

Abstract
Cyclin‐dependent kinase (CDK) 4 and CDK6 inhibitors are effective therapeutic options for hormone receptor (HR)‐positive, human epidermal growth factor receptor 2 (HER2)‐negative advanced breast cancer. Although CDK4/6 inhibitors mainly target the cyclin D‐CDK4/6‐retinoblastoma tumor suppressor protein (RB) axis, little is known about the clinical impact of inhibiting phosphorylation of other CDK4/6 target proteins. Here, we focused on other CDK4/6 targets, SMAD proteins. We showed that a CDK4/6 inhibitor palbociclib and activin‐SMAD2 signaling cooperatively inhibited cell cycle progression of a luminal‐type breast cancer cell line T47D. Palbociclib enhanced SMAD2 binding to the genome by inhibiting CDK4/6‐mediated linker phosphorylation of the SMAD2 protein. We also showed that cyclin G2 plays essential roles in SMAD2‐dependent cytostatic response. Moreover, comparison of the SMAD2 ChIP‐seq data of T47D cells with those of Hs578T (triple‐negative breast cancer cells) indicated that palbociclib augmented different SMAD2‐mediated functions based on cell type, and enhanced SMAD2 binding to the target regions on the genome without affecting its binding pattern. In summary, palbociclib enhances the cytostatic effects of the activin‐SMAD2 signaling pathway, whereas it possibly strengthens the tumor‐promoting aspect in aggressive breast cancer.
Keywords: activin, breast cancer, estrogen receptor, palbociclib, SMAD
Abbreviations
- CDK
cyclin‐dependent kinase
- ChIP, chromatin immunoprecipitation;
EMT
epithelial‐to‐mesenchymal transition
- ER
estrogen receptor
- GSEA
gene set enrichment analysis
- HER2
human epidermal growth factor receptor 2
- HR
hormone receptor
- PR
progesterone receptor
- RB
retinoblastoma tumor suppressor protein
- TGF‐β
transforming growth factor‐β
- TNBC
triple‐negative breast cancer
1. INTRODUCTION
Deregulation of cell cycle control is a hallmark of human cancer.1 Tumor‐associated cell cycle defects are often mediated by alterations in CDK activity, which are regarded as promising targets in cancer therapy.2, 3, 4 For example, CDK4/6‐selective inhibitors, such as palbociclib (PD0332991), ribociclib (LEE011), and abemaciclib (LY2835219), have shown promising clinical results in cancer patients, especially those with breast cancer.5, 6, 7, 8 CDK4 and CDK6 are highly similar serine/threonine kinases, activity of which is controlled by several mechanisms: positively by association with cyclin D (cyclins D1‐3) and negatively by binding to CDK inhibitors of the INK4 family.9 CDK4/6, in complex with cyclin D, phosphorylate the retinoblastoma tumor suppressor protein (RB, encoded by RB1) and its family members, p107 (RBL1, or RB transcriptional corepressor like 1) and p130 (RBL2). When phosphorylated, RB proteins release E2F transcription factors, resulting in the expression of genes required for cell cycle progression from G1 phase into S phase and initiation of DNA synthesis.
Breast cancer is classified into intrinsic subtypes by expression microarray data,10 and the clinicopathological surrogate definitions of the intrinsic subtypes are based on the expression of ER, PR, HER2 (also known as Erb‐B2), and Ki‐67; luminal A‐like (ER+ and/or PR+, HER2−, and low Ki‐67), luminal B‐like HER2‐negative (ER+ and/or PR+, HER2−, and high Ki‐67), luminal B‐like HER2‐positive (ER+ and/or PR+, HER2+), HER2‐positive non‐luminal (ER− and PR−, HER2+), and triple negative (ER−, PR−, HER2−).11 Recent comprehensive molecular profiling has confirmed that the cyclin D‐CDK4/6‐RB axis is frequently perturbed in breast cancer; for example, amplification of CCND1 (encoding cyclin D1) occurs, especially in luminal type (58% in luminal B).12 It indicates that luminal‐type breast cancer is a good candidate for treatment using CDK4/6 inhibitors. Presently, the inhibitors for CDK4/6 in combination with hormonal treatments have been established as effective therapeutic options for HR‐positive and HER2‐negative advanced breast cancer.
Although CDK4/6 inhibitors exert their effects mainly through RB, they still exert a partial cytostatic effect in RB1 knockdown or knockout cells before evolution of compensatory mechanisms.13, 14 This suggests that CDK4/6 inhibitors exert their effects through other substrates besides RB. Indeed, a systemic screening of 445 human nuclear proteins with at least two CDK consensus sites identified FOXM1, which plays essential roles in CDK4/6‐mediated escape from senescence.15 SMAD2 and SMAD3 are other CDK4 substrates16, 17; they are intracellular signaling components of TGF‐β and activin, which convey cytostatic signals in ER‐positive breast cancer.18, 19 SMAD proteins contain highly conserved MH1 and MH2 domains and a less conserved linker region between the two domains. The linker domain is a target for post‐translational modification including phosphorylation, which causes either activation of transcription by the SMAD complex or degradation and turnover of SMAD proteins in a context‐dependent way.20
The signaling pathways triggered by TGF‐β family members control a wide range of cellular processes.21, 22, 23 In normal or premalignant cells, TGF‐β/activin usually function as tumor suppressors by inhibiting cell proliferation and inducing apoptosis. It is widely accepted that SMAD2/3 activate the cytostatic program through induction of CDK inhibitors (p21WAF1/CIP1, p27KIP1, and p15INK4B) and inhibition of the expression of other cell cycle regulators, such as c‐Myc and Cdc25A.22 However, activation of the SMAD2/3 pathway also induces the expression of transcription factors for EMT, such as SNAIL, SLUG, ZEB1, and ZEB2, which may explain the tumor‐promoting effects of TGF‐β and activin in aggressive breast cancer.24 Linker phosphorylation of SMAD may add complexity to the bidirectional roles of TGF‐β family members.
Here, we focused on a possible crosstalk between CDK4/6 and TGF‐β/activin‐SMAD pathways. Consistent with a previous report,25 several ER‐positive breast cancer cell lines were insensitive to TGF‐β treatment; we thus used activin A (ActA) for stimulation. Our data show that CDK4/6 inhibition and the activin‐SMAD signaling pathway collectively regulate the cytostatic response. Moreover, CDK4/6 inhibitors may also strengthen the tumor‐promoting aspect of SMAD signaling in aggressive breast cancer.
