer J, Kuperman S, Nurnberger J, Porjesz B, Saccone NL, Schuckit M, Tischfield J, Wang JC, Foroud T, Rice JP & Edenberg HJ (2012) ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol Psychiatry, 17(4), 445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GM, Hoffman RS, Gold JA, Whiteman PJ, Goldfrank LR & Nelson LS (2009) Racial variations in the incidence of severe alcohol withdrawal. J Med Toxicol, 5(1), 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou A, McGhee KA, Morris SW, Thomson PA, Anderson S, McLean A, Torrance HS, Le Hellard S, Pickard BS, StClair D, Muir WJ, Blackwood DH, Porteous DJ & Evans KL (2011) Convergence of linkage, association and GWAS findings for a candidate region for bipolar disorder and schizophrenia on chromosome 4p. Mol Psychiatry, 16(3), 240–2. [DOI] [PubMed] [Google Scholar]
- CONVERGE consortium (2015) Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature, 523(7562), 588–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC & Kosobud A (1986) Sensitivity and tolerance to ethanol in mice bred to be genetically prone or resistant to ethanol withdrawal seizures. J Pharmacol Exp Ther, 239(2), 327–33. [PubMed] [Google Scholar]
- Crabbe JC & Phillips TJ (1993) Selective breeding for alcohol withdrawal severity. Behav Genet, 23(2), 171–7. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J & Kellis M (2015) Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol, 33(4), 364–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanous AH & Kendler KS (2005) Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: searching for a framework. Mol Psychiatry, 10(1), 6–13. [DOI] [PubMed] [Google Scholar]
- Fehr C, Shirley RL, Metten P, Kosobud AE, Belknap JK, Crabbe JC & Buck KJ (2004) Potential pleiotropic effects of Mpdz on vulnerability to seizures. Genes Brain Behav, 3(1), 8–19. [DOI] [PubMed] [Google Scholar]
- Fleming JD, Giresi PG, Lindahl-Allen M, Krall EB, Lieb JD & Struhl K (2015) STAT3 acts through pre-existing nucleosome-depleted regions bound by FOS during an epigenetic switch linking inflammation to cancer. Epigenetics Chromatin, 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, Anton R, Preuss UW, Ridinger M, Rujescu D, Wodarz N, Zill P, Zhao H & Farrer LA (2014a) Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry, 19(1), 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, Koesterer R, Almasy L, Zhao H & Farrer LA (2014b) Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biol Psychiatry, 76(1), 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Sherva R, Koesterer R, Almasy L, Zhao H, Kranzler HR & Farrer L (2014c) Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Mol Psychiatry, 19(6), 717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glerup S, Bolcho U, Mølgaard S, Bøggild S, Vaegter CB, Smith AH, Nieto-Gonzalez JL, Ovesen PL, Pedersen LF, Fjorback AN, Kjolby M, Login H, Holm MM, Andersen OM, Nyengaard JR, Willnow TE, Jensen K & Nykjaer A (2016) SorCS2 is required for BDNF-dependent plasticity in the hippocampus. Mol Psychiatry [DOI] [PubMed]
- Glerup S, Olsen D, Vaegter CB, Gustafsen C, Sjoegaard SS, Hermey G, Kjolby M, Molgaard S, Ulrichsen M, Boggild S, Skeldal S, Fjorback AN, Nyengaard JR, Jacobsen J, Bender D, Bjarkam CR, Sørensen ES, Füchtbauer EM, Eichele G, Madsen P, Willnow TE, Petersen CM & Nykjaer A (2014) SorCS2 regulates dopaminergic wiring and is processed into an apoptotic two-chain receptor in peripheral glia. Neuron, 82(5), 1074–87. [DOI] [PubMed] [Google Scholar]
- Heilig M, Egli M, Crabbe JC & Becker HC (2010) Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol, 15(2), 169–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen O (1998) An overview of benzodiazepines in seizure management. Epilepsia, 39(S1), S2–S6. [Google Scholar]
- Huang MC, Schwandt ML, Chester JA, Kirchhoff AM, Kao CF, Liang T, Tapocik JD, Ramchandani VA, George DT, Hodgkinson CA, Goldman D & Heilig M (2014) FKBP5 moderates alcohol withdrawal severity: human genetic association and functional validation in knockout mice. Neuropsychopharmacology, 39(8), 2029–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka M, Hayakawa M & Ingi T (2005) Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem, 280(42), 35776–83. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Aggen SH, Prescott CA, Crabbe J & Neale MC (2012) Evidence for multiple genetic factors underlying the DSM-IV criteria for alcohol dependence. Mol Psychiatry, 17(12), 1306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM & Haussler D (2002) The human genome browser at UCSC. Genome Res, 12(6), 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, Ishii S, Sugiyama T, Saito K, Isono Y, Irie R, Kushida N, Yoneyama T, Otsuka R, Kanda K, Yokoi T, Kondo H, Wagatsuma M, Murakawa K, Ishida S, Ishibashi T, Takahashi-Fujii A, Tanase T, Nagai K, Kikuchi H, Nakai K, Isogai T & Sugano S (2006) Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res, 16(1), 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke MD, Schick S, Lutz U, Willecke M, Koehnke AM, Kolb W & Gaertner I (2002) Severity of alcohol withdrawal symptoms and the T1128C polymorphism of the neuropeptide Y gene. J Neural Transm, 109(11), 1423–9. [DOI] [PubMed] [Google Scholar]
- Kohannim O, Hibar DP, Stein JL, Jahanshad N, Hua X, Rajagopalan P, Toga AW, Jack CR, Weiner MW, de Zubicaray GI, McMahon KL, Hansell NK, Martin NG, Wright MJ, Thompson PM & Alzheimer’s Disease Neuroimaging Initiative (2012) Discovery and Replication of Gene Influences on Brain Structure Using LASSO Regression. Front Neurosci, 6, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, Brown JM, Gray NE, Collavin L, Gibbons RJ, Flint J, Taylor S, Buckle VJ, Milne TA, Wood WG & Higgs DR (2012) Intragenic enhancers act as alternative promoters. Mol Cell, 45(4), 447–58. [DOI] [PubMed] [Google Scholar]
- Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, et al. (2012) A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet, 380(9859), 2224–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson AG, Al-Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, Cramp C, Cockerell OC, Cooper PN, Doughty J, Eaton B, Gamble C, Goulding PJ, Howell SJ, Hughes A, Jackson M, Jacoby A, Kellett M, Lawson GR, Leach JP, Nicolaides P, Roberts R, Shackley P, Shen J, Smith DF, Smith PE, Smith CT, Vanoli A, Williamson PR & SANAD Study group (2007a) The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet, 369(9566), 1000–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson AG, Al-Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, Cramp C, Cockerell OC, Cooper PN, Doughty J, Eaton B, Gamble C, Goulding PJ, Howell SJ, Hughes A, Jackson M, Jacoby A, Kellett M, Lawson GR, Leach JP, Nicolaides P, Roberts R, Shackley P, Shen J, Smith DF, Smith PE, Smith CT, Vanoli A, Williamson PR & SANAD Study group (2007b) The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet, 369(9566), 1016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segrè AV, Djebali S, Niarchou A, Wright FA, Consortium GTEx, Lappalainen T, Calvo M, Getz G, Dermitzakis ET, Ardlie KG & Guigó R (2015) The human transcriptome across tissues and individuals. Science, 348(6235), 660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minozzi S, Amato L, Vecchi S & Davoli M (2010) Anticonvulsants for alcohol withdrawal. Cochrane Database Syst Rev(3), CD005064. [DOI] [PubMed]
- Nykjaer A & Willnow TE (2012) Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci, 35(4), 261–70. [DOI] [PubMed] [Google Scholar]
- Ollila HM, Soronen P, Silander K, Palo OM, Kieseppä T, Kaunisto MA, Lönnqvist J, Peltonen L, Partonen T & Paunio T (2009) Findings from bipolar disorder genome-wide association studies replicate in a Finnish bipolar family-cohort. Mol Psychiatry, 14(4), 351–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL & Reich D (2006) Population structure and eigenanalysis. PLoS Genet, 2(12), e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penke Z, Morice E, Veyrac A, Gros A, Chagneau C, LeBlanc P, Samson N, Baumgärtel K, Mansuy IM, Davis S & Laroche S (2014) Zif268/Egr1 gain of function facilitates hippocampal synaptic plasticity and long-term spatial recognition memory. Philos Trans R Soc Lond B Biol Sci, 369(1633), 20130159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A, Gelernter J, Chan G, Arias A, Cubells JF, Farrer L & Kranzler HR (2007) Reliability of DSM-IV diagnostic criteria using the semi-structured assessment for drug dependence and alcoholism (SSADDA). Drug Alcohol Depend, 91(1), 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A, Gelernter J, Feinn R, Cubells JF, Pearson D, Pollastri A, Farrer L & Kranzler HR (2005) Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA). Drug and Alcohol Dependence, 80(3), 303–312. [DOI] [PubMed] [Google Scholar]
- Polimanti R, Yang C, Zhao H & Gelernter J (2015) Dissecting ancestry genomic background in substance dependence genome-wide association studies. Pharmacogenomics, 16(13), 1487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss UW, Zill P, Koller G, Bondy B, Hesselbrock V & Soyka M (2006) Ionotropic glutamate receptor gene GRIK3 SER310ALA functional polymorphism is related to delirium tremens in alcoholics. Pharmacogenomics J, 6(1), 34–41. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA & Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet, 38(8), 904–9. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, Bakker PI, Daly MJ & Sham PC (2007) PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics, 81(3), 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Gertz J, Crawford GE, Garabedian MJ & Myers RM (2012) The hypersensitive glucocorticoid response specifically regulates period 1 and expression of circadian genes. Mol Cell Biol, 32(18), 3756–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ & Myers RM (2009) Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res, 19(12), 2163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Tosto G, Vardarajan B, Rogaeva E, Ghani M, Rogers RS, Conrad C, Haines JL, Pericak-Vance MA, Fallin MD, Foroud T, Farrer LA, Schellenberg GD, George-Hyslop PS, Mayeux R & Alzheimer’s Disease Genetics Consortium (ADGC) (2013) Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP). Transl Psychiatry, 3, e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T & Kellis M (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518(7539), 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, Harte RA, Heitner S, Hickey G, Hinrichs AS, Hubley R, Karolchik D, Learned K, Lee BT, Li CH, Miga KH, Nguyen N, Paten B, Raney BJ, Smit AF, Speir ML, Zweig AS, Haussler D, Kuhn RM & Kent WJ (2015) The UCSC Genome Browser database: 2015 update. Nucleic Acids Res, 43(Database issue), D670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujescu D, Soyka M, Dahmen N, Preuss U, Hartmann AM, Giegling I, Koller G, Bondy B, Möller HJ & Szegedi A (2005) GRIN1 locus may modify the susceptibility to seizures during alcohol withdrawal. Am J Med Genet B Neuropsychiatr Genet, 133B(1), 85–7. [DOI] [PubMed] [Google Scholar]
- Schmidt LG & Sander T (2000) Genetics of alcohol withdrawal. Eur Psychiatry, 15(2), 135–9. [DOI] [PubMed] [Google Scholar]
- Schuckit MA (2014) Recognition and management of withdrawal delirium (delirium tremens). N Engl J Med, 371(22), 2109–13. [DOI] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G & Stark A (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet, 15(4), 272–86. [DOI] [PubMed] [Google Scholar]
- Skinner H & Horn J (1984) Alcohol Dependence Scale: User’s Guide Toronto, Canada: Addiction Research Foundation. [Google Scholar]
- Slutske WS, True WR, Scherrer JF, Heath AC, Bucholz KK, Eisen SA, Goldberg J, Lyons MJ & Tsuang MT (1999) The heritability of alcoholism symptoms: “indicators of genetic and environmental influence in alcohol-dependent individuals” revisited. Alcohol Clin Exp Res, 23(5), 759–69. [DOI] [PubMed] [Google Scholar]
- Speed D, Hoggart C, Petrovski S, Tachmazidou I, Coffey A, Jorgensen A, Eleftherohorinou H, De Iorio M, Todaro M, De T, Smith D, Smith PE, Jackson M, Cooper P, Kellett M, Howell S, Newton M, Yerra R, Tan M, French C, Reuber M, Sills GE, Chadwick D, Pirmohamed M, Bentley D, Scheffer I, Berkovic S, Balding D, Palotie A, Marson A, O’Brien TJ & Johnson MR (2014) A genome-wide association study and biological pathway analysis of epilepsy prognosis in a prospective cohort of newly treated epilepsy. Hum Mol Genet, 23(1), 247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA & Sellers EM (1989) Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict, 84(11), 1353–7. [DOI] [PubMed] [Google Scholar]
- Trevisan LA, Boutros N, Petrakis IL & Krystal JH (1998) Complications of alcohol withdrawal: pathophysiological insights. Alcohol Health Res World, 22(1), 61–6. [PMC free article] [PubMed] [Google Scholar]
- van Munster BC, Korevaar JC, de Rooij SE, Levi M & Zwinderman AH (2007) Genetic polymorphisms related to delirium tremens: a systematic review. Alcohol Clin Exp Res, 31(2), 177–84. [DOI] [PubMed] [Google Scholar]
- Visanji NP, Kamali Sarvestani I, Creed MC, Shams Shoaei Z, Nobrega JN, Hamani C & Hazrati LN (2015) Deep brain stimulation of the subthalamic nucleus preferentially alters the translational profile of striatopallidal neurons in an animal model of Parkinson’s disease. Front Cell Neurosci, 9, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, LaFrancois J, Padow VA, Friedman WJ, Scharfman HE, Milner TA & Hempstead BL (2014) p75NTR, but not proNGF, is upregulated following status epilepticus in mice. ASN Neuro, 6(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD & Kellis M (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res, 40(Database issue), D930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernicke C, Samochowiec J, Schmidt LG, Winterer G, Smolka M, Kucharska-Mazur J, Horodnicki J, Gallinat J & Rommelspacher H (2003) Polymorphisms in the N-methyl-D-aspartate receptor 1 and 2B subunits are associated with alcoholism-related traits. Biol Psychiatry, 54(9), 922–8. [DOI] [PubMed] [Google Scholar]
- Wetherill L, Kapoor M, Agrawal A, Bucholz K, Koller D, Bertelsen SE, Le N, Wang JC, Almasy L, Hesselbrock V, Kramer J, Nurnberger JI, Schuckit M, Tischfield JA, Xuei X, Porjesz B, Edenberg HJ, Goate AM & Foroud T (2014) Family-based association analysis of alcohol dependence criteria and severity. Alcohol Clin Exp Res, 38(2), 354–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y & Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26(17), 2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willnow TE, Petersen CM & Nykjaer A (2008) VPS10P-domain receptors - regulators of neuronal viability and function. Nat Rev Neurosci, 9(12), 899–909. [DOI] [PubMed] [Google Scholar]
- Zhou H, Polimanti R, Yang BZ, Wang Q, Han S, Sherva R, Nunez YZ, Zhao H, Farrer LA, Kranzler HR & Gelernter J (2017) Genetic Risk Variants Associated With Comorbid Alcohol Dependence and Major Depression. JAMA Psychiatry [DOI] [PMC free article] [PubMed]
- Zhou X, Long JM, Geyer MA, Masliah E, Kelsoe JR, Wynshaw-Boris A & Chien KR (2005) Reduced expression of the Sp4 gene in mice causes deficits in sensorimotor gating and memory associated with hippocampal vacuolization. Mol Psychiatry, 10(4), 393–406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.