

Abstract
Recent outstanding clinical results produced by engineered T cells, including chimeric antigen receptors, have already facilitated further research that broadens their applicability. One such direction is to explore new T cell sources for allogeneic “off‐the‐shelf” adoptive immunotherapy. Human pluripotent stem cells could serve as an alternative cell source for this purpose due to their unique features of infinite propagation ability and pluripotency. Here, we describe the current state of engineered T cell transfer with the focus on cell manufacturing processes and the potentials and challenges of induced pluripotent stem cell‐derived T cells as a starting material to construct off‐the‐shelf T‐cell banks.
Keywords: antigen specificity, iPS cell, regenerative immunotherapy, T cell differentiation, T cell rejuvenation
1. INTRODUCTION
Cancer immunotherapy has become an emerging entity for cancer therapy. Cancer immunotherapy aims to trigger, enhance, or halt the negative regulators of the cancer‐immunity cycle within the tumor microenvironment, which is postulated by Chen and Mellman (Figure 1).1 Examples include checkpoint blockers and adoptive transfer of engineered T lymphocytes expressing chimeric antigen receptors (CAR) that have shown outstanding outcomes in large‐scale clinical trials and have benefited many patients with refractory cancers. Checkpoint blockers are a good example of an inhibitor of a negative regulator of the cancer‐immunity cycle, whereas adoptive CAR‐T cell therapy is considered to initiate and/or enhance the cycle. Significant effort has been made over the past decades to evaluate the therapeutic benefit of cancer immunotherapies. The next few years to come will see more immunotherapy products with cutting‐edge technologies such as genome editing and induced pluripotent stem cell (iPSC) technologies in combination with other agents (ie, checkpoint blocker and CAR‐T cells). Major challenges for cancer immunotherapy to broaden or generalize its applicability will be the evaluation of therapeutic benefits against a broader range of solid cancers, management of off‐tumor and on‐target toxicities, and reduction of prices. Checkpoint blockers have already shown active results in different types of solid cancers and more results from clinical trials will follow. With respect to cost issues, researchers in the field of engineered T cell development have been considering allogeneic infusions instead of autologous cell manufacturing. In particular, the development of off‐the‐shelf engineered T‐cell products that are inspired by cord‐blood banks will not only reduce the cost of manufacturing, but serve as an ideal platform for synthetic T cells. In this review, we describe the current state of engineered T cell transfer with the focus on cell manufacturing processes and the potentials and challenges of iPSC‐derived T cells as a starting material to construct an off‐the‐shelf T cell bank.
Figure 1.
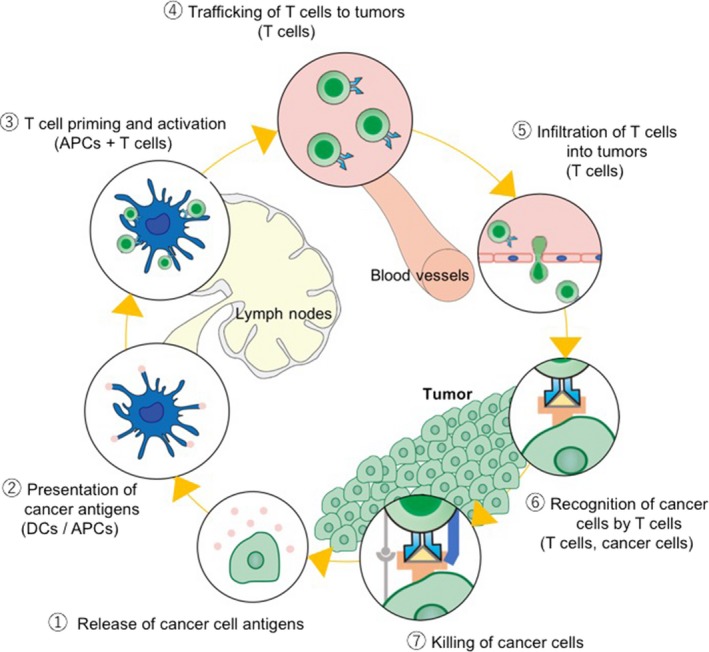
Cancer‐immunity cycle. The induction of antitumor immunity is a cyclic process that can be self‐propagating. It can amplify and extend T cell responses against cancer cells. It also contains several inhibitory factors itself to halt the cycle when the target cells (cancer cells) are eradicated. The cycle can be divided into 7 steps, as illustrated, starting with the release of cancer antigens from the cancer cells and ending with the killing of cancer cells. APC, antigen‐presenting cell; DC, dendritic cell. (Adapted from Chen and Mellman,1 with permission.)
2. ENGINEERED T CELLS FOR CANCER THERAPY
Adoptive T cell transfer for cancer immunotherapy was initially demonstrated by the Rosenberg Laboratory in the NCI, USA, using T cells isolated from a patient('s tumor on surgery, also known as tumor‐infiltrating lymphocytes (TILs). The isolated TILs are subsequently expanded ex vivo to obtain clinically relevant numbers for reinfusion.2 Since then, alternative approaches have been proposed and evaluated. Among them, Carl June's group at the University of Pennsylvania, USA, showed the feasibility of adoptive transfer of bulk T cells from peripheral blood (PBT) of patients expanded ex vivo in the presence of interleukin (IL)‐2.3 The first clinical trial using the expanded PBT was implemented on patients with relapsed/refractory non‐Hodgkin lymphoma following hematopoietic stem cell transfer and showed the safety of the procedure.4 These two seminal studies used a population of T cells with unknown antigen‐specificity.
Researchers have also explored the potentials of tumor‐antigen directed engineered T cells obtained from patient')s blood.5, 6 The manufacturing process involves isolation of PBT from patients and ex vivo expansion followed by retroviral transduction of a T‐cell receptor (TCR) or CAR. For example, CAR T cells are generated from bulk PBMCs by aphaeresis, activated with CD3/28 beads, and transduced with a viral vector harboring CAR. These production processes take approximately 10‐14 days to obtain cell doses required for reinfusion. Importantly, the potential of engineered T cells has been well demonstrated in clinical trials obtained with NY‐ESO‐1 TCR7 and CD19 CAR.8, 9, 10, 11 Last year, Novartis announced that the US FDA approved Kymriah (tisagenlecleucel), the first CAR‐T therapy product, for B‐cell precursor acute lymphoblastic leukemia. The price tag of Kymriah was determined to be $475 000 per patient, which astonished the community.
These initial encouraging results, as well as issues, call for further research and development. For instance, current procedures do not clearly define the composition of T‐cell subsets, which include CD4 helper and CD8 killer T cells. With regard to T‐cell subsets, a recent preclinical study showed the synergistic antitumor activity of CD19 CAR‐expressing CD4+ and CD8+ T cells, indicating that investigation of optimal composition might influence the therapeutic efficacy.12 In addition, each T‐cell subset also comprises naive, stem cell memory (Tscm), central memory (Tcm), effector memory (Tem), and terminal effector cells with distinct in vivo persistence and effector functions (Figure 2).13, 14 Several groups already evaluated the potential of T‐cell subsets in preclinical models and humans and found that T cells with more immature phenotypes, such as Tcm cells, sustain longer in vivo with presumably better antitumor effects.15, 16, 17, 18 In line with these findings, significant effort has been made to optimize ex vivo culture methods that promote self‐renewal of Tcm. It appeared that inclusion of IL‐15 together with IL‐2 during ex vivo culture favors self‐renewal of CD8+ Tcm cells.18
Figure 2.
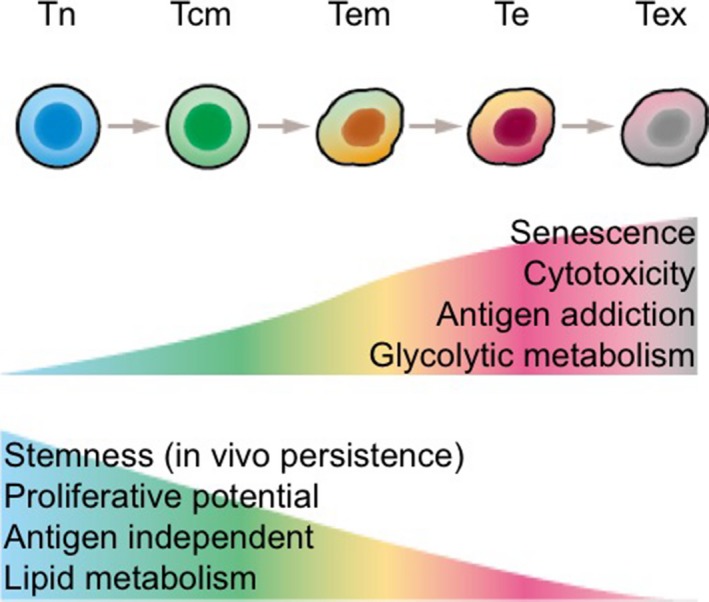
Antitumor activity and T cell subsets. After an antigen exposure, naïve T cells (Tn) vigorously expand and differentiate to central memory T cells (Tcm) and progressively become effector T cells (Te) with higher cytotoxic ability. Although Te shows the maximum cytotoxic ability within the mature T‐cell subsets, it is short‐lived and the cells readily undergo apoptosis. It is believed that Tcm or Tscm cells are the ideal T‐cell subset for T‐cell cancer immunotherapy due to its higher in vivo persistence when compared to Tem, Te, and Tex cells. Tem, T effector memory; Tex, exhausted T cell. (Part of this figure is adapted from Gattinoni et al,14 with permission.)
Although the therapeutic benefits of engineered T cells are now apparent, several issues remain to be solved.19 Clearly, cost of the products needs to be reduced in order to make it economically practical. One approach under evaluation is allogeneic therapy. Autologous T cell therapy will remain in the frontline of target validation and the production cost will be reduced with upcoming new technologies. Once the efficacy of a target is validated, then the development of banked T‐cell source that is immediately ready upon the need would contribute to broaden the applicability and cost reduction of adoptive T cell therapies. Figure 3 shows a schema of 3 different forms of engineered T cell infusions. However, the major challenges for allogeneic adoptive T cell transfer include striking the balance between rejection response from the host immunity and alloresponse to the host by infusing T cells. In particular, alloreactivity of donor T cells increases the risk of off‐tumor tissue destruction, also known as graft‐vs‐host disease (GvHD). Therefore, safety assessments would be crucial when implementing allogeneic T cell therapy. Other important issue includes poor progression‐free survival for ALL patients treated with CD19‐CART cells.20
Figure 3.
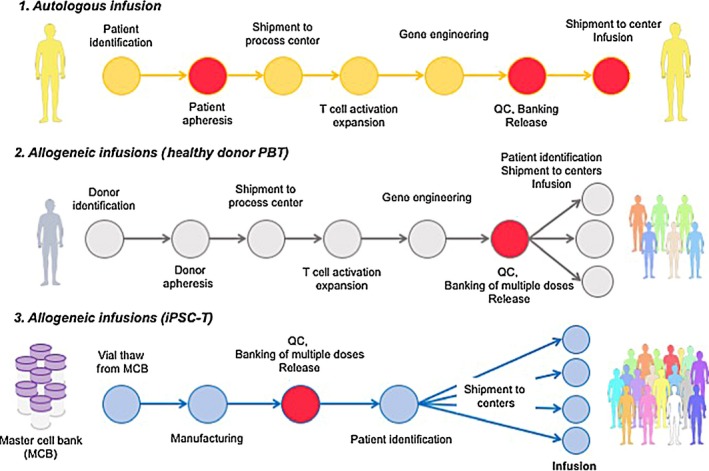
Schema of induced pluripotent stem (iPS)‐based allogeneic T‐cell immunotherapy. 1. General schema illustrating autologous T cell manufacturing. Autologous T cell manufacturing starts with patient selection followed by apheresis blood collection at a certified facility. Blood sample is shipped to a cell processing center, where the T cells are activated, selected for CD3, and subsequently transduced with a viral vector harboring a chimeric antigen receptor or a T‐cell receptor. The expanded cells are washed and cryopreserved for quality control (QC) release tests. Products are then shipped to an infusion center and infused to the patient. 2. Proposed schema of allogeneic T cell manufacturing. For this strategy, healthy donors for banking are selected and used to produce a cell bank after transduction with viral vector before patient selection. This leads to off‐the‐shelf allogeneic T‐cell immunotherapy. 3. Perspective schema of allogeneic iPSC‐derived T cell infusion. In this approach, master cell banks are first established after thorough QC and safety tests. Thereafter, a working cell bank is generated for multiple (10‐1000) doses. Following patient identification, a vial is shipped to an infusion center and the product is immediately infused into the patient. Circles in red indicate potential points of manufacturing failure. Examples of failure points for autologous infusion include failure to obtain a sufficient number of PBMCs from the patient at apheresis, unsuccessful expansion after T cell activation and/or gene transduction efficiency, and health condition of the patient at the infusion. On the contrary, allogeneic infusions of healthy donor PBT and iPSC‐T manufacturing could fail if the product cannot pass QC tests at the construction of a working cell bank (eg, too many dead cells or low gene transduction efficiency to meet the specifications). The major challenge for iPSC‐derived T cell infusion would be how to generate master cell banks (whether it is at the iPSC stage or intermediate product (s) during differentiation). (Part of this figure is modified from Clarke et al.49)
3. CELL SOURCE FOR ALLOGENEIC T CELL IMMUNOTHERAPY
The current candidates of cell sources for allogeneic T cell therapies can be divided into 2 categories: peripheral blood T cells from healthy donors and iPSC‐derived T cells. Both candidates are being actively investigated within academia and industry. With respect to donor PBT approaches, Cellectis (Paris, France) already started a phase I clinical trial last year using CD19‐directed CAR‐T cells in patients with refractory B‐cell acute lymphoblastic leukemia and so far demonstrated the feasibility of the approach. Cellectis exploits a unique approach for allogeneic CAR‐T cell manufacturing in that they disrupt the constant region of the α‐chain of the TCR gene (TRAC) by a gene editing technology, expecting to reduce the risk of GvHD. In addition, they also incorporated disruption of the CD52 gene to provide the cells with the resistance to Alemtuzumab treatment during preconditioning. The other strategy to manage the risk of GvHD is the selection of virus‐specific TCRs devoid of alloreactivity, although unpredictable cross‐reactivity would be a concern.21 Compared to the development of healthy donor PBT approaches, iPSC‐derived T cells are still in the preclinical stage of development. As iPSC‐derived somatic cells themselves are still under safety assessment in a clinical trial, functional assessment of iPSC‐derived T cells will require a few more years. For future clinical trials and subsequent commercialization to come, it is critical to establish cGMP‐compatible manufacturing process development, which includes the generation of iPSCs, differentiation of iPSCs to T cells, and expansion of iPSC‐derived T cells (Figure 3). Successful process development would require comprehensive knowledge and experts of molecular biology, developmental biology, stem cell biology, immunology, and regulatory sciences. For the rest of this review, we will summarize the current status of human PSC‐derived T cell research.
4. PLURIPOTENT STEM CELLS AS TRUE “OFF‐THE‐SHELF” T CELLS IN THE ERA OF SYNTHETIC BIOLOGY
Since reported in 1998, human ESCs have been expected to become an ultimate cell source for regenerative medicine due to the features of pluripotency; they can be propagated indefinitely while maintaining the ability to differentiate into all types of somatic cells in vitro. Within a decade from the first report of human ESC establishment, Shinya Yamanaka of Kyoto University (Kyoto, Japan) reported the successful reprogramming of mouse and later human somatic cells into pluripotency by transducing 4 transcription factors essential to ESCs.22, 23 The reprogrammed cells are termed iPSCs. Because iPSCs can be derived from a variety of somatic cells, including adult skin fibroblasts and blood cells, it is considered that iPSC technology leads to tailor‐made regenerative medicine and hence the use of otherwise harmful immunosuppressive drugs, required for allogeneic transplantation, can be avoided. These features have accelerated the research and development of regenerative medicine using PSCs.
To date, several investigators, including our laboratory, have reported the feasibility of generating T cells from human ESCs and iPSCs. The first evidence showing in vitro differentiation of T cells from ESCs was reported by Timmermans et al.24 They utilized a well‐established hematopoietic differentiation protocol using OP9 feeder layers from ESCs and a T cell differentiation protocol established for human hematopoietic stem cells.25, 26 The resulting cells expressed markers characteristic to T cells, such as CD3, and TCR and expanded and secreted γ‐interferon and tumor necrosis factor following TCR stimulation. Later in 2013, 3 groups from Japan reported the generation and redifferentiation of iPSCs from antigen‐specific T cells.27, 28, 29 In a series of papers, we and others have reported the regeneration of T cells from a T‐cell clone by reprogramming it into iPSCs and by redifferentiation into CD8+ T cells. The regenerated T cells maintained the same TCR genomic sequence to the original T cell clone. The redifferentiated T cells not only maintained the same antigen specificity, but they showed longer telomere length compared to the original T cell clones, indicating that the redifferentiated T cells had rejuvenated through the reprograming process. The proliferative ability of redifferentiated T cells was remarkably higher than those of the original T cell clone. This technique allows us to generate a large number of rejuvenated T cell clones. In addition, the feasibility of generation of CAR‐T cells from iPSCs has been reported.30 Collectively, these studies showed the proof‐of‐concept that T cells with antigen‐specific activities could be generated from pluripotent stem cells by TCRs and CARs.
Although the aforementioned studies show the potential of iPSC‐derived T cells as an alternative cell source for T cell immunotherapy, recent studies, including those at our laboratory, revealed that T cells differentiated from iPSCs using the current differentiation methods display features similar to γδT cells or innate lymphoid cells.30, 31, 32 Current differentiation culture induces T cells expressing CD56, a marker for natural killer cells, during multiple rounds of expansion. Another deviation from normal thymocyte development observed during PSC differentiation is earlier expression of TCRs at the CD4−/CD8− stage when iPSCs derived from T cells (T‐iPSCs) are used. It could be possible that prerearranged TCRs in T‐iPSCs and their earlier expression during culture skewed the differentiating cells toward innate‐like lymphocytes. Alternatively, T cells induced from fetal‐like hematopoietic stem and progenitor cells (HSPCs), which are thought to be a counterpart of iPSC‐derived HSPCs, render these properties as suggested in previous studies. These results call for further advancement and refinement of the current differentiation protocols.
Generation of T cells from PSCs requires comprehensive understanding of developmental biology and immunology to recapitulate the key events that occur during T cell commitment.33 The key events are illustrated in Figure 4. The initial event is the induction of mesoderm, which is followed by hemogenic endothelial cell derivation. The next key step is called endothelial‐to‐hematopoietic transition, the mechanism through which heterogeneous populations of HSPCs are generated.34 The other milestone is to induce T cell commitment of HSPCs. The final step represents maturation of CD4+/CD8β+ double‐positive cells into CD4 or CD8 single‐positive (SP) cells.35 A challenge in the field is to develop a chemically defined differentiation culture, as most reports use several uncharacterized components in the cultures that include FBS and xenogeneic feeder layers (ie, OP9 and OP9/DL1 feeder cells). For this purpose, Kennedy et al developed a feeder‐ and serum‐free embryoid body‐based hematopoietic differentiation method and identified a population with robust T cell potential.36 Of note, the authors showed that inhibition of a SMAD pathway during the mesoderm induction selectively induced hemogenic endothelial cells with T cell potential. The study showed the possibility that manipulation of developmental cues during culture influence the outcome of later stages (ie, HSPCs and T cells). On the contrary, differentiation of T cells from iPSC‐derived HSPCs under feeder‐ and serum‐free conditions has not been reported despite the fact that such culture conditions for bone marrow or cord blood HSPCs are available. Attempts to develop defined conditions for T cell differentiation will require further investigations, but once succeeded, it will also provide novel insights into human thymocyte differentiation as well as hematopoietic development, which is otherwise impossible to study. The other challenge for this field is to define the condition to selectively induce CD8SP and CD4SP T cells, although a recent report showed the feasibility of making both subsets by means of organoid culture from HSPCs.37
Figure 4.
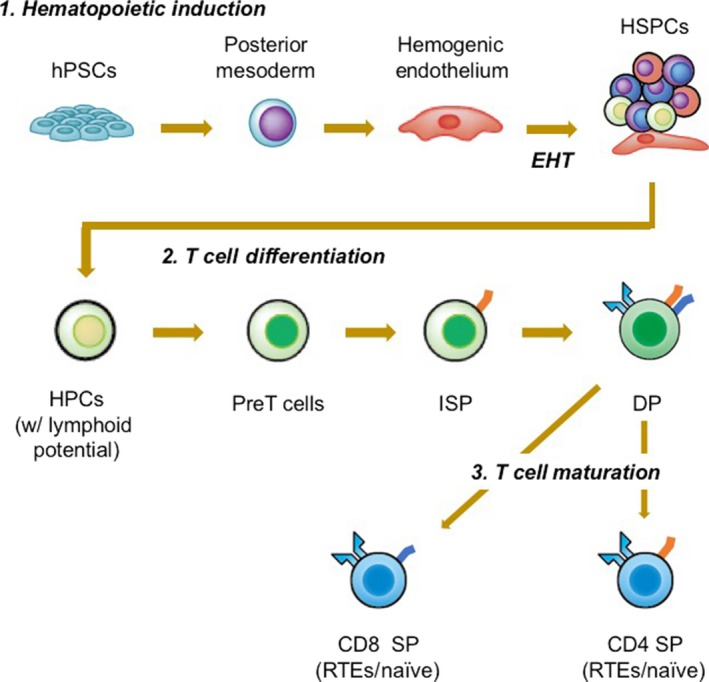
Key steps for differentiation of T cells from hematopoietic pluripotent stem cells (hPSCs). To make T cells from induced PSCs requires multiple key steps. The steps can be divided into 3 stages: 1. hematopoietic induction; 2. T cell differentiation; and 3. T cell maturation. Each stage can be subdivided into several steps. To induce hematopoietic differentiation, PSCs are first are induced to differentiate into the mesodermal cells and further instructed to hemogenic endothelium. Hemogenic endothelium cells then undergo a process known as endothelial‐to‐hematopoietic transition (EHT) where they round up in morphology and gain expression of hematopoietic markers, such as CD43 and CD45. Newly formed hematopoietic stem and progenitors (HSPCs) with lymphoid differentiation potential are further directed to T cell commitment in the presence of NOTCH signaling and become pre T cells expressing CD7, which then become CD4 expressing immature single‐positive (ISP) cells. ISPs subsequently acquire the expression of CD8β and become CD4+ and CD8αβ+ double‐positive (DP) cells, now holding a T cell receptor on the surface. Finally, DP cells mature into either CD4+ CD8− or CD4– CD8+ single‐positive (SP) T cells. HPC, hematopoietic progenitor cell; RTE, recent thymic emigrant
5. PERSPECTIVE
In this article, we have gone through the initial success of T cell immunotherapy for cancer with the focus on the development of CD19‐CART cell therapy, and explored the potential and challenges of iPSC‐derived T cells as an alternative cell source for allogeneic T cell immunotherapy. The in vivo safety and antitumor activities of iPSC‐derived T cells relative to primary T cells should be carefully and thoroughly examined in patients after undertaking in‐depth in vivo functional and safety assessments in preclinical models, such as xenograft models or non‐human primate models. Further investigation of mechanisms that underlie the differentiation of T cells from PSCs is warranted to gain more precise control to generate T cells with functions comparable to primary T cells. In parallel, efforts to scale up cell manufacturing processes and deal with regulatory requirements must not be overlooked.
Although we described the properties of iPSC T cells, other considerations need to be made when undertaking allogeneic therapy. One of them is to circumvent the risk of rejection, given that long‐term persistence correlates to therapeutic efficacy by CD19‐CART therapy. Accumulating evidence from allogeneic renal and bone marrow transplantations suggests that matching HLA‐A, ‐B, and ‐DR are beneficial to graft survival, indicating that HLA matching would be preferred in allogeneic iPSC‐T cell therapy.38, 39, 40, 41, 42 Accordingly, banking of iPSCs generated from common HLA‐homozygous donors has been proposed and is currently under development at the Center for iPS Research and Application (CiRA), Kyoto University.43 In CiRA, it has been estimated that an iPSC bank with 50 unique HLA‐homozygous iPSC lines would cover approximately 73% of the Japanese population, assuming matching at HLA‐A, ‐B, and ‐DRB1 loci.44 Similarly, another study also estimated generating 150 selected HLA homozygous lines would match 93% of the UK population.45 To date, CiRA has already accomplished the development and delivery of the first HLA‐homozygous iPSC, which will match to approximately 17% of the Japanese population and several clinical studies are underway or under planning using the iPSC grafts. For allogeneic iPSC‐T cell therapy, HLA‐homozygous iPSCs transduced with either CAR or TCR will be used as a starting material to construct an iPSC‐T cell bank. Other strategy actively considered is generation of “the universal donor” iPSC by disrupting HLA genes. In addition to HLA deletion, a recent report showed that overexpression of HLA‐E in HLA‐deficient iPSCs reduced natural killer cell‐mediated rejection, which is augmented when HLA genes are disrupted.46
The other critical issue that is worth mentioning is how we manage the risk of alloreactivity of infusing T cells. Alloreactivity would be a particular concern to allogeneic T cell therapy as it may attack and destroy host tissues. Therefore, the choice of TCRs will be critical to overall therapeutic benefits to patients. One approach to this problem would be to use Epstein‐Barr virus‐reactive TCRs. Alternatively, deletion of TCRs by the means of genome editing could minimize the alloreactivity, as reported by a recent study.47
Finally, once clinical potentials are evaluated, iPSC‐derived T cell technology could become the best platform to undertake gene manipulations to enhance functions or even add desired functions. For example, a recent study showed a proof of concept that overexpression of IL‐7 and CCL19 in CAR, designated as 7 × 19 CAR‐T cells, significantly enhanced in vivo antitumor functions.48 Significantly, depletion of host T cells prior to 7 × 19 CAR‐T cells reduced antitumor activity, indicating that injection of 7 × 19 CAR‐T cells accelerated the cancer‐immunity cycle. Combining these new technologies with iPSC‐derived T cells will generate a novel synthetic T cell. Autologous T cell therapy will remain the frontrunner to logically evaluate candidate targets. Once the therapeutic efficacy of the target is validated, allogeneic T cells derived from PSCs could play a role in the future.
CONFLICT OF INTEREST
The authors have no conflict of interest relevant to this work.
ACKNOWLEDGMENT
This work is supported in part by the Japan Society for the Promotion of Science (KAKENHI), 18033632 (S.I) and 23591413 (S.K)
Iriguchi S, Kaneko S. Toward the development of true “off‐the‐shelf” synthetic T‐cell immunotherapy. Cancer Sci. 2019;110:16–22. 10.1111/cas.13892
Funding information
Japan Society for the Promotion of Science (KAKENHI) , Grant/Award Number: 18033632 (S.I.), 23591413 (S.K.).
REFERENCES
- 1. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity. 2013;39:1‐10. [DOI] [PubMed] [Google Scholar]
- 2. Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rapoport AP, Stadtmauer EA, Aqui N, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T‐cell transfer. Nat Med. 2005;11:1230‐1237. [DOI] [PubMed] [Google Scholar]
- 4. Laport GG, Levine BL, Stadtmauer EA, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non‐Hodgkin lymphoma following CD34 + ‐selected hematopoietic cell transplantation. Blood. 2003;102:2004‐2013. [DOI] [PubMed] [Google Scholar]
- 5. Ho WY, Blattman JN, Dossett ML, et al. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431‐437. [DOI] [PubMed] [Google Scholar]
- 6. Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35‐45. [DOI] [PubMed] [Google Scholar]
- 7. Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY‐ESO‐1. J Clin Oncol. 2011;29:917‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kochenderfer JN, Dudley ME, Feldman SA, et al. B‐cell depletion and remissions of malignancy along with cytokine‐associated toxicity in a clinical trial of anti‐CD19 chimeric‐antigen‐receptor‐transduced T cells. Blood. 2012;119:2709‐2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor‐modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brentjens RJ, Davila ML, Rivière I, et al. CD19‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davila ML, Rivière I, Wang X, et al. Efficacy and toxicity management of 19‐28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric antigen receptor‐modified T cells derived from defined CD8(+) and CD4(+) subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell‐like properties. Nat Med. 2011;17:1290‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23:18‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang J, Khong HT, Dudley ME, et al. Survival, persistence, and progressive differentiation of adoptively transferred tumor‐reactive T cells associated with tumor regression. J Immunother. 2005;28:258‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berger C, Jensen MC, Lansdorp PM, et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Turtle CJ, Swanson HM, Fujii N, et al. A distinct subset of self‐renewing human memory CD8 + T cells survives cytotoxic chemotherapy. Immunity. 2009;31:834‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X, Naranjo A, Brown CE, et al. Phenotypic and functional attributes of lentivirus‐modified CD19‐specific human CD8 + central memory T cells manufactured at clinical scale. J Immunother. 2012;35:689‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heathman TRJ, Nienow AW, McCall MJ, et al. The translation of cell‐based therapies: clinical landscape and manufacturing challenges. Regen Med. 2015;10:49‐64. [DOI] [PubMed] [Google Scholar]
- 20. Park JH, Rivière I, Gonen M, et al. Long‐term follow‐up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Rivière I. Manufacture of tumor‐ and virus‐specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther. 2015;22:85‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 23. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 24. Timmermans F, Velghe I, Vanwalleghem L, et al. Generation of T cells from human embryonic stem cell‐derived hematopoietic zones. J Immunol. 2009;182:6879‐6888. [DOI] [PubMed] [Google Scholar]
- 25. Nakano T, Kodama H, Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265:1098‐1101. [DOI] [PubMed] [Google Scholar]
- 26. La Motte‐Mohs RN, Herer E, Zúñiga‐Pflücker JC. Induction of T‐cell development from human cord blood hematopoietic stem cells by Delta‐like 1 in vitro. Blood. 2005;105:1431‐1439. [DOI] [PubMed] [Google Scholar]
- 27. Nishimura T, Kaneko S, Kawana‐Tachikawa A, et al. Generation of rejuvenated antigen‐specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114‐126. [DOI] [PubMed] [Google Scholar]
- 28. Vizcardo R, Masuda K, Yamada D, et al. Regeneration of human tumor antigen‐specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31‐36. [DOI] [PubMed] [Google Scholar]
- 29. Wakao H, Yoshikiyo K, Koshimizu U, et al. Expansion of functional human mucosal‐associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:546‐558. [DOI] [PubMed] [Google Scholar]
- 30. Themeli M, Kloss CC, Ciriello G, et al. Generation of tumor‐targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31:928‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kitayama S, Zhang R, Liu T‐Y, et al. Cellular adjuvant properties, direct cytotoxicity of re‐differentiated Vα24 invariant NKT‐like cells from human induced pluripotent stem cells. Stem Cell Reports. 2016;6:213‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ueda N, Uemura Y, Zhang R, et al. Generation of TCR‐expressing innate lymphoid‐like helper cells that induce cytotoxic T cell‐mediated anti‐leukemic cell response. Stem Cell Reports. 2018;10:1935‐1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brauer PM, Singh J, Xhiku S, Zúñiga‐Pflücker JC. T cell genesis: in vitro veritas Est? Trends Immunol. 2016:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ditadi A, Sturgeon CM, Keller G. A view of human haematopoietic development from the Petri dish. Nat Rev Mol Cell Biol. 2016;18:56‐67. [DOI] [PubMed] [Google Scholar]
- 35. Spits H. Development of αβ T cells in the human thymus. Nat Rev Immunol. 2002;2:760‐772. [DOI] [PubMed] [Google Scholar]
- 36. Kennedy M, Awong G, Sturgeon CM, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722‐1735. [DOI] [PubMed] [Google Scholar]
- 37. Seet CS, He C, Bethune MT, et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods. 2017;14:521‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petersdorf EW. HLA matching in allogeneic stem cell transplantation. Curr Opin Hematol. 2004;11:386‐91. [DOI] [PubMed] [Google Scholar]
- 39. Opelz G, Döhler B. Effect of human leukocyte antigen compatibility on kidney graft survival: comparative analysis of two decades. Transplantation. 2007;84:137‐143. [DOI] [PubMed] [Google Scholar]
- 40. Opelz G, Döhler B. Pediatric kidney transplantation: analysis of donor age, HLA match, and posttransplant non‐Hodgkin lymphoma: a collaborative transplant study report. Transplantation. 2010;90:292‐297. [DOI] [PubMed] [Google Scholar]
- 41. Opelz G, Döhler B. Impact of HLA mismatching on incidence of posttransplant non‐Hodgkin lymphoma after kidney transplantation. Transplantation. 2010;89:567‐572. [DOI] [PubMed] [Google Scholar]
- 42. Kanda Y, Kanda J, Atsuta Y, et al. Impact of a single human leucocyte antigen (HLA) allele mismatch on the outcome of unrelated bone marrow transplantation over two time periods. A retrospective analysis of 3003 patients from the HLA Working Group of the Japan Society for Blood and Marrow. Br J Haematol. 2013;161:566‐577. [DOI] [PubMed] [Google Scholar]
- 43. Nakatsuji N, Nakajima F, Tokunaga K. HLA‐haplotype banking and iPS cells. Nat Biotechnol. 2008;26:739‐740. [DOI] [PubMed] [Google Scholar]
- 44. Okita K, Matsumura Y, Sato Y, et al. A more efficient method to generate integration‐free human iPS cells. Nat Methods. 2011;8:409‐412. [DOI] [PubMed] [Google Scholar]
- 45. Taylor CJ, Peacock S, Chaudhry AN, et al. Generating an iPSC bank for HLA‐matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell. 2012;11:147‐152. [DOI] [PubMed] [Google Scholar]
- 46. Gornalusse GG, Hirata RK, Funk SE, et al. HlA‐e‐expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. 2017;35:765‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eyquem J, Mansilla‐Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017:1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adachi K, Kano Y, Nagai T, et al. IL‐7 and CCL19 expression in CAR‐T cells improves immune cell infiltration and CAR‐T cell survival in the tumor. Nat Biotechnol. 2018;36:346‐351. [DOI] [PubMed] [Google Scholar]
- 49. Clarke RL, van der Stegen S, Lee T, et al. Generation of off‐the‐shelf TCR‐less CAR‐targeted cytotoxic T cells from renewable pluripotent cells for cancer immunotherapy [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14‐18; Chicago, IL. Philadelphia (PA): AACR; Cancer Res 2018;78(13 Suppl): Abstract nr LB‐108.