2. MATERIALS AND METHODS
2.1. Plasmid construction
Human SMAD2 (NM_005901) construct was as previously described.26 Human CCNG2 (NM_004354.2) was cloned by PCR, sequence‐verified, and subcloned into pcDEF3 vectors.27 Fluorescent, ubiquitination‐based cell cycle indicator (Fucci) lentiviral vectors, pCS‐EF‐mAG‐hGeminin(1‐110) and pCS‐EF‐mKO2‐hCdt1(30‐120),28 were kindly provided by Dr A. Miyawaki (RIKEN, Wako, Japan). All the constructs for lentivirus production were kindly provided by Dr H. Miyoshi (RIKEN, Wako, Japan; present address, Keio University, Japan).
2.2. Chromatin immunoprecipitation‐seq and RNA‐seq
Chromatin immunoprecipitation‐seq and RNA‐seq were carried out as described.29, 30, 31 Raw data are available at NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) (GSE117502).
2.3. Statistical analysis
Differences between two experimental groups were analyzed using Welch's t test or analysis of variance (ANOVA) followed by Tukey‐Kramer post hoc test for multiple comparison using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. Palbociclib and activin cooperatively inhibit cell cycle progression
First, we investigated TGF‐β/activin signaling activity in two representative ER‐positive human breast cancer cell lines, T47D and MCF7. Similar to TGF‐β, ActA causes cell cycle arrest through the SMAD pathway, by inducing CDK inhibitors p21WAF1/CIP1 and/or p27KIP1.19 ActA was able to phosphorylate the C‐terminal serine residues of SMAD2 and SMAD3 in both cell lines as reported previously, whereas TGF‐β could not phosphorylate the C‐terminal serine residues of SMAD2 and SMAD3 in T47D cells (Figure 1A).19, 25 We thus decided to use ActA as a ligand in the present study. ActA mainly phosphorylated SMAD2 in T47D cells, whereas it phosphorylated both SMAD2 and SMAD3 in MCF7, and HaCaT keratinocytes, which were used as a control for SMAD phosphorylation (Figure 1A).
Figure 1.
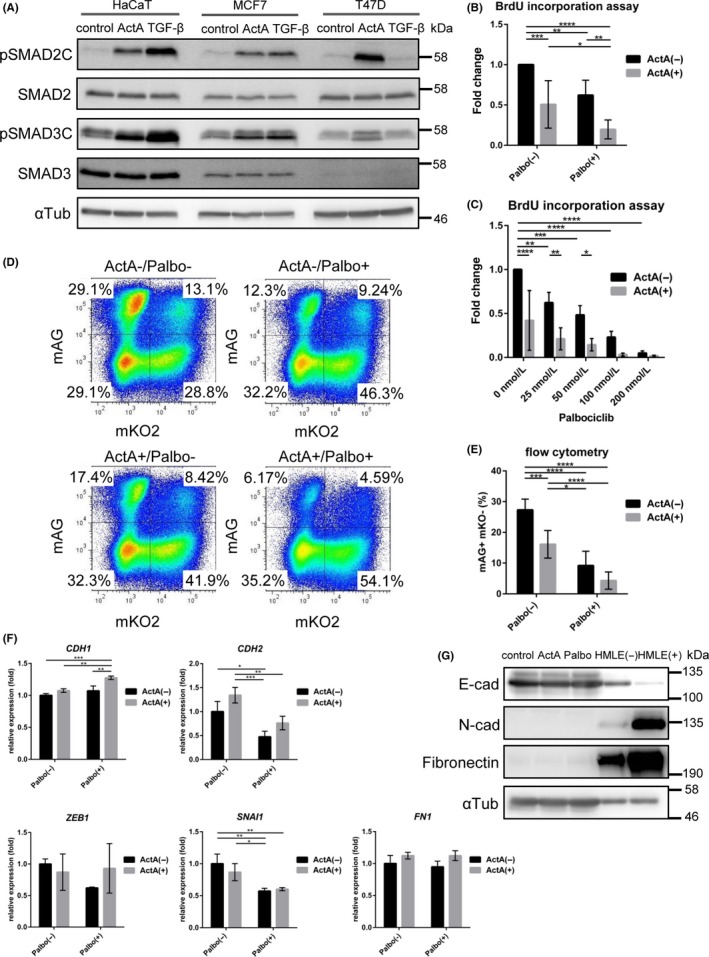
Palbociclib and activin have additive effects on cell cycle delay of a luminal‐type breast cancer cell line T47D cells. A, Immunoblot analysis in luminal breast cancer cell lines and control HaCaT keratinocytes, after 1.5 h treatment with 1 ng/mL transforming growth factor beta (TGF‐β) and 50 ng/mL activin A (ActA). B,C, BrdU incorporation assay in T47D cells. B, Cells were treated with ActA and 50 nmol/L palbociclib (Palbo). After 24 h, cells were incubated with BrdU overnight. C, Cells were treated with ActA and 0‐200 nmol/L palbociclib for 24 h. Data were normalized to the untreated condition. Data represent mean ± SD of n = 6 (B) and n = 4 (C) independent experiments. D,E, Cell cycle analysis using the Fucci system. T47D cells were treated with ActA and 50 nmol/L palbociclib for 24 h. D, Representative data of n = 7 independent experiments. E, Change in the ratio of mAG positive and mKO negative cells (S/G2/M phase). Data represent mean ± SD of n = 7 independent experiments. F, qRT‐PCR analysis of T47D cells. Cells were treated with ActA and 50 nmol/L palbociclib for 24 h. Data represent mean ± SD of n = 3 independent experiments. G, Immunoblot analysis for epithelial and mesenchymal markers in T47D cells with ActA and 1 μmol/L palbociclib treatment for 24 h. Human mammary gland epithelial HMLE cells treated with (+)/without (−) 1 ng/mL TGF‐β were used as controls. P* < .05, **P < .01, *** < .001, ****P < .0001; ANOVA with Tukey‐Kramer post hoc test in (B‐F)
A previous study showed that both cells were sensitive to palbociclib; IC50 values for T47D and MCF7 cells were reported to be 127 and 148 nmol/L,32 respectively. In another study, IC50 values calculated by BrdU incorporation assay were 15 nmol/L for both cells.33 Consistently, both palbociclib and ActA showed cytostatic effects in T47D cells in a BrdU incorporation assay (Figure 1B,C); these effects were additive in nature. A similar tendency was obtained in an EdU incorporation assay using flow cytometry (Figure S1). In addition, we used the Fucci system,28 which visualizes cell cycle activity and progression by monitoring the inverse oscillation dynamics of fluorescently tagged cell cycle fusion proteins, monomeric Azami‐Green1 (mAG)‐hGeminin and monomeric Kusabira‐Orange2 (mKO2)‐hCdt1.28 Both palbociclib and ActA decreased mAG‐positive cells, which represent cells in the S/G2/M stage, whereas they increased mKO2‐positive cells, which represent cells in G1 or G0 stage (Figure 1D,E). Again, there were additive effects between palbociclib and ActA.
To further validate these findings in an unbiased way, we conducted RNA‐sequencing (RNA‐seq) in T47D cells treated with ActA and palbociclib. We then carried out GSEA using the oncogenic signatures (C6, MSigDB) ([Link], [Link]). A gene set “RB_P107_DN.V1_UP”, which contained genes induced after RB1 and RBL1 knockout34 or genes repressed by RB/p107, was enriched in the repressed genes after palbociclib treatment ([Link], [Link]). This confirmed that palbociclib enhanced the function of RB and/or RBL1 in T47D cells. In addition, a gene set “TGFB_UP.V1_UP”, which contained the top 200 genes induced in a panel of epithelial cell lines by TGF‐β1,35 was enriched in the palbociclib‐induced genes regardless of the presence or absence of ActA ([Link], [Link]), indicating that palbociclib enhanced the ActA‐SMAD pathway. Interestingly, enrichment of the same gene set “TGFB_UP.V1_UP” was statistically significant in ActA‐treated T47D cells when the cells were also cotreated with palbociclib (Figure S2C,G). These findings suggested a crosstalk between CDK4/6 and activin‐SMAD pathways.
In contrast to the cytostatic effects of TGF‐β/activin, tumor‐promoting effects were not obvious in T47D cells. As for EMT‐like changes, ActA was not able to induce the EMT transcription factors in T47D cells (Figure 1F and Table S2). Palbociclib treatment did not affect induction of the EMT transcription factors, and it slightly induced CDH1 (which encodes E‐cadherin). Furthermore, expression of mesenchymal marker proteins, N‐cadherin (which is encoded by CDH2) and fibronectin (which is encoded by FN1), was extremely low (Figure 1G), suggesting that activation of the activin‐SMAD pathway is not sufficient to induce the EMT in T47D cells.
3.2. CDK4/6 phosphorylate Ser255 at the SMAD2 linker region
In a previous report, recombinant CDK4 was shown to phosphorylate several Ser/Thr residues of SMAD3,17 which correspond to Thr8, Thr220, and Ser255 of SMAD2. We carried out screening of the linker phosphorylation sites in T47D cells using anti‐phospho‐SMAD2 antibodies (Figure 2A,B). We found that palbociclib strongly decreased phosphorylation of SMAD2 Ser255. Another phosphorylation site, Thr8, was also weakly affected, which is also the target of extracellular signal‐regulated kinase (ERK)36 and CDK2.37 In T47D cells, phosphorylation of Thr220 and Ser250 was not affected by palbociclib treatment. The assay with anti‐phospho‐SMAD2 Ser255 antibody was validated in HEK293T cells, which ectopically express Ala (S255A) mutant instead of SMAD2 Ser255 (Figure 2C). Dose‐response experiments with palbociclib showed that phosphorylation of SMAD2 Ser255 and RB Ser780 was inhibited by a comparable concentration of palbociclib (Figure 2D).
Figure 2.
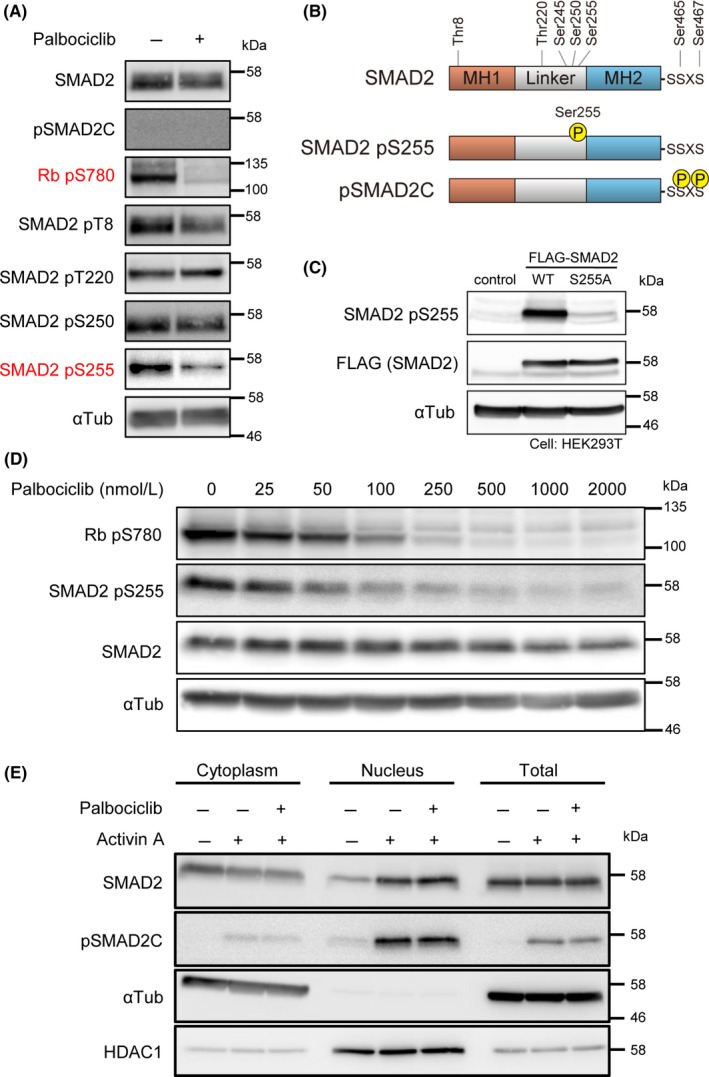
Palbociclib decreases Ser255 phosphorylation of SMAD2. A, Site‐specific phosphorylation of SMAD2 after palbociclib treatment. T47D cells were treated with 1 μmol/L palbociclib for 24 h. B, Schematic representation of representative phosphorylation sites of human SMAD2 protein. C, Validation of anti‐phospho‐SMAD2 Ser255. HEK293T cells were transfected with indicated plasmids. D, T47D cells were treated with indicated concentrations of palbociclib for 24 h to evaluate substrate phosphorylation. E, Immunoblot analysis of subcellular fractions prepared from T47D cells treated with ActA for 1.5 h and 1 μmol/L palbociclib for 24 h
Linker phosphorylation of SMAD proteins has been reported to affect the distribution or stability of SMADs.20 Cell fractionation experiments showed that C‐terminally phosphorylated SMAD2 protein was accumulated in the nucleus of the palbociclib‐treated T47D cells (Figure 2E). Thus, we were not able to detect the difference in ActA‐induced phospho‐SMAD2C abundance in the nuclei of palbociclib‐treated cells and palbociclib‐untreated cells in immunoblotting. This is in contrast to our finding that palbociclib and activin‐SMAD common target genes were cooperatively regulated. In order to analyze the crosstalk in detail especially at the level of SMAD‐DNA interaction, we carried out ChIP‐seq analyses of SMAD2 protein.
3.3. Palbociclib enhances SMAD2 binding to the genome in luminal‐type breast cancer cells
SMAD2 ChIP‐seq analyses were carried out in T47D cells treated with ActA for 1.5 hours, with or without palbociclib treatment for 24 hours (Figure 3A,B). Number of sequenced reads in the SMAD2 binding sites, which correlates with strength of SMAD2‐DNA interaction, was increased after palbociclib treatment (Figure 3A). This trend also existed in the gene loci of well‐established target genes of TGF‐β/activin and the downstream SMAD complex, PMEPA1 (also known as TEMPAI) and CDKN2B (Figure 3B). We further validated these findings using ChIP‐qPCR analysis (Figure 3C). Palbociclib treatment significantly increased the enrichment of SMAD2 binding to the PMEPA1 and CDKN2B promoters. These data suggest that palbociclib treatment affects SMAD complex‐DNA interaction at the genome level.
Figure 3.
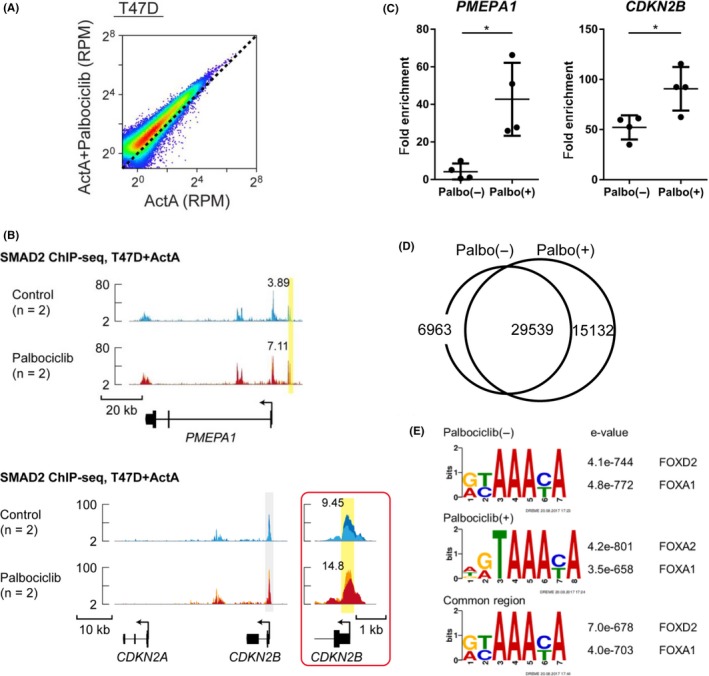
Palbociclib enhances SMAD2 binding to the genome in T47D cells. A, Scatter plot of differentially enriched sequenced reads in each SMAD2 binding region from ActA‐ and palbociclib‐treated T47D cells (n = 2) and ActA‐only control (n = 2). RPM, reads per million reads. B, PMEPA1 and CDKN2B loci are shown with the SMAD2 ChIP‐seq data of T47D cells treated with or without palbociclib (n = 2 for each condition). Direction of transcription is shown by the arrow beginning at the transcription start site. Inset shows magnification of the region marked by a gray‐colored box. Each ChIP‐seq data is presented in a different color. Values of RPM (averaged from two biological replicates) of peaks, highlighted by yellow boxes, are presented. C, SMAD2 ChIP‐qPCR analysis of T47D cells treated with ActA for 1.5 h with or without 1 μmol/L palbociclib for 24 h. The SOBP gene locus was used as a negative control region, and fold enrichment was calculated. Data represent mean ± SD of n = 4 independent experiments (*P < .05; Welch's t test). D, Venn diagram indicating the overlap of SMAD2 binding sites in T47D cells with or without treatment of palbociclib. E, Transcription factor motif analysis of SMAD2 binding regions in T47D cells
We assumed that palbociclib treatment redirected the SMAD complex to novel binding sites. However, as shown in the Venn diagram, approximately 65% of binding sites overlapped between palbociclib‐treated and ‐untreated cells, although the number of SMAD2 binding sites was greater in the palbociclib‐treated T47D cells (Figure 3D). Furthermore, de novo motif enrichment analyses showed that one of the representative motifs commonly enriched in the SMAD2 binding sites was that for the FOX family genes (Figure 3E). FOXA1 has been reported to be a master transcription factor of luminal breast epithelial cells,38 or a pioneer factor for ER‐positive breast cancer cells,39 suggesting that the SMAD complex binds to the enhancers that are already accessible by FOXA1 in ER‐positive cells.
We then validated the SMAD‐DNA interaction enhanced by palbociclib. Using a SMAD reporter construct 9 × CAGA‐luc, we confirmed that palbociclib treatment enhanced the activin‐SMAD signaling pathway (Figure 4A). In addition, TGF‐β/activin‐SMAD target genes, PMEPA1 and CDKN2B, were induced by ActA in T47D cells, and the induction was further enhanced by palbociclib treatment (Figure 4B). This enhancement by palbociclib was at least partially dependent on the SMAD pathway; an ALK4/5/7 kinase inhibitor (SB431542) attenuated the palbociclib‐mediated enhancement of the induction of PMEPA1 and CDKN2B (Figure 4C). Of note, both SMAD and FOXA1 binding motifs existed in the SMAD2 binding regions of PMEPA1 and CDKN2B genes. Moreover, by combining SMAD2 ChIP‐seq and RNA‐seq data, we identified mRNA expression of SMAD2 bound genes. GSEA analyses showed that the gene set “TGFB_UP.V1_UP” was enriched in the induced genes after palbociclib treatment (Figure 4D), which also supports the hypothesis that palbociclib regulates the set of genes through SMAD2. Thus, we concluded that palbociclib treatment enhances SMAD2 binding to the genome and induces the expression of SMAD2 target genes in T47D cells.
Figure 4.
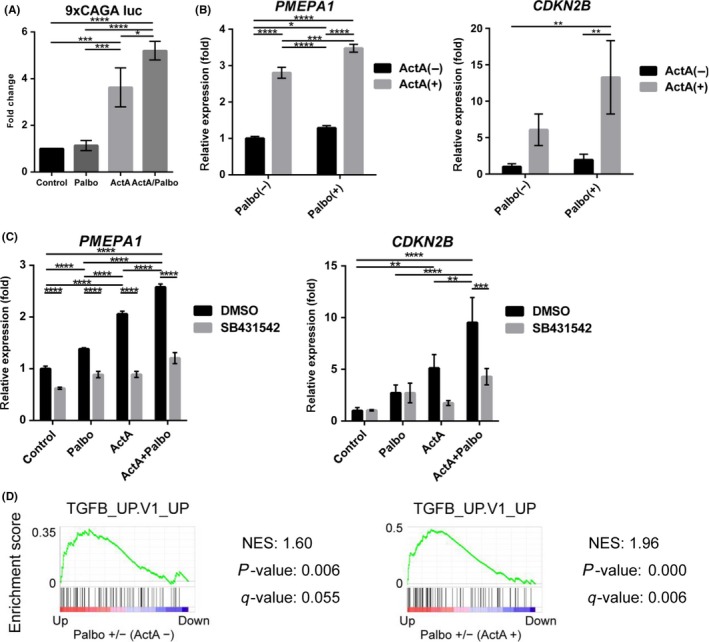
Palbociclib enhances induction of SMAD‐target genes in T47D cells. A, Activin‐SMAD signaling activity was evaluated using 9 × CAGA‐luc plasmid. Cells were treated with or without 1 μmol/L palbociclib and, 6 h later, stimulated with ActA for 24 h. Data represent mean ± SD of n = 3 independent experiments. B, qRT‐PCR analysis of T47D cells treated with or without ActA and 1 μmol/L palbociclib for 24 h. Data represent mean ± SD of n = 3 independent experiments. C, qRT‐PCR analysis of T47D cells treated with SB431542 and ActA. Cells were treated with 10 μmol/L SB431542 or DMSO for 24 h, and then treated with ActA for 24 h. Data represent mean ± SD of n = 3 independent experiments. D, Gene set enrichment analysis of SMAD2 bound genes. Genes were pre‐rank‐ordered and analyzed using the oncogenic signatures (C6, MSigDB). Gene sets with a P‐value <5% and an FDR q‐value <25% were considered significant. Enrichment score is plotted on the y‐axis. NES, normalized enrichment score. Cells were treated without (left) or with (right) 50 ng/mL ActA. *P < .05, **P < .01, ***P < .001, ****P < .0001; ANOVA with Tukey‐Kramer post hoc test in (A‐C)
3.4. Cyclin G2 plays a critical role in cell cycle regulation in luminal‐type breast cancer cells after palbociclib and activin treatment
Our data suggest that the cyclin D‐CDK4/6 axis uses the SMAD signaling pathway to regulate cell cycle progression in addition to the RB‐E2F axis. To further characterize SMAD2‐dependent cytostatic response, we analyzed RNA‐seq and ChIP‐seq data obtained in T47D cells. We first identified the genes in which the binding of SMAD2 was increased by palbociclib, and then we categorized these genes based on the effects of ActA and palbociclib treatment on their mRNA expression profile. As indicated in Figure 5A, 12 genes were isolated as palbociclib‐responsive ActA‐SMAD2 target genes. Among them, CCNG2 (encoding cyclin G2) is related to cell cycle regulation (Table Table 1 ). Both SMAD and FOXA1 binding motifs located in the SMAD2 binding region of the CCNG2 gene, and SMAD2 binding was enhanced after palbociclib treatment, as shown in ChIP‐seq data (Figure 5B); this trend was validated in ChIP‐qPCR analysis (Figure 5C). Moreover, CCNG2 mRNA was induced by ActA and further enhanced by palbociclib treatment (Figure 5D). Ectopic expression of cyclin G2 inhibited cell proliferation in T47D cells (Figure 5E). To further investigate the clinical significance of CCNG2 expression in breast cancers, we analyzed patient datasets from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC).40 Among the 12 palbociclib‐responsive SMAD2 target genes, three genes (CCNG2, CLIC3, H3F3A) could predict good prognosis of ER‐positive breast cancer patients (Figure S4). Importantly, CCNG2 was one of them, which was also consistent with a previous report41 (Figure 5F).
Figure 5.
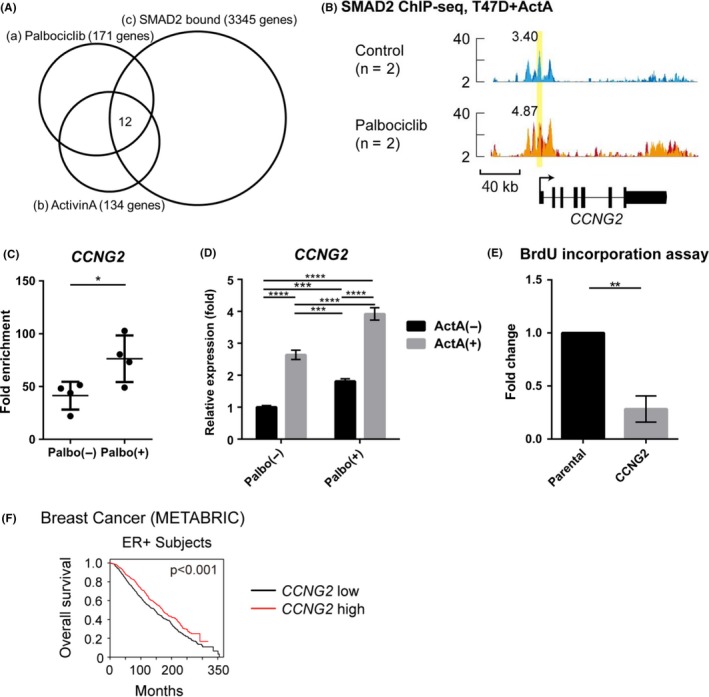
Cyclin G2 plays critical roles in cell cycle regulation in T47D cells after palbociclib and activin treatment. A, Venn diagram indicating genes that showed enhanced SMAD2 binding in T47D cells after treatment with palbociclib and induced expression by palbociclib and ActA. (a,b) Genes whose mRNA expression was induced more than 2‐fold by palbociclib (a) or by ActA (b). (c) SMAD2 target genes with enhanced SMAD2 binding by more than 2‐fold after palbociclib treatment. B, Genomic locus of CCNG2 is shown as in Figure 3B. C, SMAD2 ChIP‐qPCR analysis of T47D cells carried out as in Figure 3C. Data represent mean ± SD of n = 4 independent experiments (*P < .05; Welch's t test). D, qRT‐PCR analysis of T47D cells treated with or without ActA and 1 μmol/L palbociclib for 24 h. Data represent mean ± SD of n = 3 independent experiments (***P < .001, ****P < .0001; ANOVA with Tukey‐Kramer post hoc test). E, BrdU incorporation assay in T47D cells or T47D cells stably expressing CCNG2. Data were normalized to the control condition. Data represent mean ± SD of n = 3 independent experiments (**P < .01; Welch's t test). F, Kaplan‐Meyer analysis of overall survival of breast cancer datasets from Molecular Taxonomy of Breast Cancer International Consortium (METABRIC)40; ER+ subjects, n = 1445. Survival analysis was carried out using a log‐rank test. Data of CCNG2 are also presented together with the other genes in Figure S4
Table Table 1.
Twelve palbociclib‐responsive SMAD2 target genes with representative gene ontology terms for biological processes
| Gene name | Function | |
|---|---|---|
| (A) | CCNG2 | Regulation of cell cycle |
| (B) | CLIC3 | Chloride transport, signal transduction |
| (C) | FAM127B/RTL8A | Unknown |
| (D) | FAM25A | Unknown |
| (E) | GGT7 | Glutathione biosynthetic process, negative regulation of response to oxidative stress |
| (F) | H3F3A | DNA replication‐independent nucleosome assembly, positive regulation of cell growth, telomere organization |
| (G) | KRTCAP2 | Protein N‐linked glycosylation through arginine |
| (H) | NDUFS6 | Mitochondrial electron transport, fatty acid metabolic process |
| (I) | S100A11 | Signal transduction, negative regulation of DNA replication |
| (J) | SELM | Hormone metabolic process |
| (K) | TMSB10 | Regulation of cell migration, sequestering of actin monomers |
| (L) | TRAPPC2L | Protein complex oligomerization |
3.5. Palbociclib enhances SMAD2 binding to the genome in triple‐negative breast cancer cell lines
During malignant progression, TGF‐β/activin‐SMAD2/3 can switch its function from tumor suppressor to tumor‐promoting factor. Therefore, CDK4/6 inhibition may possibly enhance the tumor‐promoting effects of TGF‐β/activin‐SMAD2/3 in aggressive breast cancer cells. We thus decided to use TNBC cell lines, where the SMAD signaling pathway has been shown to function as a tumor promoter. Among TNBC cell lines, the RB‐proficient line Hs578T is reported to be sensitive to palbociclib (IC50 524 nmol/L).32 Hs578T cells responded to ActA and used mainly SMAD2 (Figure S5A). Notably, Hs578T cells are spindle shaped and characterized with high expression of the EMT transcription factors and reduced expression of E‐cadherin.30
We carried out dose‐response experiments with palbociclib (50‐5000 nmol/L) in Hs578T. Although phosphorylation of SMAD2 Ser255 and RB Ser780 in Hs578T was inhibited by a comparable concentration of palbociclib, the concentration was higher than that required for inhibition in T47D cells (Figures 2D,6A). We then carried out SMAD2 ChIP‐seq analysis using 1 μmol/L palbociclib and 50 ng/mL ActA in Hs578T, and compared the SMAD2 binding profile with that in T47D cells. Based on SMAD2 ChIP‐seq data, palbociclib generally enhanced SMAD2 binding to the genome in Hs578T cells (Figure 6B‐E). Palbociclib also slightly increased the responsiveness of the SMAD reporter 9 × CAGA‐luc (Figure 6F). However, mRNA expression of PMEPA1, ZEB1, or SNAI1, was not clearly affected by palbociclib treatment (Figure 6G). ActA induced CCNG2 and exerted cytostatic effects (Figure S5B,C); however, additive effects of palbociclib were not obvious. This might be caused by the fact that 1 μmol/L palbociclib was not enough to suppress phosphorylation of both RB and SMAD2 in Hs578T cells (Figure 6A). Moreover, other signaling pathways, which cause linker phosphorylation of SMAD2, such as CDK and MAPK, could be activated in TNBC. Collectively, the results of SMAD2 ChIP‐seq data of two different breast cancer cell lines showed that palbociclib enhances the SMAD signaling pathway, whereas it modulates distinct cellular programs activated by TGF‐β/activin depending on the type of breast cancer.
Figure 6.
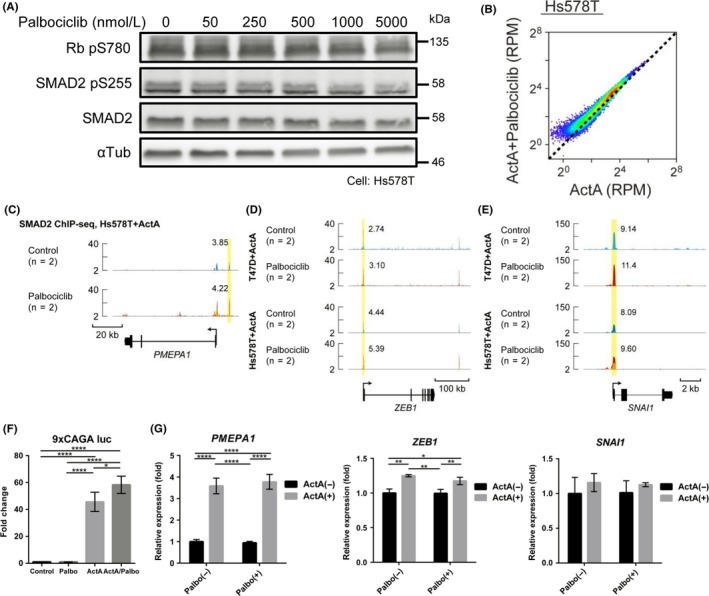
Palbociclib enhances SMAD2 binding to the genome in triple‐negative breast cancer Hs578T cells. A, Immunoblot analysis of dose‐dependent effect of palbociclib. Hs578T cells were treated with the indicated concentrations of palbociclib for 24 h. B, Scatter plot of differentially enriched sequenced reads in each SMAD2 binding region from ActA‐ and palbociclib‐treated Hs578T cells (n = 2) and ActA control (n = 2). RPM, reads per million reads. C, PMEPA1 locus is shown with the SMAD2 ChIP‐seq data of Hs578T cells treated with palbociclib or left untreated (n = 2 for each condition). Direction of transcription is shown by the arrow beginning at the transcription start site. Each ChIP‐seq data is presented in a different color. Values of RPM (averaged from two biological replicates) of peaks highlighted by yellow boxes are presented. D,E, ZEB1 and SNAI1 loci are shown as in (C), together with SMAD2 ChIP‐seq data of T47D and Hs578T cells treated with palbociclib or left untreated (n = 2 for each condition). F, Luciferase assay in Hs578T cells. Activin‐SMAD signaling activity was evaluated using pooled cells stably expressing 9 × CAGA‐Luc plasmid and CMV‐Renilla introduced by lentiviral vectors. Cells were treated with or without 1 μmol/L palbociclib and, 4‐6 h later, stimulated with ActA for 24 h. Data represent mean ± SD of n = 4 independent experiments. G, qRT‐PCR analysis of Hs578T cells treated with ActA and 1 μmol/L palbociclib for 12 h. Data represent mean ± SD of n = 3 independent experiments. *P < .05, **P < .01, ****P < .0001; ANOVA with Tukey‐Kramer post hoc test in (F‐G)
4. DISCUSSION
Cyclin‐dependent kinase 4/6 inhibitors have already proven their worth in HR‐positive, HER2‐negative advanced breast cancer. In combination with fulvestrant or aromatase inhibitors, CDK4/6 inhibitors have been approved for treatment of advanced ER‐positive, HER2‐negative luminal‐like breast cancer. Although positive ER status and RB expression are biomarkers required for CDK4/6‐mediated inhibition, other biomarkers, such as changes in the expression of genes in the cyclin D‐CDK4/6‐RB pathway, have yet to be clinically validated.3, 4 CDK4/6 inhibitors have also been reported to be active in ER‐positive and HER2‐amplified cell lines.32 Goel et al42 reported that CDK4/6 inhibitors sensitized HER2‐amplified breast cancer cells to HER2‐targeted therapies and delayed the onset of recurrence in a transgenic model of HER2‐positive breast cancer. Moreover, CDK4/6 inhibitors repress the proliferation of RB‐proficient TNBC cell lines, such as MDA‐MB‐231 and Hs578‐T.32 These data indicate that CDK4/6 inhibitors can be clinically used in the treatment of other breast cancer subtypes, at least for HER2‐positive tumors and some TNBC.
It has been shown that among the MCF‐10A series of cell lines, the TGF‐β/activin‐SMAD pathway has cytostatic effects in the premalignant and low‐grade breast carcinoma line MCF10A (M‐II and M‐III), whereas it enhances metastasis in a high‐grade breast cancer line MCF10A M‐IV.43, 44 Introduction of a SMAD3 mutant with phosphorylation‐resistant mutations in the linker was reported to inhibit primary tumor growth, but it significantly increased lung metastasis of MCF10A M‐IV.45 This was in contrast to the results reported for a SMAD3 mutant with a truncated C‐terminus (Smad3ΔC), which functions as dominant negatives.44 Smad3ΔC enhances proliferation of the low‐grade breast carcinoma line MCF10A M‐III, whereas it suppresses metastasis of the high‐grade breast cancer line MCF10A M‐IV.44 These data suggest that loss of linker phosphorylation generally strengthens SMAD‐mediated cellular responses. Consistently, our ChIP‐seq data showed that palbociclib treatment augmented SMAD2‐DNA interaction in breast cancer cell lines. In ER‐positive breast cancer T47D, it mainly enhances the cytostatic effects of the TGF‐β/activin‐SMAD signaling pathway, whereas it possibly strengthens the tumor‐promoting aspect in aggressive breast cancer.
Importantly, we found that palbociclib and the activin‐SMAD signaling pathway regulate cyclin G2 expression in T47D (Figure 5). Cyclin G2, a member of the cyclins family, and homologous to cyclin G1, negatively regulates the cell cycle and contributes to maintaining the quiescent state of differentiated cells, instead of controlling cell cycle progression.46 Xu et al47 reported that a TGF‐β family member Nodal or constitutively active ALK7 induced CCNG2 mRNA and stabilized cyclin G2 protein in ovarian cancer cells. In breast cancer cells, estradiol (E2)‐bound ER complex directly represses CCNG2.48, 49 In contrast, treatment with an ER antagonist fulvestrant, anti‐HER2 antibody trastuzumab, a PI3K inhibitor LY294002, or a mammalian target of rapamycin (mTOR) inhibitor rapamycin has been reported to increase CCNG2 mRNA.41, 50 Interestingly, knockdown of cyclin G2 was reported to dampen the cell cycle‐arrest response in fulvestrant‐treated MCF7 cells.41 In addition, cyclin G2 was identified as a target of p63, which functions as a metastatic suppressor in TNBC,51 suggesting that it serves as a central node for regulation of the cell cycle and/or metastasis of breast cancer cells.
In summary, our data indicate that CDK4/6 inhibition enhances the activin‐SMAD signaling pathway and collectively regulates cytostatic effects in T47D, without activating the tumor‐promoting functions of the TGF‐β/activin‐SMAD signaling pathway, such as the EMT program. However, this can happen in aggressive breast cancer cells where the SMAD pathway can enhance motility and invasiveness. Therefore, the indication for CDK4/6 inhibitors needs to be carefully assessed from the aspect of the TGF‐β/activin‐SMAD signaling pathway. Moreover, addition of an ALK4/5/7 kinase inhibitor to the CDK4/6 inhibitor might be beneficial in patients with high‐grade breast cancer, which may have the aggressive aspects of the SMAD signaling pathway.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We thank Dr H. Miyoshi (Keio University, Japan) for the lentivirus vector system; Dr A. Miyawaki (RIKEN, Japan) for the Fucci system; members in the Department of Molecular Pathology for technical assistance and discussion. This work was supported by KAKENHI Grants‐in‐Aid for Scientific Research (S) (15H05774) (K.M.) from the Japan Society for the Promotion of Science (JSPS). Y.T. is supported by Research Fellowship for Young Scientists (DC) and the Graduate Program for Leaders in Life Innovation from JSPS.
Harada M, Morikawa M, Ozawa T, et al. Palbociclib enhances activin‐SMAD‐induced cytostasis in estrogen receptor‐positive breast cancer. Cancer Sci. 2019;110:209‐220. 10.1111/cas.13841
Contributor Information
Kohei Miyazono, Email: miyazono@m.u-tokyo.ac.jp.
Daizo Koinuma, Email: d-koinuma@umin.ac.jp.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34:9‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417‐430. [DOI] [PubMed] [Google Scholar]
- 4. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turner NC, Ro J, Andre F, et al. Palbociclib in hormone‐receptor‐positive advanced breast cancer. N Engl J Med. 2015;373:209‐219. [DOI] [PubMed] [Google Scholar]
- 6. Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925‐1936. [DOI] [PubMed] [Google Scholar]
- 7. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first‐line therapy for HR‐positive, advanced breast cancer. N Engl J Med. 2016;375:1738‐1748. [DOI] [PubMed] [Google Scholar]
- 8. Goetz MP, Toi M, Campone M, et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35:3638‐3646. [DOI] [PubMed] [Google Scholar]
- 9. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 10. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747‐752. [DOI] [PubMed] [Google Scholar]
- 11. Goldhirsch A, Wood WC, Coates AS, et al. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011;22:1736‐1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Network . Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018‐4032. [DOI] [PubMed] [Google Scholar]
- 14. Knudsen ES, Hutcheson J, Vail P, Witkiewicz AK. Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget. 2017;8:43678‐43691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsuzaki K, Kitano C, Murata M, et al. Smad2 and Smad3 phosphorylated at both linker and COOH‐terminal regions transmit malignant TGF‐β signal in later stages of human colorectal cancer. Cancer Res. 2009;69:5321‐5330. [DOI] [PubMed] [Google Scholar]
- 17. Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin‐dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226‐231. [DOI] [PubMed] [Google Scholar]
- 18. Moses H, Barcellos‐Hoff MH. TGF‐β biology in mammary development and breast cancer. Cold Spring Harb Perspect Biol. 2011;3:a003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65:7968‐7975. [DOI] [PubMed] [Google Scholar]
- 20. Xu P, Lin X, Feng XH. Posttranslational regulation of Smads. Cold Spring Harb Perspect Biol. 2016;8:a022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morikawa M, Derynck R, Miyazono K. TGF‐β and the TGF‐β family: context‐dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miyazono K, Katsuno Y, Koinuma D, Ehata S, Morikawa M. Intracellular and extracellular TGF‐β signaling in cancer: some recent topics. Front Med. 2018;12:387‐411. [DOI] [PubMed] [Google Scholar]
- 24. Bashir M, Damineni S, Mukherjee G, Kondaiah P. Activin‐A signaling promotes epithelial‐mesenchymal transition, invasion, and metastatic growth of breast cancer. NPJ Breast Cancer. 2015;1:15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalkhoven E, Roelen BA, de Winter JP, et al. Resistance to transforming growth factor β and activin due to reduced receptor expression in human breast tumor cell lines. Cell Growth Differ. 1995;6:1151‐1161. [PubMed] [Google Scholar]
- 26. Koinuma D, Tsutsumi S, Kamimura N, et al. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol Cell Biol. 2009;29:172‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldman LA, Cutrone EC, Kotenko SV, Krause CD, Langer JA. Modifications of vectors pEF‐BOS, pcDNA1 and pcDNA3 result in improved convenience and expression. Biotechniques. 1996;21:1013‐1015. [DOI] [PubMed] [Google Scholar]
- 28. Sakaue‐Sawano A, Kurokawa H, Morimura T, et al. Visualizing spatiotemporal dynamics of multicellular cell‐cycle progression. Cell. 2008;132:487‐498. [DOI] [PubMed] [Google Scholar]
- 29. Kawasaki N, Miwa T, Hokari S, et al. Long noncoding RNA NORAD regulates transforming growth factor‐β signaling and epithelial‐to‐mesenchymal transition‐like phenotype. Cancer Sci. 2018;109:2211‐2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katsura A, Tamura Y, Hokari S, et al. ZEB1‐regulated inflammatory phenotype in breast cancer cells. Mol Oncol. 2017;11:1241‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morikawa M, Koinuma D, Mizutani A, et al. BMP sustains embryonic stem cell self‐renewal through distinct functions of different Krüppel‐like factors. Stem Cell Reports. 2016;6:64‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor‐positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raspé E, Coulonval K, Pita JM, et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med. 2017;9:1052‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lara MF, García‐Escudero R, Ruiz S, et al. Gene profiling approaches help to define the specific functions of retinoblastoma family in epidermis. Mol Carcinog. 2008;47:209‐221. [DOI] [PubMed] [Google Scholar]
- 35. Padua D, Zhang XH, Wang Q, et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin‐like 4. Cell. 2008;133:66‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Funaba M, Zimmerman CM, Mathews LS. Modulation of Smad2‐mediated signaling by extracellular signal‐regulated kinase. J Biol Chem. 2002;277:41361‐41368. [DOI] [PubMed] [Google Scholar]
- 37. Baughn LB, Di Liberto M, Niesvizky R, et al. CDK2 phosphorylation of Smad2 disrupts TGF‐β transcriptional regulation in resistant primary bone marrow myeloma cells. J Immunol. 2009;182:1810‐1817. [DOI] [PubMed] [Google Scholar]
- 38. Badve S, Turbin D, Thorat MA, et al. FOXA1 expression in breast cancer–correlation with luminal subtype A and survival. Clin Cancer Res. 2007;13:4415‐4421. [DOI] [PubMed] [Google Scholar]
- 39. Jozwik KM, Carroll JS. Pioneer factors in hormone‐dependent cancers. Nat Rev Cancer. 2012;12:381‐385. [DOI] [PubMed] [Google Scholar]
- 40. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zimmermann M, Arachchige‐Don AP, Donaldson MS, Patriarchi T, Horne MC. Cyclin G2 promotes cell cycle arrest in breast cancer cells responding to fulvestrant and metformin and correlates with patient survival. Cell Cycle. 2016;15:3278‐3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goel S, Wang Q, Watt AC, et al. Overcoming therapeutic resistance in HER2‐positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang B, Vu M, Booker T, et al. TGF‐β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tian F, DaCosta Byfield S, Parks WT, et al. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2003;63:8284‐8292. [PubMed] [Google Scholar]
- 45. Bae E, Sato M, Kim RJ, et al. Definition of Smad3 phosphorylation events that affect malignant and metastatic behaviors in breast cancer cells. Cancer Res. 2014;74:6139‐6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horne MC, Goolsby GL, Donaldson KL, Tran D, Neubauer M, Wahl AF. Cyclin G1 and cyclin G2 comprise a new family of cyclins with contrasting tissue‐specific and cell cycle‐regulated expression. J Biol Chem. 1996;271:6050‐6061. [DOI] [PubMed] [Google Scholar]
- 47. Xu G, Bernaudo S, Fu G, Lee DY, Yang BB, Peng C. Cyclin G2 is degraded through the ubiquitin‐proteasome pathway and mediates the antiproliferative effect of activin receptor‐like kinase 7. Mol Biol Cell. 2008;19:4968‐4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stossi F, Likhite VS, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen‐occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J Biol Chem. 2006;281:16272‐16278. [DOI] [PubMed] [Google Scholar]
- 49. Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up‐ and down‐regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562‐4574. [DOI] [PubMed] [Google Scholar]
- 50. Le XF, Arachchige‐Don AS, Mao W, Horne MC, Bast RC Jr. Roles of human epidermal growth factor receptor 2, c‐jun NH2‐terminal kinase, phosphoinositide 3‐kinase, and p70 S6 kinase pathways in regulation of cyclin G2 expression in human breast cancer cells. Mol Cancer Ther. 2007;6:2843‐2857. [DOI] [PubMed] [Google Scholar]
- 51. Adorno M, Cordenonsi M, Montagner M, et al. A Mutant‐p53/Smad complex opposes p63 to empower TGFβ‐induced metastasis. Cell. 2009;137:87‐98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials