

Abstract
Activation of innate immunity and deposition of blood-derived fibrin in the central nervous system (CNS) occur in autoimmune and neurodegenerative diseases, including multiple sclerosis (MS) and Alzheimer’s disease (AD). However, mechanisms linking blood-brain barrier (BBB) disruption with neurodegeneration are poorly understood, and exploration of fibrin as a therapeutic target has been limited by its beneficial clotting functions. Here we report the generation of monoclonal antibody 5B8 targeted against the cryptic fibrin epitope γ377–395 to selectively inhibit fibrin-induced inflammation and oxidative stress without interfering with clotting. 5B8 suppressed fibrin-induced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and proinflammatory gene expression. In animal models of MS and AD, 5B8 entered the CNS and bound to parenchymal fibrin, and its therapeutic administration reduced innate immune activation and neurodegeneration. Thus, fibrin-targeting immunotherapy inhibits autoimmune- and amyloid-driven neurotoxicity and may have clinical benefit without globally suppressing innate immunity or interfering with coagulation in diverse neurological diseases.
Activation of innate immunity is a key feature of neurological diseases with different etiologies, including autoimmune and neurodegenerative CNS diseases1. Increasing evidence indicates that pathogenic activation of CNS innate immunity contributes to neuronal damage and modulates the onset and progression of neurodegenerative diseases2. Oxidative injury and release of free radicals have been proposed as common mechanisms for innate immune-driven neurodegeneration and demyelination in MS and AD3–6. Chronic innate immune activation and oxidative injury are key elements driving neurodegeneration in both relapsing-remitting– and progressive MS3,7,8. In progressive MS, there is robust microglia activation, oxidative stress, and neurodegeneration3,8,9. Pathogenic activation of innate immunity contributes to oxidative stress and cognitive decline in AD5. Little is known about the pathogenic signals that activate innate immune cells toward neurotoxic phenotypes. Understanding the mechanisms of activation of CNS innate immunity is essential for deciphering how neuroinflammation contributes to neuronal damage and for designing treatments for selective suppression of pathogenic functions of innate immunity.
Innate immune activation, blood-brain barrier (BBB) disruption, and fibrin deposition are intimately linked in neurological diseases10,11. The blood coagulation factor fibrinogen extravasates into the CNS parenchyma upon BBB disruption and is converted to insoluble fibrin, a key proinflammatory matrix that activates innate immune responses11,12. Conversion of fibrinogen into fibrin exposes amino acids 377–395 in the fibrinogen γ chain (γ377–395) that bind to the CD11b I-domain of complement receptor 3 (CR3) (also known as CD11b/CD18, Mac-1, αMβ2) and induces microglia and macrophage activation13–16. Fibrin is deposited in AD and MS lesions at sites of microglial activation and macrophage infiltration (reviewed in11). Fibrin is detected in progressive MS and in active and chronic lesions (reviewed in11). In progressive MS, fibrin deposition in the cortex correlates with neuronal loss and inhibition of fibrinolysis17. BBB disruption and fibrin deposition occur also early in MS and precede demyelination18,19. Fibrinogen has been proposed as a cerebrospinal fluid and plasma biomarker for AD and mild cognitive impairment, and increased fibrinogen concentrations are considered a predictor of brain atrophy in AD (reviewed in11,20). Depletion of fibrin either genetically in fibrinogen deficient mice or by anticoagulants decreases neuroinflammation, demyelination, and axonal damage in animal models of MS and reduces microglia activation, white matter damage, and cognitive decline in animal models of AD (reviewed in11). Fibrin induces rapid and sustained microglia responses and macrophage infiltration into the CNS15,16. Although increased BBB disruption and fibrin deposition correlate with neurodegeneration, the molecular links between blood leakage into the CNS and neuronal damage are poorly understood. Furthermore, whether and how fibrin-induced activation of innate immunity is neurotoxic remains largely unknown.
Here we report an unanticipated role for fibrin as an activator of the NADPH oxidase complex that induced reactive oxygen species (ROS) release and innate immune-driven neurotoxicity in autoimmune and amyloid-driven neurodegeneration. Although innate immune activation is an attractive candidate for therapeutic intervention, selective therapies to inhibit neurotoxic effects of innate immune responses are not widely available. By targeting the γ377–395 cryptic fibrin epitope, we developed the first fibrin immunotherapy (monoclonal antibody 5B8) to selectively target the inflammatory form of fibrin without interfering with clotting or activation of innate immune cells by other ligands, such as lipopolysaccharide (LPS). 5B8 selectively bound to fibrin, but not soluble fibrinogen, and inhibited binding of fibrin to CR3 without interfering with fibrin polymerization, in vivo clotting time, or partial thromboplastin time (aPTT) in human plasma. 5B8 reduced NADPH oxidase activation, ROS release, microglial activation, and neurodegeneration in MS and AD animal models. These studies identify fibrin as a blood-derived signal that activates NADPH oxidase to promote innate immune-driven neurotoxicity, and identify fibrin-targeted immunotherapy as a novel therapeutic strategy to suppress innate immune-driven neurodegeneration at sites of increased vascular permeability without interfering with clotting or globally suppressing innate immunity.
Results
Design of fibrin-targeting immunotherapy.
The C- terminus of the fibrinogen γ chain contains two distinct non-overlapping sites at γ400–411 and γ377–395, which mediate platelet engagement and inflammation respectively (Fig. 1a). Peptide γ400–411 is the binding site for the platelet αIIbβ3 integrin receptor and is required for platelet aggregation. Peptide γ377–395 is the binding site for the CD11b I-domain of CD11b/CD1814 and is required for fibrin-induced activation of microglia and macrophages13,15,16. The γ377–395 binding site is considered “cryptic” in the soluble fibrinogen molecule and is exposed only upon conversion of fibrinogen to insoluble fibrin14 (Fig. 1a). We hypothesized that monoclonal antibodies against γ377–395 would selectively recognize fibrin and inhibit fibrin’s interaction with the CD11b I-domain without affecting blood coagulation. To control for CD11b I-domain binding specificity, we also generated mouse monoclonal antibodies against the γ190–202 fibrin epitope, which is also cryptic, but is not required for binding to the CD11b I-domain14. Using a streamlined screening strategy, we selected monoclonal antibodies that selectively bind fibrin and inhibit microglia activation (Supplementary Fig. 1a). Antibodies 5B8, 4E11, and 4F1 had the highest selectivity and specificity for γ377–395, while anti-γ190–202 1E3 did not bind γ377–395, as expected (Fig. 1b, Supplementary Fig. 1b). To discriminate between fibrinogen and fibrin in biochemical and cellular assays, we developed an in vitro fibrin substrate that binds to recombinant CD11b I-domain more strongly than fibrinogen (Supplementary Fig. 1c), suggesting γ377–395 epitope exposure in vitro. All antibodies against cryptic epitopes bound with higher affinity to fibrin than to fibrinogen; among antibodies targeting γ377–395, 5B8 bound fibrin to the greatest degree with minimal binding to soluble fibrinogen (Fig. 1c, Supplementary Fig. 1d). Competitive binding assays showed that 5B8 bound to human and mouse γ377–395 peptides, but not γ190–202 peptide (Supplementary Fig. 1e). 5B8 inhibited binding of the CD11b I-domain to fibrin (Fig. 1d), suggesting that 5B8 interferes with the ligand–receptor interaction.
Fig. 1 │. Generation and characterization of monoclonal antibody 5B8 targeting the γ377–395 fibrin epitope.

a, Structural map of cryptic epitope γ377–395 in the γC-domain of fibrinogen. The γ377–395 epitope (red) includes a core β-strand and portion of the fibrin γC-domain’s C-terminal loop and binds to the CD11b I-domain. b, Binding affinity of 5B8 to γ377–395 measured by ELISA. Data are mean ± s.e.m. of 4 independent experiments. c, Comparison of 5B8 binding to fibrin and fibrinogen by ELISA. Data are mean ± s.e.m. of 3 independent experiments. d, 5B8 competition with the CD11b I-domain for binding to fibrin by competitive ELISA. Data shown from one experiment. e, Microglia stimulated with fibrin in the presence of 5B8 (10, 20, and 40 μg/ml). Representative images are shown from unstimulated, and fibrin-treated microglia in the absence or presence of 40 μg/ml 5B8. Scale bar, 10 μm. Quantification of microglia activation (increased cell size) at 48 h after treatment is shown. Data are mean ± s.e.m. from 4 independent experiments (untreated, fibrin, fibrin + 40 μg/ml 5B8) analyzed with manual and automated quantification with similar results. ****P < 0.0001 by one-way analysis of variance (ANOVA) with Sidak’s multiple comparisons test. f, In vitro fibrin polymerization was examined in the presence of fibrinogen (FN) alone or with 5B8, IgG2b, or GPRP. Data are representative of two independent experiments with similar results. g, aPTT assay was performed in human plasma in the presence of 5B8, IgG2b, or GPRP. Data are representative of two independent experiments with similar results. h, In vivo clotting times of blood from 5B8-injected mice and uninjected controls. Data are mean ± s.e.m.; n = 8 mice per treatment. P = 0.7768 (two-tailed Mann-Whitney test); n.s., not significant.
Fibrin activates microglia, as evidenced by morphologic activation characterized by increase in cell size that is also associated with gene expression changes13,16. The morphologic activation assay, combined with automated quantification, was suitable for an unbiased and rapid comparison of multiple fibrin antibody clones in primary microglia cultures. Notably, among all antibodies generated against cryptic epitopes, only antibodies against γ377–395 inhibited fibrin-induced morphologic activation of microglia; the highest inhibition (~87%) was by 5B8 (Supplementary Fig. 2a). The anti-γ190–202 1E3 antibody also bound fibrin but did not inhibit microglial activation (Supplementary Fig. 2a), suggesting that γ377–395 must be targeted to inhibit fibrin’s proinflammatory functions. The F(ab) fragment of 5B8 also inhibited fibrin-induced microglial activation (Supplementary Fig. 2b). 5B8 inhibited fibrin-induced morphologic microglial activation in a concentration-dependent manner and reduced proinflammatory gene expression (Fig. 1e, Supplementary Fig. 2c,d). Isotype control IgG2b with endotoxin concentrations <0.002 EU/μg (Supplementary Figs. 2e) was used as control for all experiments in the study. 5B8 did not alter the polymerization time of fibrinogen to fibrin, did not inhibit activated aPTT in human plasma, and had no effect on murine plasma clotting time in vivo (Fig. 1f-h). ~80% of 5B8 was recovered 5 days after a single intraperitoneal (i.p.) injection into healthy wild-type mice (Supplementary Fig. 2f), suggesting that 5B8’s preferential binding to fibrin and minimal binding to soluble fibrinogen result in a minimal “sink-effect”.
In microglia and macrophages, fibrin induces proinflammatory gene expression associated with immune response and cell recruitment16. In bone marrow derived macrophages (BMDMs), 5B8 reduced fibrin-induced transcriptional activation of genes related to immune cell migration, adhesion, inflammatory responses, regulation of T cell proliferation, and chemotaxis, as shown by whole-genome microarray and Gene Ontology (GO) analysis (Fig. 2a-c). A heat map of the top 20 most affected transcripts showed that 5B8 inhibits fibrin-induced transcription of a group of genes that regulate innate/adaptive immune responses (Fig. 2c). Reductions in the expression of Ccl24, Tnfs18, Il12b, Cxcl3, and Ifng by 5B8 were validated by quantitative RT-PCR (Fig. 2d). 5B8 did not affect LPS-induced expression of proinflammatory genes (Fig. 2e), suggesting that the suppression of innate immune activation by 5B8 may be selective for fibrin. Further, 5B8 or its F(ab) fragment alone inhibited microglial activation and chemokine gene expression in fibrinogen-induced encephalomyelitis (Supplementary Fig. 3), an acute neuroinflammatory model resulting from fibrin-induced microglial activation16. Thus, we have devised a paradigm to develop a fibrin epitope-specific immunotherapy. The result is 5B8, a highly selective monoclonal antibody that blocks fibrin-induced, CD11b-mediated activation of innate immunity without affecting fibrin polymerization, in vivo clotting time, or aPTT in human plasma.
Fig. 2 │. Gene expression changes by 5B8 treatment of fibrin-stimulated BMDMs.
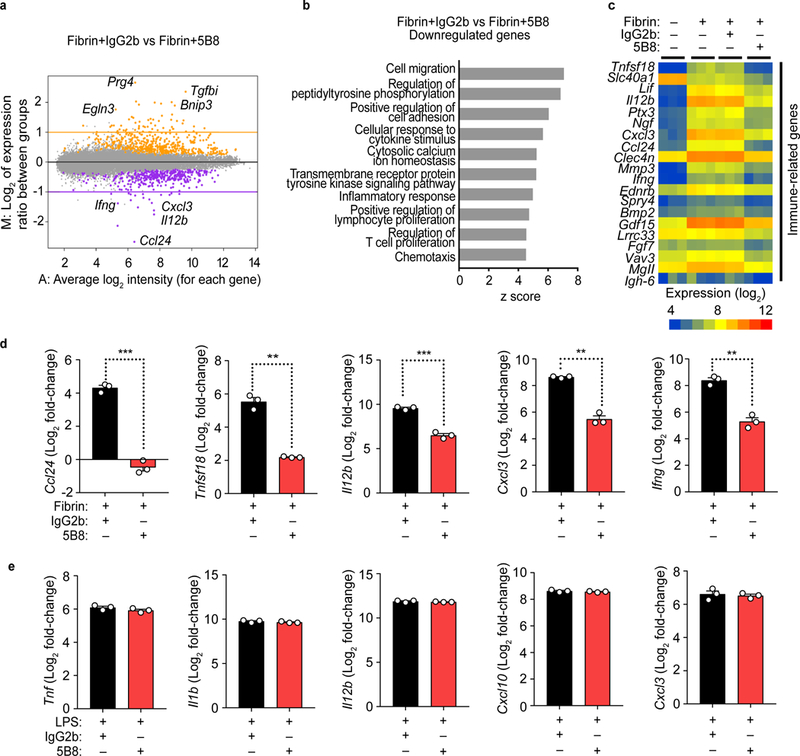
a, MA plot of whole-genome microarray analysis of 5B8- vs. IgG2b-treated BMDMs 6 h after fibrin stimulation. Colors indicate genes whose expression was significantly increased (orange) or decreased (purple) by 5B8; the remaining genes are in gray. Data are from 3 independent experiments. Linear models were fitted for each gene using the Bioconductor ‘limma’ package in R. Moderated t-statistics, fold change and the associated P values were calculated for each gene (two-tailed, raw P < 0.001). b, The top 10 biological processes relevant to 5B8-reduced innate/adaptive immune transcripts in BMDMs were identified by GO-Elite analysis (Z score >2.0, P < 0.05) c, Heat map of the top 20 transcripts that were significantly downregulated after 5B8 treatment of fibrin-stimulated BMDMs. Whole-genome microarray data from 3 independent experiments. d, Relative expression of transcripts of proinflammatory genes Ccl24, Tnfsf18, Il12b, Cxcl3, and Ifng in fibrin-stimulated BMDMs treated with 5B8 or IgG2b, measured by qRT-PCR. Y-axis indicates log2 value of relative fold changes in qRT-PCR results. Data are mean ± s.e.m. from 3 independent experiments. *** P = 0.0001 (Ccl24), ** P = 0.0052 (Tnfsf18), *** P = 0.0010 (Il12b), ** P = 0.0050 (Cxcl3), and ** P = 0.0015 (Ifng) by unpaired two-tailed t-test with Welch’s correction. e, 5B8 had no effect on LPS-induced gene expression in BMDMs analyzed by qRT-PCR. Y-axis indicates log2 value of relative fold changes in qRT-PCR results. Data are mean ± s.e.m. from 3 independent experiments. No significant difference by unpaired two-tailed t-test with Welch’s correction.
Fibrin induces NADPH oxidase-mediated neurodegeneration.
A consequence of activation of innate immunity in CNS autoimmune and neurodegenerative diseases is neurotoxicity, its successful treatment remaining a major unmet clinical need. Intriguingly, fibrin induced expression of several genes that regulate oxidative stress and ROS release (Fig. 3a) and that are implicated in neurotoxicity7,21. Ncf4, the gene that encodes the p40phox subunit of the NADPH oxidase Nox2 complex, was among the highest fibrin-upregulated genes (Fig. 3a). NADPH oxidase is a multicomponent enzyme system upregulated in human MS22 and AD23. Increased NADPH-oxidase activation and oxidative stress are common mechanisms of neurodegeneration in diseases with fibrin deposition, including MS, AD, stroke, traumatic brain injury, and brain aging6,24,25. Intriguingly, fibrin increased protein expression of the Nox2 component gp91phox, phosphorylation of p40phox, and NADPH oxidase activity; these increases were all reduced when macrophages were treated with 5B8 (Fig. 3b,c). 5B8 treatment inhibited fibrin-induced ROS release in both mouse and human macrophages (Fig. 3d,e). Fibrin-induced ROS generation was reduced in BMDMs when the p47phox subunit of NADPH oxidase was ablated genetically (p47phox–/–) and when NADPH-oxidase and CD11b were inhibited by treatment with apocynin or the anti-CD11b antibody M1/70, respectively (Fig. 3f,g and Supplementary Fig. 4a). In the culture conditions tested, fibrin stimulation or treatment with antibodies did not affect BMDM cell density (Supplementary Fig. 4b). In a co-culture of cortical neurons and macrophages, which recapitulates the non-cell-autonomous degeneration of CNS neurons, fibrin-primed macrophages induced significant loss of MAP-2+ neurons and increased fragmentation and blebbing of neurites (Fig. 3h). Notably, 5B8 effectively blocked this pathogenic cascade (Fig. 3h). These results suggest that fibrin causes neurotoxicity via a causal chain that involves CD11b-mediated innate immune cell activation and increased ROS production by NADPH oxidase.
Fig. 3 │. 5B8 blocks fibrin-induced ROS production and axonal damage.
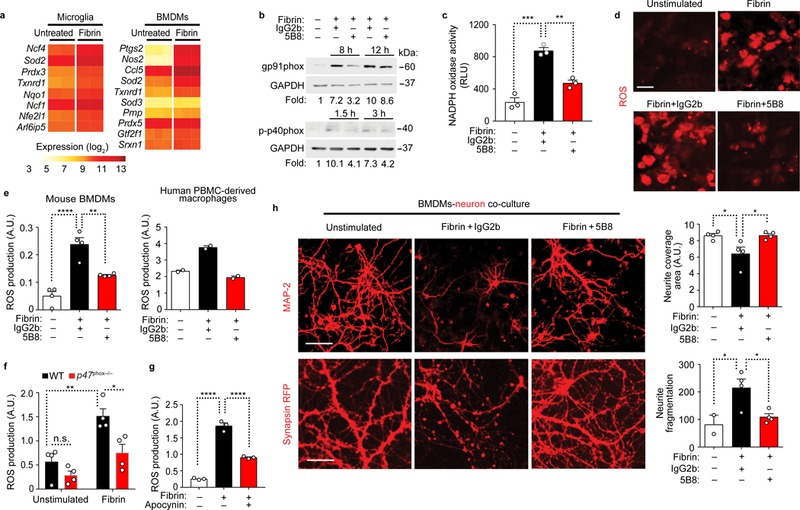
a, Heat map of significantly differentially expressed ROS-related genes in fibrin-induced microglia and BMDMs (|log2 fold change| > 0.585 and FDR < 0.05; two-tailed moderated t-test). b, Immunoblot for gp91phox and p-p40phox in BMDMs stimulated with fibrin as indicated in the presence of 5B8 or IgG2b. Representative cropped blot images from 3 independent experiments are shown. Full blots are included in Supplementary Fig. 9. c, NADPH oxidase activity in 12 h fibrin-stimulated BMDMs in the presence of 5B8 or IgG2b. Data are mean ± s.e.m. of 3 independent experiments. RLU, relative chemiluminescent light units. ** P = 0.0015 and *** P = 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. d, ROS production by fibrin-stimulated BMDMs detected with dihydroethidium (DHE, red). Scale bar, 10 μm. Data are representative of three independent experiments with similar results. e, Quantification of ROS production by DHE in BMDMs (left) and human PBMC-derived macrophages (right) 48 h after fibrin stimulation in the presence of 5B8 or IgG2b. Data are mean ± s.e.m. of 4 (left graph) and 2 (right graph) independent experiments. A.U., arbitrary units. ** P = 0.0033, **** P <0.0001 (mouse BMDMs) by one-way ANOVA with Bonferroni multiple comparisons test. f, ROS production 24 h after fibrin stimulation in BMDMs isolated from WT and p47phox–/– mice. Data are mean ± s.e.m. of n = 4 mice per group. A.U. * P = 0.0204, ** P = 0.0040 by two-way ANOVA with Sidak’s multiple comparisons test. n.s., not significant. g, ROS production measured by DHE in murine BMDMs 24 h after fibrin stimulation in the presence of apocynin. Data are mean ± s.e.m. of 3 independent experiments. A.U.-- **** P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. h, MAP-2+ (upper panel) and synapsin-RFP (lower panel) cortical neurons co-cultured with unstimulated or fibrin-stimulated BMDMs in the presence of 5B8 or IgG2b. Scale bars, 50 μm (top panels) and 30 μm (bottom panels). Quantification of MAP-2+ neurites (top) and synapsin-RFP+ neurites (bottom). Data are mean ± s.e.m. from 4 independent experiments * P = 0.0385 (unstimulated vs Fibrin+IgG2b, top), * P = 0.0370 (Fibrin+IgG2b vs Fibrin+5B8, top); * P = 0.0168 (Fibrin+IgG2b vs Fibrin+5B8, bottom) by one-way ANOVA with Bonferroni multiple comparisons test.
Fibrin immunotherapy protects from axonal damage in EAE.
Evidence from human MS has underscored the contributions of fibrin, innate immunity, and oxidative stress to disease onset, progression, and disability3,8,9,19,22,26. To examine the role of fibrin on neurotoxicity in vivo, we first tested the effects of 5B8 in three experimental autoimmune encephalomyelitis (EAE) models that simulate key aspects of MS: relapsing-remitting PLP139–151 EAE, chronic MOG35–55 EAE, and adoptive transfer of CD4+ T cells differentiated under TH1-polarizing conditions. Given prophylactically, 5B8 reduced neurologic signs in all three models compared to IgG2b isotype control (Fig. 4a). Given prophylactically, 5B8 reduced the mean maximum clinical score and delayed the first day of onset as compared to the IgG2b-injected control groups (Supplementary Tables 1, 2). Given therapeutically, 5B8 reduced the severity of relapses in PLP139–151 EAE (Fig. 4b, Supplementary Table 1). In all EAE treatment groups, 5B8 reduced the percentage of paralyzed mice (Fig. 4c). Fibrin is a cross-linked polymer of fibrinogen molecules. Consequently, anti-fibrinogen antibodies detect both fibrinogen and fibrin11. Fibrinogen is converted to fibrin at sites of increased coagulation activity11. In EAE spinal cord, coagulation activity promotes the conversion of fibrinogen to fibrin, leading to parenchymal fibrin deposition at areas of microglia activation and demyelination27. After i.p. injection of biotinylated 5B8 in mice with EAE, 5B8 spatially correlated with fibrin(ogen)-rich areas in the spinal cord (Fig. 4d), demonstrating target engagement.
Fig. 4 │. 5B8 suppresses EAE and engages fibrin target.
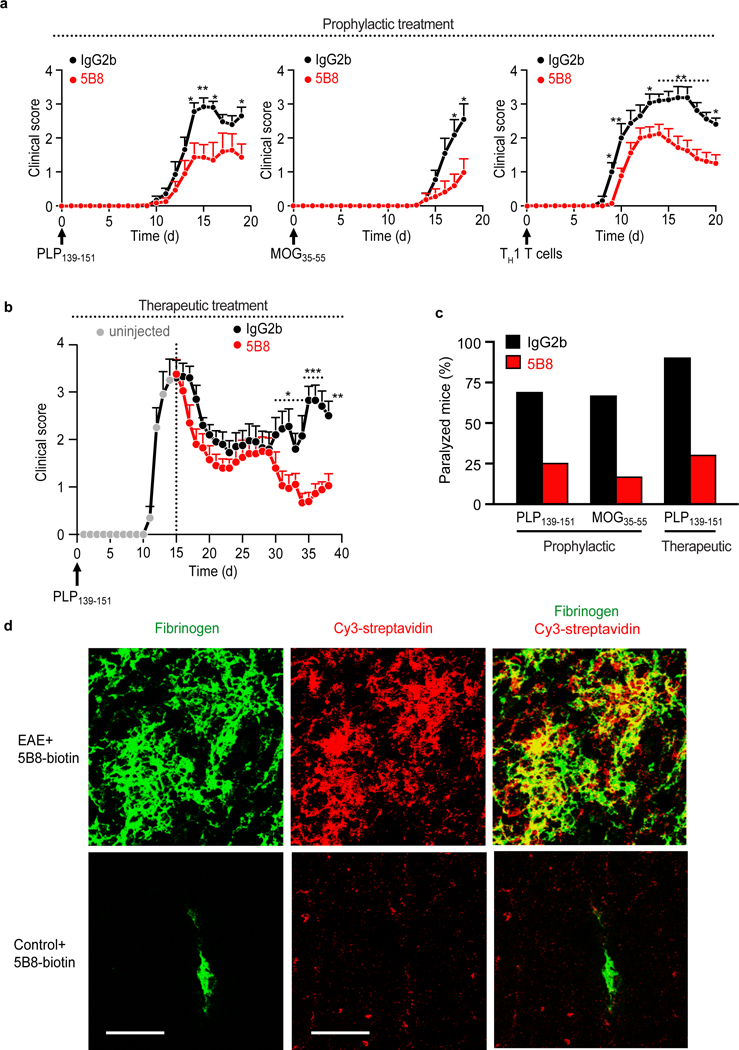
a, Reduction of clinical signs after 5B8 administration in four EAE models. Prophylactic administration of 5B8 or IgG2b isotype control in PLP139–151, MOG35–55, and adoptive transfer TH1 EAE (* P < 0.05, ** P < 0.01, linear mixed effects model with two-tailed permutation test). Mice were each given 800 μg of either 5B8 or isotype-control IgG2b every two days from day 0. Data are mean ± s.e.m. from PLP139–151 (n = 16 IgG2b and n = 16 5B8), MOG35–55 (n = 11 IgG2b and n = 12 5B8), and TH1 EAE (n = 15 IgG2b and n = 24 5B8) b, Therapeutic administration of 5B8 or IgG2b isotype control in PLP139–151 EAE after peak of disease (* P < 0.05, ** P < 0.01, *** P < 0.001, linear mixed effects model with two-tailed permutation test). For therapeutic treatment, antibodies were injected every two days starting at the peak of the initial paralytic episode. Data are mean ± s.e.m. from PLP139–151 EAE (n = 10 IgG2b and n = 10 5B8). c, Effect of 5B8 treatment on the percentage of paralyzed mice (defined as partial or complete hindlimb paralysis, score >2.5) in three EAE groups. d, Target engagement of i.p. injected biotinylated 5B8 in MOG35–55 EAE mice and healthy non-immunized control mice. Confocal microscopy of spinal cord sections from MOG35–55 EAE mice shows the spatial correlation (yellow) between biotinylated 5B8, detected with Cy3-streptavidin (red), and fibrin deposition, detected with a fibrin(ogen) antibody (green), in MOG35–55 EAE mice. Representative images are shown from n = 3 mice per group. Scale bar, 200 μm.
To determine whether 5B8 inhibits inflammatory cell recruitment into the CNS, we treated after induction of EAE in Cx3cr1GFP/+Ccr2RFP/+ mice. 5B8 inhibited both the accumulation of Cx3cr1+ microglia and the infiltration of Ccr2+ monocytes into the CNS (Fig. 5). Increased NADPH oxidase activity and excessive ROS production are implicated in MS28, and ROS generated by invading and resident CNS macrophages mediate demyelination and axonal damage in EAE7. 5B8 markedly reduced axonal damage, ROS generation, inflammation, and demyelination in EAE (Fig. 5, Supplementary Fig. 5a,b). In accordance with prior studies7,12, axonal damage correlated with ROS+ areas in EAE lesions (Supplementary Fig. 5c). The fibrin γ377–395 peptide inhibits binding of fibrin to CD11b and suppresses EAE without affecting peripheral T cell responses13. Similarly, 5B8 did not significantly alter peripheral T cell responses in PLP139–151 EAE mice (Supplementary Fig. 6), suggesting that 5B8 does not modulate peripheral adaptive immune responses. These results suggest that systemic delivery of 5B8 in vivo is well tolerated and that fibrin immunotherapy suppresses innate immune activation, oxidative stress, demyelination, and axonal damage in EAE.
Fig. 5 │. 5B8 inhibits microglial activation, monocyte recruitment, and axonal damage in EAE.
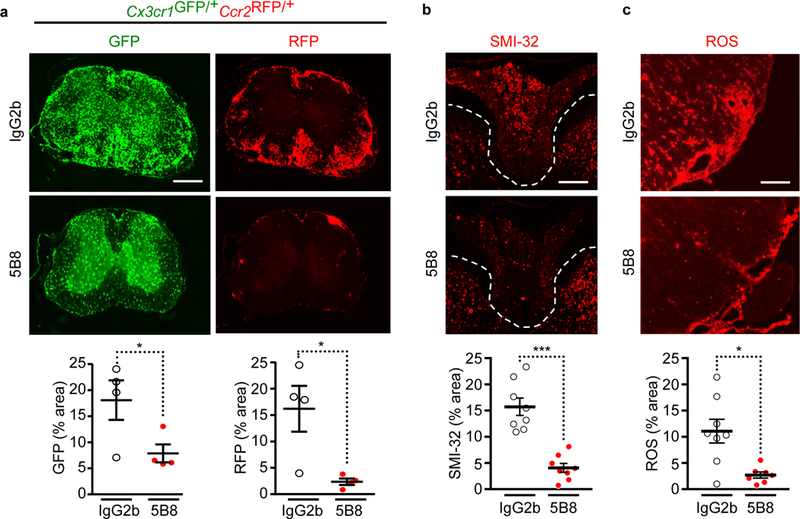
a, Spinal cord images of 5B8- or IgG2b-treated MOG35–55 EAE Cx3cr1GFP/+Ccr2RFP/+ mice. Scale bar, 500 μm. Quantification of Cx3cr1+ microglia (GFP, green) and Ccr2+ macrophages (RFP, red). Data are mean ± s.e.m.; n = 4 mice per treatment. * P = 0.0495 (GFP), * P = 0.0286 (RFP), by two-tailed Mann-Whitney test. b, SMI-32 immunoreactivity indicative of axonal damage in spinal cords of 5B8- or IgG2b-treated PLP139–151 EAE mice. Scale bar, 200 μm. Dotted lines indicate spinal cord dorsal column white matter. Quantification of SMI-32 immunoreactivity. Data are mean ± s.e.m.; n = 8 mice per group. *** P = 0.0002 , two-tailed Mann-Whitney test. c, ROS detection in the spinal cord with DHE of 5B8- or IgG2b-treated PLP139–151 EAE mice. Scale bar, 100 μm. Quantification of DHE. Data are mean ± s.e.m.; n = 8 IgG2b and n = 7 5B8. * P = 0.0140 by two-tailed Mann-Whitney test.
Fibrin immunotherapy protects AD mice.
Neurodegeneration driven by the innate immune system has emerged as a key pathogenic mechanism in AD29. We therefore tested whether blocking fibrin/CD11b interactions protects against amyloid-related neurodegeneration. In 5XFAD mice, in which transgenes encoding familial AD-mutant forms of human amyloid precursor protein and presenilin 1 are expressed in neurons, fibrin was detected in the brain as early as 3 months of age; at 5 months, fibrin accumulation was abundant at sites of amyloid deposition, which were surrounded by CD11b+ microglia (Fig. 6a, Supplementary Fig. 7a). In 5XFAD mice, fibrin deposits were detected in the vicinity of dystrophic neurites (Fig. 6b), a key feature of neurodegeneration in AD30. Systemically administered 5B8 spatially correlated with fibrin-rich areas surrounding amyloid plaques in 5XFAD mice (Fig. 6c, Supplementary Fig. 7b), demonstrating brain penetration and target engagement.
Fig. 6 │. In vivo imaging of fibrinogen leakage in 5XFAD mice.

a, Fluorescent Aβ probe methoxy-X04-positive amyloid plaque in a cortical section from a 3-month-old 5XFAD mouse co-labeled for fibrinogen (red) and CD11b (green). Scale bar: 20 μm. n = 4 mice. Representative images are shown. b, In vivo two-photon imaging of cortex from Thy1-YFP:5XFAD mice at 11 months of age shows Alexa594-conjugated fibrinogen (administered intravenously) (red), a methoxy-X04-positive Aβ plaque (blue), and dystrophic neurites (green, swollen green structures). Arrowheads indicate fibrinogen (red) surrounding a Aβ plaque (blue) at areas of neuritic loss. Arrows indicate fibrinogen extravasation at areas of dystrophic neurites (yellow) in proximity to a plaque (blue). Scale bars: 10 μm. (n = 4 Thy1-YFP:5XFAD mice and n = 2 Thy1-YFP mice. Representative images are shown. c, 5B8 in vivo target engagement in the brain of 5XFAD mice. Brain sections of 5XFAD i.p.-injected with biotinylated 5B8 were stained with Cy3-streptavidin (red), anti-fibrinogen (green), and methoxy-X04-positive Aβ plaque (blue). Orthogonal views of the y/z and x/z planes show localization of fibrinogen and 5B8 antibody around amyloid plaques. Scale bar, 10 μm. n =3 mice Representative images are shown.
To determine whether blocking fibrin/CD11b interactions protects against amyloid-related neurodegeneration, we treated 5XFAD mice with 5B8 for 2 months, starting at 3.5 months of age (1.5 months after appearance of amyloid plaques and microglial activation) (Fig. 7a). 5B8 reduced reduced loss of cholinergic neurons and microglia activation around plaques in 5XFAD mice (Fig. 7b,c) without significantly affecting amyloid plaques or the number of Mac2+ macrophages around plaques (Supplementary Fig. 8a,b). Whole-genome microarray and GO analysis of cortical gene expression in 5XFAD mice showed that 5B8 suppressed five key pathways: complement pathway, antigen presentation, cytokine response, lysozyme, and ROS (Fig. 7d). Overall, these findings suggest that blocking fibrin/CD11b signaling suppresses amyloid-induced neurodegeneration and reduces the expression of genes that promote inflammation and oxidative stress.
Fig. 7 │. 5B8 protects against neurodegeneration and inflammatory responses in 5XFAD mice.
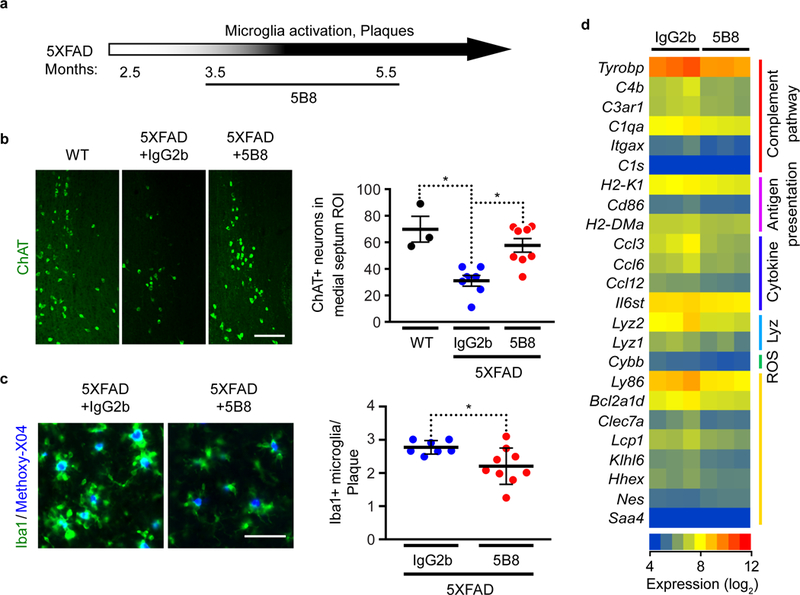
a, Schematic representation of 5B8 administration after appearance of plaques and microglia activation in 5XFAD mice (800 μg i.p. every other day for 2 months starting at 3.5 months of age). b, ChAT-positive cholinergic neurons in the medial septum of non-transgenic littermate control (WT), 5XFAD mice treated with IgG2b or 5B8. Scale bars 80 μm. Quantification of ChAT+ neurons. Data are mean ± s.e.m.; n = 3 WT, n = 7 IgG2b-treated and n = 8 5B8-treated 5XFAD mice. * P = 0.0211 (WT vs IgG2b), * P = 0.0157 (IgG2b vs 5B8) by Kruskal–Wallis with Dunn’s multiple comparisons test. c, Iba-1+ cells (green) around Methoxy-X04+ plaques (blue) in the cortex of 5XFAD mice treated with IgG2b or 5B8. Quantification of plaque-associated Iba-1+ microglia. Scale bar: 50 μm. Data are mean ± s.e.m.; n = 7 IgG2b-treated and n = 9 5B8-treated 5XFAD mice. * P = 0.0337 by two-tailed Mann-Whitney test. d, Affymetrix microarray gene expression analysis in the cortex of 5B8- or IgG2b-treated 5XFAD mice. Linear models were fitted for each gene using the Bioconductor ‘limma’ package in R. Moderated t-statistics, fold change and the associated P values were calculated for each gene (two-tailed, raw P < 0.05). Heatmap of select genes with differential expression patterns from GO terms were identified by GO-Elite analysis.
Fibrin immunotherapy suppresses TYROBP network genes.
Gene network analysis has broadened our understanding of AD pathogenesis by revealing immune gene networks as likely causal contributors to AD pathology31. Targeting these networks could culminate in the development of novel therapeutics. To identify gene networks that might be regulated by 5B8, we interrogated our transcriptomic data using biological network analysis. First, we performed genetic network modeling of genes downregulated by 5B8. We found a highly connected hub of co-expressed genes primarily within the complement pathway, including TYROBP (DAP12), C4b, and C1q (Fig. 8a). The 5B8-downregulated cluster of co-expressed genes shows a striking overlap with gene networks implicated in the pathogenesis of human AD, including those encoding the TYROBP-related microglial module31. Indeed, TYROBP, encoding a co-receptor for CD11b and the human AD-associated receptor Trem2, was one of the top 5B8-downregulated genes (Fig. 7d, Fig. 8a). Overlay of gene expression data obtained in 5B8-treated 5XFAD mice with neuroinflammatory gene networks from human AD brains showed a reduction of human AD-related inflammatory networks by 5B8. By overlaying our data on a mouse version of the human AD TYROBP module31, we found that 5B8 downregulated 65% of the Tyrobp-related network at various levels of significance, including Tyrobp at the nexus of the network (Fig. 8b). Several components of the TYROBP signaling pathway were also downregulated (Supplementary Fig. 8c). This bioinformatic approach allowed us to gain insights into the mechanisms of action of 5B8 treatment, as we identified downregulation of co-expressed genes primarily within the complement pathway that were relevant to AD pathogenesis. Integrated gene network analysis could be a useful tool to better understand the mechanism of action of new drugs and assess their efficacy in suppressing inflammatory gene networks relevant to human disease. Thus, fibrin inhibition by 5B8 may suppress pathogenic activation of innate immune pathways that mediate amyloid-related neurodegeneration.
Fig. 8 │. 5B8 suppresses the complement/TYROBP microglial module in 5XFAD mice.
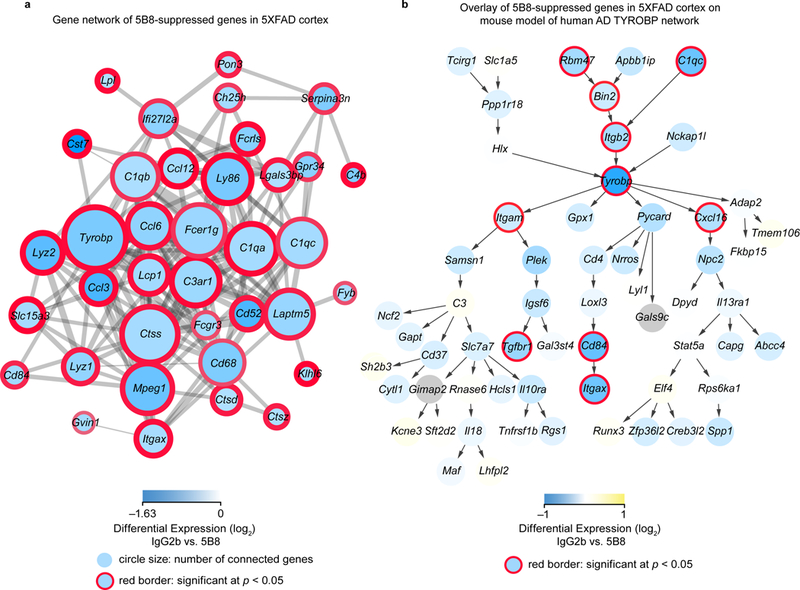
a, Co-expression analysis revealed 5B8-downregulated genes to be densely interconnected, with Tyrobp forming a major hub. Blue shading indicates the level of reduction of gene expression by 5B8 compared to IgG2b treatment. The thickness of the red border around the circles indicates the statistical significance of the differential expression, and the size of the circles indicates the number of connected genes in this co-expression network. Downregulated genes of interest were selected from those with log2 fold change of −0.5 or less and raw p-value < 0.05 (two-tailed moderated t-test). b, Mouse model of the human AD TYROBP network with data overlay of the 5B8-downregulated genes in the 5XFAD mice. Yellow-Blue gradient fill color indicates differential expression and red border thickness indicates significance of P < 0.05 (two-tailed moderated t-test).
Discussion
Our study revealed a previously unknown neurodegenerative pathway in which fibrin activates NADPH oxidase in innate immune cells to promote ROS production and neurotoxicity. By developing an antibody-based approach to selectively target a key inflammatory fibrin domain, we showed here that fibrin-targeting immunotherapy can be selective and efficacious in suppressing neuroinflammation and neurodegeneration. Consequently, preventing fibrin from engaging this mechanism may lead to the development of therapeutic strategies for neurodegenerative diseases with neurovascular dysfunction. This discovery may have implications beyond the CNS, since fibrin deposition is a common thread in inflammatory pathologies, such as rheumatoid arthritis, colitis, and Duchenne muscular dystrophy (reviewed in11,12). Fibrin/CD11b signaling induces chemokine release and macrophage recruitment16 and increased NADPH oxidase activation culminating in ROS release and oxidative stress (this study). Therefore, inhibitors of fibrin/CD11b interaction, such as 5B8, have the potential to affect both inflammatory processes and oxidative stress. Inhibiting fibrin formation with anticoagulants can cause adverse effects by increasing bleeding risk32. Epitope-selective targeting of fibrin may overcome this challenge by selective suppression of its damaging functions without adverse anticoagulant effects. Overall, fibrin immunotherapy could represent a selective multi-indication therapy to suppress fibrin-induced chronic inflammation by inhibiting release of proinflammatory factors and oxidative stress at sites of increased vascular permeability in neurological and other inflammatory diseases.
Our study revealed a novel function of fibrin/CD11b signaling as an activator of NADPH oxidase-dependent ROS production. CD11b integrin-dependent respiratory bursts require DAP12 receptor signaling33, . CD11b and DAP12 are co-expressed in microglia and act synergistically to promote the production of superoxide ions and induce neuronal death33. Intriguingly, DAP12 was a major pathway downregulated by fibrin immunotherapy in the 5XFAD mouse brain. Fibrin—via activation of CD11b/CD18 signaling—might increase DAP12 expression and potentiate CD11b/DAP12-dependent oxidative damage by promoting the phosphorylation and assembly of the NADPH oxidase complex34,35 and activating kinase pathways13. In addition to fibrin, several other immune-mediated mechanisms can activate NADPH oxidase, such as Aβ binding to CD36 scavenger receptor, or activation of CR3 or TLR4 signaling by α-synuclein or LPS25. Aβ-induced activation of NADPH oxidase in innate immune cells contributes to neurovascular dysfunction, neurodegeneration, and cognitive decline in AD mice5,36. Fibrin and Aβ may have an additive effect on NADPH oxidase activation via CD11b/CD18 and CD36, respectively. Genome-wide association studies, converged linkage analysis, and gene expression data have pointed to immune regulation of AD pathology. These studies, in combination with limited success of clinical trials targeting amyloid, have led to a reassessment of AD pathogenesis and the need for developing new treatments against different targets37. Vascular and amyloid pathologies are independent and additive predictors of cognitive decline in the elderly38,39, suggesting that they may need to be targeted independently for maximal therapeutic benefit. Fibrin immunotherapy did not reduce Aβ deposition after 2 months of administration, suggesting that the neuroprotective effects of inhibiting fibrin/CD11b interactions occur in the presence of Aβ. Future studies will determine whether fibrin/CD11b signaling and 5B8 also affect cognitive performance in AD mice.
Fibrin immunotherapy represents a novel approach to selectively suppress pathogenic innate immune responses with potential clinical implications. In MS, anti-inflammatory medications have little effect in the progressive phase of disease, which is mediated by innate immune mechanisms3,8. Other modulators of innate immunity, such as complement, CR3, TNF, NF-κB, PI3K, and TLR4, are ubiquitously expressed in the brain and the periphery and have reported roles in regulating physiological CNS functions. In contrast, fibrin is not present in the normal brain, but is abundantly deposited in the CNS after BBB disruption11. 5B8 is selective for inhibiting innate immune activation by fibrin-CR3 interactions and does not affect activation of macrophages by other ligands, such as LPS, suggesting that innate immune activation by other ligand/receptor interactions can proceed normally. Phagocytic pathways in innate immune cells are required for debris clearance and remyelination40. Fibrin-primed macrophages inhibit the differentiation of oligodendrocyte precursor cells41, which are susceptible to oxidative stress42. Fibrin immunotherapy could suppress fibrin-induced chronic inflammation and potentially promote repair by inhibiting oxidative stress and release of proinflammatory factors. Microglia and macrophages are functionally distinct populations with diverse functions in neurodegenerative diseases43–46. Future studies using CyToF and large transcriptomic profiling in microglia and macrophages will be required to determine the potential differential effects of fibrin on innate immune populations. The fibrin γ377–395 inflammatory epitope has been validated genetically in inflammatory models in the brain and periphery with findings replicated in numerous laboratories (reviewed in11,12). It is possible that pharmacologic targeting of fibrin-CR3 interactions could represent a selective treatment to add to the toolbox of anti-innate immune therapies that may be particularly efficacious for suppressing neuroinflammation at sites of BBB leakage and vascular damage with fibrin deposition.
The available genetic and pharmacologic tools used to study the γ377–395 epitope (fibrinogen gamma chain Fgg390−396A mutant mice, γ377–395 peptide, 5B8 antibody in this study) have primarily revealed pathogenic functions for the γ377–395 epitope in inflammation. Fibrinogen alpha chain-deficient (Fga–/–) and Fgg390−396A mice13,47,48, mice treated with either 5B8 (this study) or γ377–395 peptide13, or anticoagulants13,26,41 are not immunocompromised and can be housed in conventional animal facilities without opportunistic infections. In humans, congenital afibrinogenemia, a genetic disorder characterized by complete absence of fibrinogen, is associated with excessive bleeding but no increase in opportunistic infections49. Fibrin inhibits repair as persistent fibrin deposition inhibits wound healing50, while fibrinogen depletion increases remyelination41,51. These studies do not exclude the possibility that the fibrinogen γ377–395 epitope may have physiological functions that have not yet become apparent using the genetic and pharmacologic tools available. Collectively, prior genetic studies on the γ377–395 epitope and our findings support that fibrin is inducing, in a ligand-selective manner, a specific gene signature in immune cells that produces primarily pathogenic functions. Consequently, targeting fibrin-induced activation of innate immunity has the potential to suppress proinflammatory and neurotoxic pathways without significant interference with protective functions of the innate immune system. Future immunotoxicology studies will also be required to fully test potential immune dysfunction in the context of fibrin immunotherapy.
Fibrinogen is a coagulation factor essential for blood clotting. Studies in Fgg390−396A mice have shown that the γ377–395 epitope is not required for fibrin polymerization, platelet aggregation, and does not increase bleeding risk48. Fgg390−396A mice have normal fibrinogen concentrations, fibrin polymerization, platelet aggregation, normal thrombus formation in FeCl3-injured carotid arteries, pregnancy and birth rates. Furthermore, they do not develop spontaneous hemorrhagic events and tolerate major surgical procedures (e.g., abdominal surgeries), without inordinate bleeding48. In contrast to Fgg390−396A mice, Fga–/– mice that lack fibrinogen exhibit broad hemostatic abnormalities secondary to the lack of clotting function and failure of fibrinogen-supported platelet aggregation, and uniformly cannot sustain pregnancies due to hemostatic requirements at the placenta-maternal interface47, a phenotype also observed in mice lacking fXIIIA52. Consistent with experimental findings in Fgg390−396A mice, the fibrin γ377–395 peptide does not inhibit normal clotting time in vivo or fibrin polymerization in vitro13. Our study shows that 5B8 does not affect normal clotting time in vivo, fibrin polymerization in vitro, or aPTT in human plasma. Similar to the Fgg390−396A mice, mice treated with 5B8 even for prolonged periods up to ~2 months did not develop any spontaneous bleeding. The 5B8 γ377–395 epitope does not overlap with the γ398–406 residues known to be enzymatically cross-linked by transglutaminase fXIII53. Nevertheless, comparative studies of fXIII-mediated crosslinking in vitro and in vivo will be of interest in future studies of 5B8. This is could be of particular interest in light of studies of Fgg390−396A mice in a model of venous thrombosis54. Here, Fgg390−396A mice were shown to maintain fXIII-mediated γ chain cross-linking but at a reduced rate, and this resulted in smaller venous thrombi. Overall, studies in Fgg390−396A mice48, data obtained with the fibrin γ377–395 peptide13 and in this study suggest that the γ377–395 epitope and 5B8 do not interfere with fibrin clotting and presents limited risk of spontaneous hemorrhagic events. Future toxicology studies will be required to determine whether 5B8 may interfere with other features of hemostasis and thrombosis.
Together, data from human pathology, genetic and pharmacologic studies in mice, transcriptomic and signal transduction studies establish fibrin as a dual inflammatory and oxidative stress signal in the CNS and suggest that fibrin immunotherapy could prevent neurodegeneration and other complications in conditions associated with fibrin deposits at sites of BBB disruption. Fibrin immunotherapy may hold promise as a new therapeutic strategy for neurological and inflammatory disorders with vascular damage.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available at <insert link here>
Animals.
SJL/J, C57BL/6, Cx3cr1GFP/+ 55, Thy1-YFP (Tg(Thy1-YFP)HJrs)56, 5XFAD (B6SJL-Tg (APPSwFlLon,PSEN1*M146L*L286V) 6799 Vas/Mmjax)57, and p47phox–/–58 mice were purchased from The Jackson Laboratory and Sprague-Dawley rat P0 litters from Charles River Laboratories. 5XFAD were crossed with Thy1-YFP to generate 5XFAD:Thy1-YFP mice. Ccr2RFP/RFP on a C57BL/6 background provided by I. F. Charo (Gladstone Institutes), and bred to generate Cx3cr1GFP/+Ccr2RFP/+ mice59. Mice were housed in groups of five under standard vivarium conditions and a 12 h light/dark cycle. All animal protocols were approved by the Committee of Animal Research at the University of California, San Francisco, and in accord with the National Institutes of Health guidelines.
Mouse mAb production.
Synthesis of human fibrinogen γ190–202 and γ377–395 peptides and mouse monoclonal antibody production was performed by A&G Pharmaceuticals. Screening of 480 hybridomas was performed by ELISA against peptides or the carrier protein. Positive clones were expanded and retested to confirm peptide epitope reactivity to either γ190–202 or γ377–395. Target candidates were identified as outlined in Supplementary Fig. 1a. The structural map of the γC-domain of fibrinogen was generated using the PyMOL Molecular Graphics System, Version 1.6 Schrödinger, LLC with PDB ID 1FIC .
Peptide Binding assays.
Human γ377–395 peptide (Genscript) in carbonate buffer (0.1 M NaHCO3 and 0.15 M NaCl) was coated onto MaxiSorp ELISA plates (Thermo Fisher Scientific) overnight at 4 °C. Wells were incubated with blocking buffer (3% bovine serum albumin (BSA) and 0.1% NP-40 in PBS) for 2 h at 25 ˚C, washed with PBS 0.05% Tween-20. Antibodies 5B8, 4F1, 4E11, 1E3 were diluted in blocking buffer and added to the wells for 2 h at 25 ˚C followed by secondary polyclonal goat anti-mouse IgG/HRP (#P0447, DAKO) in blocking buffer for 2 h at 25 ˚C and developed by TMB substrate (Sigma-Aldrich) with 450nm absorbance measured with an Envision Microplate reader (Perkin Elmer). For competition ELISA, the 5B8 antibody was diluted to 2 nM in blocking buffer and preincubated with increasing concentrations of human or mouse γ377–395 and γ190–202 peptides for 3 h at 25 ˚C. Competition of 5B8 to γ377–395 or γ190–202 was assessed by incubating on plates coated with human γ377–395 peptide.
Fibrin/Fibrinogen binding.
Human plasminogen-free fibrinogen (#341578, EMD Millipore) was IgG depleted using Pierce albumin/IgG removal kit (Thermo Fisher Scientific). 25 μg/ml IgG-depleted human plasminogen-free fibrinogen in 20 mM HEPES pH 7.4 buffer was treated with 0.3 U/ml bovine thrombin (Sigma-Aldrich) and 7 mM CaCl2 in MaxiSorp ELISA plates (Thermo Fisher Scientific) for 1.5 h at 37 °C. Formed fibrin was dried onto the wells at 37 °C overnight. 25 μg/ml IgG-depleted human fibrinogen was coated onto the wells at 37 °C for 2.5 h. Plates were washed with binding buffer (10 mM HEPES, pH 7.4 150 mM NaCl, 3 mM EDTA, and 0.05% Tween-20) and incubated with blocking buffer (binding buffer containing 3% BSA) for 1 h at 25 ˚C. Monoclonal antibodies in blocking buffer were incubated for 1 h at 25 ˚C. Following washing with binding buffer, polyclonal goat anti-mouse IgG/HRP (#P0447, DAKO) in blocking buffer was added for 1 h at 25 ˚C and signal was developed by Lumi-Phos HRP (Lumigen) measured with an Envision Microplate reader. Experiments of antibody binding to peptides, fibrin, and fibrinogen performed at the Gladstone Institutes were independently reproduced at the UCSF Small Molecule Discovery Center and at Lundbeck, US with similar results.
CD11b I-domain production.
pET15-His-TEV-CD11b was constructed by cloning the CD11b I-domain (residues Q130-S326) into a pET15b-derived plasmid at the NdeI and XhoI sites, preceded by an N-terminal 6xHis tag and followed by a TEV protease cleavage site (pET15-His-TEV-CD11b), then transformed into Rosetta2(DE3) cells. Protein was expressed and purified as described60, with the exception that cells were lysed using a pressure driven microfluidizer and His-containing proteins were trapped using TALON cobalt affinity resin. Protein purity was assessed to be > 90% by LC/MS and gel electrophoresis.
CD11b I-domain ELISA.
96-well ELISA plates (Greiner) were coated with 25 μg/ml fibrin or fibrinogen and incubated in blocking buffer as indicated for binding assays for 1 h prior to addition of 50 μl per well of biotinylated CD11b I-domain in PBS with 0.5% BSA and 0.05% Tween-20 for 2 h at 37 °C, followed by incubation with 1 μg/ml HRP-coupled Streptavidin (BD Pharmingen, 1:1000) for 1 h at 25 ˚C and developed by incubation with TMB/E substrate (Chemicon-Millipore) and absorbance measured at 450nm with a Synergy H4 plate reader (BioTek). For 5B8 competition ELISA, after incubation in blocking buffer, 5B8 antibody was diluted at 2-fold concentrations from 0.02 μM to 10 μM and incubated for 2 h at 37 °C, followed by an incubation of 6.88 μM CD11b I-domain for 2 h at 25 ˚C.
Cell Culture.
Primary microglia and BMDMs were isolated, cultured, and treated with fibrin or LPS as previously described16. PBMCs (AllCells) (2×106 cells/mL) were plated in RPMI-1640 media supplemented with 10% heat-inactivated FBS, 1% penicillin-streptomycin, and 50 ng/ml human M-CSF (#300–25, Peprotech) in tissue-culture treated dishes (Corning). After 24 h, non-adherent cells were removed, and adherent cells were cultivated for 7–8 additional days at 37 °C in 5% CO2.
Morphometry.
Primary microglia were stimulated with fibrin for 48 has described16. Microglia were fixed with 4% PFA, permeabilized with 0.1% Triton X-100 and immunostained with FITC-labeled Isolectin B4 (1:300; Sigma-Aldrich) and/or CellMask™ Red Stain (Thermo Scientific). Blocking involved pre-incubation with 80 μg/ml antibodies or mouse IgG2b (clone MPC-11, BioXCell BE0086 distributed by the UCSF Monoclonal Antibody Core, endotoxin concentrations < 0.002 EU/μg) or IgG2b (eBiosciences endotoxin level < 0.01 EU/μg) for 2 h at 37 °C before plating of cells for a final concentration of antibodies at 40 μg/ml. Manual Analysis: Images were collected using an Axioplan 2 Zeiss microscope with an Axiocam HRc camera and analyzed using ImageJ. 250 cells per condition were counted for each experiment. Activated microglia were classified based on a surface area > 2,000 μm2. Automated Image Acquisition and Analysis: Images of microglia cells were acquired with the GEHC IN-Cell Analyzer 2000™, using a 10× lens and excitation/emission filter pairs of 350nm/455nm (CellMask™ Red Stain) and 579nm/624nm (Hoechst dye). Images were analyzed with the GEHC IN-Cell Developer Toolbox version 1.9. The Hoechst-stained nuclei were segmented using a “nuclear” segmentation type, with a set minimum target area of 30 μm2 and set sensitivity of 75%. To minimize artifacts, segmentation with less than 120 intensity units or area greater than 1000 μm2 were excluded. The CellMask™ Red-stained whole microglia cells were segmented using an “intensity” segmentation type, with a set threshold between 200 – 4095 intensity units. The borders of adjacent contacting cells were resolved using the “clump breaking” segmentation post-processing, which utilizes discrete nuclei as seeds. For segmented by size cells > 800 μm2 were classified as activated microglia. Only cells containing a nucleus within the cell body area were accepted. All segmented nuclei and cells were then recorded as individual counts.
Fibrin polymerization assay.
Human fibrinogen was diluted to 0.15 mg/ml in 20 mM HEPES (pH 7.4) containing 0.15 M NaCl and 5 mM ε-amino caproic acid and combined with 0.3 U/ml thrombin (Sigma-Aldrich), and 10 mM CaCl2. Antibodies (50 μg/ml) or GPRP peptide (Bachem) were incubated with fibrinogen for 2 h at 37 °C before mixing with CaCl2/thrombin. Absorbance at 350nm over intervals of 60 s was measured using a SpectraMax M5 microplate reader (Molecular Devices) with SoftMax Pro 5.2 software (Phoenix Technologies Ltd.).
Plasma clotting time and aPTT assay.
Plasma-clotting times were measured as described13 by combining equal volumes of citrated plasma and 2 U/ml bovine thrombin (Enzyme Research Laboratories) and 40 mM CaCl2 at 37 °C. aPTT assay was performed in pooled normal human plasma (Innovative Research) using 50 μg/ml 5B8 or IgG2b or 0.5 mM GPRP peptide (Bachem). Plasma was incubated with 5B8 or IgG2b at 37 °C for 2 h. Clotting was initiated by aPTT-XL (Ellagic acid activator) and 0.02 M CaCl2 (Thermo Fisher Scientific) and monitored at 405nm in a SpectraMax M5 microplate reader.
Real time qPCR, multi-plex qPCR and microarray analysis.
RNA isolation, real-time PCR, and microarray analysis were performed as described16. Primers are listed in Supplementary Table 3. Microarray data from GEO GSE71084 were used to select ROS genes with significant log2 expression to generate heat maps for primary microglia and BMDMs. For multiplex qPCR, primary microglia were plated in 96-well black μ-clear-bottom microtiter plates (Greiner Bio-One) in DMEM, 10 % FBS, 1 % penicillin/streptomycin and allowed to adhere for 24 h prior to being treated for 4 h with 1 μM fibrin D-dimer (HyTest) as described12. 20 μg/ml 5B8 or IgG2b were pre-incubated with fibrin D-dimer in DMEM for 3 h at 37 ºC before cell treatment. Gene expression was assessed using the RT2 Profiler PCR Array (Rat Inflammatory & Autoimmunity; Qiagen), and only differentially expressed genes (fold > 2, P < 0.05) were reported as the mean fold change. Gene expression antibody inhibition experiments performed at the Gladstone Institutes were independently reproduced at Lundbeck, US with similar results.
Oxidant detection with DHE.
BMDMs or PBMCs were incubated in RPMI containing 5 μM DHE (Invitrogen) for 30 min. 100,000 cells/well were plated on 96-well black μ-clear-bottom microtiter plates (Greiner Bio-One) precoated with 25 μg/ml fibrin. 20 μg/ml 5B8 or IgG2b (UCSF Monoclonal Antibody Core clone LTF-2) were added in fibrin-coated wells 2 h before plating cells. BMDMs were incubated with 300 μM apocynin (Calbiochem) for 1 h or 5 μg of anti-CD11b (M1/70, eBioscience) for 30 min before plating. Cells were incubated on fibrin for 24–48 h, fixed with 4% PFA for 10 min, and DHE fluorescence was detected at 518nm/605nm using SpectraMax M5 microplate reader. Cells were quantified by DAPI counterstaining. In mice, in vivo DHE administration and detection was performed as described15.
NADPH oxidase activity.
BMDMs were plated on 25 μg/ml fibrin-coated 6-well plates (Greiner Bio-One). 20 μg/ml 5B8 or IgG2b were added to the fibrin plates 2 h before plating cells. NADPH oxidase activity was assayed by the lucigenin-enhanced chemiluminescence method61. Cells were collected by a cell scraper and homogenized in ice-cold Krebs buffer pH 7.4 (119 mM NaCl, 2.5 mM KCl, 1mM NaH2PO4, 1.3 mM MgCl2, 2.5 mM CaCl2, 11 mM glucose, and 20 mM HEPES, pH 7.4). The cell homogenate was centrifuged at 1000g, and the pellet was resuspended with luminescence buffer (Krebs buffer containing 10 μM lucigenin, Cayman Chemicals), before adding 100 μM NADPH substrate (Sigma-Aldrich). Luminescence was detected by an EnSpire microplate reader.
Immunoblots.
BMDMs cultured on 25 μg/ml fibrin-coated plates were lysed in RIPA lysis buffer (EMD Millipore) with protease/phosphatase inhibitor cocktail (EMD Millipore). Protein extracts (20 μg) were separated by electrophoresis on NuPAGE 4–12% Bis-Tris Gel (Life Technologies) and transferred onto nitrocellulose. Blots were blocked in 5% milk in TBST and incubated with gp91phox (1:1000; clone EPR6991, Abcam), Phospho-p40phox (1:1000; Thr154, Cell Signaling Technology), and GAPDH (1:5000; clone 14C10, Cell Signaling Technology) using horseradish peroxidase-conjugated secondary antibody (1:5000; Cell Signaling Technology #7074S) and enhanced chemiluminescence (ECL) kit (GE Healthcare) for detection. Densitometry analysis was performed using ImageJ.
Cortical neuron-macrophage co-culture.
Cortices from P1 rats were finely minced and digested for 30 min at 37 °C in DPBS (Gibco) containing papain (Worthington Biochemicals) and DNase (Sigma-Aldrich). Papain was inhibited by the addition of ovomucoid (Worthington Biochemicals). Neurons were plated at a density of 60,000 cells/cm2 on poly-D-lysine pre-coated 8-well Permanox chamber slides (Sigma-Aldrich) in Neurobasal/B27 medium (Invitrogen) and cultured for 10 days. Rat BMDMs were cultured in RPMI-1640 with 10% heat-inactivated FBS (Invitrogen), 1% penicillin-streptomycin (Corning), and 10 ng/ml rat M-CSF (#400–28, Peprotec). BMDMs were plated on 25 μg/ml fibrin-coated plates with 20 μg/ml 5B8 or IgG2b for 24 h and were lifted with PBS-EDTA as described16 and added to cortical neuron cultures for two days, fixed with 4% PFA and immunostained with anti-MAP-2 (1:1000; clone AP20, EMD Millipore) and thresholded images were quantified with the NeurphologyJ plug-in in ImageJ. 2.5 × 1010 GC/ml of AAV1.hSyn.TurboRFP (University of Pennsylvania Vector Core) was used to transduce primary cortical neurons for 8 d prior to the addition of fibrin-stimulated BMDMs for 12 h. RFP images were thresholded and the neurite fragments were analyzed using the ImageJ plugin ‘Analyze Particles’. Quantification was performed by an observer blinded to the experimental treatments.
Pharmacokinetics.
C57BL/6 mice were injected with 400 μg 5B8 i.p. Blood samples were collected by micro-accusampler and centrifuged at 2500 rpm for 15 min. Plasma was stored at −80 °C until analysis. Plasma was diluted 1:1000 in blocking buffer (3% BSA and 0.1% NP-40 in PBS). 5B8 was diluted in blocking buffer to create a concentration curve. Plasma and 5B8 were added to wells coated with human γ377–395 peptide and incubated for 2 h at 25 ˚C. After washing, polyclonal goat anti-mouse IgG-HRP (#P0447, DAKO) in blocking buffer was added for 2 h at 25 ˚C and the assay was developed with TMB Liquid substrate (Sigma-Aldrich) with absorbance measured using an Envision Microplate reader.
EAE.
EAE was induced in 8–9-week-old female SJL/J, C57BL/6, or Cx3cr1GFP/+Ccr2RFP/+ mice by subcutaneous immunization with 100 μg PLP139–151 (HSLGKWLGHPDKF; Auspep Pty Ltd) or 50 μg MOG35–55 (MEVGWYRSPFSRVVHLYRDGK; Auspep Pty Ltd) in complete Freund’s adjuvant (Sigma-Aldrich) supplemented with 400 μg of heat-inactivated mycobacterium tuberculosis H37Ra (Difco Laboratories). Two days after immunization, mice were injected i.p. with 200 ng (C57BL/6) or 75 ng (SJL/J) pertussis toxin (Sigma-Aldrich). For adoptive transfer, donor SJL/J mice were immunized as described above, and on day 10, cells from draining lymph nodes and spleen were isolated. Lymphocytes were re-stimulated with 20 μg/ml PLP139–151 peptide and 10 ng/ml of IL-12 (eBioscience) for 4 d, and 3 × 107 cells were transferred into healthy SJL/J recipients. Frequency of donor CD4+ T cells producing IFNγ by FACS was assessed before transfer. For prophylactic treatment, mice were each given 800 μg of either 5B8 or isotype-control IgG2b every two days from day 0. For therapeutic treatment, antibodies were injected every two days starting at the peak of the initial paralytic episode. Mice were observed daily and clinical scores were assessed by observers blinded to treatments as follows: 0, no symptoms; 1, loss of tail tone; 2, ataxia; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis; 5, moribund.
Histology and immunohistochemistry.
For histological analysis, spinal cords and brains were processed as described13,62. Antibodies used were: mouse anti-neurofilament H non-phosphorylated (1:100; BioLegend clone SMI-32), rabbit anti-Iba-1 (1:1000; Wako #019–19741), and mouse anti-myelin basic protein (1:100; BioLegend clone SMI-99) and Alexa Fluor 488 or 594 (1:500; Jackson ImmunoResearch) for detection. Immunohistochemistry for anti-MBP was performed with the Mouse on Mouse (M.O.M.) kit (Vector Labs). Ccr2RFP/+Cx3cr1GFP/+ mice were perfused with 4% PFA and spinal cord segments were frozen in OCT, then cut into 10-μm sections and stained with DAPI for 5 min. DHE labeling was performed as described 15. For analysis of 5XFAD mice, mice were perfused with 4% PFA and brains were postfixed in 4% PFA overnight at 4 °C, except for CD11b staining, which was postfixed for 4 h. Immunohistochemistry in coronal brain sections (30 μm) were performed using antibodies against ChAT (1:1000, EMD Millipore #AB144P), CD11b (1:150; eBioscience clone M1/70), Iba-1 (1:1000; Wako), Mac-2 (1:1000; Cedarlane clone M3/38), fibrinogen (1:1000; J. L. Degen, Cincinnati Children’s Hospital Medical Center), and human Aβ (1:500; IBL-America #18584). Amyloid plaques were labeled with methoxy-XO4 (fluorescent probe for Aβ, Tocris) as described63. Images were acquired with an Axioplan II epifluorescence microscope (Zeiss) equipped with dry Plan-Neofluar objectives (10 × 0.3 NA, 20 × 0.5 NA, or 40 × 0.75 NA) or all-in-one BZ-X700 fluorescence microscope (Keyence) or a Fluoview FV1000 (Olympus) confocal microscope and FluoView Software v3.1b. Excitation lasers were set to 405, 488, and 543nm and Olympus 20 × 1.00 NA, Olympus 25 × 1.05 NA, or an Olympus 40 × 0.8 NA water immersion lens with 1.0 μm z-step and Kalman filtering using line mode of 2 were used. Iba1, Aβ, Mac2+ quantification was averaged from 9 field of views of high Aβ plaque density throughout the cortex. Images were quantified using ImageJ by observers blinded to the experimental conditions.
Flow cytometry.
Primary mouse splenocytes were isolated from 5B8- and IgG2b-treated mice 10 days after immunization with PLP139–151. Cell suspensions were stained with combinations of antibodies: CD45 (BioLegend clone 30-F11), CD3 (BioLegend clone 17A2), CD4 (BioLegend clone GK1.5), CD11b (BioLegend clone M1/70), B220 (BD Bioscience clone RA3–6B2), Ly6G (eBioscience clone RB6–8C5), and CD11c (BioLegend clone N418). For cytokine analysis, cells were incubated for 4 h with Cell Activation Cocktail (BioLegend) and surface stained for CD3, CD4, and CD8 (BioLegend clone 53–6.7). Cells were then fixed with Cytofix/Cytoperm solution (BD Biosciences), and intracellular cytokine staining was performed with anti-IFN-γ (BioLegend clone XMG1.2) and anti-IL-17-FITC (BioLegend clone TC11–18H10.1). All antibodies were used at 1:300. Flow cytometry was performed on an LSR II (BD Biosciences). Data were analyzed using FlowJo (Tree Star).
Stereotactic fibrinogen injection and drug treatment.
Stereotactic injection of fibrinogen into the corpus callosum of Cx3cr1GFP/+ or C57BL/6 mice and analysis were performed as described15,16. 5B8, or 5B8-F(ab) fragment, or isotype-control IgG2b (clone MPC-11, eBioscience) was injected (10 μg at 0.2 μl/min) into the cerebral ventricle (AP, −2.0 mm; ML, 0 mm, DV, −2.0 mm) with a 10-μl syringe attached to a 33-gauge needle 30 min before fibrinogen injection.
5B8 penetration in the CNS and target engagement.
Mice were injected i.p. with 800 μg/mouse of biotinylated 5B8 every 2 days (3 total doses). Mice were perfused with saline, and spinal cord or brain were processed for fresh frozen sections as described. Sections were fixed with 4% PFA for 10 min and biotinylated 5B8 was detected using Cy3-conjugated streptavidin (1:100; Invitrogen) for 30 min at 25 ˚C. Sections were incubated for 1h with fibrinogen antibody (1:2000), followed by FITC donkey anti-rabbit (1:500; Jackson ImmunoResearch) for 30 min at 25 ˚C. For amyloid plaque staining, sections were counterstained with Methoxy-X04 (Tocris; 4% vol of 10 mg/ml).
Two-photon in vivo imaging.
Thy1-YFP and Thy1-YFP:5XFAD mice were injected with Alexa594-conjugated fibrinogen (Invitrogen) intravenously daily for 3 days as described15. Mice were also injected i.p. with Methoxy-X04 24 h prior to imaging. Methoxy-X04 was solubilized with DMSO:propylene glycol:PBS pH 7.5 (ratio 2:9:9) at 5 mg/ml. On day of imaging, a small craniotomy was made and a custom-made metal plate affixed to a stage to stabilize the skull. Alexa594-fibrinogen solution was injected retro-orbitally prior to imaging. The anesthetized animal was placed on a heated pad under an Ultima-IV multiphoton microscope (Prairie) equipped with MaiTai DeepSee-eHP lasers (Spectra Physics). The excitation wavelength was 820nm to simultaneously visualize fibrinogen, methoxy-X04 and YFP dendrites. Imaging was performed from 20 to 150 μm below the dura, using a Nikon 40 × 0.8 NA -immersion lenses with a 1.0 μm z-step. z-stacks of images were projected along the z-axis to recreate two-dimensional representations of the 3D structures within the imaged volumes. Images were adjusted for brightness, contrast and background noise with ImageJ. Spectral unmixing plugin in ImageJ was used to separate overlapping signals.
Network analyses.
The TYROBP network and the Microglia Pathogen Phagocytosis Pathway were translated from human-specific, static figures to mouse-specific data models using WikiPathways73 and HomoloGene, The coexpression network was constructed by extracting the dominant cluster of downregulated genes of interest (selected from those with log2 fold change of −0.02 or less and raw P-value < −0.05) based on coexpression interaction data provided by GeneMANIA74. Subsequent network visualization, layout, data overlays and subnetwork extraction were performed in Cytoscape75.
Statistical analyses.
Data are presented as mean ± s.e.m. Statistical calculations were performed with GraphPad Prism (Version 6.03). Sample sizes were based on previous experiments and not predetermined by statistical methods. Statistical significance was determined with non-parametric two-sided Mann-Whitney test, Kruskal-Wallis test, Welch’s two-sample t-test, one-way or two-way analysis of variance followed by Bonferroni or Tukey’s post-test (multiple comparisons). Linear models were fitted for each gene using the Bioconductor ‘limma’ package in R64. Moderated t-statistics, fold change and the associated P values were calculated for each gene. Mice were randomly assigned to experimental groups for EAE and 5XFAD experiments. Scoring of EAE mice was performed in a blinded fashion. All animals survived until the end of the study and all data points were included in analysis. All histopathological analysis and quantification was performed blinded.
To compare EAE clinical scores, the linear mixed effects models were performed using the R statistical package. Means of maximum clinical scores were compared using a two-sample t-test as implemented in R using the Welch’s t-test and the Wilcoxon rank sum test. P-values were corrected for multiple testing using the Holm procedure. The statistical significance of the changes in the mean clinical score for each day of the experiment was estimated using permutation tests. The corresponding p-values were estimated using 1000 permutations in all prophylactic antibody treatment and 10000 permutations for therapeutic antibody treatment. In each permutation, mice were randomly permuted. To compare the first day of onset, the log-rank test was used as implemented by the survdiff function in the survival package in R.
Supplementary Material
Acknowledgements
We thank J.L. Degen for expert advice and critical reading of the manuscript and S. Pintchovski, I. Kadiu, J. Palop, and J. Egebjerg for insightful discussions. We thank B. Cabriga, R. Meza Acevedo, L. Ta, and A. Williams for expert technical assistance, G. Maki for graphics, G. Howard and K. Claiborne for editorial. The Gladstone Center for In Vivo Imaging Research was supported in part by grants from H. Lundbeck A/S, the S.D. Bechtel, Jr. Foundation, and the Conrad N. Hilton Foundation 17348 to K.A. The microscopy studies were carried out in part at facilities adapted for this project at the National Center for Microscopy and Imaging Research, which is supported by grant P41 GM10341 awarded to M.H.E. Gladstone Institutes was supported by NIH/NCRR grant RR18928. The Mouse Pathology Core of the UCSF Helen Diller Family Comprehensive Cancer Center was supported by CA082103. J.K.R, D.D., and A.S.M. were supported by National Multiple Sclerosis Society (NMSS) Postdoctoral Fellowships; J.K.R. and D.D by Race to Erase MS Young Investigator Awards and American Heart Association (AHA) Scientist Development Grants; V.A.R. by postdoctoral fellowships from AHA and NIH/NINDS F32 NS096920; A.S.M. by NIAID T32AI733429 and NMSS FG-1708–28925, K.K.H. by NSF predoctoral fellowship DGE-0648991/1144247; M.A.P. by a NIH/NICHD K12-HD072222; L.M. by a gift from the Dolby Family; S.S.Z. by NIH R01 NS092835; R21 NS108159, NMSS RG1701–26628, RG 5179A10/2, the Weill Institute, and the Maisin Foundation; R.A.S by NIH R01 NS081149. This work was supported by grants from NMSS RG3782, H. Lundbeck A/S, the Conrad N. Hilton Foundation 17348, a gift from the Levine Family, and NIH/NINDS R01 NS052189, R21 NS082976, and R35 NS097976 to K.A.
H. Lundbeck A/S sponsored research in K.A.’s laboratory at the Gladstone Institutes. K.A. is a co-founder and scientific advisor of MedaRed, Inc. K.A. is an inventor on University of California issued patents US7807645, US8569242, US8877195, and US8980836 covering fibrin antibodies. K.A. and J.K.R are co-inventors on Gladstone Institutes issued patent US9669112 covering fibrin in vivo models and Gladstone Institutes pending US20160320370 patent application covering in vitro fibrin assays. K.A., A.M.F., M.R.A., and K.K.A. are co-inventors on the Gladstone Institutes and University of California pending US20170003280 patent application covering assays for inhibition of microglia activation. Their interests are managed by the Gladstone Institutes in accordance with its conflict of interest policy. J.B.S., R.B.N., S.B.P, L.O.P, V.M., S.H.Z. were Lundbeck employees during the time the work was performed.
Footnotes
Competing Interests
The authors declare competing financial interests.
Additional information
Supplementary Information accompanies this paper and includes nine Supplementary Figures and three Supplementary Tables.
Reporting Summary. Further information on research design is available in the Nature research Reporting Summary linked to this article.
Data Availability. GEO data supporting the findings of this study have been deposited in the GEO depository under accession numbers GSE118920 and GSE118921. Networks are permanently referenced in two Wikipathways entries http://wikipathways.org/index.php?title=Pathway:WP3625&oldid=85694, http://wikipathways.org/index.php?title=Pathway:WP3626&oldid=85691. The authors declare that all other data supporting the findings of this study are available within the paper. Any additional data can be made available from the corresponding author upon request.
References
- 1.Rivest S Regulation of innate immune responses in the brain. Nat Rev Immunol 9, 429–439 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM & Brown MA Innate immunity in the central nervous system. J Clin Invest 122, 1164–1171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lassmann H, van Horssen J & Mahad D Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8, 647–656 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Schuh C et al. Oxidative tissue injury in multiple sclerosis is only partly reflected in experimental disease models. Acta Neuropathol 128, 247–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heppner FL, Ransohoff RM & Becher B Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Lassmann H Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer’s disease. J Neural Transm 118, 747–752 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Nikic I et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17, 495–499 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Chitnis T & Weiner HL CNS inflammation and neurodegeneration. J Clin Invest 127, 3577–3587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frischer JM et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132, 1175–1189 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Z, Nelson AR, Betsholtz C & Zlokovic BV Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163, 1064–1078 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen MA, Ryu JK & Akassoglou K Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci 19, 283–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davalos D & Akassoglou K Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol 34, 43–62 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Adams RA et al. The fibrin-derived gamma377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 204, 571–582 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ugarova TP et al. Sequence gamma 377–395(P2), but not gamma 190–202(P1), is the binding site for the alpha MI-domain of integrin alpha M beta 2 in the gamma C-domain of fibrinogen. Biochemistry 42, 9365–9373 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Davalos D et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3, 1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryu JK et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun 6, 8164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yates RL et al. Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann Neurol 82, 259–270 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Marik C, Felts PA, Bauer J, Lassmann H & Smith KJ Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain 130, 2800–2815 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee NJ et al. Spatiotemporal distribution of fibrinogen in marmoset and human inflammatory demyelination. Brain 141, 1637–1649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strickland S Blood will out: vascular contributions to Alzheimer’s disease. J Clin Invest 128, 556–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nathan C & Cunningham-Bussel A Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol 13, 349–361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer MT et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimohama S et al. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun 273, 5–9 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Ma MW et al. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener 12, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haslund-Vinding J, McBean G, Jaquet V & Vilhardt F NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease. Br J Pharmacol 174, 1733–1749 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han MH et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Davalos D et al. Early detection of thrombin activity in neuroinflammatory disease. Ann Neurol 75, 303–308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haider L et al. Oxidative damage in multiple sclerosis lesions. Brain 134, 1914–1924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heneka MT et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeKosky ST & Scheff SW Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27, 457–464 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Zhang B et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoeb M & Fang MC Assessing bleeding risk in patients taking anticoagulants. J Thromb Thrombolysis 35, 312–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mocsai A et al. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol 7, 1326–1333 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bedard K & Krause KH The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87, 245–313 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Turnbull IR & Colonna M Activating and inhibitory functions of DAP12. Nat Rev Immunol 7, 155–161 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Park L et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc Natl Acad Sci U S A 108, 5063–5068 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cummings J Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci 11, 147–152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vemuri P et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 138, 761–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabin JS et al. Interactive Associations of Vascular Risk and beta-Amyloid Burden With Cognitive Decline in Clinically Normal Elderly Individuals: Findings From the Harvard Aging Brain Study. JAMA Neurol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miron VE et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16, 1211–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen MA et al. Fibrinogen Activates BMP Signaling in Oligodendrocyte Progenitor Cells and Inhibits Remyelination after Vascular Damage. Neuron 96, 1003–1012 e1007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oka A, Belliveau MJ, Rosenberg PA & Volpe JJ Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci 13, 1441–1453 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butovsky O et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ajami B et al. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat Neurosci 21, 541–551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Mrdjen D et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 380–395 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Suh TT et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 9, 2020–2033 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Flick MJ et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113, 1596–1606 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lak M, Keihani M, Elahi F, Peyvandi F & Mannucci PM Bleeding and thrombosis in 55 patients with inherited afibrinogenaemia. Br J Haematol 107, 204–206 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Bugge TH et al. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 87, 709–719 (1996). [DOI] [PubMed] [Google Scholar]
- 51.Akassoglou K, Yu W-M, Akpinar P & Strickland S Fibrin inhibits peripheral nerve regeneration by arresting Schwann cell differentiation. Neuron 33, 861–875 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Koseki-Kuno S, Yamakawa M, Dickneite G & Ichinose A Factor XIII A subunit-deficient mice developed severe uterine bleeding events and subsequent spontaneous miscarriages. Blood 102, 4410–4412 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Chen R & Doolittle RF - cross-linking sites in human and bovine fibrin. Biochemistry 10, 4487–4491 (1971). [DOI] [PubMed] [Google Scholar]
- 54.Aleman MM et al. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest 124, 3590–3600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung S et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng G et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Oakley H et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jackson SH, Gallin JI & Holland SM The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 182, 751–758 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saederup N et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5, e13693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jensen MR et al. Structural Basis for Simvastatin Competitive Antagonism of Complement Receptor 3. J Biol Chem 291, 16963–16976 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abid MR, Spokes KC, Shih SC & Aird WC NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J Biol Chem 282, 35373–35385 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Davalos D et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3, 1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klunk WE et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61, 797–805 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Gentleman RC et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.