

Abstract
Potent androgen receptor pathway inhibition (ARPI) therapies have given rise to a lethal, aggressive subtype of castration‐resistant prostate cancer (CRPC) called treatment‐induced neuroendocrine prostate cancer (t‐NEPC). Now, t‐NEPC poses a major clinical problem as approximately 20% of CRPC cases bear this subtype—a rate of occurrence that is predicted to rise with the widespread use of ARPI therapies. Unfortunately, there are no targeted therapies currently available to treat t‐NEPC as the origin and molecular underpinnings of t‐NEPC development remain unclear. In the present study, we identify that RNA splicing of the G protein‐coupled receptor kinase‐interacting protein 1 (GIT1) gene is a unique event in t‐NEPC patients. Specifically, upregulation of the GIT1‐A splice variant and downregulation of the GIT1‐C variant expressions are associated with t‐NEPC patient tumors, patient‐derived xenografts, and cell models. RNA‐binding assays show that RNA splicing of GIT1 is directly driven by SRRM4 and is associated with the neuroendocrine phenotype in CRPC cohorts. We show that GIT1‐A and GIT1‐C regulate differential transcriptomes in prostate cancer cells, where GIT1‐A regulates genes associated with morphogenesis, neural function, environmental sensing via cell‐adhesion processes, and epigenetic regulation. Consistent with our transcriptomic analyses, we report opposing functions of GIT1‐A and GIT1‐C in the stability of focal adhesions, whereby GIT1‐A promotes focal adhesion stability. In summary, our study is the first to report that alternative RNA splicing of the GIT1 gene is associated with t‐NEPC and reprograms its function involving FA‐mediated signaling and cell processes, which may contribute to t‐NEPC development.
Keywords: alternative RNA splicing, castration resistant, GIT1, neuroendocrine prostate cancer, SRRM4
Abbreviations
- AdNC
adenocarcinoma prostate cancer with neuroendocrine cells
- AdPC
prostate adenocarcinoma
- ARPI
androgen receptor pathway inhibition
- CI
confidence interval
- CRPC
castration‐resistant prostate cancer
- CRPC‐Ad
castration‐resistant adenocarcinoma
- Ctrl
control
- FA
focal adhesion
- GIT1
G‐protein‐coupled receptor kinase‐interacting protein 1
- GO
gene ontology
- GSEA
gene set enrichment analysis
- IHC
immunohistochemistry
- LTL
living tumor laboratory
- NE
neuroendocrine
- NEPC
neuroendocrine prostate cancer
- NES
normalized enrichment score
- PDX
patient‐derived xenograft
- RISH
RNA in situ hybridization
- RNA‐ChIP
RNA‐chromatin immunoprecipitation
- SCLC
small‐cell lung cancer
- SCNC
small‐cell neuroendocrine prostate cancer
- TMA
tissue microarray
- t‐NEPC
treatment‐induced neuroendocrine prostate cancer
- VPC
Vancouver Prostate Centre
1. INTRODUCTION
Although de novo NEPC represents <2% of all prostate cancer incidences, the increasing prevalence of t‐NEPC as an outcome of selection pressures exerted by potent ARPI treatments is becoming a paramount clinical problem.1, 2 Currently, t‐NEPC accounts for 16%‐25% of all cases of CRPC.1, 3 Patients with t‐NEPC have a mean survival rate of approximately 7 months post‐diagnosis, as the disease is already very aggressive and more resistant to contemporary chemo‐ and radiation therapies.4 Presently, there are no targeted therapies available to treat t‐NEPC effectively due to the limited understanding of the mechanisms initiating its origin and development. Furthermore, t‐NEPC is predicted to become even more prevalent considering the extensive clinical applications of ARPI therapies.1, 2 This highlights the importance of delineating the molecular underpinnings of t‐NEPC to inform future therapies that can prevent or mitigate its development.
Emerging evidence suggests that t‐NEPC is derived from AdPC, and this transition can occur as a result of NE differentiation. Several studies have reported that AdPC and t‐NEPC tumors have similar genotypes (ie, somatic copy number, point mutations, and polyploidy), while their transcriptome, epigenome, and cellular morphologies differ.5, 6, 7, 8 In recent studies, we have shown that a pre‐mRNA splicing factor, SRRM4, can drive NE differentiation to transform LNCaP AdPC cells into t‐NEPC tumors by reprogramming the transcriptome through alternative RNA splicing under ARPI.9 This report established a new NEPC cell model, called LnNE, which uses SRRM4‐overexpressing LNCaP cells to create five generations of xenografts and cell models.10 Interestingly, this SRRM4‐directed RNA splicing profile shares a similar pattern to the diverse alternative splicing patterns seen in the neural system during development, where SRRM4‐spliced target genes have recognized functions that are crucial for neural programs early in development.2 Furthermore, our studies have reported that this reprogrammed RNA splicing signature is unique to NEPC patient tumors, PDX models, and cell models, indicating a clear variance in the phenotype of NEPC and AdPC tumors.9, 10, 11 The SRRM4‐driven t‐NEPC‐specific reprogramming of the transcriptome modifies anti‐apoptotic factors (eg, Bif‐1), epigenetic modifiers (eg, MEAF6‐1), and transcriptional regulators (eg, REST, FOXA1) that are important for regulating cell survival,12 proliferation and tumorigenesis,13 and neural differentiation, respectively.14, 15, 16, 17 Together, these studies propose a model in which SRRM4 drives t‐NEPC development through alternative splicing of downstream genes.2, 10, 11
Among the genes alternatively spliced by SRRM4, the GIT1 gene is differentially spliced into GIT1‐A and GIT1‐C, where the GIT1‐A splice variant is uniquely found in t‐NEPC.9, 11 GIT1 is a multifaceted signaling scaffold protein within the ArfGAP family of proteins that contains a conserved architecture, including an N‐terminal ArfGAP domain, three ankyrin repeats, a Spa2‐homology domain, a coiled‐coil domain, and a FA targeting domain.18 GIT1 has a variety of canonical biological functions, such as endocytosis regulation of receptors, FA regulation, cell motility, morphogenesis, angiogenesis, and neural functions (such as synapse and dendritic spine morphogenesis).18 One of the key functions of GIT1 in epithelial cells is its recruitment of FA proteins, which is important for the formation of stable invadopodia structures, which use stable FAs to degrade the ECM and enable efficient invasion of cancer cells.18 In neural systems, GIT1‐mediated recruitment of FA proteins activate signaling pathways important for regulating directed spine morphogenesis, cell‐to‐cell communication, and stability of synapses.19, 20 FA‐mediated signaling can involve other fundamental aspects of cell biology including survival, proliferation, and environmental sensing of the ECM, mechanical stress, growth hormones, and hypoxic conditions (ie, oxygen and pH alteration).21, 22 Additionally, dysregulation or malfunction of GIT1 has been associated with various neural‐associated diseases, such as Huntington's disease and glioblastoma.18 Increased GIT1 expression has also been reported to promote the migration, invasion, and metastasis of liver, colon, melanoma, lung, renal, and non‐SCLC cells.18 However, the function of GIT1 in the progression of prostate cancer, as well as the role of GIT1 RNA splicing in any cancers, is unknown and has not been previously studied. Here, we investigate the role of GIT1 gene splicing during t‐NEPC progression and the function of its alternative splice isoforms, GIT1‐A and GIT1‐C.
2. MATERIALS AND METHODS
2.1. Clinical RNA‐seq datasets and sample collection
RNA‐seq datasets from the Beltran 2011,23 Beltran 2016,6 and Vancouver Prostate Centre (VPC) 201824 clinical cohorts were used. Cohort composition for the Beltran 2011, Beltran 2016, and VPC 2018 was seven NEPC and 30 AdPC, 15 NEPC and 34 CRPC‐Ad, and five NEPC and 24 AdPC, respectively. RNA‐seq datasets of PDX from the LTL and from the LnNE cell model were previously reported.7 RNA samples from the LTL PDX models were shared by Dr Yuzhuo Wang from the VPC (Vancouver, BC, Canada). All specimen collection protocols and RNA extraction are described in their associated publications.
2.2. RNA‐seq analysis pipeline
Implemented in‐house and published with the VPC 2018 cohort, the RNA‐seq analysis pipeline and algorithms have previously been described.24 However, in the present study, we updated the pipeline to use the Hg38 human genome build and Ensembl GRCh38.86 gene tracks. Cufflinks were also used to identify and quantify alternative splice variants present within all annotated genes.
2.3. Prostate cancer cell models and culture
22Rv1, C4‐2, DU145, LNCaP, PC‐3, VCaP, and NCI‐H660 cell lines were purchased from ATCC (Manassas, VA, USA). LNCaP95 (LN95) cells were kindly provided by Dr Alan Meeker (Johns Hopkins University, MD, USA). SCLC cell line NCI‐H82 was kindly provided by Dr Yuzhuo Wang from the VPC. HEK293T cells were generously provided by Dr Ralph Buttyan from the VPC. The BPH‐1 cell line was provided by Dr Simon Hayward (Vanderbilt University, TN, USA). The LnNE and DuNE cell models, along with their control cells, were previously established by our group.9, 11 All cell‐culturing conditions have been previously reported.2, 13
2.4. Castration‐resistant prostate cancer tissue microarray
The CRPC TMA containing 64 tissue cores from 32 patients that had undergone hormonal therapy, chemotherapy, or radiotherapy was obtained from the tissue bank at VPC. Histopathology of typical AdPC (n = 44), AdNC (atypical AdPC with ≥10% NE cells; n = 6), and SCNC (small‐cell NEPC with NE cells only; n = 6) has previously been reported and characterized.11
2.5. RNA in situ hybridization and immunohistochemistry assays and scoring
RNA in situ hybridization probes targeting GIT1‐A (944‐985 bp; NM_001085454.1) and GIT1‐C (941‐983 bp; NM_014030.3) were designed by Advanced Cell Diagnostics (Hayward, CA, USA). Optimization of RISH probes and RISH probes targeting SRRM4 has previously been characterized.11, 25 RISH results are available in Table S1. RISH assays were carried out using the BaseScope assay kit (Advanced Cell Diagnostics) following the manufacturer's protocols. Red dots indicate positive RISH signals under 10× and 40× magnification. IHC assays on the CRPC TMA and scoring methods for RISH or IHC were carried out, as previously described.11, 24 Briefly, RISH scores of 0, 1, and 2 indicate no positive signal, ≤20% positive signal, and >20% positive signal of cells within a tissue core, respectively. IHC scores were calculated by the signal intensity (no, low, medium, and high as 0‐3, respectively) multiplied by the percentage of positive cells—scores of ≥0.3 were considered to be positive. The SL801 autoloader and Leica SCN400 scanning system (Leica Microsystems; Concord, ON, Canada) were used to digitize the slides at a magnification of 40×. All antibodies used are listed in Table S2.
2.6. DNA and siRNA constructs and transfections
All transfections were carried out using Lipofectamine 3000 (Invitrogen; Waltham, MA, USA), according to the manufacturer's protocols. HnRNP I, U1A, p54nrb, PTB, ASF1/SF2, SRRM4, and U2AF64 expression plasmids and siRNA targeting SRRM4 have been previously described.11, 13
2.7. Real‐time qPCR and immunoblotting
Real‐time qPCR and immunoblot assays were carried out as previously described.26, 27, 28 Antibodies and primers used are listed in Tables S2 and S3, respectively. Three biological repeats were carried out for each experiment.
2.8. RNA‐chromatin immunoprecipitation assays
RNA‐chromatin immunoprecipitation assays were carried out as previously described.9, 27 Briefly, cells were transfected with either an empty vector (control) or Flag‐SRRM4 plasmid. RNA bound to protein was cross‐linked with formaldehyde and immunoprecipitated with anti‐Flag antibody. Eluted RNA fragments were used as templates for RT‐qPCR.
2.9. Construction of stable cell lines by a lentiviral approach
The GIT1‐A‐flag plasmid (#15225) was purchased from Addgene (Cambridge, MA, USA) and was used as a template to clone cDNA of the GIT1‐C gene, as previously described.27 Plasmids were used to create lentiviral expression vectors, using a pFUGWBW backbone, to establish the DU145(Ctrl), DU145(GIT1‐A), and DU145(GIT1‐C) stable cell lines, as previously described.27 Cell lines were cultured under blasticidin selection (5 μg/mL) and overexpression of the GIT1 splice variants was confirmed by both RT‐qPCR and immunoblotting assays.
2.10. AmpliSeq transcriptome sequencing and gene set enrichment analysis
DU145(Ctrl), DU145(GIT1‐A), and DU145(GIT1‐C) stable cell lines were processed using the mirVana RNA Isolation kit (Ambion, Burlington, ON, Canada) following the manufacturer's protocol. Next, AmpliSeq transcriptome library preparation, sequencing, and primary analyses were completed by the UBC‐DMCBH Next Generation Sequencing Centre (Vancouver, BC, Canada), as previously detailed.29 Transcriptome data are available in Table S4. GSEA (www.broadinstitute.org/gsea/index.jsp) was carried out using the latest MSigDB GO collection.
2.11. Focal adhesion assays, immunofluorescence, and microscopy
Cells were seeded on coverslips and serum‐starved the next day. Following serum‐starvation, cells were treated with 10 μmol/L nocodazole (Sigma‐Aldrich; St Louis, MO, USA) for 4 hours, subsequently washed away, and replenished with serum‐containing medium. Cells were fixed at 0 or 120 minutes after the washout for immunofluorescence (IF), costained against GIT1 (Alexa Fluor‐594; Invitrogen) and vinculin (Alexa Fluor‐488; Invitrogen), and then mounted with DAPI (Vector Labs; Burlingame, CA, USA). Antibody information is listed in Table S2. Cells were imaged using the Zeiss AxioObserver Z1 microscope (Carl Zeiss AG; Oberkochen, Germany), and the ZEN program profiled the overlapping GIT1 and vinculin signals in the FA complexes. Three biological repeats were carried out for each experiment.
2.12. Statistics
For VPC and Beltran cohorts, all values were log2‐transformed prior to statistical testing. All clinical groupwise comparisons were calculated using a standard Student's t test. Multiple test corrections were applied to P‐values using the Bonferroni and Hochberg correction method to minimize false discovery rate. Analyses between two groups were compared using unpaired Student's t test. Pearson's χ2 test was carried out to compare GIT1‐A and GIT1‐C RISH scores to the different tumor groups. One‐way ANOVA and Newman‐Keuls multiple comparison test were carried out to compare between multiple groups. Person r correlation analyses were done to compare between RISH scores and the expression of NE positive markers, as well as GIT1‐A or GIT1‐C expression with NEPC and AdPC marker expression or SRRM4 expression. Fisher's exact test was used to calculate the sensitivity and specificity of GIT1‐A to predict the NEPC phenotype. Levels of significance were set at P‐values of 0.05, 0.01, and 0.001, denoted by *, **, and ***, respectively.
3. RESULTS
3.1. Expression profiling of GIT1 splice variants in NEPC patients, PDX, and cell models
Whole‐transcriptome datasets of Beltran 2011,23 Beltran 2016,6 and VPC 201824 patient cohorts, LTL PDX models,7 and LnNE10 RNA‐sequenced datasets were analyzed to determine the expression of GIT1 splice variants in the various prostate cancer models. The software Integrative Genomics Viewer was used to visualize the coverage of RNA‐seq reads corresponding to the GIT1 gene. We confirmed the presence of GIT1‐A, which differs from GIT1‐C by an alternatively spliced 9‐amino acid microexon (exon 8), in the NEPC models, but not the AdPC models (Figure 1A). All three patient cohorts confirmed a robust increase in GIT1‐A expression in NEPC tumors (P < 0.0001) when the expression was compared to that of AdPC or CRPC‐Ad (Figure 1B). Interestingly, GIT1‐C expression was decreased by six‐ (P < 0.05), 11‐ (P < 0.01), and 13‐fold (P < 0.0001) in NEPC tumors when the expression was compared to AdPC subtypes in the VPC 2018, Beltran 2011, and Beltran 2016 cohorts, respectively. Although total GIT1 levels were increased in the Beltran 2011 NEPC tumors, neither VPC 2018 nor Beltran 2016 cohorts showed significant changes in their total GIT1 expression (P = 0.91 and P = 0.077, respectively). This inverse relationship between the expressions of the two splice variants was validated by RT‐qPCR in the LTL PDX models (Figure 1C), as well as in the NEPC cell models LnNE and DuNE (a unique SRRM4‐driven transformation of DU145 AdPC cells to NEPC tumors11; Figure 1D). Here, NEPC PDX and cell models expressed increased levels of GIT1‐A, but decreased levels of GIT1‐C, when compared to the levels in AdPC models. Noteworthily, the 331‐7‐R PDX and LnNE cells are t‐NEPC models that differentiated from their AdPC phenotypes (331‐7 and LNCaP, respectively) after ARPI. Furthermore, profiling GIT1‐A and GIT1‐C expression in a panel of different prostate cell lines (ie, benign, AdPC, NEPC, SCLC) showed that, overall, NEPC and SCLC cell lines expressed higher copy numbers of GIT1‐A and lower copy numbers of GIT1‐C when compared to expression in benign and AdPC cell lines (Figure 1E). Together, these data suggest that the alternative splicing of the GIT1 gene may be associated with the progression to t‐NEPC.
Figure 1.
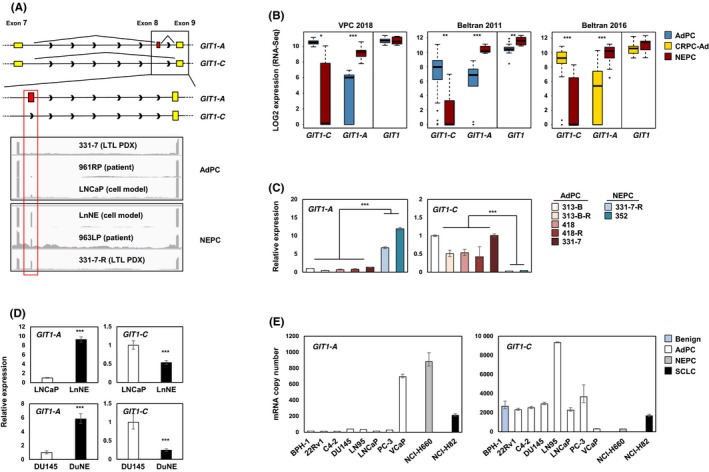
Expression profiling of GIT1 splice variants in NEPC patient, PDX, and cell models. A, Illustration of the GIT1‐C and GIT1‐A exonic regions, where yellow represents constitutive exons and red represents the nine‐amino acid alternatively spliced microexon (exon 8). Integrative Genomics Viewer was used to visualize the coverage of GIT1 by RNA‐seq reads. Grey peaks represent the sequencing depth of each respective exon. B, RNA‐seq data of total GIT1 and splice variant expressions from VPC 2018 (AdPC n = 24 and NEPC n = 5), Beltran 2011 (AdPC n = 30 and NEPC n = 7), and Beltran 2016 (CRPC‐Ad n = 34 and NEPC n = 15) cohorts are shown. C‐D, Relative RNA levels of GIT1 splice variants in the (C) LTL PDX models and (D) NEPC cell models (LnNE and DuNE) was validated. E, GIT1 splice variant expressions were profiled from a benign prostate cell line (BPH‐1), a panel of AdPC cell lines (22Rv1, C4‐2, DU145, LN95, LNCaP PC‐3, and VCaP), a NEPC cell line (NCI‐H660) and a SCLC cell line (NCI‐H82) by RT‐qPCR for absolute quantification using a standard curve. All results are presented as mean ± SD (Student's t test; *, **, and *** represent P < 0.05, P < 0.01, and P < 0.0001, respectively). AdPC, prostate adenocarcinoma; CRPC‐Ad, castration‐resistant adenocarcinoma; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; LTL, living tumor laboratory; NEPC, neuroendocrine prostate cancer; PDX, patient‐derived xenograft; SCLC, small‐cell lung cancer; VPC, Vancouver Prostate Centre
3.2. RNA splicing of GIT1 is associated with clinical NEPC tumors
Currently, there are no commercially available GIT1 antibodies to specifically detect each splice variant. As a result, we used RISH, a well‐established alternative technique,11, 24 on a TMA to study and confirm the expression of GIT1‐A and GIT1‐C. We created probes to target the unique exon 7/8 junction of GIT1‐A, as well as the exon 7/9 junction of GIT1‐C, to detect RNA expression levels on the TMA. Human CRPC TMA contains 64 cores: 52 AdPC, six AdNC, and six SCNC. Details of these prostate cancer subtype classifications have been previously published by our group.11, 24 Briefly, AdPC is classified by the presence of glandular structures, large cells with a prominent nucleolus, and no NE cells. Next, SCNC tumors contain only NE cell populations, grow as solid sheets, and show typical NE cell features, such as a scant cytoplasm, salt‐and‐pepper nuclei, and a high nucleus‐to‐cytoplasm ratio. Finally, AdNC subtypes are histologically similar to AdPC; however, they are atypical tumors comprising a mixed‐cell population containing ≥10% NE cells and are positive for at least two NE markers.
We found that GIT1‐A is expressed in 21 out of 64 tissue cores (Table S1). The 21 cores positive for GIT1‐A included all six SCNC cores (five of six had a RISH score of 2) and five AdNC cores (Figure 2A). Conversely, 51 of 52 AdPC cores were positive for GIT1‐C, whereas only two AdNC and two SCNC cores were positive (RISH score of 1). Correlation studies indicate that GIT1‐A expression levels increase in AdNC, and even further in SCNC, when compared to the expression in AdPC. Moreover, GIT1‐C expression levels decrease in AdNC and SCNC when compared to the expression levels in AdPC (Figure 2B,C). GIT1‐A expression showed significant positive correlation (P < 0.0001) with all three NE markers (CHGA, r = 0.7209; SYP, r = 0.7152; and CD56, r = 0.7148) and negative correlation with markers of AdPC (androgen receptor [AR], r = −0.3937, P = 0.0013; and prostate‐specific antigen [PSA], r = −0.3259, P = 0.0086). Inversely, GIT1‐C expression was negatively correlated with CHGA (r = −0.6193, P < 0.0001), SYP (r = −0.6353, P < 0.0001), and CD56 (r = −0.4158, P < 0.001) expression, but positively correlated with AR expression (r = 0.4105, P < 0.001; Table 1). Furthermore, GIT1‐A expression was positively correlated with the number of positive NE markers in a tissue core (r = 0.7172, P < 0.001), whereas GIT1‐C expression was negatively correlated (r = −0.6473, P < 0.001; Figure 2D).
Figure 2.
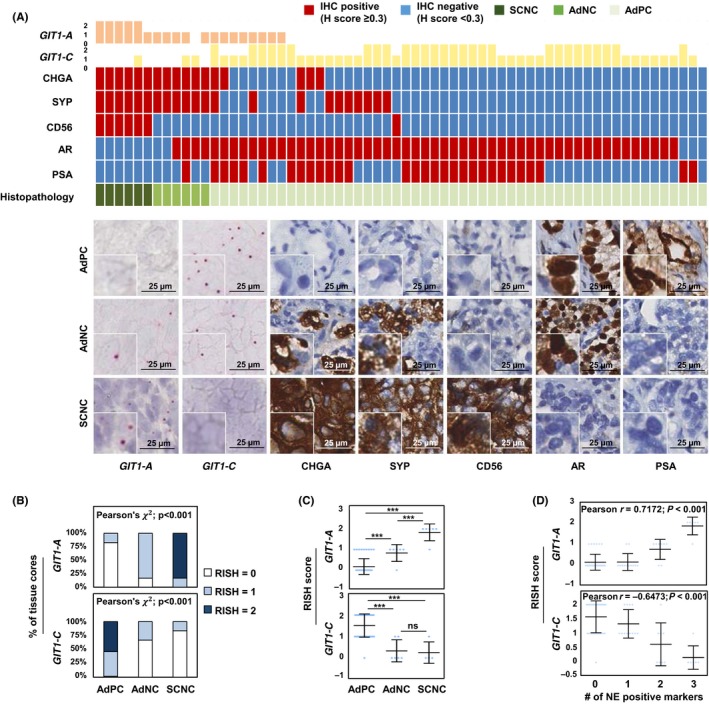
RNA splicing of GIT1 is associated with clinical NEPC tumors. A, RISH probes targeting the exons 7/8 or exons 7/9 junction were created to detect GIT1‐A or GIT1‐C, respectively, in a human CRPC TMA (n = 64 cores). TMA was stained against CHGA, SYP, CD56, AR and PSA by immunohistochemistry (IHC). Columns in the heatmap represent one of 64 cores. One representative core from each of the histologically diagnosed AdPC (n = 52), AdNC (n = 6), and SCNC (n = 6) cores is shown. Scale bars represent 25 μm. B‐C, Cores were grouped according to their histopathology report, and their respective RISH scores were plotted to present the (B) percentage of cores containing the same RISH score (Pearson's χ2 test) or (C) average RISH score within each tumor subtype (one‐way ANOVA; ***P < 0.001; ns, non‐significant). D, RISH scores from each core were plotted with respect to the number of positive NE markers within the same core (Pearson's r correlation). AdNC, adenocarcinoma prostate cancer with neuroendocrine cells; AdPC, prostate adenocarcinoma; CRPC, castration‐resistant prostate cancer; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; NE, neuroendocrine; RISH, RNA in situ hybridization; SCNC, small‐cell neuroendocrine prostate cancer; TMA, tissue microarray
Table 1.
Correlation of GIT1 splice variants with clinical diagnostic markers of AdPC and NEPC
| Correlation with GIT1‐A | Pearson r value | P value |
|---|---|---|
| CHGA | 0.7209 | <0.0001 |
| SYP | 0.7152 | <0.0001 |
| CD56 | 0.7148 | <0.0001 |
| AR | −0.3937 | 0.0013 |
| PSA | −0.3259 | 0.0086 |
| Correlation with GIT1‐C | Pearson r value | P value |
|---|---|---|
| CHGA | −0.6193 | <0.0001 |
| SYP | −0.6353 | <0.0001 |
| CD56 | −0.4158 | <0.001 |
| AR | 0.4105 | <0.001 |
| PSA | 0.04045 | ns |
Pearson's r correlation was carried out between the expression of GIT1‐A or GIT1‐C and expressions of NEPC (ie, CHGA, SYP, and CD56) or AdPC (ie, AR and PSA) diagnostic markers used in the clinic. AdPC, prostate adenocarcinoma; AR, androgen receptor; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; NEPC, neuroendocrine prostate cancer; ns, not significant; PSA, prostate‐specific antigen.
To determine the diagnostic reliability of GIT1‐A to detect a NEPC phenotype, we defined AdNC and SCNC as NEPC. Fisher's exact test showed that the GIT1‐A sensitivity of correctly identifying the NEPC phenotype was 0.9773 (95% CI: 0.8798‐0.9994) and the specificity was 0.55 (95% CI: 0.3153‐0.7694; Table 3). These results indicate that although GIT1‐A is detectable in NEPC cores, the low specificity suggests that AdPC tumors may also express GIT1‐A. However, it is unknown whether these AdPC GIT1‐A‐positive cores progressed to NEPC. Overall, these collective findings indicate a strong positive association between GIT1 splice variant expression and t‐NEPC development, namely that GIT1‐A expression manifests largely in NEPC tumors.
Table 3.
Sensitivity and specificity of GIT1‐A as a biomarker for NEPC
| GIT1‐A | AdPC | NEPC | Sensitivity | Specificity | P value |
|---|---|---|---|---|---|
| + | 1 | 11 | 0.9773 | 0.55 | <0.0001 |
| − | 43 | 9 | (0.8798‐0.9994) | (0.3153‐0.7694) |
Fisher's exact test was carried out to determine the sensitivity and specificity of GIT1‐A as a diagnostic biomarker for NEPC prediction. SCNC and AdNC are defined as NEPC in this case. AdNC, adenocarcinoma prostate cancer with neuroendocrine cells; AdPC, prostate adenocarcinoma; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; NEPC, neuroendocrine prostate cancer; SCNC, small‐cell neuroendocrine prostate cancer.
3.3. SRRM4 regulates RNA splicing of GIT1
Based on our previous report, SRRM4 predominately drove a t‐NEPC‐unique RNA splicing signature, promoting the transformation of AdPC to a NEPC phenotype.9, 11 RISH probes targeting SRRM4 were hybridized on identical CRPC TMA cores (Table S1). SRRM4 expression was strongly correlated with GIT1‐A (r = 0.7994, P < 0.0001) and negatively correlated with GIT1‐C expression (r = −0.6067, P <0.0001; Figure 3A and Table 2). To investigate how SRRM4 mediates GIT1 splicing, we overexpressed SRRM4 in LNCaP cells and detected a five‐fold increase in GIT1‐A expression and a ~40% reduction in GIT1‐C expression (Figure 3B). Transfection with siRNA in the LnNE model was used to knock down SRRM4, which reduced GIT1‐A expression by ~20% and increased GIT1‐C expression by six‐fold (Figure 3C). Within the panel of splicing factors tested, SRRM4 was unique for its GIT1‐splicing activity (Figure 3D). We designed a ChIP probe specific to the 3′ intron splicing site upstream of the alternatively spliced microexon 8 of GIT1. Using an RNA‐ChIP binding assay, we determined that SRRM4 directly binds to GIT1 and facilitates its splicing at exon 8 but does not bind to the negative control region on GAPDH (Figure 3E). Collectively, these results show that SRRM4 directly splices the GIT1 gene to promote the neural‐specific GIT1‐A splice product.
Figure 3.
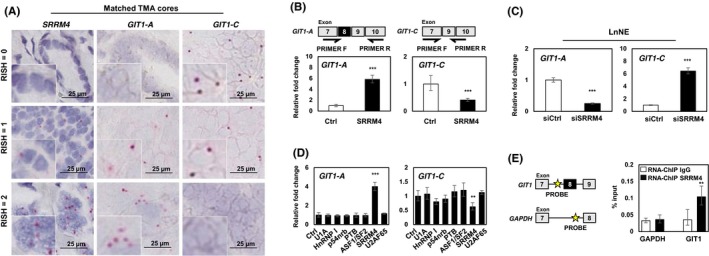
SRRM4 regulates RNA splicing of GIT1. A, Matched TMA cores are shown to represent the associations of the expressions of SRRM4 with GIT1‐A and GIT1‐C. Scale bars represent 25 μm. B, LNCaP cells were transfected with 4 μg flag‐SRRM4 and subsequently extracted for RNA. Relative expressions of GIT1‐A or GIT1‐C were compared to 18S by RT‐qPCR. Primer pairs designed for unique exon junctions in GIT1‐A and GIT1‐C variants are illustrated. C, NEPC cell model, LnNE, was transfected with 20 μmol/L siRNA targeting SRRM4 or negative control siRNA to determine the splicing activity of GIT1. D, Various RNA splicing factors (4 μg) or control (empty vector) were transfected into LNCaP cells and subsequently extracted for RNA. RT‐qPCR was carried out to compare GIT1 splice variant expression compared to 18S (one‐way ANOVA; n = 3; ** and *** denotes P < 0.01 and P < 0.001, respectively). E, RNA‐ChIP probes for the intron sequence upstream of the alternatively spliced microexon (exon 8) of GIT1 and probes for negative‐control gene GADPH were created, as indicated by the yellow star. RNA‐ChIP was carried out with LNCaP cells transfected with 10 μg Flag‐SRRM4 and immunoprecipitated with anti‐Flag antibody. RNA fragments were eluted and used as templates for the antisense primers/probes by RT‐qPCR. All experiments were repeated three times. Unless otherwise indicated, results are presented as mean ± SD (Student's t test, n = 3; **P < 0.01; ***P < 0.001). Ctrl, control; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; NEPC, neuroendocrine prostate cancer; RISH, RNA in situ hybridization; RNA‐ChIP, RNA‐chromatin immunoprecipitation; TMA, tissue microarray
Table 2.
SRRM4 expression is associated with GIT1 splice variant expressions in CRPC
| GIT1‐A RISH score | ||||
|---|---|---|---|---|
| 0 | 1 | 2 | ||
| SRRM4 RISH score | 0 | 42 | 5 | 0 |
| 1 | 2 | 7 | 1 | |
| 2 | 0 | 3 | 4 | |
| r = 0.7994; P < 0.0001 | ||||
| GIT1‐C RISH score | ||||
|---|---|---|---|---|
| 0 | 1 | 2 | ||
| SRRM4 RISH score | 0 | 0 | 21 | 26 |
| 1 | 3 | 4 | 3 | |
| 2 | 6 | 1 | 0 | |
| r = −0.6067; P < 0.0001 | ||||
Pearson's r correlation was applied between the expressions of GIT1‐A or GIT1‐C and SRRM4 expression. CRPC, castration‐resistant prostate cancer; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; RISH, RNA in situ hybridization.
3.4. Transcriptome and cellular functions of GIT1 splice variants
To determine the functional significance of GIT1‐A and GIT1‐C, we transduced DU145 cells to overexpress GIT1‐A (DU145(GIT1‐A)) or GIT1‐C (DU145(GIT1‐C)) and confirmed their expression by immunoblotting and RT‐qPCR (Figure 4A). Using Ion AmpliSeq Transcriptome analyses, we profiled and compared the DU145(GIT1‐A) or DU145(GIT1‐C) transcriptome to DU145(Ctrl) (n = 524 and 755 genes, respectively; Figure 4B). The transcriptomes were mostly distinct, with the exception of 177 genes in common. To further investigate the biological differences between the GIT1‐A and GIT1‐C transcriptomes, we carried out GSEA and compared the two phenotypes, showing that the GIT1‐A transcriptome was enriched with gene sets related to neural function, morphogenesis, environmental sensing, and epigenetic regulation (Figure 4C). The GIT1‐C transcriptome was enriched with gene sets associated with general immune function and metabolism (Figure S1). Within the four subgroups enriched in the GIT1‐A transcriptome, we extracted the leading‐edge genes from the “GO regulation of cell adhesion mediated by integrin,” “GO cardiac epithelial to mesenchymal transition,” and “GO neuron recognition” MSigDB gene sets and generated a heatmap using GSEA software (Figure 4D). Among these genes identified, we validated the expression of nine using RT‐qPCR (Figure 4E). These results suggest that GIT1‐A and GIT1‐C show differential functions, whereby GIT1‐A facilitates progression to a neuronal transcriptome indicative of NEPC. Consistent with the GSEA predictions, we confirmed that the GIT1 splice variants differentially regulate FA (Figure 5). We observed that GIT1‐A promoted, whereas GIT1‐C compromised the stability of FAs, indicated by vinculin (a universal FA marker), when DU145 cells were treated with nocodazole. Collectively, our transcriptomic and FA assays indicate distinctive functional roles of GIT1 splice variants in regulating FA stability, which may contribute to NEPC development.
Figure 4.
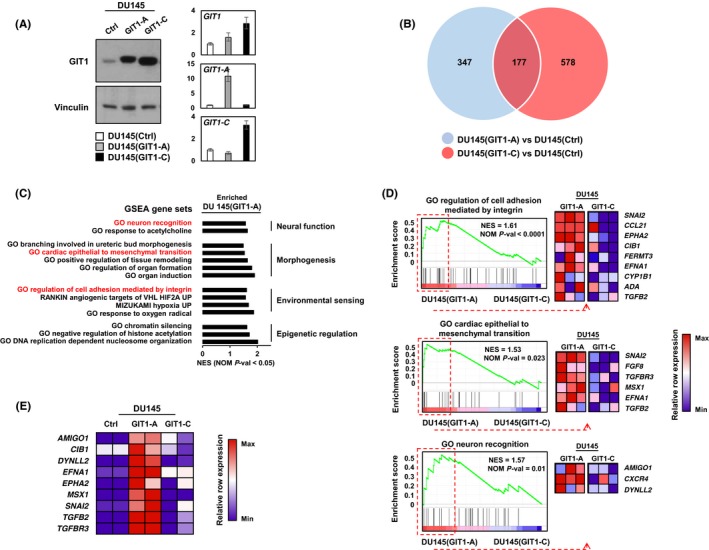
Transcriptome profiling of GIT1 splice variants. A, DU145 stable cell lines overexpressing GIT1‐A, GIT1‐C, or empty vector (Ctrl) were created by lentiviral transduction and expressions were validated by immunoblot and RT‐qPCR. B‐C, Transcriptomes of these cell lines (Ctrl, n = 2; GIT1‐A, n = 3; GIT1‐C, n = 3) were profiled by Ion AmpliSeq Transcriptome. B, Compared to control, genes unique to the transcriptomes of GIT1‐A (n = 524) or GIT1‐C (n = 755) overlapped (n = 177) using a fold change threshold of 1.2 and P < 0.05 cut‐off. C, The transcriptomes of GIT1‐A and GIT1‐C were analyzed by GSEA based on the latest MSigDB database for each collection. GSEA showed enrichment of genes associated with neural function, morphogenesis, environmental sensing, and epigenetic function in the DU145(GIT1‐A) cells. D, GSEA enrichment plots from these categories are presented where differential expressions of the leading‐edge genes are shown in the heatmaps created by the GSEA software. E, Expression of nine of these genes was validated and confirmed by RT‐qPCR relative to the DU145(Ctrl) cell line (n = 2 per cell line). Heatmap was created using the normalized z‐scores of each row. All experiments were repeated three times. GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; GO, gene ontology; GSEA, gene set enrichment analysis; NES, normalized enrichment score; NOM P‐val, nominal P‐value
Figure 5.
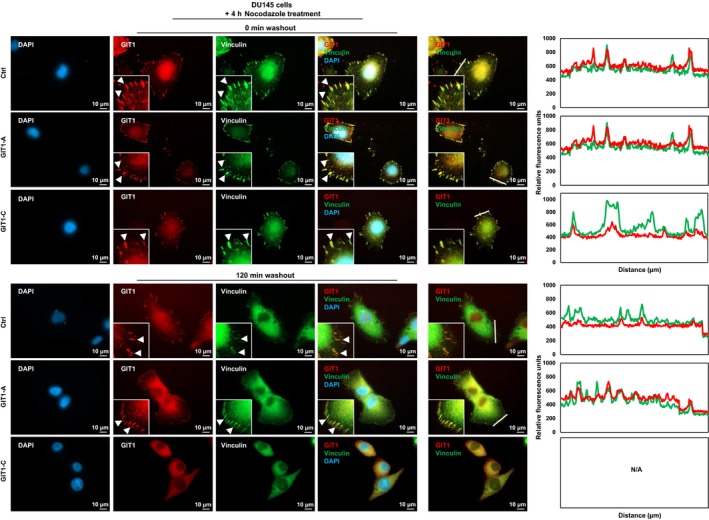
Differential functions of the GIT1 splice variants in FA stability. DU145 stable cell lines overexpressing GIT1‐A, GIT1‐C, or empty vector were seeded on coverslips and serum‐starved. They were treated with 10 μmol/L nocodazole for 4 h, subsequently washed away, and replaced with serum‐containing medium. Cells were fixed at 0 or 120 min after the washout, costained against GIT1 and vinculin, and then mounted with DAPI staining mount. Cells were imaged using a Zeiss AxioObserver Z1 (Carl Zeiss AG; Oberkochen, Germany) microscope, where the scale bar represents 10 μm. Arrowheads indicate FA complexes. Overlapping signals between GIT1 and vinculin appear yellow. Overlapping of the two signals in a cross‐section (indicated by white line) of FA complexes were profiled by the ZEN program. All experiments were repeated three times. FA, focal adhesion; GIT1, G‐protein‐coupled receptor kinase‐interacting protein 1; IF, immunofluorescence; ZEN, ZEISS efficient navigation
4. DISCUSSION
Using patient tumor samples, PDX models, and cell models, we are the first to characterize the alternative RNA splicing of the GIT1 gene in its association with NEPC progression. We found that an inverse correlation of high GIT1‐A and low GIT1‐C expression is associated with the NEPC phenotype, when compared to GIT1 splice variant expression in AdPC subtypes (Figures 1 and 2, Tables 1 and 3). We demonstrate that SRRM4 is an important regulator of GIT1 post‐translational modifications, whereby SRRM4 expression in NEPC tumors is associated with the splicing of the GIT1 gene (Figure 3, Table 2). From whole‐transcriptome analyses, we report differential transcriptomes of GIT1‐A and GIT1‐C, where GIT1‐A regulates gene sets that are associated with morphogenesis, neural function, environmental sensing, and epigenetic regulation (Figure 4). Consistent with our transcriptomic analyses, we report opposing functions of GIT1‐A and GIT1‐C in the stability of FA, whereby GIT1‐A‐overexpressing cells promote FA stability (Figure 5). Altogether, our study shows that SRRM4‐mediated RNA splicing of the GIT1 gene reprograms its function involving FA‐mediated signaling and cell processes, which may contribute to t‐NEPC development.
Our current understanding of SRRM4‐driven NEPC progression involves multi‐complex mechanisms, including NE differentiation, apoptosis evasion, cell proliferation, and tumorigenesis. Subsequent studies have found that these processes involved in NEPC progression are facilitated by REST, Bif‐1, and MEAF6 which are functionally reprogrammed through SRRM4‐driven alternative splicing to mediate NE differentiation,9 apoptosis evasion,12 and cell proliferation and tumorigenesis,13 respectively. However, there are many other cellular functions of SRRM4 waiting to be defined. In fact, this study is the first to report that SRRM4 regulates RNA splicing of GIT1 to reprogram its function, whereby the neural‐specific GIT1‐A splice variant regulates genes associated with cell‐adhesion processes (Figure 4C) and enhances FA stability (Figure 5). In support of this, DuNE is an SRRM4‐driven NEPC model that expresses genes related to FA processes, suggesting that SRRM4 may modulate FA‐mediated signaling and cell processes through the GIT1 splice variants. This understanding demonstrates that multiple gene networks and cellular processes are altered during NEPC progression, by which cell‐adhesion gene networks are only a part of this multifaceted process. Moreover, SRRM4 splices genes that are canonical components of many epigenetic complexes, such as MEAF6 and PHF21A.9 GIT1 has also been suggested to regulate epigenetic modifications through its interaction with MAT2B, which synthesizes methyl donors for DNA and histone methylation during cancer progression.18 Additionally, our transcriptomic analyses show that the GIT1‐A transcriptome is enriched with gene sets related to epigenetic regulation (Figure 4C). Although specific epigenetic regulation of GIT1 has yet to be reported, we hypothesize that SRRM4‐driven RNA splicing mechanisms may interplay with epigenetic mechanisms to reprogram AdPC cells to promote and establish t‐NEPC development.
In conclusion, our research suggests that RNA splicing of the GIT1 gene is associated with the progression of NEPC, evident by our comprehensive transcriptomic data, which show distinct molecular changes regulated by GIT1‐A and GIT1‐C. In addition, we confirm that GIT1 splice variants differentially regulate the stability of FAs. As SRRM4 is a demonstrated driver of NEPC progression, our studies show a novel function of SRRM4 in regulating FA‐mediated processes through GIT1 gene splicing.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank Dr James Manley (Columbia University), Dr Gourisankar Ghosh (UC San Diego), Dr Allain Frédéric (Institute for Molecular Biology and Biophysics Eidgenössiche Technische Hochshule, Switzerland), and Dr Benjamin Blencowe (University of Toronto) for their expression plasmids used in Figure 3D. Thank you to Sahil Kumar for editing assistance. This study was supported by the Canadian Institutes of Health Research (MOP137007 & PTJ156150; X. Dong).
Lee AR, Gan Y, Xie N, Ramnarine VR, Lovnicki JM, Dong X. Alternative RNA splicing of the GIT1 gene is associated with neuroendocrine prostate cancer. Cancer Sci. 2019;110:245–255. 10.1111/cas.13869
REFERENCES
- 1. Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol. 2018;15(5):271‐286. [DOI] [PubMed] [Google Scholar]
- 2. Lee AR, Che N, Lovnicki JM, Dong X. Development of neuroendocrine prostate cancers by the Ser/Arg repetitive matrix 4‐mediated RNA splicing network. Front Oncol. 2018;8:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bluemn EG, Coleman IM, Lucas JM, et al. Androgen receptor pathway‐independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32(4):474‐489.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akamatsu S, Inoue T, Ogawa O, Gleave ME. Clinical and molecular features of treatment‐related neuroendocrine prostate cancer. Int J Urol. 2018;25(4):345‐351. [DOI] [PubMed] [Google Scholar]
- 5. Aparicio AM, Shen L, Tapia EL, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22(6):1520‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration‐resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin D, Wyatt AW, Xue H, et al. High fidelity patient‐derived xenografts for accelerating prostate cancer discovery and drug development. Can Res. 2014;74(4):1272‐1283. [DOI] [PubMed] [Google Scholar]
- 8. Park JW, Lee JK, Sheu KM, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science. 2018;362(6410):91‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Donmez N, Sahinalp C, et al. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur Urol. 2017;71(1):68‐78. [DOI] [PubMed] [Google Scholar]
- 10. Li Y, Chen R, Bowden M, et al. Establishment of a neuroendocrine prostate cancer model driven by the RNA splicing factor SRRM4. Oncotarget. 2017;8(40):66878‐66888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee AR, Gan Y, Tang Y, Dong X. A novel mechanism of SRRM4 in promoting neuroendocrine prostate cancer development via a pluripotency gene network. EBioMedicine. 2018;35:167‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gan Y, Li Y, Long Z, et al. Roles of alternative RNA splicing of the Bif‐1 gene by SRRM4 during the development of treatment‐induced neuroendocrine prostate cancer. EBioMedicine. 2018;31:267‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee AR, Li Y, Xie N, et al. Alternative RNA splicing of the MEAF6 gene facilitates neuroendocrine prostate cancer progression. Oncotarget. 2017;8(17):27966‐27975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen R, Li Y, Buttyan R, Dong X. Implications of PI3K/AKT inhibition on REST protein stability and neuroendocrine phenotype acquisition in prostate cancer cells. Oncotarget. 2017;8(49):84863‐84876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lapuk AV, Wu C, Wyatt AW, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol. 2012;227(3):286‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shimojo M, Shudo Y, Ikeda M, Kobashi T, Ito S. The small cell lung cancer‐specific isoform of RE1‐silencing transcription factor (REST) is regulated by neural‐specific Ser/Arg repeat‐related protein of 100 kDa (nSR100). Mol Cancer Res. 2013;11(10):1258‐1268. [DOI] [PubMed] [Google Scholar]
- 17. Zhang X, Coleman IM, Brown LG, et al. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration‐resistant prostate cancer. Clin Cancer Res. 2015;21(20):4698‐4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou W, Li X, Premont RT. Expanding functions of GIT Arf GTPase‐activating proteins, PIX Rho guanine nucleotide exchange factors and GIT‐PIX complexes. J Cell Sci. 2016;129(10):1963‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS. Regulation of postsynaptic structure and protein localization by the Rho‐type guanine nucleotide exchange factor dPix. Neuron. 2001;32(3):415‐424. [DOI] [PubMed] [Google Scholar]
- 20. Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25(13):3379‐3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petrova V, Annicchiarico‐Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. 2018;7(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10(1):21‐33. [DOI] [PubMed] [Google Scholar]
- 23. Beltran H, Rickman DS, Park K, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramnarine VR, Alshalalfa M, Mo F, et al. The long noncoding RNA landscape of neuroendocrine prostate cancer and its clinical implications. GigaScience. 2018;7(6):1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y, Zhang Q, Lovnicki J, et al. SRRM4 gene expression correlates with neuroendocrine prostate cancer. Prostate. 2018;1‐9. [DOI] [PubMed] [Google Scholar]
- 26. Lee AR, Hung W, Xie N, Liu L, He L, Dong X. Tyrosine residues regulate multiple nuclear functions of P54nrb. J Cell Physiol. 2017;232(4):852‐861. [DOI] [PubMed] [Google Scholar]
- 27. Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33(24):3140‐3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu Y, Liu L, Xie N, et al. Expression and function of the progesterone receptor in human prostate stroma provide novel insights to cell proliferation control. J Clin Endocrinol Metab. 2013;98(7):2887‐2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li W, Turner A, Aggarwal P, et al. Comprehensive evaluation of AmpliSeq transcriptome, a novel targeted whole transcriptome RNA sequencing methodology for global gene expression analysis. BMC Genom. 2015;16:1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials