

Abstract
Vasoactive intestinal peptide (VIP) is a modulator of inflammatory responses. VIP receptors are expressed in several tumor types, such as colorectal carcinoma. The study described herein was conducted to confirm the presence of VIP and its receptors (VPAC1 and VPAC2) in surgically resected hepatocellular carcinoma (HCC) tissues and in the HCC cell line Huh7. The mechanism responsible for apoptosis of HCC cells was then examined because VIP treatment (10−10 M) significantly suppressed proliferation of Huh7 cells. In examining apoptosis‐related proteins, we found caspase‐3 to be significantly increased and Bcl‐xL and cyclic AMP (cAMP) response element‐binding protein (CREB) to be significantly decreased in Huh7 cells cultured with VIP. Furthermore, the CREB level and phosphorylation were reduced. These effects were reversed by the addition of VIP receptor antagonist or cAMP antagonist Rp‐cAMPS. Pretreatment with cAMP analogue blocked the increased apoptosis, suggesting that VIP induces apoptosis via a PKA‐independent signaling mechanism. Our data indicate that VIP prevents the progression of HCC by apoptosis through the cAMP/Bcl‐xL pathway.
Keywords: apoptosis, Bcl‐xL, CREB, hepatocellular carcinoma, vasoactive intestinal peptide
1. INTRODUCTION
Hepatocellular carcinoma (HCC) is a primary cancer of the liver. Although many management and therapeutic strategies have been established, recurrence of HCC is not uncommon.1 Surgical resection, the main treatment for HCC, offers some hope for long‐term cure, but for prevention of both metastasis and recurrence, a new chemotherapeutic strategy is needed. The process of recurrence begins long before signs or symptoms appear. Stress has been implicated in tumor growth and progression.2, 3, 4 The sympathetic nervous system and hypothalamic‐pituitary‐adrenal axis regulate the release of stress‐related hormones such as cortisol, catecholamines and neuropeptides.5 These mediators can trigger tumor antigenicity and modulate the immune response to tumor cells. There is some evidence that glucocorticoids and sympathetic neurotransmitters directly affect tumor cell growth and survival.2 Several neuropeptides are known to regulate liver function not only through the central nervous system but also through the autonomic nervous system.6, 7 Vasoactive intestinal peptide (VIP) is one of these neuropeptides. VIP is a 28‐amino acid neuroendocrine mediator. It has 68% identity at the amino acid level with pituitary adenylate cyclase‐activating polypeptide (PACAP) and belongs to the secretin peptide family.8 Although VIP has a wide range of biological activity, it is present mainly in the gastric mucosa and is involved, as a hormone, in the regulation of gastric function. VIP receptors are of 2 subtypes: vasoactive intestinal polypeptide receptors 1 and 2 (VPAC1 and VPAC2, respectively). VPAC1 is found in the liver, lung, kidney and prostate,9 whereas VPAC2 is found mainly in smooth muscle and in vessels.9 Studies have revealed expression of VIP receptors in several human carcinomas, including HCC, and in vitro studies have shown that VIP inhibits proliferation of hepatoma and glioma cells.10, 11, 12, 13, 14 However, the mechanisms by which VIP modulates HCC pathogenesis remain unknown. In the study described herein, we confirmed expression of VIP in HCC tissues, and we showed the effects of VIP in the Huh7 HCC cell line in vitro.
2. MATERIALS AND METHODS
2.1. Reagents
Vasoactive intestinal peptide (VIP), VIP receptor antagonist [D‐p‐Cl‐Phe6, Leu17]‐VIP and cyclic AMP (cAMP) analogue N6,2′‐O‐Dibutyryladenosine 3′,5′‐cyclic monophosphate sodium salt (dibutyryl‐cAMP) were obtained from Sigma‐Aldrich (St. Louis, MO, USA), and cAMP antagonist Rp‐cAMPS was obtained from BIOLOG Life Science Institute (Bremen, Germany). A list of the used primary antibodies is included in Table 1.
Table 1.
List of primary antibodies used for immunohistochemistry and western blot analysis
| Antibody | Dilution | Source |
|---|---|---|
| Anti‐VIP rabbit polyclonal Ab | 1:100 | Santa Cruz Biotechnology (Santa Cruz, CA, USA) |
| Anti‐VPAC1 rabbit polyclonal Ab | 1:100 | Santa Cruz Biotechnology |
| Anti‐VPAC2 rabbit polyclonal Ab | 1:100 | Santa Cruz Biotechnology |
| Anti‐CREB rabbit polyclonal Ab | 1:200 | Abcam, Tokyo, Japan |
| Anti‐Caspase‐3 mouse monoclonal Ab | 1:400 | Santa Cruz Biotechnology |
| Anti‐Bcl‐xL rabbit polyclonal Ab | 1:200 | Santa Cruz Biotechnology |
| Anti‐Bcl‐2 mouse monoclonal Ab | 1:400 | Santa Cruz Biotechnology |
| Anti‐Bax rabbit polyclonal Ab | 1:200 | Santa Cruz Biotechnology |
| Anti‐Bad rabbit polyclonal Ab | 1:200 | Santa Cruz Biotechnology |
| Anti‐NFκB p65 rabbit polyclonal Ab | 1:200 | Santa Cruz Biotechnology |
| Anti‐TRADD rabbit polyclonal Ab | 1:200 | Santa Cruz Biotechnology |
| Anti Cyclin D rabbit polyclonal Ab | 1:200 | Santa Cruz Biotechnology |
| Anti‐α‐Tubulin mouse monoclonal Ab | 1:500 | Wako, Tokyo, Japan |
Ab, antibody; CREB, cAMP element‐binding protein; NFκB, nuclear factor kappa‐light‐chain‐enhancer of B cells; TRADD, TNF receptor type 1‐associated death domain protein; VIP, vasoactive intestinal peptide; VPAC, VIP G‐protein coupled receptor.
2.2. Human hepatocellular carcinoma and noncancer liver tissues
Samples of human hepatocellular carcinoma (HCC) tissue were obtained from 12 patients undergoing partial hepatectomy at St. Marianna University Hospital. Written informed consent for use of the resected tissues for research purposes had been provided by each of the patients prior to the surgery. The tissue samples were stored in the internal human tissue bank and were managed using anonymized numbers. Some tissues were from patients with hepatitis B virus‐related HCC (n = 3), some were from patients with hepatitis C virus‐related HCC (n = 3), some were from patients with nonvirus‐related HCC (n = 3), and some were from patients with liver metastases from colorectal cancer (n = 3). All of HCC specimens were primary liver cancer. Once obtained, the surgical specimens were immediately frozen in liquid nitrogen. Samples of noncancer liver tissue that had been obtained from Caucasian and Hispanic transplantation donors were provided by the National Disease Research Interchange (Philadelphia, PA, USA) through the Biomedical Research Institute, Human and Animal Bridging Research Organization (Chiba, Japan). Noncancer liver tissues and HCC tissues had been fixed in 10% formalin and embedded in paraffin.
2.3. Human hepatocellular carcinoma cells, primary hepatocytes and cell cultures
Human hepatocellular carcinoma cell lines Huh7 and HepG2 were obtained from the Riken Gene Bank (Tsukuba, Japan). The Huh7 cells were cultured in RPM1‐1640 medium (Sigma) supplemented with 10% FCS (Invitrogen, Grand Island, NY, USA), 100 μg/mL streptomycin, and 100 U/mL penicillin at 37°C under a humidified atmosphere of 5% CO2 and 95% O2. The cells were seeded on a culture dish and incubated for 24 hours for use in the experiments described below.
Primary cells of cryopreserved human hepatocyte were obtained from Xeno Teck (Kansas City, KS, USA). The donor of No. H1000.H15B+ was a Hispanic 35‐year‐old man who died of head trauma. The donor of No. 1500. H15B+ was a Caucasian 40‐year‐old woman who died of a cardiovascular accident. Primary hepatocytes were seeded on a 24‐well Cell‐able flat plate (Toyo Gosei, Tokyo, Japan) and cultured with Ranford's medium (Nissui Pharmaceutical, Tokyo, Japan) for 24 hours.
2.4. Assay of Huh7 cell proliferation
Huh7 cells (4 × 104) were cultured with VIP at concentrations of 10−8‐10−12 M, for 4, 16 and 24 hours on 96‐well flat11‐bottom plates (Costar, Corning, NY, USA) in 200 μL of RPM1‐1640 medium. Cell proliferation was evaluated with a CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. After incubation, 20 μL of MTS (3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt) substrate included in the kit was added to each well. Absorbance at 490 nm was measured with a microplate reader (Multiscan, Thermo Labsystems, Ventaa, Finland). Proliferation of the Huh7 cells was expressed as the ratio of the optical density of the VIP‐treated cells to that of untreated cells.
2.5. Western blot analysis
Whole proteins were extracted from the Huh7 cells with cell lysing buffer (Tris‐HCl 50 mmol/L, pH 7.4; EGTA 1 mmol/L; .001% leupeptin) and the nuclear proteins were prepared with an NE‐PER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology, Rockford, IL, USA). The total protein concentration was measured with a Bio‐Rad Protein Assay Kit (Bio‐Rad, Hercules, CA, USA). The samples (30 μg) were resolved by 10% SDS‐polyacrylamide gel electrophoresis and then transferred to a Hybond‐ECL membrane (Roche Diagnostics, Indianapolis, IN, USA). The membrane was blocked overnight at 4°C with 5% skim milk in TBS containing Tween 20 (NaCl 150 mmol/L, Tris‐HCl 10 mmol/L, pH 7.5, Tween .5%). The membrane was then incubated with primary antibody for 2 hours at room temperature. Reactive proteins were detected by means of enhanced chemiluminescence (ImmnoStar, Wako, Tokyo). Signal intensities of the detected bands were analyzed using a C‐DiGit Chemiluminescent Western Blot Scanner (LI‐COR Bioscience, Lincoln, NE, USA). Protein expression was quantified by densitometric analysis. The expression level of α‐tubulin was used as a control.
2.6. Immunohistochemistry
Immunohistochemical staining for VIP, VPAC1 and VPAC2 was performed in normal liver tissues, in tissues from various HCC (hepatitis B virus‐related, hepatitis C virus‐related and nonvirus‐related HCC), in liver metastases from colorectal cancer and in adjacent nontumorous tissues. Paraffin‐embedded tissues were deparaffinized in xylene and rehydrated in a graded ethanol series. Endogenous peroxidase activity in the tissue sections was blocked with .1% hydrogen peroxide. Immunoreactivity in sections was visualized with the use of an EnVision detection system (Dako, Carpinteria, CA, USA) according to the manufacturer's instructions. Tissues were then counterstained with hematoxylin.
2.7. Immunostaining
Huh7 (2 × 104) cells were cultured on a microchamber slide and allowed to adhere for 24 hours. The slide was fixed in cold acetone for 15 minutes and blocked with 2% skim milk for 30 minutes. Immunoreactivity in sections was visualized with the use of an LSAB alkaline phosphatase kit (Dako) according to the manufacturer's instructions. Tissues were then counterstained with hematoxylin.
2.8. Assay of apoptosis of human hepatocellular carcinoma cells
For assay of apoptosis of HCC cells, Huh7 cells (2 × 104) were incubated with VIP at concentrations of 10−9‐10−11 M for 24 hours on a 96‐well plate. Apoptosis was measured by means of the Cell Death Detection ELISA (Roche Diagnostics). The cells were homogenized with lysis buffer and incubated for 1 hour at room temperature. The cells were washed for removal of all unbound components, and the ABTS substrate was reacted for quantitative determination of the number of nucleosomes. Optical density was measured with a microplate reader (Multiscan) at 405 nm to quantitate the relative substrate of the samples.
2.9. Assay of intracellular cAMP
For assay of intracellular cAMP, Huh7 cells (3 × 104) were seeded on a 96‐well plate and incubated for 24 hours with VIP at concentrations of 10−9, 10−10 and 10−11 M, and intercellular cAMP was quantified in these cells by means of ELISA (cAMP XP Assay Kit, Cell Signaling Technology, Danvers, MA, USA) with VIP for 24 hours. The cells were homogenized with lysis buffer and incubated at room temperature for 3 hours on a horizontal orbital plate shaker. The plate was washed, tetramethylbenzidine (TMB) substrate was added to the samples, and the cells were incubated for 30 minutes. Optical density was measured at 450 nm.
2.10. Assay of phosphorylated CREB
For assay of phosphorylated CREB (pCREB), Huh7 cells (3 × 104) were seeded onto a 96‐well plate and incubated for 24 hours with VIP at concentrations of 10−9‐10−11 M. pCREB (ser133) signal was evaluated with the use of a PathScan p‐CREB (Ser133) Sandwich ELISA Kit (Cell Signaling Technology). The cells were then homogenized with cell lysis buffer, and the lysates were incubated for 2 hours at 37°C. The cells were incubated in detection antibody at 37°C for 1 hour and then in HRP‐conjugated secondary antibody. TMB substrate was added, and optical density was measured at 450 nm.
2.11. Statistical analysis
Data are shown as mean ± SEM. The Steel‐Dwass test for multiple comparisons was used to evaluate differences between respective groups. All statistical analyses were performed with JMP 13 (SAS Institute, Cary, NC, USA), and P < .05 was considered significant.
3. RESULTS
3.1. Localization of vasoactive intestinal peptide, VPAC1 and VPAC2 in human noncancer liver tissues, human hepatocellular carcinoma tissues, and Huh7 cells
Staining for VIP was not prominent in noncancer liver tissues, but staining for VIP was strong in the various HCC tissues. Staining for VPAC1 and VPAC2 was prominent in the various HCC tissues and adjacent nontumorous tissues. Mostly hepatocytes in nontumorous regions of HCC tissues were expressed in VIP and VPAC. Furthermore, the expressions of VIP and VPAC were also observed in cancer cells. Fewer VPAC1‐positive and VPAC2‐positive cells were found in the noncancer liver and liver metastases (Figure 1A). Strong staining for VIP and the 2 VIP receptors was observed in the nonvirus‐related HCC tissues and adjacent nontumorous tissues; staining for VIP and its receptors was also strong in the Huh7 cells (Figure 1B).
Figure 1.
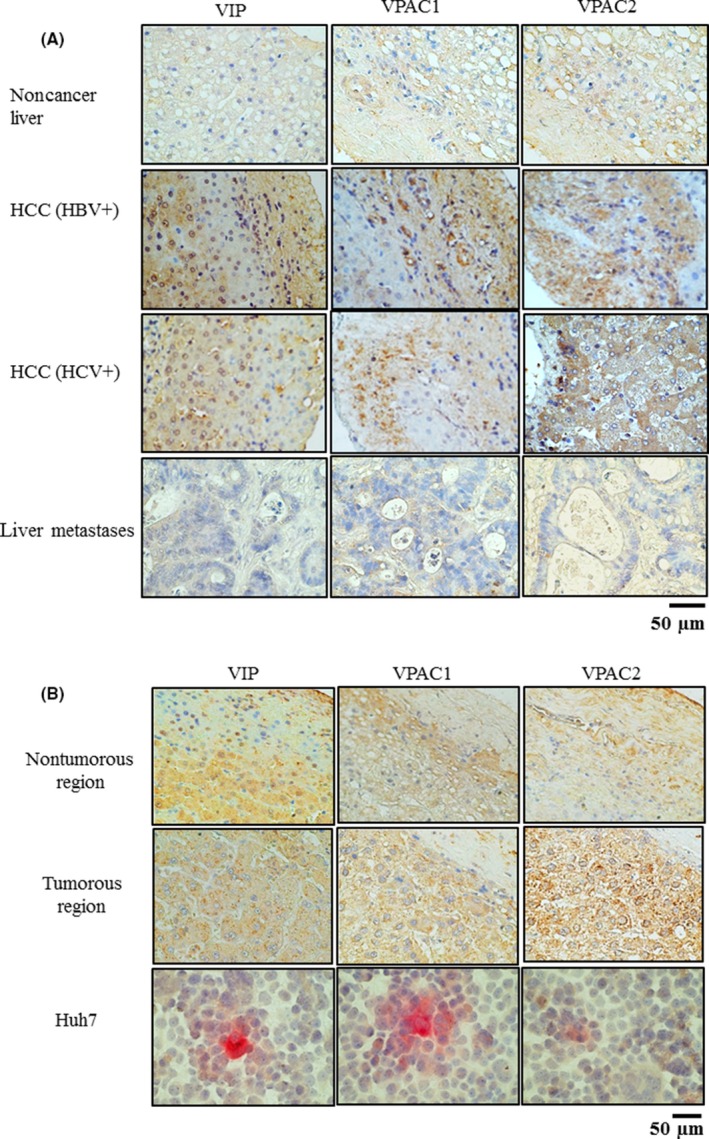
A, Immnohistochemical staining for vasoactive intestinal peptide (VIP), VIP G protein‐coupled receptor (VPAC)1 and VPAC2 in noncancer liver, liver metastases of colorectal cancer and hepatocellular carcinoma (HCC) of various etiologies, and adjacent nontumorous tissues (from at least 10 cm away from the HCC). The data represent 3 individual patients. Original magnification is ×200. B, Immunohistochemical staining for VIP, VPAC1 and VPAC2 in non‐HBV‐related HCC, non‐HCV‐related HCC and Huh7 cells. Original magnification of HCC tissues is ×200, and that of Huh7 cells is ×400. The nontumorous tissues were free of tumor cells. HBV+, hepatitis B virus‐positive; HCV+, hepatitis C virus‐positive
3.2. Proliferation and apoptosis of human hepatocellular carcinoma cells cultured with vasoactive intestinal peptide
Proliferation of Huh7 cells was significantly decreased in cells treated for 24 hours with 10−11 M, 10−10 M and 10−9 M VIP (87.7 ± 9.1%, 91.3 ± 8.8% and 90.3 ± 8.1%, respectively; P < .05) in comparison to cell proliferation in untreated cells (100.0 ± 3.6%) Proliferation of HepG2 cells was also significantly decreased in cells treated for 24 hours with 10−10 M, and 10−9 M VIP (94.8 ± 1.2% and 95.5 ± .9%, respectively; P < .05) in comparison to cell proliferation in untreated cells (100.0 ± 1.0%; Fig. 2A). Because Huh7 cells are derived from Japanese patients and differentiated human liver cancer functions, Huh7 cells were used for detailed mechanism of cell proliferation in the experiment.15 Proliferation was increased in the untreated cells. Conversely, proliferation was slightly decreased at 4 hours and significantly decreased at 24 hours after treatment of Huh7 cells with10−11 M, 10−10 M and 10−9 M VIP (89.1 ± 1.4%, 85.8 ± 2.5% and 83.9 ± 5.7%, respectively; P < .05) in comparison to that of untreated cells (108.0 ± 1.05%; Figure 2B). Furthermore, apoptosis was significantly increased in the Huh7 cells treated with 10−11 M, 10−10 M and 10−9 M VIP (138.1 ± 4.1%, 155.7 ± 5.9% and 144.4 ± 6.1%, respectively; P < .01) in comparison to that of untreated cells (100.0 ± 5.4%) (Figure 2C).
Figure 2.

Graphical representations of proliferation and apoptosis of hepatocellular carcinoma (HCC) cells treated with vasoactive intestinal peptide (VIP) at various concentrations. A, Huh 7 or HepG2 Cell proliferations are expressed as the ratio of the optical density of VIP‐treated cells to that of untreated (control) cells. The data represent 13 independent experiments. Values are mean ± SEM, *P < .05, **P < .01 vs control. B, Proliferation of Huh7 cells (4 × 104) treated with VIP was evaluated for 24‐h periods. The data represent 7 independent experiments, and values are mean ± SEM. **P < .01 vs control. C, Apoptosis of Huh7 cells treated with VIP. Apoptosis is shown relative to that of control cells and the data represent 9 independent experiments and expressed as mean ± SEM percentages. **P < .01 vs control. D, Apoptosis in the 2 types of primary cells. Apoptosis is shown relative to that of control cells, and the data represent 7 independent experiments. No significant difference was found between the 2 groups
3.3. Apoptosis of primary hepatocytes cultured with vasoactive intestinal peptide
Apoptosis was induced in the 2 types of primary HCC cells cultured with 10−10 M VIP. Apoptosis was not induced in the untreated primary HCC cells (Figure 2D).
3.4. cAMP concentrations and CREB and pCREB levels in Huh7 cells cultured with vasoactive intestinal peptide
The concentration of intracellular cAMP (.5 ± .1 nmol/L) was significantly decreased in Huh7 cells cultured with VIP (Figure 3A). In addition, expression of CREB was significantly decreased in the nuclei of Huh7 cells cultured with VIP 10−11 M, VIP 10−10 M and VIP 10−9 M (48.3 ± 6.6%, 45.1 ± 4.9% and 53.7 ± 5.4%, respectively; Figure 3B). The pCREB levels in Huh7 cells cultured with VIP 10−11 M and VIP 10−9 M were significantly decreased (Figure 3C). We next examined whether the VIP‐induced inhibition of the cAMP/CREB activity was mediated by the VIP receptors. When Huh7 cells were pretreated with VIP receptor antagonist, there was no reduction in the cAMP concentration (Figure 3D).
Figure 3.
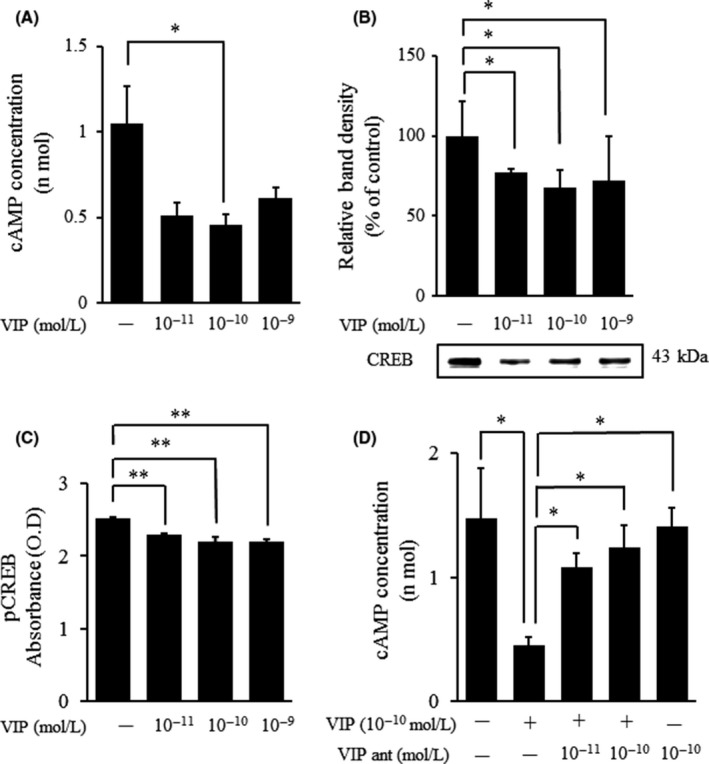
Graphical representations of ELISA‐determined expression of cAMP, cAMP response element‐binding protein (CREB) and phosphorylated CREB (pCREB) in Huh7 cells cultured with vasoactive intestinal peptide (VIP) at various concentrations for 15 min. A, Concentrations of intracellular cAMP in Huh7 cells (2 × 104) treated with VIP for 15 min. Huh7 cells were 80% confluent at the time of treatment. The data represent 6 independent experiments and expressed as mean ± SEM. *P < .05 vs control. B, Western blot analysis showed that VIP significantly decreased CREB protein levels in the nucleus of Huh7 cells treated with VIP. The data represent 5 independent experiments and expressed as mean ± SEM. *P < .05 vs control. C, pCREB was measured using ELISA in VIP‐induced Huh7 cells. VIP‐treated Huh7 cells for 15 min after VIP addition. The data represent 8 independent experiments. Data are expressed as mean ± SEM, **P < .01 vs the control. D, Effects of [D‐p‐Cl‐Phe6, Leu17]‐VIP on the concentration of cAMP in Huh7 cells. Huh7 cells were precultured with [D‐p‐Cl‐Phe6, Leu17]‐VIP for 1 . The data represent 7 independent experiments and expressed as mean ± SEM. *P < .05 vs the control
3.5. Levels of apoptosis‐related proteins in Huh7 cells cultured with vasoactive intestinal peptide
Expressions of caspase‐3, Bcl‐2 family and TNF receptor type 1‐associated death domain protein (TRADD) as apoptosis‐related proteins and of NFκB p65 and cyclin D were examined. Caspase‐3 protein levels (140.7 ± 10.6%) were significantly increased in cells cultured with VIP 10−10 M in comparison to levels in nontreated cells (100.0 ± 7.2%) (Fig. 4A), whereas Bcl‐xL protein levels were significantly decreased in cells cultured with 10−11 M, 10−10 M and 10−9 M VIP (43.3 ± 5.6%, 41.5 ± 6.4% and 28.0 ± 4.1%, P < .05, respectively) in comparison to levels in untreated cells (100 ± 20.4%; Figure 4B). Levels of other Bcl‐2 family, Bax and Bad were unchanged in VIP‐treated Huh7 cells (Figure 4C). Levels of TRADD, NFκB and cyclin‐D were also unchanged in VIP‐treated Huh7 cells (Figure 4D).
Figure 4.

Graphical representations and results of western blotting of protein levels of caspase‐3, Bcl‐xL, nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NFκB p65), TNF receptor type 1‐associated death domain protein (TRADD) and cyclin‐D in Huh7 cells treated with vasoactive intestinal peptide (VIP) at various concentrations for 24 h. A, B, Caspase‐3 and Bcl‐xL protein levels in Huh7 cells treated with VIP for 24 h. VIP (10−10 M) significantly increased caspase‐3 protein levels, and VIP significantly decreased Bcl‐xL protein levels. α‐tubulin was used as a control. The data represent 7 independent experiments. Values are mean ± SEM, *P < .05 vs control. C, Expression of Bax and Bad in Huh7 cells treated with VIP. D, Expression of NFκB, TNF receptor type 1‐associated death domain protein (TRADD) and cyclin‐D in Huh7 cells treated with VIP did not differ significantly
3.6. Effects of [D‐p‐Cl‐Phe6, Leu17]‐VIP and Rp‐cAMPS, dibutyryil‐cAMP on apoptosis and cell proliferation in vasoactive intestinal peptide‐treated Huh7 cells
To clarify whether the decrease in cell proliferation and the Bcl‐xL protein level in VIP‐treated Huh7 cells mediates the VIP receptor and cAMP/CREB pathway, Huh7 cells were preincubated with VIP receptor antagonist [D‐p‐Cl‐Phe6, Leu17]‐VIP or cAMP inhibitor Rp‐cAMPS for 1 hour, and this was followed by addition of VIP. The significant decreases in cell proliferation and Bcl‐xL protein in Huh7 cells treated with VIP 10−10 M were prevented by preculture with [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMPS (Figure 5A, B). In addition, [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMPS blocked the increase in apoptosis in VIP‐treated Huh7 cells. Preculture with [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMPS alone was shown to have no effect.
Figure 5.

Graphical representations of the effects of [D‐p‐Cl‐Phe6, Leu17]‐VIP and Rp‐cAMPS on cell proliferation (A) and Bcl‐xL protein levels (B) in Huh7 cells. Huh7 cells were pretreated with [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMPS for 1 h before 24‐h incubation with vasoactive intestinal peptide (VIP). Cell proliferation was evaluated by MTS assay. The data represent 18 independent experiments. Bcl‐xL protein levels were evaluated by western blotting. The data represent 7 independent experiments, and values are shown as mean ± SEM. *P < .05 vs control
We initially expected pretreatment with Rp‐cAMP to further decrease the cell proliferation and the Bcl‐xL protein levels in VIP‐treated Huh7 cells. However, the results were opposite what we had expected. We considered that pretreatment with cAMP analogue dibutyryl‐cAMP would yield similar results. Pretreatment with Rp‐cAMPS or dibutyryl‐cAMP significantly blocked the increased apoptosis in VIP‐treated Huh7 cells (Figure 6B).
Figure 6.

Graphical representations of the effects of [D‐p‐Cl‐Phe6, Leu17]‐VIP and Rp‐cAMPS on vasoactive intestinal peptide (VIP)‐induced apoptosis in Huh7 cells treated with VIP. A, Huh7 cells were precultured with [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMPS for 1 h. The data represent 7 independent experiments. Apoptosis levels in Huh7 cells precultured with Rp‐cAMPS or dibutyryl cAMP. B, The data represent 7 independent experiments and shown as mean ± SEM. *P < .05 vs control
4. DISCUSSION
The liver is indispensable for the maintenance of life and has a wide range of functions including, for example, detoxification, glycogen storage, protein and fat synthesis, and protein metabolism.16 It is abundantly innervated by the sympathetic and parasympathetic branches of the autonomic nervous system. Physiological control of the liver is sustained by secreted neurotransmitters: not only adrenaline but also various peptides such as substance P, VIP, calcitonin gene‐related peptide (CGRP) and somatostatin.6, 7 Studies have shown association between release of neural transmitters and both tumor development and tumor metastasis.17, 18, 19 For example, substance P promotes tumor cell proliferation and angiogenesis.20 Alternatively, somatostatin inhibits the tumor growth, and, thus, somatostatin analogue is used effectively for treatment of liver or pancreatic cancer.21, 22 Overexpression of VIP receptors VPAC1 and VPAC2 is seen in many human cancers.9, 23, 24 Although, these receptors are known to be expressed in HCC, their roles in tumor growth and progression remain undetermined. Thus, we investigated the expression of both VIP and its receptors in noncancer liver and in HCC and their influence on the pathogenesis of HCC.
Although VIP and its receptors were not prominent in noncancer liver tissue, they were clearly present in all HCC tissues, whether or not the HCC was derived from hepatitis. VIP, VPAC1 and VPAC2 were localized to both tumorous and nontumorous regions. That is, expression of VIP and its receptors might not be related to hepatitis virus infection.
These findings indicate that VIP is produced in hepatocytes and in tumor cells and that VIP can influence HCC cells through its receptors.
Persistent inflammation is known to contribute to carcinogenesis and tumor progression.25 Neurotransmitter secretion increases in response to injury‐induced inflammation and to the inflammation that accompanies carcinogenesis. Thus, the prominent expression of VIP and of its receptors may be related to the inflammation associated with HCC development.
To clarify whether VIP is involved in the development of HCC, we investigated the effect of VIP on HCC cell proliferation. We used Huh7 cells derived from Japanese patients with HCC to verify the expression of VIP and its receptors. Proliferation of Huh7 cells was inhibited by 24‐hour culture with VIP at 10−9 M, 10−10 M and 10−11 M. VIP has been shown to inhibit proliferation of several types of cells,6, 17, 18, 19 and it had a similar influence on our HCC cells.
The maximum inhibitory effect of VIP was produced when the blood concentration reached the physiological range by treatment with VIP 10−10 M. We have found that VIP 10−10 M significantly inhibited proliferation of synovial cells obtained from patients with rheumatoid arthritis.26 Thus, the physiological and pathological effects of neuropeptides may depend on the blood concentrations reached. We found that VIP 10−8 M did not significantly inhibit cell proliferation, perhaps because of receptor desensitization due to normalization and depletion of the second messenger.
Inhibition of HCC cell proliferation can be explained by apoptosis and cell cycle arrest. When we cultured Huh7 cells with VIP 10−11 M, 10−10 M or 10−9 M for 24 h, apoptosis increased significantly. If VIP were to induce apoptosis in normal hepatocytes, hepatic function would be severely affected. We analyzed apoptosis in 2 species of primary cultured human hepatocytes and found that VIP did not induce apoptosis in these primary cultured hepatocytes, indicating that VIP did not affect normal hepatocytes due to absence of the VIP receptors.
The action of neuropeptides is mediated by their binding to specific membrane‐associated receptors. VIP belong to the G protein‐coupled receptor family, and VIP acts by coupling with VIP receptors and modulating the cAMP/PKA signaling pathway, which leads to activation of CREB, a PKA target.27, 28, 29, 30 We found that VIP significantly reduced cAMP concentrations and CREB protein levels. VIP also inhibited the phosphorylation of ser133 on CREB in Huh7 cells. To examine whether the inhibition of cAMP/CREB by VIP was mediated by the VIP receptors, Huh7 cells were precultured with VPAC receptor antagonist [D‐p‐Cl‐Phe6, Leu17]‐VIP before incubation with VIP10−10 M. Pretreatment with [D‐p‐Cl‐Phe6, Leu17] VIP (at 10−11 M and 10−10 M) blocked the inhibition of CREB and its phosphorylation, suggesting that VIP inactivates CREB via VIP receptors in HCC cells.
We evaluated whether VIP‐induced apoptosis of HCC cells contributes to death receptor, cell cycle arrest and transcription factors. When VIP‐induced apoptosis of Huh7 cells was analyzed, no effects were seen on expression of death receptor TRADD, the Fas ligand pathway, or anti‐apoptotic factors cyclin‐D1 and NFκB p65. Although BAX and Bad expressions were unchanged, a decrease in apoptosis‐inhibiting factor Bcl‐xL and an elevation in caspase‐3 protein were observed in VIP 10−10 M‐cultured Huh7 cells. These results indicate that VIP‐induced apoptosis is mediated through the mitochondrial pathway.
CREB and Bcl‐xL proteins play important roles in the resistance of cancer cells to apoptosis induction and, thus, contribute to tumor growth and metastasis. Expression of dominant‐negative CREB has been shown to decrease the carcinogenic and metastatic potential of cancer cells.31 Decreased CREB phosphorylation and subsequent downregulation of Bcl‐xL under PKCκ suppression contribute to the hepatic apoptosis during sepsis.32 These results suggest that VIP‐induced apoptosis is mediated via a mitochondrial death pathway and is caused by downregulation of the cAMP/CREB pathway.
We speculated that pretreatment with cAMP/PKA inhibitor Rp‐cAMP would further increase the VIP‐induced apoptotic effect and inhibition of HCC cell proliferation. Interestingly, pretreatment with Rp‐cAMP 10−7 M inhibited the increase in apoptosis of Huh7 cells treated with VIP 10−10 M. The preculture with [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMPS significantly prevented the inhibition of cell proliferation and the decrease in Bcl‐xL protein levels in VIP‐treated Huh7 cells. In addition, the VIP‐induced increase in apoptosis of Huh 7 cells decreased under pretreatment with [D‐p‐Cl‐Phe6, Leu17]‐VIP or Rp‐cAMP. To confirm the ability of cAMP analogue to suppress Huh7 cell apoptosis, pretreatment with cAMP analogue dibutyryl cAMP was used. Presence of the PKA inhibitor and cAMP analogue yielded similar changes in apoptosis. The reason for the discrepant result was that Rp‐cAMP had the same effect as a partial agonist for cAMP activation. Gerdin et al showed that a PKA‐independent signaling mechanism is mediated by another cAMP‐binding protein and a subsequent signaling pathway, and such a protein was later identified as an exchange protein directly activated by cAMP (Epac).33, 34 PACAP, which shows high sequence homology to VIP, leads to neuronal differentiation and CREB activation via the Epac signaling pathway.35, 36 Epac signaling (ie, PKA‐independent signaling) may contribute to VIP‐induced increase in apoptosis of HCC cells. Further studies are needed to clarify the mechanism.
In conclusion, we showed that VIP and VIP receptors are expressed in HCC cells and that they act to decrease cell proliferation and increase apoptosis. Cytokines, antiviral agents and anticancer agents are often used in combination to treat HCC. VIP should be noted as a neuropeptide capable of inducing apoptosis of malignant tumor cells. The action of VIP alone is weak, having decreased HCC cell proliferation by only 20%, but the combined use of VIP or VIP analogue and anticancer drugs is suggested and has, in fact, been proposed previously.37 Our findings point to new pharmacological strategies that can be used to inhibit cancer progression.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
ETHICAL APPROVAL
The study was approved by the St. Marianna University School of Medicine Ethics Committee (Approval No. 1207).
Hara M, Takeba Y, Iiri T, et al. Vasoactive intestinal peptide increases apoptosis of hepatocellular carcinoma by inhibiting the cAMP/Bcl‐xL pathway. Cancer Sci. 2019;110:235–244. 10.1111/cas.13861
REFERENCES
- 1. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118‐1127. [DOI] [PubMed] [Google Scholar]
- 2. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;128:939‐944. [DOI] [PubMed] [Google Scholar]
- 3. Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer. 2009;115:3379‐3391. [DOI] [PubMed] [Google Scholar]
- 4. Shi M, Liu D, Yang Z, Guo N. Central and peripheral nervous systems: master controllers in cancer metastasis. Cancer Metastasis Rev. 2013;32:603‐621. [DOI] [PubMed] [Google Scholar]
- 5. Tang J, Li Z, Lu L, Cho CH. β‐Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol. 2013;23:533‐542. [DOI] [PubMed] [Google Scholar]
- 6. Ito Y, Magari S, Sakanaka M. Immunoelectron‐microscopic localization of peptidergic nerve fibers around lymphatic capillaries in the rat liver. Arch Histol Cytol. 1990;53:199‐208. [DOI] [PubMed] [Google Scholar]
- 7. Stoyanova II, Gulubova MV. Peptidergic nerve fibres in the human liver. Acta Histochem. 1998;100:245‐256. [DOI] [PubMed] [Google Scholar]
- 8. Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;562:249‐290. [DOI] [PubMed] [Google Scholar]
- 9. Dickson L, Finlayson K. VPAC and PAC receptors: from ligands to function. Pharmacol Ther. 2009;1213:294‐316. [DOI] [PubMed] [Google Scholar]
- 10. Absood A, Hu B, Bassily N, Colletti L. VIP inhibits human HepG2 cell proliferation in vitro. Regul Pept. 2008;146:285‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'Amico AG, Scuderi S, Saccone S, Castorina A, Drago F, D'Agata V. Antiproliferative effects of PACAP and VIP in serum‐starved glioma cells. J Mol Neurosci. 2013;512:503‐513. [DOI] [PubMed] [Google Scholar]
- 12. Dufes C, Alleaume C, Montoni A, Olivier JC, Muller JM. Effects of the vasoactive intestinal peptide (VIP) and related peptides on glioblastoma cell growth in vitro. J Mol Neurosci. 2003;212:91‐102. [DOI] [PubMed] [Google Scholar]
- 13. Horvath G, Brubel R, Kovacs K, et al. Effects of PACAP on oxidative stress‐induced cell death in rat kidney and human hepatocyte cells. J Mol Neurosci. 2011;431:67‐75. [DOI] [PubMed] [Google Scholar]
- 14. Cochaud S, Meunier AC, Monvoisin A, Bensalma S, Muller JM, Chadéneau C. Neuropeptides of the VIP family inhibit glioblastoma cell invasion. J Neurooncol. 2015;122:63‐73. [DOI] [PubMed] [Google Scholar]
- 15. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858‐3863. [PubMed] [Google Scholar]
- 16. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13:267‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang EV, Sood AK, Chen M, et al. Norepinephrine up‐regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)‐2, and MMP‐9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006;66:10357‐10364. [DOI] [PubMed] [Google Scholar]
- 18. Tilan J, Kitlinska J. Sympathetic neurotransmitters and tumor angiogenesis‐link between stress and cancer progression. J Oncol. 2010;2010:539706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Magnon C, Hall SJ, Lin J, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. [DOI] [PubMed] [Google Scholar]
- 20. Muñoz M, Coveñas R. Involvement of substance P and the NK‐1 receptor in cancer progression. Peptides. 2013;48:1‐9. [DOI] [PubMed] [Google Scholar]
- 21. Borbath I, Leclercq IA, Abarca‐Quinones J, et al. Inhibition of early preneoplastic events in the rat liver by the somatostatin analog lanreotide. Cancer Sci. 2007;98:1831‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li M, Wang X, Li W, et al. Somatostatin receptor‐1 induces cell cycle arrest and inhibits tumor growth in pancreatic cancer. Cancer Sci. 2008;99:2218‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szilasi M, Buglyo A, Treszl A, Kiss L, Schally AV, Halmos G. Gene expression of vasoactive intestinal peptide receptors in human lung cancer. Int J Oncol. 2011;39:1019‐1024. [DOI] [PubMed] [Google Scholar]
- 24. Moody TW, Nuche‐Berenguer B, Jensen RT. Vasoactive intestinal peptide/pituitary adenylate cyclase activating polypeptide, and their receptors and cancer. Curr Opin Endocrinol Diabetes Obes. 2016;23:38‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539‐545. [DOI] [PubMed] [Google Scholar]
- 26. Takeba Y, Suzuki N, Kaneko A, Asai T, Sakane T. Evidence for neural regulation of inflammatory synovial cell functions by secreting calcitonin gene‐related peptide and vasoactive intestinal peptide in patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:2418‐2429. [DOI] [PubMed] [Google Scholar]
- 27. Valdehita A, Bajo AM, Fernández‐Martínez AB, et al. Nuclear localization of vasoactive intestinal peptide (VIP) receptors in human breast cancer. Peptides. 2010;31:2035‐2045. [DOI] [PubMed] [Google Scholar]
- 28. Dostmann WR. (RP)‐cAMPS inhibits the cAMP‐dependent protein kinase by blocking the cAMP‐induced conformational transition. FEBS Lett. 1995;375:231‐234. [DOI] [PubMed] [Google Scholar]
- 29. Vacas E, Fernandez‐Martinez AB, Bajo AM, et al. Vasoactive intestinal peptide (VIP) inhibits human renal cell carcinoma proliferation. Biochim Biophys Acta. 2012;1823:1676‐1685. [DOI] [PubMed] [Google Scholar]
- 30. Yang J, Shi QD, Song TB, et al. Vasoactive intestinal peptide increases VEGF expression to promote proliferation of brain vascular endothelial cells via the cAMP/PKA pathway after ischemic insult in vitro. Peptides. 2013;42:105‐111. [DOI] [PubMed] [Google Scholar]
- 31. Aggarwal S, Kim SW, Ryu SH, Chung WC, Koo JS. Growth suppression of lung cancer cells by targeting cyclic AMP response element‐binding protein. Cancer Res. 2008;68:981‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsieh YC, Chen YH, Jao HC, Hsu HK, Huang LJ, Hsu C. Role of CREB phosphorylation in hepatic apoptosis under PKCa suppression during sepsis. Shock. 2005;24:357‐363. [DOI] [PubMed] [Google Scholar]
- 33. Gerdin MJ, Eiden LE. Regulation of PC12 cell differentiation by cAMP signaling to ERK independent of PKA: do all the connections add up? Sci STKE. 2007;382:pe15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown LM1, Rogers KE, McCammon JA, Insel PA. Identification and validation of modulators of exchange protein activated by cAMP (Epac) activity: structure‐function implications for Epac activation and inhibition. J Biol Chem. 2014;289:8217‐8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kiermayer S, Biondi RM, Imig J, et al. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol Biol Cell. 2005;16:5639‐5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi GX, Rehmann H, Andres DA. A novel cyclic AMP‐dependent Epac‐Rit signaling pathway contributes to PACAP38‐mediated neuronal differentiation. Mol Cell Biol. 2006;26:9136‐9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gelber E, Granoth R, Fridkin M, et al. A lipophilic vasoactive intestinal peptide analogue enhances the antiproliferative effect of chemotherapeutic agents on cancer cell lines. Cancer. 2001;92:2172‐2180. [DOI] [PubMed] [Google Scholar]