

SUMMARY
The dynamic process by which nuclear RNAi engages a transcriptionally active target, before the repressive state is stably established, remains largely a mystery. Here, we found that the onset of exogenous dsRNA-induced nuclear RNAi in C. elegans is a transgenerational process, and it requires a putative histone methyltransferase (HMT), SET-32. By developing a CRISPR-based genetic approach, we found that silencing establishment at the endogenous targets of germline nuclear RNAi also requires SET-32. Although SET-32 and two H3K9 HMTs, MET-2 and SET-25, are dispensable for the maintenance of silencing, they do contribute to transcriptional repression in mutants that lack the germline nuclear Argonaute protein HRDE-1, suggesting a conditional role of heterochromatin in the maintenance phase. Our study indicates that (1) establishment and maintenance of siRNA-guided transcriptional repression are two distinct processes with different genetic requirements and (2) the rate- limiting step of the establishment phase is a transge-nerational, chromatin-based process.
Graphical Abstract
In Brief
Deciphering mechanisms of transgenerational epigenetic gene regulation is critical for understanding of development, aging, and disease. In this study, Kalinava et al. examine the establishment of RNAi-mediated epigenetic silencing. The identification of the bottleneck step provides critical insight into the regulation of this pathway.
INTRODUCTION
The term RNAi originally refers to the phenomenon of exogenous dsRNA-triggered gene silencing (Fire et al., 1998; Kalinava et al., 2017; Kennerdell and Carthew, 1998), in which target mRNA is degraded by a small interfering RNA (siRNA)-associated Argonaute (AGO) protein, resulting in post-transcriptional gene silencing (PTGS) (Elbashir et al., 2001; Hammond et al., 2000, 2001; Tuschl et al., 1999). In addition to this mechanism, siRNAs can also target a gene for transcriptional gene silencing (TGS) in plants, fungi, and animals (Martienssen and Moazed, 2015; Pezic et al., 2014; Sienski et al., 2012; Wassenegger, 2000). We will use the terms classical RNAi and nuclear RNAi to refer to PTGS and TGS, respectively. Classical RNAi results in rapid degradation of exogenous dsRNA and homologous single-stranded transcripts, while long-term, stable silencing of transposons and other types of repetitive genomic elements is facilitated by nuclear RNAi.
In C. elegans, nuclear RNAi effects include histone modifications (H3K9me3 and H3K27me3) and transcriptional repression at endo-siRNA-targeted loci (Buckley et al., 2012; Gu et al., 2012; Guang et al., 2010; Mao et al., 2015). The study of the endogenous silencing events, which provide a rich source of targets to study the physiological functions and underlying mechanisms, is further complemented by highly manipulatable approaches using exogenous triggers such as dsRNA or transgenes (Ashe et al., 2012; Gu et al., 2012; Guang et al., 2010; Leopold et al., 2015; Minkina and Hunter, 2017; Shirayama et al., 2012). Exogenous dsRNA-induced nuclear RNAi relies on the upstream steps of classical RNAi for siRNA biogenesis (Grishok et al., 2000; Gu et al., 2012), but also requires nuclear RNAi-specific protein factors, including the germline-specific nuclear AGO protein WAGO-9/HRDE-1 (Akay et al., 2017; Ashe et al., 2012; Buckley et al., 2012; Guang et al., 2010; Shirayama et al., 2012; Spracklin et al., 2017; Weiser et al., 2017). The germline nuclear RNAi pathway is essential for various transgenerational silencing phenomena in C. elegans (Alcazar et al., 2008; Ashe et al., 2012; Bagijn et al., 2012; Burkhart et al., 2011; Gu et al., 2012; Leopold et al., 2015; Minkina and Hunter, 2017; Shirayama et al., 2012). In the case of exogenous dsRNA-induced heritable RNAi, the heterochromatin response, as well as the silencing effect, can persist for multiple generations after the initial dsRNA exposure (administered by feeding or injection) has been ceased (Alcazar et al., 2008; Ashe et al., 2012; Buckley et al., 2012; Grishok et al., 2000; Gu et al., 2012; Mao et al., 2015; Vastenhouw et al., 2006), providing a highly tractable system to study the transgenerational epigenetic inheritance of silencing.
The germline nuclear RNAi pathway plays an important role in maintaining genome stability (Bagijn et al., 2012; McMurchy et al., 2017; Ni etal., 2014) and is essential for germline development when C. elegans is under heat stress (Ashe et al., 2012; Buckley et al., 2012; Ni et al., 2016; Weiser et al., 2017). Sequencing study of HRDE-1-associated endogenous siRNAs (endo-siRNAs) suggest that a diverse set of genomic regions can be targeted by the germline nuclear RNAi pathway (Buckley et al., 2012). We previously refined the putative endogenous targets by identifying ones that lose H3K9me3, transcriptional repression, or both in mutant animals that lack HRDE-1 (Ni et al., 2014). We refer to these targets as the exemplary endogenous targets of germline nuclear RNAi, which primarily consist of long terminal repeat (LTR) retrotransposons, but also include other types of repetitive DNA and some protein-coding genes. Interestingly, regions with germline nuclear RNAi-dependent heterochromatin (GRH) and regions with germline nuclear RNAi-dependent transcription silencing (GRTS) only partially overlap, raising the question whether the nuclear RNAi-dependent transcriptional silencing can be caused by a heterochromatin-independent mechanism (Kalinava et al., 2017; Ni et al., 2014).
To investigate the function of heterochromatin in this pathway, we and others found that germline nuclear RNAi-dependent H3K9 methylation requires multiple putative histone methyl-transferases (HMTs), MET-2, SET-25, and SET-32 (Kalinava et al., 2017; Mao et al., 2015; Spracklin et al., 2017). All three proteins contain the SET domain, an evolutionarily conserved moiety that methylates the lysine residues of histone proteins. Based on the H3K9 methylation levels in the embryos of various mutant strains, MET-2 has been proposed to be a mono- and di-methylase for H3K9, and SET-25 a tri-methylase for H3K9 (Garrigues et al., 2015; Towbin et al., 2012). Mutant adults that lack SET-32 also show significant loss in the H3K9me3 chromatin immunoprecipitation (ChIP) signal (Kalinava et al., 2017). None of the three proteins have been biochemically characterized for the HMT activity. Studies using different experimental setups and target genes have reported that these putative H3K9 HMTs are required for the siRNA-guided epigenetic silencing in some cases (Ashe et al., 2012; Minkina and Hunter, 2017; Spracklin et al., 2017), but are dispensable in others (Kalinava et al., 2017; Lev et al., 2017; Minkina and Hunter, 2017). met-2 mutant animals even exhibit the phenotype of enhanced heritable RNAi (Levet al., 2017). We recently showed that H3K9me3 can be decoupled from transcriptional repression in nuclear RNAi (Kalinava et al., 2017). Abolishing the H3K9me3 deposition by mutating all three putative HMT genes (met-2, set-25, and set-32) did not lead to any de-repression at the endogenous HRDE-1 targets (the ones targeted by endo-siRNAs), nor did it cause any defects in transcriptional silencing or heritable silencing at a gene targeted by exogenous dsRNA. This argues against a simple model in which H3K9me3 plays a direct or dominant role in transcriptional repression at the nuclear RNAi targets. However, for the endogenous targets, our previous study only examined the requirement of these proteins in maintenance of silencing. It is unknown whether any of the MET-2, SET-25, and SET-32 is required for de novo silencing of actively transcribed endogenous targets. For studies using exogenous dsRNA-induced nuclear RNAi, worms were first exposed to dsRNA for several generations to achieve the steady-state level of repression before the silencing effects were examined (Kalinava et al., 2017). Therefore, the roles of putative H3K9 HMTs in the onset of nuclear RNAi were unknown.
In principle, a genome surveillance mechanism should consist of at least two distinct phases: establishment and maintenance of silencing. The establishment phase involves the recognition of foreign genetic material and the onset of silencing. Once initiated, the silencing state is then inherited or reinforced in the maintenance phase. In the case of exogenous dsRNA-induced nuclear RNAi, the establishment phase can be defined as the period in which dsRNA leads to the maximal level of transcriptional repression at the target gene, while the maintenance phase can be defined as the period in which a steady-state-level transcriptional repression has been reached in the presence of dsRNA. The maintenance phase is followed by the inheritance phase, in which the transcriptional repression persists, but eventually dissipates, in the progeny that are no longer exposed to dsRNA. Both the maintenance and inheritance phases of nuclear RNAi have been actively investigated. However, the establishment of the nuclear RNAi has not been carefully characterized. Previous studies observed a one-generation delay between the initial dsRNA treatment and the nuclear RNAi effects (Buckley et al., 2012; Burton et al., 2011; Gu et al., 2012), suggesting that the establishment of nuclear RNAi is a transgenerational process. It is still unknown how soon the transcriptional repression can occur after the initial dsRNA exposure. Most of the published studies rely on phenotype or protein expression to measure the silencing state. As such, a lack of transcriptional repression can be masked by an active classical RNAi (post-transcriptional silencing).
In the case of the endogenous nuclear RNAi targets (guided by endo-siRNAs), the establishment phase likely occurs soon after the first invasion of an LTR retrotransposon or other types of repetitive DNA into the C. elegans genome. We and others previously found that the transcription of these endogenous targets are highly active in the hrde-1 and other nuclear RNAi mutants (Buckley et al., 2012; Ni et al., 2014, 2016), indicating that these targets still contain their transcriptional activating cis-regulatory elements. It remains an open question how soon a transposable element is silenced after its initial integration into the genome. Transgene has a strong tendency to be silenced in the C. elegans germline (Kelly et al., 1997). Interestingly, the silencing does not occur immediately after the transformation but gradually reaches the highest level over a course of multiple generations (Kelly et al., 1997), suggesting that the establishment of silencing at a transgene is a transgenerational process. The establishment process at the endogenous nuclear RNAi targets is highly important to our knowledge of genome surveillance. However, this process is largely elusive due to a lack of experimental system. Published work on the endogenous targets have been limited on the maintenance phase. In this study, we developed different strategies to examine the establishment process of silencing for an exogenous dsRNA target and endogenous targets. In these experiments, both types of targets started with active transcription. We monitored the onset of repression for multiple generations, and, for the endogenous targets, at the whole-genome level. We found that the establishment of nuclear RNAi is a transgenerational process and requires SET-32 and SET-25, although at different degrees.
RESULTS
The Onset of Nuclear RNAi-Mediated Silencing in Wild- Type Animals Occurs One Generation after the Exogenous dsRNA Exposure
To investigate the transgenerational onset of nuclear RNAi, we performed a five-generation oma-1 RNAi experiment by feeding worms with oma-1 dsRNA-expressing E. coli (Figure 1A). oma-1 is a non-essential germline-specific gene (Lin, 2003) and has been routinely used to study nuclear RNAi and epigenetic inheritance of silencing. In this experiment, young adult worms that were exposed to oma-1 dsRNA for one to five generations were collected (referred to as F1[dsRNA+] to F5[dsRNA+] animals). Adult worms fed on E. coli OP50, which does not express any worm-specific dsRNA, were used as the control (dsRNA− control).
Figure 1. Multigenerational Analysis of Exogenous dsRNA-Triggered RNAi at oma-1.

(A) A schematic of the experiment. F1–F5(dsRNA+), adult animals fed on oma-1 dsRNA-expressing E. coli for one to five generations, were used to test the establishment of silencing. F1’ and F2’(dsRNA−), the first and second generation after shifting from oma-1 dsRNA+ to dsRNA−, were used to test the inheritance of silencing.
(B) oma-1 mRNA RT-qPCR analysis of the control (dsRNA−), F1(dsRNA+), and F2(dsRNA+) samples.
(C) oma-1 pre-mRNA RT-qPCR analysis of the control (dsRNA−) and F1–F5(dsRNA+) samples. This is one of two biological replicates for this experiment. The second one is shown in Figure S1.
(D) H3K9me3 ChIP-seq coverage plots for the oma-1 locus (top panel) and LTR retrotransposon Cer3 (bottom panel). Wild-type animals of control (dsRNA−), F1(dsRNA+), and F2(dsRNA+) were used. Cer3, used as a control locus here, is an endogenous HRDE-1 target with a high level of H3K9me3, which is not affected by oma-1 RNAi. All profiles are normalized by total reads aligned to the whole genome.
(E) oma-1 pre-mRNA RT-qPCR analysis of the progeny (F1’[dsRNA−] and F2’[dsRNA−]) of the F1(dsRNA+) animals.
(F) oma-1 pre-mRNA RT-qPCR analysis of the progeny (F1’[dsRNA−]) of the F2(dsRNA+) animals. The values for the control, F1(dsRNA+), and F2(dsRNA+) samples from (C) are also used in (E) and (F) for comparison. Each RT-qPCR result in (B), (C), (E), and (F) represent the mean value of n = 3; whiskers represent the SD.
To examine the nuclear RNAi-mediated silencing at oma-1, we measured the oma-1 pre-mRNA levels by using RT-qPCR, with random hexamer oligoes as the RT primer and an oma-1 intron-specific and an exon-specific primer for the qPCR. To examine the combined effects of classical RNAi and nuclear RNAi at oma-1, we measured the oma-1 mature mRNA levels using the oligo-dT as the RT primer and two exon-specific primers for the qPCR. The same strategy has been used in our previous study (Kalinava et al., 2017).
The wild-type animals exhibited robust repression of oma-1 at the mRNA level in F1(dsRNA+), with approximately 78% reduction compared to the dsRNA− control animals (Figure 1B). Further reduction in oma-1 mRNA was observed in the F2(dsRNA+) animals.
Compared to the repression measured at the oma-1 mRNA level, the one at the pre-mRNA level was delayed by one generation in wild-type animals. The level of oma-1 pre-mRNA was reduced by 23% or remained unchanged in F1(dsRNA+) animals compared to the dsRNA− control (two biological replicates; Figures 2C and S1). The levels of oma-1 pre-mRNAs in F2–F5(dsRNA+) samples were relative stable, at approximately 40% of the one in the dsRNA− control. This result suggests a difference in the speed of onset between classical RNAi and nuclear RNAi in wild-type animals: a robust silencing mediated by the classical RNAi begins within the first generation of dsRNA exposure, and a robust silencing mediated by the nuclear RNAi begins at the second generation.
Figure 2. Silencing Establishment Assay of Endogenous HRDE-1 Targets.

(A) A schematic of the silencing establishment assay using CRISPR-Cas9-mediated gene conversion of the hrde-1(tm1200) mutation to the wild-type sequence.
(B and C) RT-qPCR analysis for two endogenous HRDE-1 targets at pre- and post-conversion generations, as well as samples with the identical genotypes as the post-conversion generations but without experiencing the hrde-1 mutation (naive samples). RNA expressions in different samples were normalized to the ones in the wild-type strain (N2). Two biological replicates were shown in (B) and (C). Each RT-qPCR value in (B) and (C) represents the mean value of n = 3; whiskers represent the SD. The GRH locus used in this analysis is chrV:5471001–5479000. The GRTS locus used in this analysis is LTR retrotransposon Cer8, located at chrV:5179680–5191222. (GRH, germline nuclear RNAi-mediated heterochromatin; GRTS, germline nuclear RNAi-mediated transcriptional silencing. GRH and GRTS regions, as well as the annotated genes located in these regions, were from Tables S1–S3 of Ni et al., 2014).
We also examined the level of H3K9me3 at the oma-1 locus in the same samples of the F1(dsRNA+) and F2(dsRNA+) wild-type animals by performing H3K9me3 chromatin immunoprecipitation sequencing (ChIP-seq) analysis. We found that both samples exhibited high levels of H3K9me3 in oma-1 gene (Figure 1D), and the levels were similar to our previously reported level in animals treated with oma-1 dsRNA for three to four generations (Kalinava et al., 2017). Therefore, the onset of robust H3K9me3 at oma-1 begins at the first generation of dsRNA exposure, one generation earlier than the transcriptional repression.
As a control for the requirement of nuclear RNAi for the observed transcriptional repression of oma-1, we examined the oma-1 pre-mRNA and mRNA levels in hrde-1 mutant animals fed with oma-1 dsRNA. Similar to N2, oma-1 expression at the pre-mRNA level was not repressed in the F1(dsRNA+) hrde-1 animals (Figure 1C), confirming the requirement of nuclear RNAi for the transcriptional repression. Interestingly, oma-1 RNAi led to a progressive increase in oma-1 pre-mRNA level in the F2–F5(dsRNA+) hrde-1 animals. The same effect was observed in our previous study (Kalinava et al., 2017). Here, we also performed the oma-1 RNAi experiment in nrde-2 mutant strain for three generations, which is also defective in nuclear RNAi (Guang et al., 2010), and again observed progressively increased oma-1 pre-mRNA levels compared to dsRNA− control nrde-2 mutant animals (Figure S1A). These results suggest that exogenous dsRNA can affect transcription or co-transcriptional RNA processing in a nuclear RNAi-independent manner. Future studies are required to investigate the nature of such effect.
Despite the increased pre-mRNA, we observed approximately 40% reduction in oma-1 mRNA in both the F1(dsRNA+) and F2(dsRNA+) hrde-1 animals compared to the dsRNA− control hrde-1 animals (Figure 1B). The reduction is consistent with the previous finding that the classical RNAi is active in the hrde-1 mutant (Buckley et al., 2012; Kalinava et al., 2017). However, the oma-1 mRNA levels in the F1(dsRNA+) and F2(dsRNA+) hrde-1 animals were much higher than the F1(dsRNA+) and F2(dsRNA+) wild-type animals (Figure 1B). This may due to the active transcription of oma-1, a partial defect of classical RNAi in the hrde-1 mutant, or both. Further study is required to distinguish these possibilities.
set-32 Mutation Results in a More Gradual, Multigenerational Establishment of exo-dsRNA-Induced Transcriptional Repression
To investigate whether any of the putative H3K9 HMTs plays a role in the establishment of transcriptional repression, we performed multigenerational oma-1 RNAi experiments using set-32, met-2, and set-25 single-mutant strains, as well as the met-2 set-25 double mutant. Two different set-32 mutant alleles were used in this study. One is the set-32(ok1457) allele (C. elegans Deletion Mutant Consortium, 2012) which has an in-frame deletion of 156 aa (position 50–205), located before the SET domain, and has been used in previous studies (Ashe et al., 2012; Kalinava et al., 2017). In this study, we also used clustered regularly interspaced short palindromic repeats (CRISPR) to generate a second allele, set-32(red11), which lacks the SET domain. We measured the dsRNA-induced H3K9me3 at oma-1 using this new allele (three to four generations of dsRNA+), and found that, same as set-32(ok1457), set-32(red11) caused a significant reduction in, but did not completely abolish, the RNAi-induced H3K9me3 (Figure S2C). Also, the same as previously observed for the set-32(ok1475) mutant (Kalinava et al., 2017), set-32(red11) mutant animals exhibited no defect in heritable RNAi after three to four generations of exo-dsRNA exposure (Figures S2A and S2B). For the onset of silencing, oma-1 mRNA was robustly silenced by RNAi in both set-32 mutant alleles at the first generation of the dsRNA exposure, similar to wild-type animals (Figure 1B). Interestingly, the onset of nuclear RNAi, measured by pre-mRNA, was much slower in set-32 mutant than the wild-type animals. Both set-32 mutant alleles generally had higher oma-1 pre-mRNA than wild-type animals in the first four dsRNA+ generations (Figures 1C and S1). It was at the fifth generation when the repression reached the steady-state level observed in the wild-type animals (Figures 1C and S1). Therefore, set-32 mutation causes a multigenerational delay in dsRNA-triggered transcriptional repression. We also tested met-2, set-25, and met-2 set-25 mutant animals. All three mutants exhibited wild-type-like profiles in pre-mRNA repression at the first and second generations of the dsRNA exposure (Figure S1B). These results suggest that, although both SET-32 and MET-2 SET-25 contribute to the H3K9me3 at the RNAi target, their functions are not equivalent. SET-32-dependent activity, but not MET-2 SET-25-dependent activity, promotes the onset of exogenous dsRNA-induced transcriptional repression. The SET domain of SET-26 can methylate H3K9 in vitro (Greer et al., 2014). We previously showed that neither SET-26 nor SET-9, which is 96% identical to SET-26 at the amino acid level, is required for the dsRNA-induced H3K9me3 at oma-1 (Kalinava et al., 2017). Here, we found that the set-9 set-26 double mutant had no defect in the onset of the dsRNA-induced transcriptional repression at oma-1 (Figures S1C and S1D).
To examine inheritance of silencing induced by a shorter duration of dsRNA exposure (one or two generations as opposed to four), we cultured the progeny of dsRNA-exposed animals on OP50 plates that lacked oma-1 dsRNA for one or two generations (referred to as inheritance generations) (Figure 1A). For wild-type animals, both one-generation and two-generation dsRNA exposures resulted in robust repression at the pre-mRNA level in the inheritance generations (Figures 1E and 1F). Interestingly, after only one generation of dsRNA exposure, the first and second inheritance generations showed progressively stronger oma-1 repression at the pre-mRNA levels (Figure 1E), suggesting that the transgenerational establishment of transcriptional repression does not require continuous dsRNA exposure. For set-32 mutants, we again observed delayed nuclear RNAi in the inheritance generations (Figures 1E and 1F), confirming the role of SET-32 in promoting the establishment of nuclear RNAi-mediated silencing.
Developing a CRISPR-Based Approach to Study the Establishment of Silencing at the Endogenous Targets of Germline Nuclear RNAi
One challenge of studying the establishment of silencing at the endogenous targets (ones targeted by endo-siRNAs) is to begin with an actively transcribed target for the experiment. This can be achieved at the whole-genome level by using a nuclear RNAi-defective mutant, such as hrde-1, in which a defined set of endogenous targets are actively transcribed (Buckley et al., 2012; Ni et al., 2014, 2016). To examine the establishment of silencing at these endogenous targets, we used CRISPR-mediated gene conversion to repair the loss-of-function hrde-1(tm1200) mutation in worms that had carried the hrde-1 mutation for more than 20 generations. We wanted to know how fast the repressive states can be established at the endogenous nuclear RNAi targets when the HRDE-1 activity is restored. Besides the CRISPR method, a wild-type hrde-1 allele can also be introduced to the hrde-1 mutant strain via a genetic cross. We did not choose this method because genetic crosses will introduce epigenetically repressed alleles of the endogenous targets, as well as a foreign siRNA population.
To repair the hrde-1 mutation using CRISPR, a mixture of hrde-1-targeting Cas9 ribonuclease complex and repair template DNA was injected into the gonads of hrde-1 mutant hermaphrodite adults (Figure 2A). The wild-type hrde-1 allele generated by gene conversion was annotated as hrde-1(+R), and the naive wild-type hrde-1 allele (i.e., an allele that never experienced any mutation) as hrde-1 (+). Progeny of the injected animals were produced by self-fertilization throughout the experiment. The F1 hrde-1(+R/−) and F2 hrde-1(+R/+R) individuals were identified by PCR and Sanger sequencing. We then collected later generations of the homozygous hrde-1 (+R) adults and examined the abundance of the transcripts of the endogenous targets. The F3 was the earliest generation that can be collected for the assay because the F1 and F2 progeny were used for genotyping. We measured the abundance of the transcripts for two endogenous targets by RT-qPCR. Both established their repressive states in the F3 hrde-1(+R) and later generations (two biological replicates; Figures 2B and 2C). We also identified the F2 hrde-1(−/−) individuals and collected their progeny (F3) and performed RT-qPCR analysis (Figure S3D). The endogenous targets remained de-repressed in the F3 hrde-1(−/−) progeny (Figure S3D), as expected. These results indicate that silencing of endogenous targets can be established at the F3 generation after repairing the hrde-1 mutation by gene conversion.
SET-32 Is Required for the Establishment of Silencing for Some of the Endogenous Targets
To test whether the putative H3K9 HMTs are required for establishment of silencing at the endogenous targets, we generated set-32;hrde-1 and met-2 set-25 hrde-1 mutant strains. Two different alleles (ok1457 and red11) of set-32 mutations were used this experiment. For set-32(ok1457), we collected F3, F5, F8, and F20 populations after the gene conversion. For set-32(red11), we collected F3, F4, F5, and F20 populations. Both set-32 alleles exhibited a delay in the establishment of silencing for eight or five generations. Full repressions of these targets were reached at the F20 generation (Figures 2B and 2C). We confirmed that the early generations of the hrde-1(+R) animals had similar hrde-1 mRNA expressions as the hrde-1 (+) animals (Figure S3). Therefore, the delayed establishment of silencing is not due to a lower hrde-1 mRNA expression in the early post-conversion generations. For the met-2 set-25 hrde-1 mutant strain, two independent lines of met-2 set-25 hrde-1 [+R/+R] worms were followed and both fully established the repressive states by the F3 generation for the two tested targets (Figure 2B).
To examine the establishment of silencing at the whole-genome level, we performed RNA sequencing (RNA-seq) for the post-conversion populations (hrde-1 [+R/+R], set-32;hrde-1 [+R/+R], and met-2 set-25 hrde-1 [+R /+R]), as well as animals of the identical genotypes but with the naive wild-type hrde-1 allele (hrde-1[+/+], set-32;hrde-1 [+/+], and met-2 set-25 hrde-1[+/+]). We sequenced rRNA-depleted total RNA of each sample in order to include both polyadenylated RNA and non-polyadenylated RNA. For the post-conversion samples, we performed RNA-seq for the F4, F5, and F20 populations for hrde-1(+R), F3, F5, and F20 for met-2 set-25 hrde-1(+R), F5, F8, and F20 for set-32(ok1457) hrde-1(+R), and F4, F5, and F20 for set-32(red11) hrde-1(+R). Our whole-genome analysis indicated that repairing hrde-1 mutation fully silenced all of the endogenous targets as early as in the F4 post-conversion population of the animals carrying the wild type met-2, set-25, and set-32 genes (Figures 3A and 3E). In contrast, some of the endogenous targets remained actively expressed even in the F20 post-conversion populations of animals carrying either the set-32(red11) or set-32(ok1457) mutation (Figures 3C–3E). These targets are referred to as putative irreversible targets. After examining the RNA-seq profiles individually, we identified four exemplary irreversible targets, which include two LTR retrotransposons (Cer9 and Cer12), a putative protein-coding gene c38d9.2 in chromosome V, and an approximately 5-kb region in chromosome II (containing two uncharacterized putative protein-coding genes f15d4.5 and f15d4.6) (Figures 3F and S5–S8). All of these loci became transcriptional activated in hrde-1 and set-32;hrde-1 mutants, but remained silent in set-32 mutant (with naive wild-type hrde-1), despite the losses of H3K9me3 at these regions (Figures 3F and S5–S8). These data confirm our previous finding that SET-32 is dispensable for the maintenance of silencing at the native HRDE-1 targets (Kalinava et al., 2017). These irreversible targets represent a bona fide epigenetic phenomenon in which two genetically identical populations (cultured in the same conditions and harvested at the same developmental stage) exhibit different gene expression profiles.
Figure 3. RNA-Seq Analysis of Silencing Establishment at Endogenous HRDE-1 Targets.

(A–D) Scatterplot analysis of normalized RNA-seq counts for all 1-kb genomic regions comparing animals in the post-conversion generations with animals of the same genotype but without experiencing hrde-1 mutation (naive samples). (A) Wild-type set-32;met-2 set-25 background, (B) met-2 set-25 double-mutant background, (C) set-32(ok1457) background, and (D) set-32(red11) background. GRTS and GRH regions (see Figure 1 legend for their definitions) are colored in red and blue, respectively.
(E) Boxplot analysis of the changes in RNA expression of GRTS or GRH regions between post-conversion animals and the corresponding naive samples.
(F) An exemplary endogenous HRDE-1 target (LTR retrotransposon Cer9) that is defective in silencing establishment in the set-32 mutant for at least 20 generations, but not in the met-2 set-25 mutant. Normalized total RNA-seq signals were plotted. Columns from left to right are the naive samples (hrde-1[+]), pre-conversion samples (hrde-1[−]), and F5 and F20 post-conversion samples (hrde-1[+R]). Different genetic backgrounds (wild-type [WT] or various H3K9 HMT mutants) are in different rows as indicated. All results represented here are normalized by total reads aligned to the whole genome.*p<7 ×10−5 (Wilcoxon singed-rank test).
We found that met-2 set-25 are required for the establishment of silencing in only one of the four irreversible targets, c38d9.2 (Figure S7). Our global analysis indicated that most of the endogenous targets established the silencing states as early as in the F3 met-2 set-25 hrde-1(+R) populations (Figures 3B and 3E).
We also performed the silencing establishment assay using the set-32;met-2 set-25 hrde-1 mutant strain. All four irreversible targets identified in the set-32 mutant ground failed to initiate the silencing the F5 set-32;met-2 set-25 hrde-1(+R) population (Figures S4–S8). The expression levels of these targets in F5 set-32;met-2 set-25 hrde-1(+R) and F5 set-32; hrde-1(+R) were similar.
We note that majority of the irreversible targets in set-32 mutant belong to the GRTS regions. In contrast, most of the GRH regions restored the transcriptional repression by the F4–F5 post-repair generation in the set-32 mutant background (Figures 3C–3E). Taken together, our results indicate that SET-32 is required for the establishment of the repression states of some endogenous HRDE-1 targets. Although met-2 set-25 mutant animals had a much weaker phenotype than set-32 in this assay, we cannot rule out that MET-2 and SET-25 are required for the establishment of silencing in the first and second post-conversion generations, which were precluded in the assay due to the screening procedure after CRISPR.
To explore the role of endo-siRNAs in the establishment of silencing, we performed small RNA-sequencing (sRNA-seq) analysis for the pre-conversion samples, the F5 and F20 post-conversion generations, and the matching samples with the naive wild-type hrde-1 allele. We found that the endo-siRNA profiles at the irreversible targets in set-32 hrde-1 (+) was similar to the ones in the wild-type (WT) animals, arguing against an active role of SET-32 in promoting the endo-siRNA levels at these targets. The set-32 hrde-1 (−) and the post-conversion set-32 hrde-1 (+R) animals had abundant endo-siRNAs at the irreversible targets. Their profiles were different from the ones in the set-32 hrde-1(+) but similar to the ones in the hrde-1(−) animals (Figures S5–S8). These results argue against the possibility that the delayed establishment of silencing observed in set-32 mutant is caused by a lag in endo-siRNA expression. The changes in the endo-siRNA expressions in the set-32 hrde-1(−) and the post-conversion set-32 hrde-1 (+R) animals, in comparison to WT animals, are likely due to the increased transcripts of the irreversible targets, which template the endo-siRNA synthesis.
H3K9 HMTs Are Required to Maintain the Repressive States for Some Endogenous HRDE-1 Targets in hrde-1 Mutant Animals
We performed Pol II ChIP-seq and RNA-seq analyses for various compound mutants including set-32;hrde-1, met-2 set-25 hrde-1, and set-32;met-2 set-25 hrde-1. As expected, the endogenous HRDE-1 targets were de-repressed in these compound mutants (Figures S9A and S9B). Intriguingly, for a small subset of the targets, the hmt; hrde-1 compound mutants exhibited much higher RNA expression and Pol II occupancy than the hrde-1 single mutant (Figures 4A and S5), indicating that the loss of H3K9me3 further enhances the hrde-1 mutation’s defects in transcriptional repression for these targets. Such enhancement is unexpected because set-32, met-2 set-25, set-32;met-2 set-25 mutant animals have no silencing defects at the endogenous targets, as reported previously (Kalinava et al., 2017) and shown again using the new data generated in this study (Figure S9). An exemplary endogenous target with such enhanced de-repression is the LTR retrotransposon Cer3 (Figures 4B–4D). Either set-32 single or met-2 set-25 double mutations, when combined with the hrde-1 mutation, drastically increased the Pol II occupancy at Cer3 and Cer3 transcripts. Therefore, the enhanced de-repression occurs at the transcriptional level. The enhanced de-repression caused by different H3K9 HMTs appears to be additive because the quadruple mutant of set-32;met-2 set-25 hrde-1 showed the highest Cer3 expression (Figures 4C and 4D). Interestingly, Cer3 is not an irreversible target (Figure 4E). These results indicate that, for a small subset of the endogenous targets, H3K9me3 HMTs are required for the maintenance of repression. However, such requirements are only apparent when HRDE-1 activity is compromised. We note that, for the majority of the endogenous HRDE-1 targets, the combined mutations in H3K9 HMT genes and hrde-1 had no additive effect of de-repression compared to the hrde-1 single mutant (Figure S9).
Figure 4. Enhanced Silencing Defects in the Endogenous HRDE-1 Targets Caused by the Combination of H3K9 HMT and hrde-1 Mutations.

(A) Scatterplots with changes in RNA-seq signals between a compound mutant and hrde-1 single mutant plotted in the x axis and changes in Pol II ChIP-seq signals plotted in the y axis for all 1-kb genomic regions. Regions with ≥ 2-fold increases in both RNA expression and Pol II occupancy in all three H3K9 hmt;hrde-1 compound mutants over hrde-1 single mutant were marked with red dots.
(B–D) The H3K9me3 ChIP-seq (data from Kalinava et al., 2017) (B), Pol II ChIP-seq (S2 phosphorylated) (C), and RNA-seq (D) coverage plots at the LTR retrotransposon Cer3 in WT and various mutant animals. Panels marked with an asterisk (*) use a different scale to accommodate the enhanced expression levels observed for the genotype.
(E) RNA-seq profiles at Cer3 for post-conversion samples (hrde-1[+R/+R]) with WT H3K9 HMT genes or various H3K9 HMT mutations. All results represented in this figure are normalized by total reads aligned to the whole genome.
DISCUSSION
The Establishment and Maintenance Phases of Germline Nuclear RNAi in C. elegans
H3K9me3 is an evolutionarily conserved nuclear RNAi effect in fungi, plants, and animals. In C. elegans, this effect is highly specific and prominent at target loci. Furthermore, it is transgenerationally heritable and linked to the germline immortality (Ashe et al., 2012; Buckley et al., 2012; Burton et al., 2011; Gu et al., 2012; Shirayama et al., 2012; Weiser et al., 2017). H3K9me3 at nuclear RNAi targets requires three putative HMTs: MET-2, SET-25, and SET-32 (Kalinava et al., 2017; Mao et al., 2015; Spracklin et al., 2017). Strikingly, we previously found that a complete loss of H3K9me3 in the mutant worms that lack the three HMTs did not cause any defect in transcriptional repression at the endogenous targets (Kalinava et al., 2017). The experimental setup in the previous study was designed to examine the maintenance of silencing and precluded us from studying the establishment phase. For a newly inserted transposon that is transcriptionally active, the establishment phase precedes the maintenance phase and two phases may involve different mechanisms.
Exo-dsRNA-triggered nuclear RNAi provides an inducible system in which to study the establishment, maintenance, and inheritance of epigenetic silencing at a gene that is normally actively expressed. The previous discovery of a transgenerational delay in the onset of H3K9me3 at the target gene (Burton et al., 2011; Gu et al., 2012) suggests that the establishment of nuclear RNAi is a gradual process. The kinetics of transcriptional repression during the early generations of dsRNA exposure have not been examined before this study.
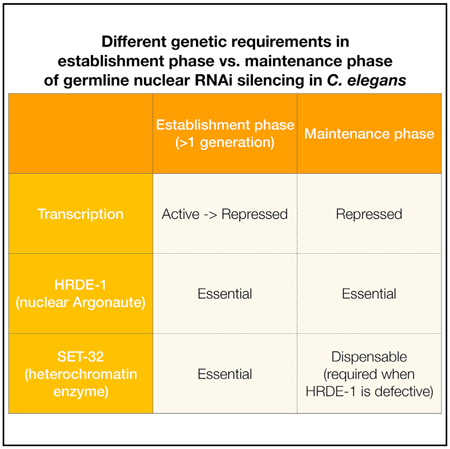
In this study, we examined the establishment phase for both endo-siRNA-guided and exogenous dsRNA-triggered nuclear RNAi. Based on the results of this and our previous work (Kalinava et al., 2017), we propose a model in which the establishment and maintenance phases of nuclear RNAi have the following distinctions (Figure 5).
Figure 5. Functional and Mechanistic Differences between Classical RNAi and Nuclear RNAi, and Different Phases of Nuclear RNAi in C. elegans.

(A) Classical RNAi provides an immediate silencing response at the same generation when animals are exposed to dsRNA. Classical RNAi silencing is at the post-transcriptional level, and is not transgenerationally heritable. In contrast, nuclear RNAi silencing takes more than one generation to be established. Nuclear RNAi represses target loci at the transcriptional level and is heritable. It provides long-term transgenerational silencing.
(B) Establishment and maintenance phases of nuclear RNAi involve different transcriptional activities and genetic requirements.
-
(1)
The transcriptional state of the target locus. At the maintenance phase, the target is stably repressed and kept in a heterochromatic state. In contrast, the establishment phase begins with an actively transcribed and euchromatic state at the target locus. Converting from an active state to a stably repressed state requires removal of euchromatic marks and the deposition of heterochromatic marks. Therefore, the speed of the silencing establishment may depend on the relative strengths of the two opposing forces.
-
(2)
Silencing mechanism. We propose that nuclear RNAi consists of multiple distinct silencing mechanisms. One of these mechanisms is an H3K9me3-independent silencing mechanism, which likely is the main source of silencing during the maintenance phase. The active transcription during the establishment phase may pose a strong force against the H3K9me3-independent silencing mechanism. Such antagonistic interactions between euchromatin (H3K36me3) and heterochromatin have been shown to occur in the C. elegans germline (Gaydos et al., 2012; Weiser et al., 2017). It is conceivable that SET-32-depedent H3K9me3 promotes the onset of silencing, either by providing an additional force of repression (H3K9me3-dependent in this case) or by enhancing the efficacy of H3K9me3-independent silencing mechanism. However, given enough time, H3K9me3-independent mechanisms can eventually establish silencing in the absence of SET-32. Although the prolonged lag in silencing is tolerated by the mutant worms at the organismal level in the laboratory condition, this molecular defect may create a window for the transposition of a newly invaded mobile DNA, and therefore it is likely to be selected against in a wild population.
The establishment phase of an epigenetic silencing phenomenon is often difficult to study because of its transient nature. The set-32 mutation slows down the onset of silencing and therefore provides an expanded window to dissect the extraordinarily complex process.
Although MET-2, SET-25, and SET-32 all contribute to the H3K9me3 at the nuclear RNAi targets, SET-32 and MET-2 SET-25 are not equivalent in their role in silencing establishment. This may due to differential expression of these proteins. Alternatively, the functional difference could be due to certain unknown H3K9me3-independent activities associated with individual enzymes. Future study is required to investigate these possibilities. In agreement with our finding, the study by Woodhouse et al. (2018) (in this issue of Cell Reports) found that SET-32 is required for the establishment of transge-nerational silencing induced by dsRNA at a gfp reporter gene. The Ashe group found that SET-25 is also required in the establishment. We found that SET-25 is required for the silencing establishment for one of the endogenous targets, but not the other SET-32-dependent ones, including the exogenous dsRNA-targeted oma-1. This difference is perhaps due to different chromatin structures and transcription activities at the target genes.
What Is the Functional Significance of the One-Generation Delay in Exo-dsRNA-Induced Nuclear RNAi?
Although exo-dsRNA was used as an artificial means to induce RNAi in this study, it is akin to a situation in which host animals encounter viral nucleic acids. We speculate that such a delay is a consequence of the specialization of classical RNAi and nuclear RNAi: the former eliminates the immediate threat by degrading the target RNA, and the latter provides long-term silencing at the chromatin level, which is critical for the control of retroviruses or retrotransposons. One possible benefit of the delay of nuclear RNAi is to allow the biogenesis of secondary siRNAs (Pak and Fire, 2007; Sijen et al., 2007), which requires mRNA as template for the RdRP activity. If both nuclear RNAi and classical RNAi are fully active at the initial encounter of dsRNA, germ cells may not be able to produce sufficient siRNAs to facilitate silencing inheritance.
CRISPR Provides a Unique Advantage for Studying Transgenerational Epigenetics
In this study, we used CRISPR-mediated gene editing to repair a mutation in the hrde-1 gene, which allowed us to capture the graduate changes in the endogenous targets after the nuclear RNAi machinery is turned on in the subsequent generations. This approach is analogous to heat or small molecule-inducible gene expression, but without the need to change the native promoter or protein sequence of the inducible gene. This approach also avoids crossing two strains carrying different genetic and epigenetic backgrounds, which makes this approach highly tractable. (For example, there is no need to distinguish different epialleles.)
Our study showed that, after repairing hrde-1, WT animals fully restored silencing at the endogenous targets in the third generation. Our current implementation of CRISPR does not allow us to examine the first and second generations after the HRDE-1 activity is restored. The one-generation delay observed for the exo-dsRNA-triggered nuclear RNAi raises a possibility that the establishment of silencing at the endogenous targets may also have a transgenerational delay even in a WT H3K9 HMT background.
CRISPR-mediated genome editing provides a powerful tool for biomedical research and curing genetic diseases. Our study provides a cautionary example that repairing a mutated gene may not immediately restore the normal function expected for the WT allele. Therefore, epigenetic effects must be considered when editing genes, especially when chromatin factors and epigenetic pathways are involved.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests and further information for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Sam G. Gu (sam.gu@rutegers.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All C.elegans strains used in this study are listed in Key Resources Table. C. elegans was cultured with OP50 E. coli as the food source for all non-RNAi experiments (Brenner, 1974). Worms were cultured at 20°C for all of the experiments conducted in this study. Synchronized young adult animals were ground in liquid nitrogen by mortar and pestle and stored at −80°C and used for all data analysis. Experiments involving WT and mutant C. elegans are approved by Rutgers Environmental Health and Safety under “recombinant DNA” protocol.
METHOD DETAILS
oma-1 RNAi experiments
oma-1 RNAi experiments were performed as described previously (Kalinava et al., 2017; Timmons et al., 2001) with the following modifications. Two schemes of oma-1 RNAi were used in this study. To examine the onset of RNAi, synchronized L1 larvae were released onto oma-1 RNAi plates which contained oma-1-dsRNA-expressing E. coli. The young adult animals are referred to as F1(dsRNA+). To obtain worms of F2(dsRNA+) or with extended generations of dsRNA exposure, eggs from dsRNA+ adult animals were collected and hatched in M9 buffer. L1s were released onto oma-1 RNAi plates for another generation of dsRNA exposure. Young adult animals (59-60 hours after L1 release at 20°C, before egg laying starts, but embryos are visible inside the uterus) were harvested for various assays throughout the experiments. For the oma-1 heritable RNAi and H3K9me3 ChIP experiments in Figure S2, worms of mixed developmental stages were first cultured on oma-1 RNAi plates continuously for 9-10 days starting with approximately 5 L3-L4 worms, followed by one round of synchronized culture on oma-1 RNAi plates to collect young adults. Samples used in the same figure panel were prepared in parallel.
Gene conversion to repair hrde-1 mutation using CRISPR-Cas9
We repaired the hrde-1(tm1200) mutation to the WT sequence by injecting a plasmid Cas9 plasmid and hrde-1-targeting a hrde-1 targeting Cas-9 ribonuclease complex and PCR fragment as the repair template. A dpy-10(cn64) co-conversion marker was used (Arribere et al., 2014; Paix et al., 2015). 10-15 adult hermaphrodite animals (P0s) of each strain were injected. We screened 48-96 F1s animals using single-worm PCR from broods of injected animals with high frequencies of roller F1s. This yielded with 2-8 F1s bearing potential gene conversion events (heterozygotes) identified by the size of PCR products. For each putative F1 hit, 12-24 F2 worms a WT copy of dpy-10 were individually transferred to plates. After laying eggs, the F2 worms with homozygous WT hrde-1 sequences were identified by PCR and Sanger sequencing. We refer to the CRISPR/Cas-9-generated WT hrde-1 sequence as hrde-1(+R) to distinguish it from the naive WT hrde-1 sequence, referred to as hrde-1(+). F3, F4, F5, F10, and F20 post-conversion adults were collected either by hand picking (F3 and F4) or synchronized culture (F5 and later generations). The control hrde-1(+) samples (WT, set-32, met-2 set-25, set-32; met-2 set-25) and hrde-1(−) samples (hrde-1, set-32; hrde-1, met-2 set-25 hrde-1 and set-32; met-2 set-25 hrde-1) were collected by picking adult animals, identically to the F3 and F4 samples of the hrde-1(+R) samples.
Total RNA extraction
For samples of pooled whole worms, TRIzol reagent (Life Technologies) was added to the frozen sample of approximately 20 worms in the M9 buffer. To ensure break down of worm bodies, we used 3-4 cycles of freeze-thawing in TRIzol, then performed total RNA extraction according to the manufacture’s protocol. This procedure yielded 1-3 μg of total RNA for each sample.
mRNA and pre-mRNA RT-qPCR
Total RNA extraction was performed using the TRIzol Reagent. 1 μg of total RNA was used for the first strand cDNA synthesis with Superscript III RT kit (Life Technologies) and oligo-dT as the primer for mRNA RT-qPCR and the random hexamers as the primer mix for pre-mRNA RT-qPCR.
qPCR was performed using KAPA SYBR FAST Universal 2 × PCR Master Mix (KAPA Biosystems) on a Mastercycler EP Realplex realtime PCR system (Eppendorf) according to the manufacturer’s instructions. qPCR primers are listed in Key Resources Table. Each sample was processed in triplicate. Reported values for the fold change of mRNA and pre-mRNA were calculated using ΔΔCT analysis. tba-1 was used as a reference gene.
High-throughput sequencing
Chromatin immunoprecipitation (ChIP): Pol II and H3K9me3 ChIP-seq was performed as described previously (Ni et al., 2014). The anti-RNA Pol II S2 antibody (ab5095, Abcam) and anti-H3K9me3 antibody (ab8898, Abcam) were used.
RNA-seq: Ribosomal RNA (rRNA) was removed from the total RNA using RNase H and anti-rRNA oligo mixture (total of 110, listed in Table S1). The rRNA-removal procedure was adopted from (Frøkær-Jensen et al., 2016). Briefly 1.25 μg of anti-rRNA oligos were mixed with 0.5 μg total RNA in 1x Hybridization Buffer (100 mM Tris-HCl pH 7.4, 200mM NaCl) in a final volume of 8 μl. The sample was denatured at 95°C for 2 minutes, and then cooled at −0.1°C /sec to 45°C. Equal volumes of Hybridase Thermostable RNase H (5U/μl,Epicenter) and 10x RNaseH Reaction Buffer (500 mM Tris-HCl pH 7.4, 1 M NaCl, 200 mM MgCl2) were mixed and preheated to 45°C before use. 2 μl of the enzyme mix was added to each reaction mix, which was then incubated at 45°C for 1 hr. To remove DNA oligos, 5 μl TURBO DNase buffer, 3 μl TURBO DNase (Invitrogen), and 32 μl diH2O were added to the reaction mix, followed by 37°C incubation for 30 minutes. To purify RNA, 300 μl STOP solution (1 M ammonium acetate, 10 mM EDTA) was added to the reaction mix, followed by phenol/chloroform (1:1) extraction and ethanol precipitation. The resulting RNA (without poly(A) selection) was used to prepare the RNA-seq library as described previously (Ni et al., 2014).
siRNA-seq: small RNA isolation and RNA-seq library construction were performed as described previously (Ni et al., 2014).
All libraries were sequenced using Illumina HiSeq 2500 platform, with a 50-nt single-end run and dedicated index sequencing. All libraries used in this study are listed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Whole genome alignment of the sequencing reads to the C. elegans genome (WS190 version) was done using Bowtie (0.12.7) (Langmead et al., 2009). Only perfectly aligned reads were used for data analysis. Reads that aligned to N different loci were scaled by a factor of 1/N. Custom R and python scripts were used in this study. Normalization based on total reads aligned to the whole genome for each library was used for all data analysis, except for the ΔPol II boxplot analysis in Figure S9C, where total reads count of the top 5th percentile of the Pol II signal in corresponding library was used as the normalization factor. The three-region Venn diagram was generated using a web-based software (http://www.benfrederickson.com/venn-diagrams-with-d3.js/). Wilcoxon signed-rank test was performed to calculate the p values for all box-plot analysis in Figures 3E, S9C, and S9D. Welch Two Sample t test was used to calculate all other p values.
DATA AND SOFTWARE AVAILABILITY
The accession number for the raw sequencing data in the fastq format reported in this paper is NCBI GEO: GSE117662.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Histone H3 (tri methyl K9) antibody - ChIP Grade | Abcam | Cat# 8898, Lot# GR285794-2;RRID: AB_306848 |
| Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S2) antibody - ChIP Grade | Abcam | Cat# ab5095, Lot# GR231750-1; RRID: AB_304749 |
| Bacterial and Virus Strains | ||
| E. coli OP50 | CGC | NA |
| E. coli HT115(DE3) | CGC | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FORMALDEHYDE 37% MICROFILTERED | Electron Microscopy Sciences | Cat# 15686 |
| TRIzol-REAGENT | Invitrogen | Cat# 15596-018 |
| RNA Fragmentation Reagents | Invitrogen | Cat# AM8740 |
| T4 PNK | NEB | Cat# M0201L |
| T4 RNA Ligase 1 | NEB | Cat# M0204S |
| T4 RNA ligase 2, truncated | NEB | Cat# M0242L |
| Critical Commercial Assays | ||
| KAPA Hyper Prep Kit | KAPABIOSYSTEM | Cat# KK8505 |
| KAPA SYBR FAST Universal 2 × PCR Master Mix | KAPABIOSYSTEM | Cat# KK4602 |
| Deposited Data | ||
| NGS data | This study | NGS: GSE117662 |
| Experimental Models: Organisms/Strains | ||
| C. elegans Bristol strain | CGC | N2 |
| set-32(ok1457) I | (C. elegans Deletion Mutant Consortium, 2012) | VC967 |
| set-32(red11) I | This study | CSS186 |
| nrde-2(gg091) II | (Guang et al., 2010) | YY186 |
| met-2(n4256) III | (Andersen and Horvitz, 2007) | MT13293 |
| hrde-1(tm1200) III | (C. elegans Deletion Mutant Consortium, 2012) | TM1300 |
| set-25(n5021) III | (Andersen and Horvitz, 2007) | MT17463 |
| set-26(tm3526) IV | (C. elegans Deletion Mutant Consortium, 2012) | TM3526 |
| set-9(red8) IV | (Kalinava et al., 2017) | CSS164 |
| set-26(tm3526) set-9(red8) | this study | CSS412 |
| set-32(red11); hrde-1(tm1200) | this study | CSS425 |
| met-2(n4256) set-25(n5021) | (Kalinava et al., 2017) | CSS95 |
| set-32(red11);met-2(n4256) set-25(n5021) | (Kalinava et al., 2017) | CSS419 |
| met-2(n4256) set-25(n5021) hrde-1(tm1200) | this study | CSS400 |
| set-32(red11);met-2(n4256) set-25(n5021) hrde-1(tm1200) | this study | CSS415 |
| Oligonucleotides | ||
| forward qPCR primer for oma-1 mRNA: ctacaggactctgcccatacg | IDT | SG-1016 |
| reverse qPCR primer for oma-1 mRNA: ggcctggcaaacatttctaa | IDT | SG-1019 |
| forward qPCR primer for oma-1 pre-mRNA:ttaatgacc tctgttttaggtg | IDT | SG-1022 |
| reverse qPCR primer for oma-1 pre-mRNA:gaacccaaaa ccatcgaatc | IDT | SG-1023 |
| forward qPCR primer for oma-1 ChIP: acatgtatttttgctcactgtaa | IDT | SG-1018 |
| reverse qPCR primer for oma-1 ChIP: ggcctggcaaacatttctaa | IDT | SG-1019 |
| forward ChIP qPCR primer for an H3K9me3 negative control locus: agccatggcacaaaaagaag | IDT | SG-1044 |
| reverse ChIP qPCR primer for an H3K9me3 negative control locus: tgtggcctgagaagacaaaa | IDT | SG-1045 |
| forward qPCR primer for tba-1 mRNA: TCCAAGCGAG ACCAGGCTTCAG | IDT | SG-1108 |
| reverse qPCR primer for tba-1 mRNA: TCAACACTGCCAT CGCCGCC | IDT | SG-1109 |
| HR oligo for set-32 (red11) CRISPR allele): tcttcgaatatacagacacg aatgttttgaacccgGCTAGCAtgctccgcaagaaacattgcgaaagcatgta ttatgtctcaaaaggaagagaat | IDT | SG-1173 |
| T7 in vitro transcription template oligo for crRNA targeting hrde-1 tm1200 mutation (seed sequence: taatcgtatgTggaaaccg): AAAACAGCATAGCTCTAAAAC cggtttccAcatacgatta CCCTATAGTGAGTCGTATTA | IDT | SG-1285 |
| upstream PCR primer used to generated a 424 bp genomic DNA fragment as template DNA to repair the hrde-1 (tm1200) mutation by CRISPR: aaggagaagttgcccaggag | IDT | SG-1292 |
| downstream PCR primer used to generated a 424 bp genomic DNA fragment as template DNA to repair the hrde-1(tm1200) mutation by CRISPR: atctcggtacctgtcgttgc | IDT | SG-1293 |
| Recombinant DNA | ||
| oma-1 dsRNA feeding plasmid | (Kalinava et al., 2017) | pSG42 |
| Software and Algorithms | ||
| Bowtie 0.12.7 | (Langmead et al., 2009) | NA |
Highlights.
The onset of nuclear RNAi is a transgenerational process
A putative histone methyltransferase, SET-32, promotes silencing establishment
MET-2, SET-25, and SET-32 are involved for the maintenance of silencing
For endogenous targets, their requirements are conditional on hrde-1 mutation
ACKNOWLEDGMENTS
We thank Elaine Gavin, Shobhna Patel, Monica Driscoll, Lamia Wahba, and Andy Fire for help and suggestions. Research reported in this publication was supported by the Busch Biomedical Grant and the National Institute of General Medical Sciences of the NIH, United States, under Award R01GM111752. Some strains were provided by the CGC, United States, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes nine figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.086.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akay A, Di Domenico T, Suen KM, Nabih A, Parada GE, Larance M, Medhi R, Berkyurek AC, Zhang X, Wedeles CJ, et al. (2017). The helicase aquarius/EMB-4 is required to overcome intronic barriers to allow nuclear RNAi pathways to heritably silence transcription. Dev. Cell 42, 241–255.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcazar RM, Lin R, and Fire AZ (2008).Transmission dynamics of heritable silencing induced by double-stranded RNA in Caenorhabditis elegans. Genetics 180, 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, and Horvitz HR (2007). Two C. elegans histone methyltransferases repress lin-3EGF transcription to inhibit vulval development. Development 134, 2991–2999. [DOI] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe A, Sapetschnig A, Weick EM, Mitchell J, Bagijn MP, Cording AC, Doebley AL, Goldstein LD, Lehrbach NJ, Le Pen J, et al. (2012). piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell 150, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagijn MP, Goldstein LD, Sapetschnig A, Weick EM, Bouasker S, Lehrbach NJ, Simard MJ, and Miska EA (2012). Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science 337, 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, and Kennedy S (2012). A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature 489,447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart KB, Guang S, Buckley BA, Wong L, Bochner AF, and Kennedy S (2011). A pre-mRNA-associating factor links endogenous siRNAs to chromatin regulation. PLoS Genet. 7, e1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NO, Burkhart KB, and Kennedy S (2011). Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 108,19683–19688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Deletion Mutant Consortium (2012). Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 (Bethesda) 2, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, and Tuschl T (2001). RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, and Mello CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Frøkæ-Jensen C, Jain N, Hansen L, Davis MW, Li Y, Zhao D, Rebora K, Millet JRM, Liu X, Kim SK, et al. (2016). An abundant class of noncoding DNA can prevent stochastic gene silencing in the C. elegans germline. Cell 166, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigues JM, Sidoli S, Garcia BA, and Strome S (2015). Defining heterochromatin in C. elegans through genome-wide analysis of the heterochromatin protein 1 homolog HPL-2. Genome Res. 25, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaydos LJ, Rechtsteiner A, Egelhofer TA, Carroll CR, and Strome S (2012). Antagonism between MES-4 and Polycomb repressive complex 2 promotes appropriate gene expression in C. elegans germ cells. Cell Rep. 2, 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Beese-Sims SE, Brookes E, Spadafora R, Zhu Y, Rothbart SB, Aristizábal-Corrales D, Chen S, Badeaux AI, Jin Q, et al. (2014). A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Rep. 7, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Tabara H, and Mello CC (2000). Genetic requirements for inheritance of RNAi in C. elegans. Science 287, 2494–2497. [DOI] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, and Fire A (2012). Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat. Genet. 44, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, and Kennedy S (2010). Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465, 1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, and Hannon GJ (2000). An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Boettcher S, Caudy AA, Kobayashi R, and Hannon GJ (2001). Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293, 1146–1150. [DOI] [PubMed] [Google Scholar]
- Kalinava N, Ni JZ, Peterman K, Chen E, and Gu SG (2017). Decoupling the downstream effects of germline nuclear RNAi reveals that H3K9me3 is dispensable for heritable RNAi and the maintenance of endogenous siRNA-mediated transcriptional silencing in Caenorhabditis elegans. Epigenetics Chromatin 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WG, Xu S, Montgomery MK, and Fire A (1997). Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics 146, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerdell JR, and Carthew RW (1998). Use of dsRNA−mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell 95, 1017–1026. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold LE, Heestand BN, Seong S, Shtessel L, and Ahmed S (2015). Lack of pairing during meiosis triggers multigenerational transgene silencing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 112, E2667–E2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev I, Seroussi U, Gingold H, Bril R, Anava S, and Rechavi O (2017). MET-2-dependent H3K9 methylation suppresses transgenerational small RNA inheritance. Curr. Biol. 27, 1138–1147. [DOI] [PubMed] [Google Scholar]
- Lin R (2003). A gain-of-function mutation in oma-1, a C. elegans gene required foroocyte maturation, results in delayed degradation of maternal proteins and embryonic lethality. Dev. Biol. 258, 226–239. [DOI] [PubMed] [Google Scholar]
- Mao H, Zhu C, Zong D, Weng C, Yang X, Huang H, Liu D, Feng X, and Guang S (2015). The Nrde pathway mediates small-RNA-directed histone H3 lysine 27 trimethylation in Caenorhabditis elegans. Curr. Biol. 25, 2398–2403. [DOI] [PubMed] [Google Scholar]
- Martienssen R, and Moazed D (2015). RNAi and heterochromatin assembly. Cold Spring Harb. Perspect. Biol. 7, a019323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurchy AN, Stempor P, Gaarenstroom T, Wysolmerski B, Dong Y, Aussianikava D, Appert A, Huang N, Kolasinska-Zwierz P, Sapetschnig A, et al. (2017). A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. eLife 6, e21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkina O, and Hunter CP (2017). Stable heritable germline silencing directs somatic silencing at an endogenous locus. Mol. Cell 65, 659–670.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Chen E, and Gu SG (2014). Complex coding of endogenous siRNA, transcriptional silencing and H3K9 methylation on native targets of germline nuclear RNAi in C. elegans. BMC Genomics 15, 1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Kalinava N, Chen E, Huang A, Trinh T, and Gu SG (2016). A transgenerational role of the germline nuclear RNAi pathway in repressing heat stress-induced transcriptional activation in C. elegans. Epigenetics Chromatin 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, and Seydoux G (2015). High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR- Cas9 ribonucleoprotein complexes. Genetics 201, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J, and Fire A (2007). Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science 315, 241–244. [DOI] [PubMed] [Google Scholar]
- Pezic D, Manakov SA, Sachidanandam R, and Aravin AA (2014). piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev. 28, 1410–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D Jr., and Mello CC (2012). piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell 150, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Dönertas D, and Brennecke J (2012).Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151, 964–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Steiner FA, Thijssen KL, and Plasterk RH (2007). Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science 315, 244–247. [DOI] [PubMed] [Google Scholar]
- Spracklin G, Fields B, Wan G, Vijayendran D, Wallig A, Shukla A, and Kennedy S (2017). Identification and characterization of Caenorhabditis elegans RNAi inheritance machinery. Genetics, Published online May 22, 2017. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, and Fire A (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. [DOI] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meis- ter P, Askjaer P, and Gasser SM (2012). Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150, 934–947. [DOI] [PubMed] [Google Scholar]
- Tuschl T, Zamore PD, Lehmann R, Bartel DP, and Sharp PA (1999). Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 13, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Brunschwig K, Okihara KL, Müller F, Tijsterman M, and Plasterk RH (2006). Gene expression: long-term gene silencing by RNAi. Nature 442, 882. [DOI] [PubMed] [Google Scholar]
- Wassenegger M (2000). RNA-directed DNA methylation. Plant Mol. Biol. 43, 203–220. [DOI] [PubMed] [Google Scholar]
- Weiser NE, Yang DX, Feng S, Kalinava N, Brown KC, Khanikar J, Freeberg MA, Snyder MJ, Csankovszki G, Chan RC, et al. (2017). MORC-1 integrates nuclear RNAi and transgenerational chromatin architecture to promote germline immortality. Dev. Cell 41, 408–423.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse RM, Buchmann G, Hoe M, Harney D, Larance M, Boag PR, and Ashe A (2018). The chromatin modifiers SET-25 and SET-32 are required for initiation but not long-term maintenance of transgenerational epigenetic inheritance. Cell Reports 25, this issue, 2259–2272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.