

Abstract
For efficacy of peptide vaccination immunotherapy for patients with cancer, endogenous expression of the target peptide/human leukocyte antigen (HLA) on cancer cells is required. However, it is difficult to evaluate the expression status of a peptide/HLA complex because of the lack of a soluble T‐cell receptor (TCR) that reacts with tumor‐associated antigen (TAA) with high avidity. In the present study, we developed two soluble TCR‐multimers that were each directed to TAA, survivin‐2B (SVN‐2B) and PBF in the context of HLA‐A24 (SVN‐2B TCR‐multimer and PBF TCR‐multimer, respectively), from CTL clones that were established from peptide‐vaccinated patients. Both TCR multimers could recognize cognate peptide‐pulsed antigen‐presenting cells, C1R‐A24 cells, in a CD8‐independent method. Moreover, the PBF TCR‐multimer successfully recognized a PBF peptide naturally presented on HLA‐A24+ PBF + osteosarcoma cells. Taken together, the results indicated that a TCR‐multimer might be useful for detection of a TAA‐derived peptide presented by HLA in patients receiving immunotherapy.
Keywords: HLA‐A24, multimer, PBF, survivin, T‐cell receptor
1. INTRODUCTION
Peptide vaccination immunotherapy using human leukocyte antigen (HLA) class I‐binding short peptides has been used to induce tumor‐specific cytotoxic T lymphocytes (CTL) in patients with various cancers, because various tumor‐associated antigens (TAA) have been identified.1, 2 Generally, TAA‐derived peptide vaccination induces antigen‐specific CTL in cancer patients. We have also observed antigen‐specific CTL responses in patients with various carcinomas and sarcomas.3, 4 For the efficacy of peptide vaccination immunotherapy, endogenous expression of the target peptide/HLA on cancer cells is required. However, it is difficult to evaluate the expression status of a peptide/HLA complex because of the lack of a soluble T‐cell receptor (TCR) reacting with TAA with high avidity.
We previously reported an artificial monoclonal antibody (mAb), D12, directed to osteosarcoma antigen PBF‐derived peptide/HLA‐A2 complex on osteosarcoma cells.5 mAb D12 showed high affinity and characteristics of TCR‐like specificity, but some cross‐reactivity to other antigens was observed when using a mAb D12 multimer. Generally, the advantage of TCR‐like mAbs is their high affinity. However, regarding the mAb D12, the specificity of TCR might be superior to that of TCR‐like mAb.
In the present study, we successfully developed two soluble TCR‐multimers that were each directed to TAA, survivin‐2B and PBF in the context of HLA‐A24 (SVN‐2B TCR‐multimer and PBF TCR‐multimer, respectively) from CTL clones that were established form peptide‐vaccinated patients. Both TCR multimers could recognize cognate peptide‐pulsed antigen‐presenting cells, C1R‐A24 cells. Although SVN‐2B TCR‐multimer failed to recognize an endogenous peptide/HLA complex, PBF TCR‐multimer successfully recognized the PBF peptide naturally presented on HLA‐A24+PBF+ osteosarcoma cells.
2. MATERIALS AND METHODS
The present study was carried out in accordance with the guidelines established by the Declaration of Helsinki and was approved by the Ethics Committee of Medical and Biological Laboratories Company, Limited, Sapporo Medical University and Higashi‐Sapporo Hospital. The patients, their families, and healthy donors provided informed consent for the use of blood samples in our research.
2.1. Peptides
Survivin‐2B (SVN‐2B) peptide (AYACNTSTL),6 PBF peptide (AYRPVSRNI)7 and HIV peptide (RYLRDQQLL) were synthesized (>95% purity) by Sigma‐Aldrich Japan (Ishikari, Japan).
2.2. Cells, antibodies, tetramers and flow cytometry analysis
The following cells, antibodies, and tetramers were used for staining and cell sorting. C1R‐A24 cells, lymphoblastoid C1R cells transfected with HLA‐A*24:02 cDNA, were a kind gift from Dr M. Takiguchi (Kumamoto University School of Medicine, Japan). Sup‐T1 cell lines were purchased from the Riken Cell Bank (Tsukuba, Japan). Jurkat/MA was kindly donated by Dr Yoshinobu Ichiki (University of Occupational and Environmental Health, Japan) and Dr Sjoerd H. van der Burg (Leiden University Medical Centre, Netherlands).8 Osteosarcoma cell lines KIKU, HOS and HOS‐A24 were established at Sapporo Medical University. CD8‐FITC, 7‐AAD, and IOTest Beta Mark were purchased from Beckman Coulter Inc. (Miami, FL, USA). HLA‐A*24:02 survivin‐2B tetramer (amino acids 80‐88; AYACNTSTL), HLA‐A*24:02 PBF A24.2 tetramer (amino acids 145‐153; AYRPVSRNI), HLA‐A*24:02 WT1 modified tetramer (amino acids 235‐43; CYTWNQMNL), and HLA‐A*24:02 HIV‐negative tetramer (amino acids 584‐592; RYLRDQQLL) were purchased from Medical and Biological Laboratories Co., Ltd (Nagoya, Japan). DAPI was purchased from Vector Laboratories Inc. (Burlingame, CA, USA). Cells were first stained with tetramers at 4°C for 15 minutes and then stained with an anti‐CD8 mAb at 4°C for 15 minutes. FACSCanto and FACSCalibur (BD Biosciences, San Diego, CA, USA) were used for flow cytometry analysis. CellQuest software (BD Biosciences) and FlowJo software (Tree Star, Inc., San Carlos, CA, USA) were used for data analysis.
2.3. CTL clones
Survivin‐2B or PBF‐specific CTL lines were established in our laboratory using PBMC from SVN‐2B or PBF peptide‐vaccinated HLA‐A24+ donors, respectively, by a mixed lymphocyte peptide culture (MLPC) according to the method described by Karanikas et al.9 Briefly, antigenic peptides were directly added to PBMC at 10 μg/mL suspended in a 2‐mL medium in a 24‐well plate (BD Biosciences), and cultures were maintained at 37°C in 5% CO2. On day 2, recombinant human interleukin (IL)‐2 (50 U/mL; Shionogi Pharmaceutical Institute Co., Osaka, Japan) was added. Starting on day 5, half‐medium change and supplementation of IL‐2 were carried out every other day until day 14. Resultant cells were stained by the tetramer at 4°C for 30 minutes at 1:20 dilution and then stained for a further 20 minutes with FITC‐conjugated anti‐CD8 mAb. Tetramer‐positive CTL were detected by FACSAriaII (BD Biosciences). To establish CTL clones, tetramer‐positive T cells were isolated by single cell sorting using FACSAriaII. Cells were seeded with irradiated allogeneic PBMC (1 × 105) in 100 μL AIM‐V containing 10% Human AB Serum (HS), IL‐2 (200 U/mL) and phytohemagglutinin‐P (PHA; 7.5 μg/mL; Wako Chemicals, Osaka, Japan) in 96‐well round‐bottom microplates. On day 7, 100 μL AIM‐V containing 10% HS and IL‐2 was added. On day 14, all of the cells that proliferated were collected and washed, and the medium was replaced with fresh AIM‐V containing 10% HS and IL‐2, followed by maintenance in a 48‐well microplate at 0.5‐1 × 106 cells/well. Finally, ITG‐MT3 and FKS‐D11P CTL clones were established.
2.4. Cytotoxicity assay
Antigen‐specific lytic activity of CTL clone ITG‐MT3 was evaluated by using a 51Cr release assay. HLA‐A24‐positive C1R‐A24 cells and HLA‐negative K562 cells in 100 μL complete medium were labeled with 3.7 MBq 51Cr for 1 hour at 37°C. For peptide reconstitution assays, 1 μmol/L of a synthetic peptide was added 1 hour before introducing effector cells. After 4 hours of incubation with effector cells, radioactivities of supernatants were analyzed with a gamma counter. Percentage specific lysis was determined by: ([experimental release − spontaneous release]/[maximum release − spontaneous release]) × 100. Antigen‐specific lytic activity of CTL clone FKS‐D11P was evaluated by using a carboxyfluorescein diacetate succinimidyl ester (CFSE)‐based cytotoxicity assay.10 Target C1R‐A24 cells were labeled with 1 μmol/L 6‐CFSE (Wako Pure Chemical Industry, Osaka, Japan) for 10 minutes at 25°C. After two washes, CFSE‐labeled target cells were cocultured with graded numbers of effector T cells for 5 hours at 37°C in 5% CO2 in the presence or absence of peptides in 96‐well microtiter plates. Whole cells were harvested and stained with Kusabira Orange‐labeled Annexin‐V (MBL, Nagoya, Japan) for 15 minutes at 25°C according to the manufacturer's instructions, and the absolute number of surviving cells was determined using a FACSCalibur (BD Biosciences) with the aid of CellQuest software (BD Biosciences). Percentage lysis was calculated as follows: ([ET − T0]/[100 − T0]) × 100. ET indicates the percentage of CFSE+ Annexin‐V+ target cells cocultured with effector cells, and T0 indicates the percentage of CFSE+ Annexin‐V+ target cells without effector cells.
2.5. Repertoire analysis of TCR β chains and PCR cloning of antigen‐specific TCR α/β genes
An IOTest Beta Mark TCR Vβ Repertoire Kit was used for the analysis of TCR β chains with antibodies against the following TCR Vβ regions: Vβ1, Vβ2, Vβ3, Vβ4, Vβ5.1, Vβ5.2, Vβ5.3, Vβ7.1, Vβ7.2, Vβ8, Vβ9, Vβ11, Vβ12, Vβ13.1, Vβ13.2, Vβ13.6, Vβ14, Vβ16, Vβ17, Vβ18, Vβ20, Vβ21.3, Vβ22, and Vβ23. The TCR α chain is composed of a TCR alpha chain variable region (TRAV), a joining region (TRAJ), and a constant region (TRAC). The TCR β chain is composed of a TCR beta chain variable region (TRBV), a diversity region (TRBD), a joining region (TRBJ), and a constant region (TRBC). Cloning and sequencing of TCR α/β genes using PCR were carried out as previously described.11 Briefly, total RNA from sorted CTL was prepared with an RNeasy Mini Kit (QIAGEN, Hilden, Germany), and an aliquot (1 μg) was subjected to reverse transcription using an oligo (dT) primer and SuperScript III (Thermo Fisher Scientific, Waltham, MA, USA). First‐strand cDNA was amplified by PCR with coding region‐specific primers for TRBV12‐3/4 and TRBC1/2 (TCR β gene of ITG‐MT3), TRBV9 and TRBC1/2 (TCR β gene of FKS‐D11P), and various TRAV primers and TRAC (TCR α chain).
2.6. Transfection of TCR genes in lymphoma cells
T‐cell receptor α chain/pCDNA3.1 and TCR β chain/pEF6/Myc‐His plasmids (10 μg each) were transduced into lymphoma cells by electroporation using the Neon Transfection system (Thermo Fisher Scientific), and selection was done in a medium containing 0.5 mg/mL G418 (Roche Diagnostics, Basel, Switzerland) and 5 μg/mL blasticidin (Thermo Fisher Scientific). Expression levels of the transduced genes were assessed by flow cytometry with tetramer staining.
2.7. Analysis of cytokine production and reporter gene assay
An IFN‐γ ELISPOT assay was carried out as described previously using a human IFN‐γ ELISPOT set (BD Biosciences).12 Briefly, nitrocellulose‐bottomed 96‐well plates were coated with an anti‐IFN‐γ antibody. Wells were washed with PBS and blocked for 2 hours by 10% FBS/RPMI1640 medium. After washing with PBS, TCR‐transfected cells were added. Peptide‐pulsed C1R‐A24 cells were then added to each well, and the plates were incubated overnight. The following day, media were discarded, and the wells were washed prior to addition of a biotinylated secondary antibody. The plates were incubated for 2 hours and washed, and avidin‐enzyme conjugate was added to each well. The plates were incubated at room temperature for 1 hour, and a 3‐amino‐9‐ethylcarbazole (AEC) substrate (BD Biosciences) was added to each well and incubated at room temperature for approximately 15 minutes. The reaction was terminated by washing with water, and detectable spots were counted. The NFAT reporter gene assay was described previously.8 An NFAT‐luciferase reporter gene‐transduced Jurkat/MA cell line was kindly donated by Dr Yoshinobu Ichiki (University of Occupational and Environmental Health, Japan) and Dr Sjoerd H. van der Burg (Leiden University Medical Centre, Netherlands). To measure the activation of TCR‐transduced Jurkat/MA cells by peptide‐pulsed C1R‐A24 or K562 cells loaded with 0, 0.01, 0.1, 1, 10, 100 μg/mL of each of the peptides or 100 μg/mL of HIV peptide, Jurkat/MA cells were incubated overnight with target cells in a 96‐well plate. After incubation with various stimuli, the cells were analyzed for luciferase activity. Luminescence was subsequently measured in a Lumat LB 9506 luminometer (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany).
2.8. Recombinant TCR‐multimer
DNA sequences of the extracellular domain of TCR α or β chains were inserted into the expression vector pXC17.4 or pXC18.4 (Lonza Ltd, Basel, Switzerland) using T4 DNA ligase. Constructs of the expression vector of TCR α or β chains were tagged with a fos‐6xHis‐tag or a jun‐BirA‐site, respectively. CHO‐K1SV GSKO cells (Lonza) were transfected with these constructs by electroporation using a Neon Transfection system (Thermo Fisher Scientific), followed by limiting dilution. Soluble recombinant TCR α and β proteins were dimerized by a leucine zipper in the fos‐jun interaction (TCR‐monomer). The soluble TCR‐monomer was purified by a Ni‐sepharose excel column (GE Healthcare, Chicago, IL, USA). After purification, TCR‐monomer was fractioned by liquid chromatography (AKTA25; GE Healthcare), followed by analysis of purity using HPLC (Superdex 200; GE Healthcare) (Figures S1 and S2). TCR‐monomer was biotinylated at BirA‐site and multimerized with PE‐leveled streptavidin.
2.9. Human leukocyte antigen‐A24 blocking assay
Cells were first treated with mouse IgG1 isotype control or anti‐HLA‐A24 mAb clone C7709A2.6 (kindly provided by Dr P. G. Coulie, Ludwig Institute for Cancer Research, Brussels Branch). After incubation at 4°C for 30 minutes, cells were washed once and stained with TCR‐multimers at 4°C for 1 hour. After TCR‐multimer staining, cells were washed once and analyzed by flow cytometry using FACSCalibur. CellQuest software and FlowJo software were used for data analysis.
2.10. Surface plasmon resonance analysis
Surface plasmon resonance analysis was carried out using a ProteOn XPR36 (Bio‐Rad Laboratories, Hercules, CA, USA) according to the protocol described by Nahshol et al.13 Briefly, 1 μg/mL of a biotinylated monomer (HLA‐A*24:02/SVN‐2B peptide, HLA‐A*24:02/PBF peptide or HLA‐A*24:02/HIV peptide) in PBS supplemented with 0.005% Tween‐20 (PBST) was injected at 30 μL/min at 25°C to be captured on a NeutrAvidin‐immobilized NLC sensor tip (Bio‐Rad Laboratories, Hercules, CA, USA). Subsequently, 1 mmol/L biotin in PBST was injected for blocking. Serially diluted TCR‐multimer in PBST was injected at 50 μL/min at 25°C. All binding sensorgrams were collected and analyzed using ProteOn Manager software (Bio‐Rad).
2.11. T‐cell receptor‐multimer staining and immunofluorescence microscopy
Cells were first stained with TCR‐multimers at 4°C for 1 hour. The cells were fixed with 4% paraformaldehyde and stained with DAPI. Immunofluorescence microscopy was carried out using BZ‐9000 (Keyence Corporation, Osaka, Japan) and AxioObserver.Z1 (Carl Zeiss, Oberkochen, Germany).
2.12. Statistical analysis
Results are given as means and standard deviations. Statistical comparisons were made using two‐tailed Student's t tests; P‐values of .05 were considered significant.
3. RESULTS
3.1. Induction of antigen‐specific CTL clones with high avidity
We first attempted to establish SVN‐2B‐ or PBF‐specific CTL as the source of TCR genes. CTL were induced using PBMC from A24+ peptide‐vaccinated patients. After mixed lymphocyte peptide culture (MLPC), PBF or SVN‐2B tetramer‐positive T cells were induced (Figure 1A). After single cell sorting and cell expanding, we established eight SVN‐2B‐specific CTL clones and twelve PBF‐specific CTL clones, respectively. As shown in Figure 1A, the CTL clones ITG‐MT3 and FKS‐D11P were recognized by SVN‐2B tetramer and PBF tetramer, respectively. Percentages and absolute numbers of tetramer‐positive T cells among ITG‐MT3 and FKS‐D11P cells were much higher than those among the other CTL clones (data not shown).
Figure 1.
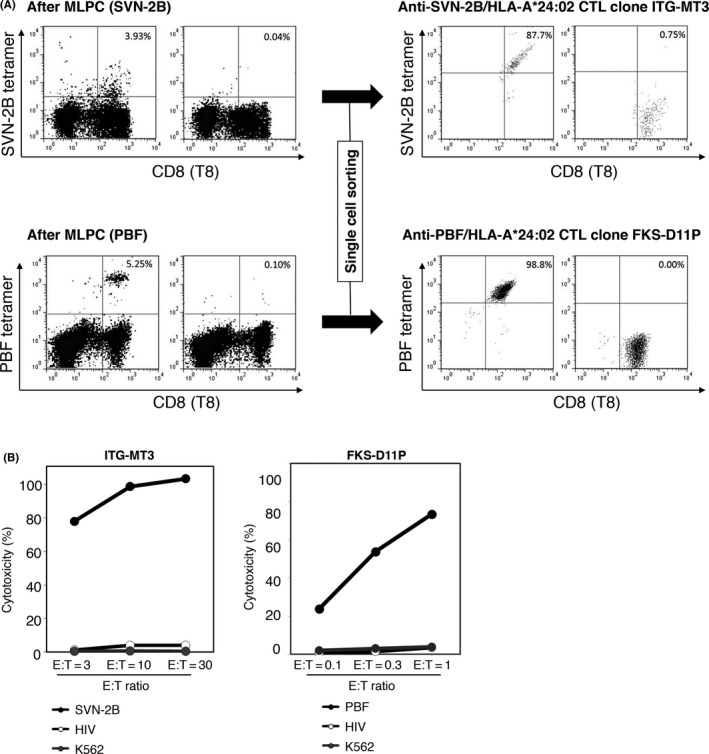
Establishment of anti‐survivin‐2B (SVN‐2B) and anti‐PBF CTL clones, ITG‐MT3 and FKS‐D11P. A, Results of FACS analysis of tetramer‐positive CD8+ T cells after mixed lymphocyte peptide culture (MLPC) using PBMC of a vaccinated patient (left panel) and CTL clones (ITG‐MT3 for SNV‐2B and FKS‐D11P for PBF) after single cell sorting (right panel) are shown. Human leukocyte antigen (HLA)‐A*24:02‐HIV‐negative tetramer was used as a control. Proportions of tetramer‐positive cells among CD8+ T cells are indicated. B, Cytotoxicity of CTL clones against peptide‐pulsed C1R‐A24 cells at 1 μmol/L or K562 cells at the indicated effector : target ratio (E:T)
ITG‐MT3 cells showed strong and specific cytotoxicity against C1R‐A24 cells that were pulsed with A24‐SVN‐2B peptides (Figure 1B). Moreover, FKS‐D11P cells showed strong and specific cytotoxicity against C1R‐A24 cells that were pulsed with A24‐PBF peptides at a lower effector : target (E:T) ratio (Figure 1B). These results indicated that FKS‐D11P TCR could recognize these A24/epitope peptide complexes with higher avidity than that of ITG‐MT3 TCR.
3.2. Clonotyping of TCR α/β repertoires and cloning TCR genes
Next, we identified the TCR Vβ repertoire of ITG‐MT3 and FKS‐D11P cells using a TCR Vβ Repertoire Kit, which could account for about 70% of the variations in TCR Vβ. We confirmed that the TCR β chains of ITG‐MT3 and FKS‐D11P cells were recognized by anti‐TCR Vβ8 and Vβ1, respectively (Figure 2A).
Figure 2.
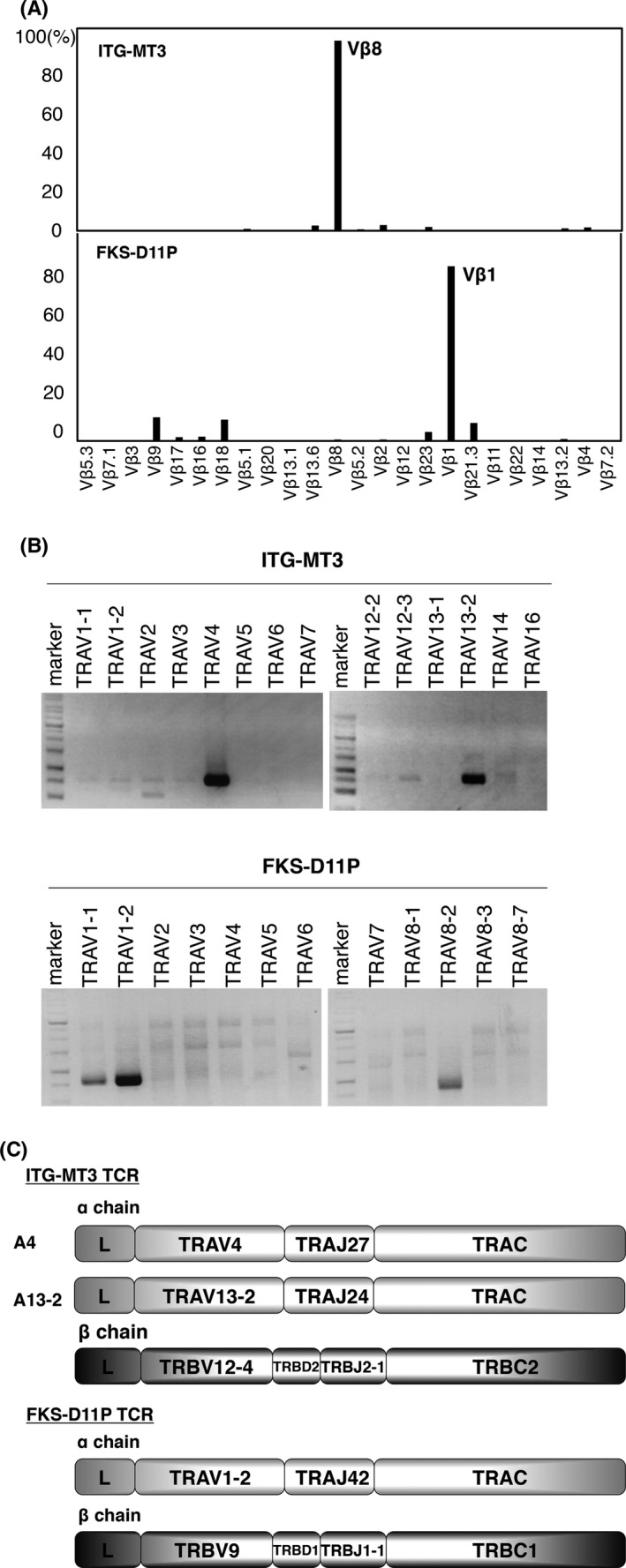
Cloning of T‐cell receptor (TCR) genes of ITG‐MT3 and FKS‐D11P. A, Reactivity of anti‐TCR Vb antibodies of ITG‐MT3 (upper panel) and FKS‐D11P (lower panel) analyzed by FACS. B, Clonotype PCR of the TCR Va repertoires of ITG‐MT3 and FKS‐D11P. TCR Va genes were amplified using coding region‐specific primer pairs for various TCR α chains. C, Construction of TCR α and β chains of ITG‐MT3 and FKS‐D11P
ITG‐MT3 TCR α and β genes were amplified by PCR with specific primers for TRAC and various TRAV, TRBV12‐3/4 and TRBC1/2. As a result, we found that the TCR Vα chains of ITG‐MT3 cells comprised TRAV4 and TRAV13‐2 (Figure 2B). Because of the high homology, the sequences for the TRBV12‐3/TRBC1, TRBV12‐3/TRBC2 and TRBV12‐4/TRBC1 PCR products were the same as that for TRBV12‐4/TRBC2.
FKS‐D11P TCR α and β genes were amplified by PCR with specific primers for TRAC and various TCR α chains and TRBV9 and TRBC1/2. As a result, we found that the TCR Vα chains of FKS‐D11P cells comprised TRAV1‐1, TRAV1‐2 and TRAV8‐2 (Figure 2B). The TRAV1‐1 PCR product was the same as that for TRBV1‐2, and TRAV8‐2 showed a frame shift mutation. These results suggested that ITG‐MT3 cells had two types of TCR α chains (A4: TRAV4/TRAJ27/TRAC; A13‐2: TRAV13‐2/TRAJ24/TRAC) and one TCR β chain (B12‐4: TRBV12‐4/TRBD2/TRBJ2‐1/TRBC2) and that FKS‐D11P cells had one TCR α chain (A1‐2: TRAV1‐2/TRAJ42/TRAC) and one TCR β chain (B9: TRBV9/TRBD1/TRBJ1‐1/TRBC1) (Figure 2C).
3.3. T‐cell receptor‐transduced T‐cell lymphoma cell lines specifically recognized antigenic peptide‐presented C1R‐A24 cells
To evaluate TCR reactivity to SVN‐2B or PBF tetramers, we transiently transduced the TCR α/β genes from ITG‐MT3 cells or FKS‐D11P cells into three T‐cell lymphoma cell lines, Sup‐T1 (Figure 3A). Only TCR TRAV4 and TRBV12‐4 (A4/B12‐4) on Sup‐T1 cells could react with the SVN‐2B tetramer (Figure 3A). Transduced TCR of FKS‐D11P on Sup‐T1 cells could react with the PBF tetramer. Stable TCR‐transduced Sup‐T1 and Jurkat/MA cells were also established by drug selection (Figure 3B‐E). Surface expression of transduced TCR was continuously maintained. Moreover, ITG‐MT3 TCR‐transduced Jurkat/MA cells and FKS‐D11P TCR‐transduced Jurkat/MA cells recognized specific peptide‐pulsed C1R‐A24 cells and could activate NFAT‐Luc reporter (Figure 3F,G), followed by IFN‐γ release (Figure 3H,I). These results indicated that both transduced TCR were functional.
Figure 3.
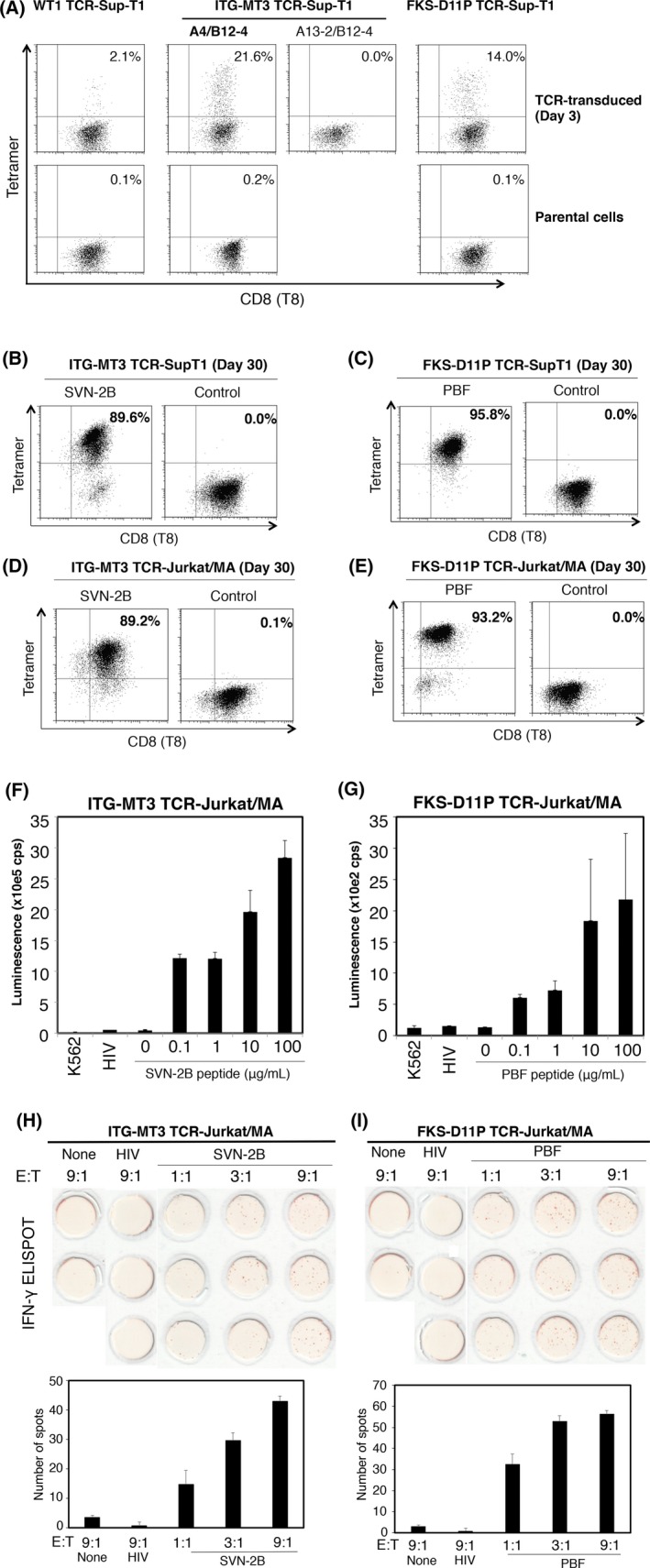
T‐cell receptor (TCR)‐transduced T‐cell lymphoma cell lines showed reactivity with a specific peptide/human leukocyte antigen (HLA)‐A*24:02 tetramer. A‐C, Tetramer reactivity to TCR‐transduced cell lines. Sup‐T1 cells (A‐C) and Jurkat/MA cells (D,E) were used as exogenous TCR‐transduced cell lines. Tetramer staining was carried out on the indicated days. WT1 TCR was used as a positive control. FACS was carried out on indicated days after TCR gene electroporation. (F‐I) Reactivity of TCR‐transduced Jurkat/MA cells against peptide‐pulsed C1R‐A24 cells. (F,G) NFAT‐luciferase reporter assay of ITG‐MT3 TCR‐ and FKS‐D11P TCR‐transduced Jurkat/MA cells. (H,I) Cytokine production capacity of interferon (IFN)‐γ ELISPOT of ITG‐MT3 TCR‐ and FKS‐D11P TCR‐transduced Jurkat/MA cells. Assays were carried out at the indicated effector (TCR‐transduced cells) and target (C1R‐A24 cells) ratio. E : T, effector : target ratio
3.4. CD8 dependency of HLA‐TCR interaction
CD8 molecule binds to the nonpolymorphic aspect of the HLA class I molecule and sometimes augments TCR‐HLA interaction.14 To investigate CD8 dependency of TCR‐HLA interaction, ITG‐MT3 TCR and FKS‐D11P TCR genes were transfected to J.RT3‐T3.5 cells, CD8 and TCR β chain‐deficient mutant Jurkat cells.15 As a result, both transduced TCR could react with specific tetramers, but the CD8‐dependent WT1 TCR11 could not react with the WT1 tetramer (Figure 4A). These results suggested that both ITG‐MT3 and FKS‐D11P TCR could react with the peptide/HLA complex in a CD8‐independent way.
Figure 4.
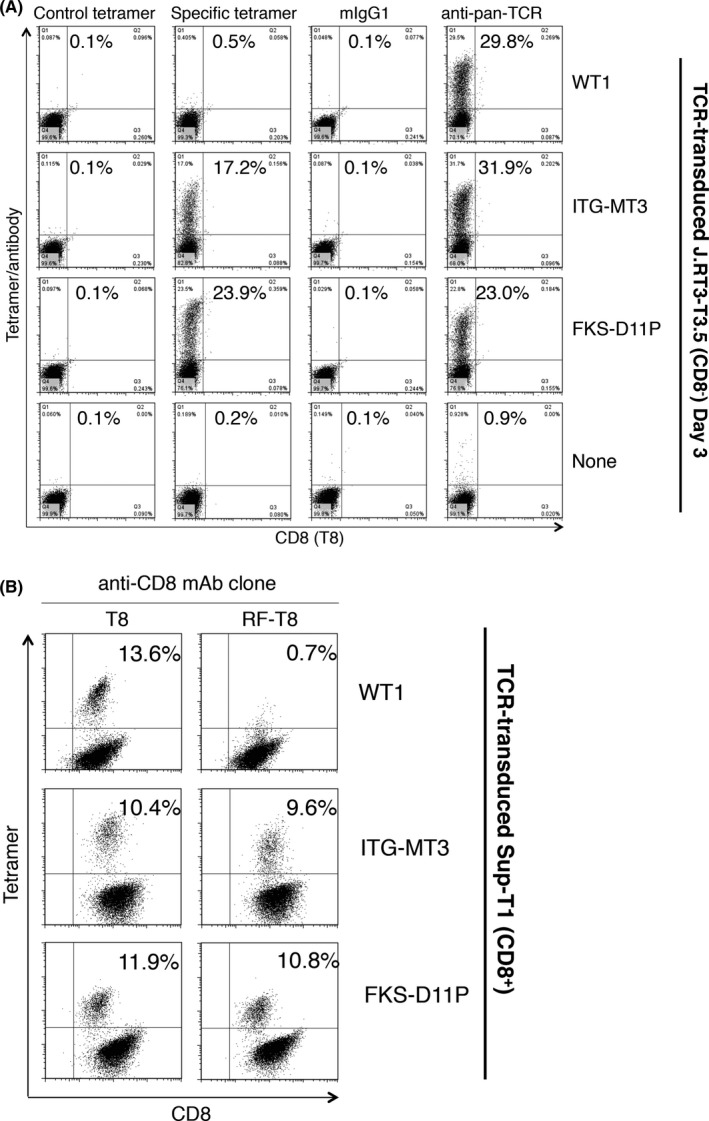
Both ITG‐MT3 T‐cell receptor (TCR) and FKS‐D11P TCR showed CD8‐independent interaction between TCR and peptide/human leukocyte antigen (HLA) complex. A, Tetramer reactivity of TCR‐transduced J.RT3‐T3.5 cells lacking CD8 expression. Negative tetramer (HLA‐A*24:02/HIV), mIgG1 and anti‐pan TCR mAb were used as controls. B, Interference in tetramer reactivity caused by anti‐CD8 antibody. Sup‐T1 (CD8+) cells and specific TCR‐expressed cells were mixed at a ratio of 9:1 and costained with the tetramer and anti‐CD8 antibody as indicated
In addition, two anti‐CD8 clones were used to assess the inhibitory effects of tetramer staining. Clone RFT‐8 has an HLA‐CD8 interaction‐inhibition effect and clone T8 did not have any HLA‐CD8 interaction‐inhibition effect. As shown in Figure 4B, reactivities with specific tetramers of both ITG‐MT3 TCR and FKS‐D11P TCR were not influenced by the anti‐CD8 mAb clone RFT‐8. In contrast, CD8‐dependent WT1 TCR decreased reactivity with the WT1 tetramer by clone RFT‐8. The other anti‐CD8 mAbs showed compatible reactivities (Figure S3 and Table S1). These results strongly suggested that both ITG‐MT3 TCR and FKS‐D11P TCR could react with HLA in a CD8‐independent way.
3.5. Generation of a recombinant TCR‐multimer
To detect specific HLA/peptide complexes without viable cells, we tried to produce a TCR‐multimer using the extracellular domain of recombinant TCR α/β proteins. The construct of TCR‐monomers is shown in Figure 5A. Recombinant proteins of TCR α/β chains were dimerized with the fos‐jun leucine zipper, followed by BirA site biotinylation (TCR‐monomer). A TCR‐monomer was multimerized with streptavidin‐PE by biotin‐avidin reaction (TCR‐multimer). TCR‐multimers of ITG‐MT3 and FKS‐D11P were designated as SVN‐2B TCR‐multimer and PBF TCR‐multimer, respectively. SVN‐2B and PBF TCR‐multimers could detect more than 1 μg/mL of several specific peptide‐pulsed C1R‐A24 cells by FACS (Figure 5B,C). Reactivities of TCR‐multimers also depended on their concentrations (Figure S4) and on the concentrations of the pulsed peptides (Figures S5 and S6). In addition, these reactivities of TCR‐multimers were blocked by anti‐HLA‐A24 mAb C7709A2.6 (Figure 5D,E). These results indicated the both TCR‐multimers could react with each specific peptide in an HLA‐A24 restriction method similar to TCR expressed on CTL clones and T‐cell lymphoma cell lines. The reactions of TCR‐multimers against specific peptides could also be detected by immunofluorescence microscopy (Figure 5F,G). Positive staining was observed in the cytoplasmic membrane as a diffuse circulation pattern. These results also suggested that numerous peptides were presented by HLA‐A24 molecules on such target cells pulsed with peptides at high concentrations.
Figure 5.
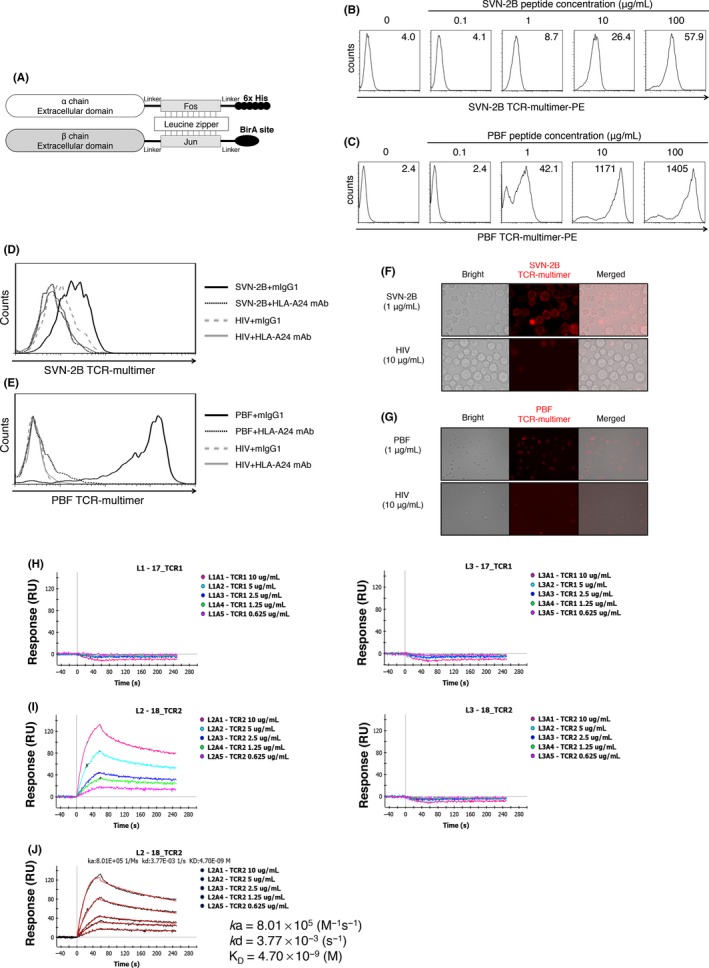
Survivin‐2B (SVN‐2B) T‐cell receptor (TCR)‐ and PBF TCR‐multimers showed specific reactivity against peptide‐pulsed antigen‐presenting cells. A, Schema of the TCR‐multimer. B,C Reactivity of TCR‐multimer against peptide‐pulsed C1R‐A24 cells at the indicated concentrations. SVN‐2B TCR‐multimer (B) and PBF TCR‐multimer (C) were constructed from ITG‐MT3 and FKS‐D11P, respectively. D,E Blocking assay using anti‐human leukocyte antigen (HLA)‐A24 mAb. Peptide‐pulsed C1R‐A24 cells were incubated with the TCR‐multimer and anti‐HLA‐A24 mAb (C7709A2.6.) or control mIgG1. (F,G) Immunofluorescence microscopy. Peptide‐pulsed C1R‐A24 cells were incubated with the TCR‐multimer. Interaction between SVN‐2B TCR and HLA‐A*24:02/SNV‐2B peptide complex (H) and between PBF TCR and HLA‐A*24:02/PBF peptide complex (I). J, Analysis of the kinetics between PBF TCR and HLA‐A*24:02/PBF peptide complex
To determine the molecular affinity between the TCR and HLA/peptide complex, we carried out surface plasmon resonance analysis (Figure 5H‐J). PBF TCR showed high and specific affinity against the HLA‐A*24:02/PBF peptide complex (KD value of 4.70 × 10−9 mol/L). SVN‐2B TCR did not show any interaction with HLA‐A*24:02/SVN‐2B peptide complex.
3.6. PBF TCR‐multimer with high avidity could detect naturally presented PBF peptide on osteosarcoma cell lines in the context of HLA‐A24
Finally, we assessed whether the PBF TCR‐multimer could detect naturally presented HLA‐A24/PBF peptide complexes by using immunofluorescence microscopy. It was found that the PBF TCR‐multimer could detect the PBF peptide presented on PBF+HLA‐A24+ osteosarcoma cell lines, KIKU and HOS‐A24 but not on HOS (PBF+HLA‐A24−) cells (Figure 6A‐C). Expression of HLA‐A24 was confirmed by staining with an anti‐HLA‐A24 mAb. Positive staining was observed as a few bright spots on osteosarcoma cytoplasmic membranes, in contrast to peptide‐pulsed target cells. The results suggested that the number of naturally presented HLA‐A24/PBF peptide complexes might be very small.
Figure 6.
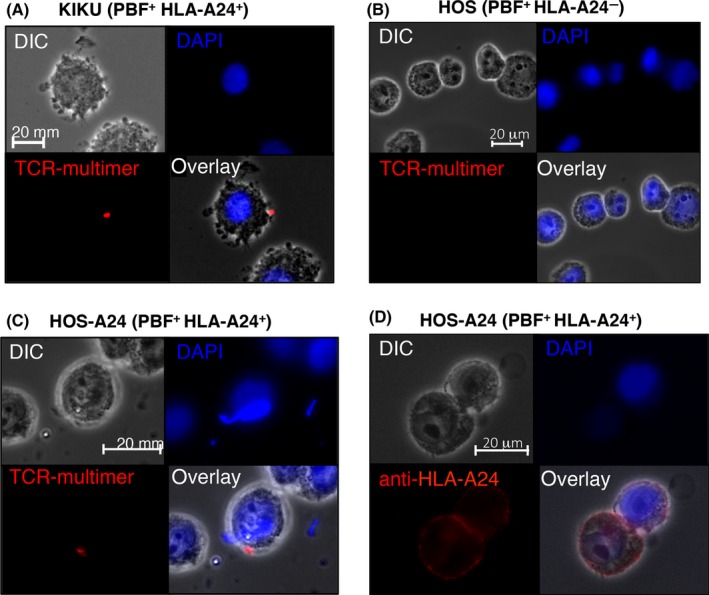
PBF T‐cell receptor (TCR)‐multimer could detect naturally presented PBF peptide/human leukocyte antigen (HLA)‐A*24:02 complex on osteosarcoma cell lines. Immunofluorescence microscopy of PBF + HLA‐A24+ osteosarcoma cell lines KIKU (A), HOS (B), and HOS‐A24 (C) stained with PBF TCR‐multimer. D, HOS‐A24 cells stained with anti‐HLA‐A24 antibody, clone 17A10
4. DISCUSSION
In the present study, we established CTL clones with high avidity directed to TAA‐derived peptides, SVN‐2B and PBF, in the context of HLA‐A*24:02 as a source of the TCR gene. Next, we constructed TCR‐multimers reacting with exogenously pulsed cognate peptides presented by C1R‐A24 cells. Moreover, the PBF TCR‐multimer successfully detected a naturally presented PBF peptide on HLA‐A24+PBF+ osteosarcoma cell lines.
For constructing a probe with TCR‐like specificities, single‐chain variable fragment (scFv) phage display library technology is very useful.16 We previously established a scFv clone reacting with a PBF peptide naturally presented by HLA‐A2+ osteosarcoma cells using phage display scFv libraries.5 However, we failed to obtain scFv clones directed to the HLA‐A24/PBF peptide complex and directed to HLA‐A24/SVN‐2B peptide complex (Tsukahara et al, unpublished observation, 2014). Therefore, both scFv phage display library technology and a TCR‐multimer are still required for constructing probes with TCR‐like specificity for various TAA.
So far, results of several studies aimed at the development of a TCR multimer have been reported. However, most of the TCR were directed to viral antigens, human T‐cell leukemia virus type 1 tax, HIV‐gag and HBV env.17, 18, 19 Regarding a TAA‐specific TCR, only Bethune et al reported soluble TCR tetramers and dextramers directed to MART1 26‐35(A27L) and NY‐ESO‐1 157‐167(C165V) in the context of HLA‐A221. Generally, induction of CTL directed to viral antigens with high avidity TCR might be easy because immunogenicity of viral antigens is higher than that of TAA. Therefore, it is more difficult to obtain TAA‐specific TCR with high avidity sufficient for the construction of functional TCR‐multimers. Affinity of FKS‐D11P TCR (KD value of 4.70 × 10−9 mol/L) was much higher than that of naturally occurring TCR binding to HLA class I/peptides (K D value of 1–50 × 10−6 mol/L).20 In previous studies on a soluble TCR‐multimer, K D values of TCR for HLA‐A2/HTLV type 1 Tax11‐19, HLA‐A2/HBV env183‐191, HLA‐A2/MART126‐35(A27L) and HLA‐A2/NY‐ESO‐1157‐167(C165V) were 1.06 × 10−6, 0.6 × 10−6, 5.4 × 10−6 and 4.3 × 10−6 mol/L, respectively.17, 19, 21 These results suggested that the affinities of successfully constructed soluble TCR were also higher than the general affinity of TCR. In addition, both ITG‐MT3 TCR and FKS‐D11P TCR showed CD8‐independent interactions between the TCR and HLA/peptide complex. This might be because peptide vaccination had successfully elicited high‐affinity TCR with CD8‐independency in vivo. Our observations suggested that CD8‐independency of TCR is also important for construction of a TCR‐multimer with the same high affinity as that of TCR.
In the present study, we used only established CTL clones recognizing vaccinated epitopes as the source of TCR to construct a TCR multimer. By using this strategy, immunodominant TCR directed to unknown antigens including neoantigens among TIL (Tumor‐infiltrating lymphocyte) in primary cancer tissues could not be obtained. Deep sequencing of TRAV and TRBV combined with 5′RACE of dominant repertoires might be useful to construct immunodominant TCR multimers reacting with unknown antigens.
In a clinical setting, primary biopsy specimens might be used to evaluate the HLA/peptide complex on osteosarcoma cells. However, the expression status of target antigens in metastatic or recurrent lesions might be different from that in the primary lesion. Difficulty in assessing lesions of solid cancer including osteosarcoma is an important problem. In the future, if circulating osteosarcoma cells can be isolated as a liquid biopsy, adequate evaluation of the HLA/peptide complex on osteosarcoma cells might be easier.
In conclusion, we successfully generated TCR‐multimers reacting with HLA/TAA‐derived peptides, HLA‐A24/SVN‐2B peptide and HLA‐A24/PBF peptide complexes. Moreover, the PBF TCR‐multimer with high avidity could react with a naturally presented PBF peptide on osteosarcoma cell lines. Taken together, the results indicated that a TCR‐multimer might be useful for the detection of target antigens in immunotherapy.
CONFLICTS OF INTEREST
T. Torigoe received financial support through collaboration with Medical Biological Laboratories Co., Ltd. The remaining authors declare no commercial or financial conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors thank Mr Li. Dongliang, Ms Masae Itoh and Ms Hitomi Nakamura (MBL, Ina, Japan) for kindly preparing the HLA‐tetramers. The authors also thank the staff of Japanese Red Cross Hokkaido Block Blood Center for kind donation of human sera. This work was supported by grants from Japan Society for the Promotion of Science Grants‐in‐aid for Scientific Research (KAKENHI) 16H05451 (to T. Tsukahara) and 17H01540 (to T. Torigoe), the Takeda Science Foundation (2018‐Kenkyu‐Shorei to T. Tsukahara), the Cell Science Research Foundation (2016‐Kenkyu‐Zyosei to T. Tsukahara) and a grant‐in‐aid of Ono Cancer Research Fund (2017‐3 to T. Tsukahara).
Watanabe K, Tsukahara T, Toji S, et al. Development of a T‐cell receptor multimer with high avidity for detecting a naturally presented tumor‐associated antigen on osteosarcoma cells. Cancer Sci. 2019;110:40–51. 10.1111/cas.13854
REFERENCES
- 1. Coulie PG, Connerotte T. Human tumor‐specific T lymphocytes: does function matter more than number? Curr Opin Immunol. 2005;17:320‐325. [DOI] [PubMed] [Google Scholar]
- 2. Hirohashi Y, Torigoe T, Tsukahara T, Kanaseki T, Kochin V, Sato N. Immune responses to human cancer stem‐like cells/cancer‐initiating cells. Cancer Sci. 2016;107:12‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsukahara T, Hirohashi Y, Kanaseki T, et al. Peptide vaccination therapy: towards the next generation. Pathol Int. 2016;66:547‐553. [DOI] [PubMed] [Google Scholar]
- 4. Tsukahara T, Emori M, Murata K, et al. The future of immunotherapy for sarcoma. Expert Opin Biol Ther. 2016;16:1049‐1057. [DOI] [PubMed] [Google Scholar]
- 5. Tsukahara T, Emori M, Murata K, et al. Specific targeting of a naturally presented osteosarcoma antigen PBF peptide using an artificial monoclonal antibody. J Biol Chem. 2014;289:22035‐22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirohashi Y, Torigoe T, Maeda A, et al. An HLA‐A24‐restricted cytotoxic T lymphocyte epitope of a tumor‐associated protein, survivin. Clin Cancer Res. 2002;8:1731‐1739. [PubMed] [Google Scholar]
- 7. Tsukahara T, Kawaguchi S, Torigoe T, et al. Prognostic impact and immunogenicity of a novel osteosarcoma antigen, papillomavirus binding factor, in patients with osteosarcoma. Cancer Sci. 2008;99:368‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aarnoudse CA, Kruse M, Konopitzky R, Brouwenstijn N, Schrier PI. TCR reconstitution in Jurkat reporter cells facilitates the identification of novel tumor antigens by cDNA expression cloning. Int J Cancer. 2002;99:7‐13. [DOI] [PubMed] [Google Scholar]
- 9. Karanikas V, Lurquin C, Colau D, et al. Monoclonal anti‐MAGE‐3 CTL responses in melanoma patients displaying tumor regression after vaccination with a recombinant canarypox virus. J Immunol. 2003;171:4898‐4904. [DOI] [PubMed] [Google Scholar]
- 10. Watanabe K, Suzuki S, Kamei M, et al. CD137‐guided isolation and expansion of antigen‐specific CD8 cells for potential use in adoptive immunotherapy. Int J Hematol. 2008;88:311‐320. [DOI] [PubMed] [Google Scholar]
- 11. Watanabe K, Toji S, Ohtake J, et al. Establishment of a stable T lymphoma cell line transduced with HLA‐A*24:02‐restricted WT1‐specific TCR genes and its application to antigen‐specific immunomonitoring. Biomed Res. 2013;34:41‐50. [DOI] [PubMed] [Google Scholar]
- 12. Morita R, Nishizawa S, Torigoe T, et al. Heat shock protein DNAJB8 is a novel target for immunotherapy of colon cancer‐initiating cells. Cancer Sci. 2014;105:389‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nahshol O, Bronner V, Notcovich A, Rubrecht L, Laune D, Bravman T. Parallel kinetic analysis and affinity determination of hundreds of monoclonal antibodies using the ProteOn XPR36. Anal Biochem. 2008;383:52‐60. [DOI] [PubMed] [Google Scholar]
- 14. Norment AM, Salter RD, Parham P, Engelhard VH, Littman DR. Cell‐cell adhesion mediated by CD8 and MHC class I molecules. Nature. 1988;336:79‐81. [DOI] [PubMed] [Google Scholar]
- 15. Ohashi PS, Mak TW, Van den Elsen P, et al. Reconstitution of an active surface T3/T‐cell antigen receptor by DNA transfer. Nature. 1985;316:606‐609. [DOI] [PubMed] [Google Scholar]
- 16. Dahan R, Reiter Y. T‐cell‐receptor‐like antibodies – generation, function and applications. Expert Rev Mol Med. 2012;14:e6. [DOI] [PubMed] [Google Scholar]
- 17. Laugel B, Boulter JM, Lissin N, et al. Design of soluble recombinant T cell receptors for antigen targeting and T cell inhibition. J Biol Chem. 2005;280:1882‐1892. [DOI] [PubMed] [Google Scholar]
- 18. Anikeeva N, Mareeva T, Liu W, Sykulev Y. Can oligomeric T‐cell receptor be used as a tool to detect viral peptide epitopes on infected cells? Clin Immunol. 2009;130:98‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Low JL, Naidoo A, Yeo G, et al. Binding of TCR multimers and a TCR‐like antibody with distinct fine‐specificities is dependent on the surface density of HLA complexes. PLoS ONE. 2012;7:e51397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bethune MT, Comin‐Anduix B, Hwang Fu YH, Ribas A, Baltimore D. Preparation of peptide‐MHC and T‐cell receptor dextramers by biotinylated dextran doping. Biotechniques. 2017;62:123‐130. [DOI] [PubMed] [Google Scholar]
- 21. van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659‐684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials