

Abstract
Gliomas are the most common central nervous system tumors. They show malignant characteristics indicating rapid proliferation and a high invasive capacity and are associated with a poor prognosis. In our previous study, p68 was overexpressed in glioma cells and correlated with both the degree of glioma differentiation and poor overall survival. Downregulating p68 significantly suppressed proliferation in glioma cells. Moreover, we found that the p68 gene promoted glioma cell growth by activating the nuclear factor‐κB signaling pathway by a downstream molecular mechanism that remains incompletely understood. In this study, we found that dual specificity phosphatase 5 (DUSP5) is a downstream target of p68, using microarray analysis, and that p68 negatively regulates DUSP5. Upregulating DUSP5 in stably expressing cell lines (U87 and LN‐229) suppressed proliferation, invasion, and migration in glioma cells in vitro, consistent with the downregulation of p68. Furthermore, upregulating DUSP5 inhibited ERK phosphorylation, whereas downregulating DUSP5 rescued the level of ERK phosphorylation, indicating that DUSP5 might negatively regulate ERK signaling. Additionally, we show that DUSP5 levels were lower in high‐grade glioma than in low‐grade glioma. These results suggest that the p68‐induced negative regulation of DUSP5 promoted invasion by glioma cells and mediated the activation of the ERK signaling pathway.
Keywords: DUSP5, glioma, invasion, migration, p68/DDX5
1. INTRODUCTION
Gliomas are the most common primary tumors in the central nervous system (CNS). Glioblastoma (GBM), the most malignant type of glioma, is characterized by necrosis, endothelial proliferation, and aggressive invasiveness. The major cause of the high lethality observed in malignant glioma is diffuse invasion into healthy brain parenchyma, which precludes complete surgical resection.1 These invading cells are extremely resistant to radiation and chemotherapy and are not currently treatable by any available anti‐invasive therapies. Although many key signaling pathways have been found to facilitate glial tumorigenesis, the mechanisms by which glioma cells infiltrate surrounding normal brain tissue remain unclear.2, 3 Therefore, increasing our understanding of the molecular mechanisms underlying the invasive behavior of glioma is essential for developing novel therapeutic approaches to treat aggressive glioma invasiveness.
P68 (also known as DDX5) is considered a member of the prototypic DEAD‐box family of RNA helicases.4, 5 Because it is an ATP‐dependent RNA helicase, p68 can manipulate RNA structures, including pre‐mRNA, rRNA, and microRNA.6, 7, 8, 9, 10, 11 P68 could also act as a transcriptional coactivator of several cancer‐associated transcription factors, such as p53 tumor suppressor,11 β‐catenin,12 and the androgen receptor,13 which plays an essential role in cell proliferation, oncogenesis, and early stage organ development.14 Indeed, p68 is aberrantly expressed or modified in different types of tumors, including breast, prostate, and colon cancer.13, 15, 16 Our previous results showed that p68 protein levels were significantly higher in high‐grade glioma and that a high expression level of p68 was associated with poor survival in glioma patients. Moreover, we found that p68 induced glioma tumor growth by binding to nuclear factor‐κB (NF‐κB) p50, indicating that p68 plays a significant role in gliomagenesis.17 However, the downstream molecular mechanism by which p68 regulates the invasive behavior of glioma remains unclear.
In mammalian cells, dual specificity phosphatases (DUSPs, also known as mitogen‐activated protein kinase phosphatases) include 16 catalytically active enzymatic family members. Dual specificity phosphatase 5 (DUSP5) is a growth factor‐inducible phosphatase that possesses a functional nuclear localization signal that targets and anchors ERK1 and ERK2 to the nucleus.18, 19 It plays an essential role in cellular proliferation and differentiation by negatively regulating members of the MAPK superfamily (MAPK/ERK, SAPK/JNK, and p38).20 Because DUSP5 performs such comprehensive functions, studies aimed at exploring the role of DUSP5 in glioma are of great interest.
Here, we use a microarray gene expression analysis to reveal that the DUSP5 gene acts as a downstream target of p68. Both p68 knockdown and DUSP5 upregulation suppressed proliferation, invasion, and migration in glioma cells in vitro. Moreover, we found that upregulating DUSP5 impaired p68‐induced glioma proliferation, invasion, and migration. In addition, we found that DUSP5 could negatively regulate ERK phosphorylation, indicating the potential existence of a p68/DUSP5/ERK signaling‐mediated mechanism in glioma. These results suggest that p68 induces invasiveness in glioma cells by negatively regulating DUSP5 and that DUSP5 acts as a negative regulator of glioma cell motility and the ERK pathway.
2. MATERIALS AND METHODS
2.1. Cell line, cell culture, and cell transfection
Human glioma cells (lines U251, A172, Hs683, LN‐229, and U87; ATCC, Manassas, VA, USA) were cultured at 37°C in 5% CO2. The OL glioma cell line was generously provided by Professor Kazuyoshi Ikuta (Microbiology Research Institute, Osaka University, Osaka, Japan). U87 and LN‐229 cells were transiently transfected with DUSP5 plasmids (Gene Pharma, Hai Shang, China) using Effectene (Qiagen, Valencia, CA, USA) or with p68 siRNA (no. sc‐37141; Santa Cruz Biotechnology, Dallas, TX, USA) and/or DUSP5 siRNA (no. sc‐60554; Santa Cruz Biotechnology) using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) as recommended by the manufacturer's protocol.
2.2. Reagents and chemicals
Rabbit anti‐p68 antibodies (Santa Cruz Biotechnology), rabbit anti‐DUSP5 antibodies (Abcam, Cambridge, MA, USA), and rabbit anti‐GAPDH antibodies (Abcam) were purchased for western blot, immunofluorescent, or immunohistochemical assays. Alexa Fluor‐488 goat anti‐rabbit IgG and DAPI and MTT assay reagents were purchased from Invitrogen and DingGuo Biotech (Beijing, China), respectively.
2.3. Clinical samples and histology
Fresh centers of human diffuse astrocytoma and GBM samples were obtained from patients undergoing no‐chemotherapy or radiation therapy and classified and characterized according to the 2016 WHO CNS tumor classification. Tissue samples were obtained and immediately frozen in liquid nitrogen. All patients provided informed consent for tissue samples to be used for scientific purposes, and this study was approved by the ethics committee of Harbin Medical University (Harbin, China).
2.4. Quantitative real‐time PCR
Total tissue RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Approximately 1 μg RNA was used to synthesize cDNA. The gene expression levels of p68 and DUSP5 were determined by quantitative real‐time PCR (qRT‐PCR) and analyzed using LightCycler analysis software (Roche, Basel, Switzerland), and GAPDH was used as the endogenous control.21 Quantitative RT‐PCR was carried out with the following primers: p68 sense (GenBank accession no. NM_001320595.1), 5′‐TTTATGAAGCCAATTTCCCTGC‐3′; and antisense, 5′‐CCACTCCAACCATATCCAATCC‐3′; and DUSP5 (NM_004419.3) sense, 5′‐CAATGAGGTAGTTGGTTGAAGTAG‐3′; and antisense, 5′‐CTGAGAAGAGGTGGAATGA‐GA‐3′.
2.5. Gene expression profiling
The mRNA expression levels of genes of interest were analyzed in U87 glioma cells after p68 knockdown in a Human Twin Chip Human 44 K (Genocheck, Ansan, Korea) microarray analysis. Total cell RNA was isolated using TRIzol reagent after cells were transfected with p68 siRNA or an siRNA negative control (si‐NC). Gene expression was normalized and differential expression analyzed using GeneSpring GX 7.3 (Agilent Technology, Folsom, CA, USA). All microarray data have been submitted to the Gene Expression Omnibus database (GEO accession no. GSE103981).
2.6. Western blot analysis
Total U87 and LN‐229 cell protein was extracted and lysed in RIPA buffer (Thermo, Shanghai, China) PMSF (Beyotime, Beijing, China). Lysates (15 μg) were then separated on 12.5% SDS‐PAGE gels, transferred to PVDF membranes (Millipore, Danvers, MA, USA), blocked in 5% skim milk containing 0.05% Tween 20‐TBS for 1 hour, and then incubated with primary Abs at 4°C overnight. After the membranes were incubated with anti‐rabbit IgG‐HRP secondary Abs (1:5000; Santa Cruz Biotechnology), the resulting immunoreactive complexes were visualized using SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA, USA).
2.7. Immunofluorescence
Transfected cells were fixed in 4% paraformaldehyde, permeabilized, blocked with 2% BSA and 3% goat serum for 30 minutes, incubated with primary Abs diluted in blocking buffer and secondary Abs for 1 hour, and then incubated with goat anti‐rabbit Abs (1:100; Alexa Fluor) or goat anti‐mouse Abs (1:100; Invitrogen) for 1 hour. Finally, the sections were washed with TBS with Tween‐20 and mounted with mounting medium containing DAPI in preparation for fluorescence. Images were captured using Biorevo BZ‐9000 fluorescence microscopy (Keyence, Osaka, Japan) and inverted LSM510 confocal laser microscopy (Carl Zeiss, Oberkochen, Germany).
2.8. Immunohistochemical staining
Human brain tumor tissue sections (5‐μm thick) were dewaxed, quenched with 3% H2O2 in methanol for 30 minutes, incubated with pre‐immune IgG or Abs in PBS containing 1% BSA at 48°C overnight, and then incubated with HRP‐conjugated Abs and processed with an ImmunoPure Metal Enhanced Diaminobenzidine Substrate Kit (Pierce Biotechnology, Waltham, MA, USA).
2.9. Cell proliferation assay
In the MTT assays, transfected cells were seeded in 96‐well plates at a density of 2000 cells/well, cultured at 37°C in 5% CO2 for 48 hours, treated with 20 μL MTT at 37°C for 4 hours, stopped with 100 mL DMSO, and analyzed at 570 nm by a multiwell plate reader.
2.10. Wound‐healing assay
Transfected cells were seeded in 6‐well plates (1 × 105 cells/well), washed with serum‐free medium, and then incubated with serum‐free DMEM at 37°C for 8 hours. Each sample was captured, and relative cell mobility was calculated by the following formula: relative mobility = (1 − distance between the edges of invaded scratches/distance between the edges of initial scratches) × 100%.
2.11. Transwell assay
Transfected cells were suspended in serum‐free medium at a density of 1 × 105 cells/mL. Subsequently, 200 μL of a cell suspension and 800 μL DMEM media supplemented with 30% FBS were added to the upper chambers of Transwell chambers (Corning Life Sciences, New York, NY, USA) that were pretreated with Matrigel (BD Biosciences, San Jose, CA, USA) and incubated at 37°C for 8 hours. Then the cells on the lower surface of the membrane were fixed in 4% paraformaldehyde (Sinopharm, Shanghai, China) for 20 minutes, stained with 0.5% crystal violet (Amresco, Solon, OH, USA), and subsequently captured and counted under a light microscope (AE31; Motic, Xiamen, China) at 200× magnification.
2.12. Statistical analysis
Statistical comparisons of the results obtained between 2 different groups were analyzed by Student's t test using GraphPad Prism software (GraphPad Software, San Diego, CA, USA). The data are presented as the mean ± SD, and P < .05 was considered statistically significant.
3. RESULTS
3.1. Expression of p68 in glioma cell lines and tissues
The mRNA expression level of p68 was significantly different among these 6 glioma cell lines. P68 was expressed at higher levels in LN‐229 and U87 cells than in U251, A172, Hs683, and OL cells (Figure 1A). An approximately 68‐kDa single band representing p68 was clearly detected in all cell lines but was most strongly detected in LN‐229 and U87 glioma cells (Figure 1B), consistent with the qRT‐PCR analysis. To assess the efficiency of the p68 siRNA for knocking down p68 in glioma cell lines, we selected the LN‐229 and U87 cell lines because they expressed the highest levels of p68.
Figure 1.
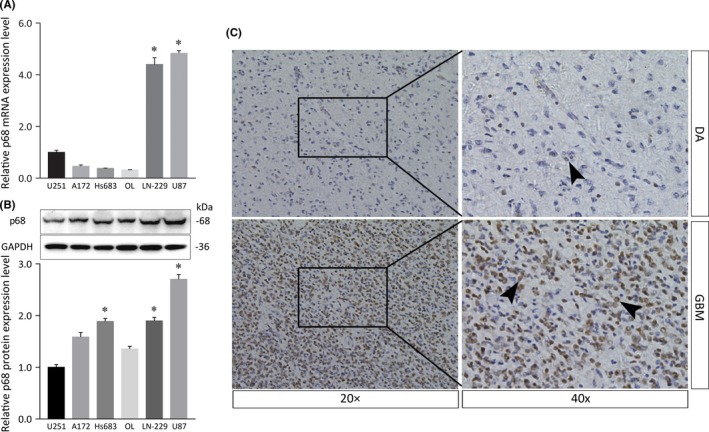
Expression of p68 in various glioma cells and immunolocalization of p68 in diffuse astrocytoma and glioblastoma. A, Relative mRNA expression levels of the p68 gene (p68 mRNA:GAPDH mRNA ratios) in U251, A172, Hs683, OL, LN‐229, and U87 glioma cell lines were calculated by quantitative real‐time PCR. B, Western blot analysis showing the p68 protein in glioma cell lines. Histogram of relative p68 protein expression with relative ratios shown as percentages of the p68/GAPDH band. Bars represent SD; *P < .05. C, Immunolocalization of p68 in diffuse astrocytoma (DA; upper panels) and glioblastoma (GBM; lower panels) tissues. Note that p68 immunolocalizes to neoplastic astrocytes (right panels, arrowheads). Strong staining was detected in GBM tissues (lower panels), whereas faint staining was observed in DA tissues (upper panels)
In immunohistochemistry specimens obtained in diffuse astrocytoma and GBM, p68 was predominantly immunolocalized in cell nuclei in malignant GBM, whereas only faint staining was detected in neoplastic astrocytes in diffuse astrocytoma (Figure 1C). The density of p68‐positive cells was significantly higher in GBM than in diffuse astrocytoma when random microscopic fields were analyzed at the same magnification (Figure 1C, left panels).
3.2. DUSP5 acts downstream of p68
To gain a global view of the signaling pathways engaged by the p68 gene in glioma cell lines, we undertook a microarray analysis to verify the gene profiling results obtained after p68 silencing (Figure 2). Whole‐genome expression profiling of U87 cells transfected with p68 siRNA revealed that DUSP5 was expressed at 2‐fold or higher levels in U87 cells transfected with p68 siRNA than in those transfected with an si‐NC, suggesting that DUSP5 acts downstream of p68 (Figure 2D).
Figure 2.
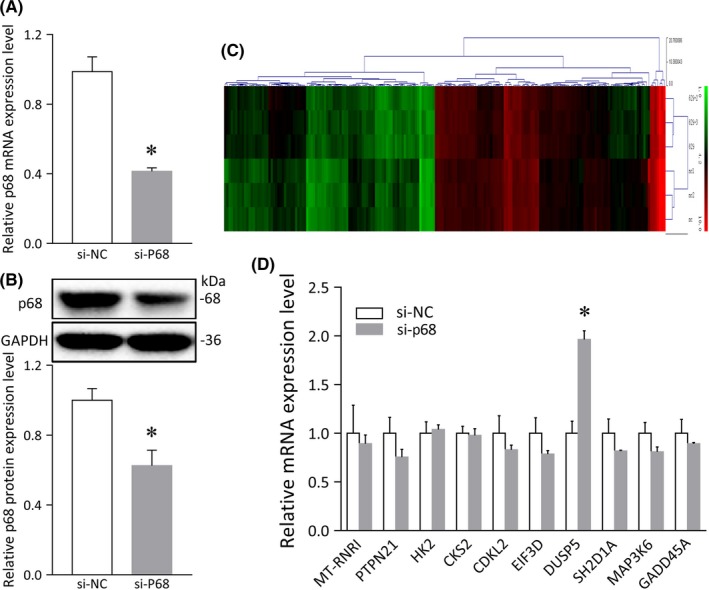
Differentially expressed genes in U87 cells transfected with p68 siRNA. A, p68 mRNA levels detected by quantitative real‐time PCR after p68 knockdown. B, Histogram of relative p68 protein expression levels after p68 downregulation with relative ratios shown as percentages of the p68/GAPDH band. C, Hierarchical clustering heat map of genes in a microarray analysis (red, upregulated genes vs green, downregulated genes). D, mRNA profiling and statistical analysis of U87 cells transfected with p68 siRNA and the siRNA negative control (si‐NC). Error bars represent SD, *P < .05
To further verify this result, we downregulated the p68 gene in both U87 and LN‐229 cells and found that the DUSP5 mRNA was expressed at high levels in both U87 and LN‐229 cells following treatment with p68 siRNA (Figure 3A) and that DUSP5 protein levels were higher in cells transfected with the p68 siRNA than in cells transfected with the siRNA control and si‐NC groups (Figure 3B). As expected, immunofluorescence analysis showed that immunofluorescent staining for DUSP5 was increased after p68 was silenced and that p68 colocalized with DUSP5 at the subcellular level (Figure 3C). To evaluate the expression of the DUSP5 protein in human glioma tissues, we used immunohistochemical analysis with a DUSP5 Ab to detect the protein in tissue samples obtained from patients with diffuse astrocytoma and GBM. The results showed that DUSP5 immunolocalized in more neoplastic astrocyte nuclei in diffuse astrocytoma than in GBM (Figure 3D). These results indicated that DUSP5, as a downstream target of p68, potentially negatively regulates the malignancy grade in gliomas.
Figure 3.
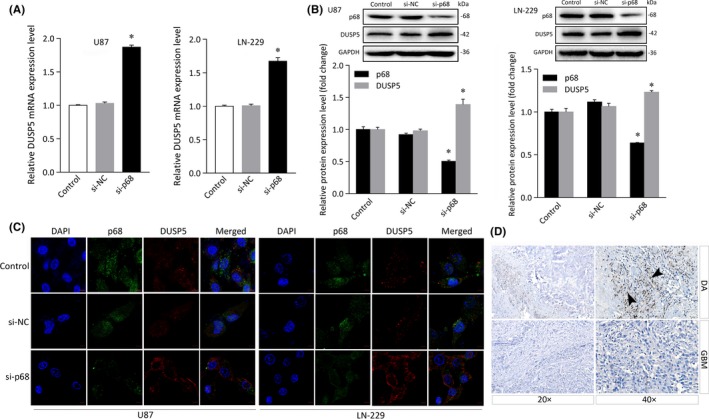
Expression of dual specificity phosphatase 5 (DUSP5) in glioma cells after p68 knockdown and immunolocalization of DUSP in diffuse astrocytoma and glioblastoma. A, DUSP5 mRNA expression after p68 knockdown. B, Histogram of relative p68 or DUSP5 protein expression levels with relative ratios shown as percentages of the p68/GAPDH or DUSP5/GAPDH band. C, Immunofluorescence assay of the cellular localization of p68 and DUSP5. Scale bar = 50 μm. D, Immunohistochemical staining for DUSP5 shows its protein levels in diffuse astrocytoma (DA, upper panels) and glioblastoma (GBM, lower panels) tissues
3.3. Knockdown of p68 suppresses proliferation, invasion, and migration in glioma cells
Next, to investigate whether p68 affects the biological behaviors of U87 and LN‐229 cells, we undertook a p68 knockdown function assay. First, proliferation assays were carried out to assess the effects of p68 knockdown on glioma proliferation. Knockdown of p68 effectively suppressed proliferation in U87 cells (mean ± SD, 0.55 ± 0.03; P < .05) compared to the results obtained in the control and si‐NC groups (0.92 ± 0.001; 0.93 ± 0.01). Similar results were obtained in LN‐229 cells; proliferation was significantly lower in cells treated with the p68 siRNA (0.45 ± 0.02; P < .05) than in the control‐ and si‐NC‐treated groups (0.96 ± 0.01 and 0.95 ± 0.01, respectively; Figure 4A).
Figure 4.
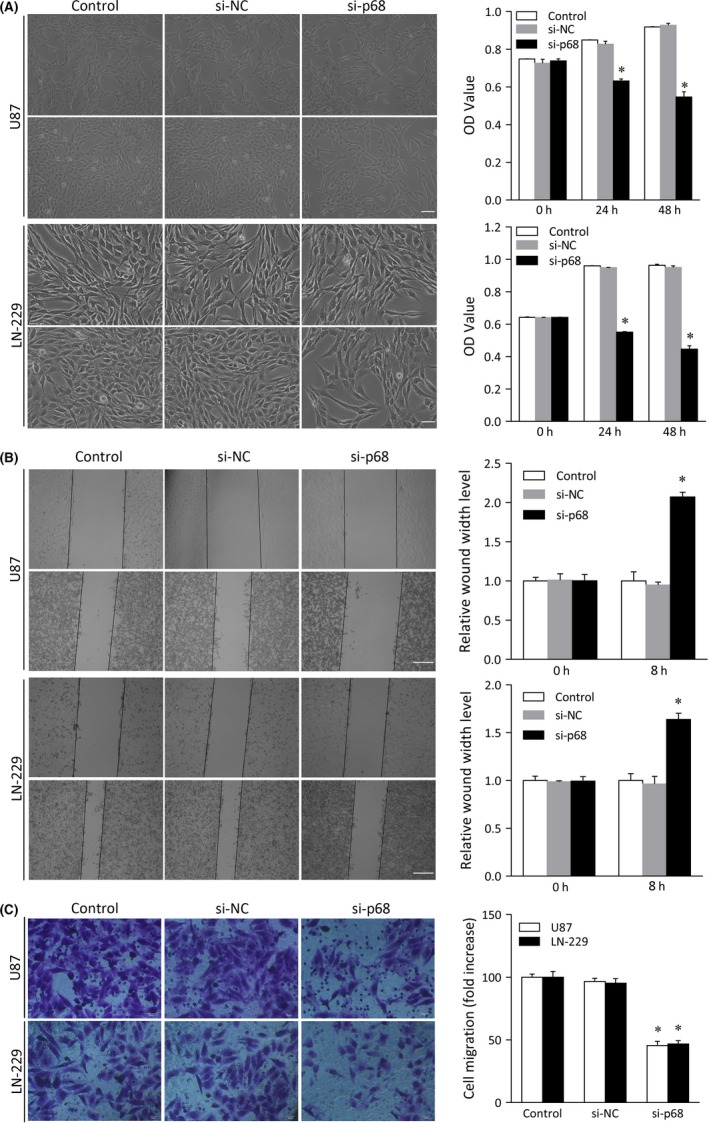
Analysis of cell proliferation, invasion, and migration in glioma cell lines transfected with p68 siRNA. U87 and LN‐229 cells were transfected with control, siRNA negative control (si‐NC), or p68 siRNA. A, MTT assay of U87 and LN‐229 cell proliferation. Scale bar = 50 μm. OD, optical density. B, Wound healing assay of the invasive capacity of U87 and LN‐229 cells and quantitative histogram analysis. Left panels, micrographs of invaded cells; right panels, relative quantitative analysis of the scratch width of invaded cells. Scale bar = 50 μm. C, Transwell assay of U87 and LN‐229 cell migration and quantitative analysis. Bars represent SD; *P < .05
The results of invasion and migration assays also indicated that p68 knockdown significantly suppressed the invasiveness and migratory capacity of U87 cells (2.07 ± 0.06 and 45.50 ± 3.37, respectively; P < .01) compared to results obtained in control‐transfected cells (1.00 ± 0.03 and 1.00 ± 0.11 and 100.00 ± 2.57 and 95.32 ± 3.65, respectively). The invasiveness and migratory capacity of LN‐229 cells were also suppressed by p68 siRNA (1.64 ± 0.06 and 46.78 ± 2.68, respectively; P < .05) compared to the results obtained in the control and si‐NC groups (1.00 ± 0.02 and 1.01 ± 0.08, respectively), whereas there was no detectable effect in the control and negative transfection groups (100.00 ± 4.64 and 95.32 ± 3.65, respectively; Figure 4B,C). These results indicate that p68 knockdown suppressed proliferation, invasion and migration in glioma cells in vitro.
3.4. DUSP5 suppresses proliferation, invasiveness, and migration in glioma cells
We next investigated whether DUSP5 overexpression regulates cell proliferation, migration, and invasion in U87 and LN‐229 cells transfected with DUSP5 plasmids, both of which showed relatively lower expression levels of DUSP5 (Figure 3B). These high mRNA and protein levels of DUSP5 were validated by qRT‐PCR and immunoblotting, whereas transfection with a control plasmid vector or an empty plasmid vector had no effect on DUSP mRNA and protein levels (Figure 5A,B). To further evaluate the subcellular localization of DUSP5, we undertook an immunofluorescence analysis of U87 and LN‐229 cells transfected with DUSP5. Immunofluorescence staining results showed that, in these cells, DUSP5 was expressed at higher levels and mainly localized in the nuclear membrane and cytoplasm compared to the results observed in the control plasmid vector‐ or empty plasmid vector‐treated groups (Figure 5C). The cytological localization of DUSP5 was consistent with the membrane‐anchored behavior associated with a nuclear protein.
Figure 5.
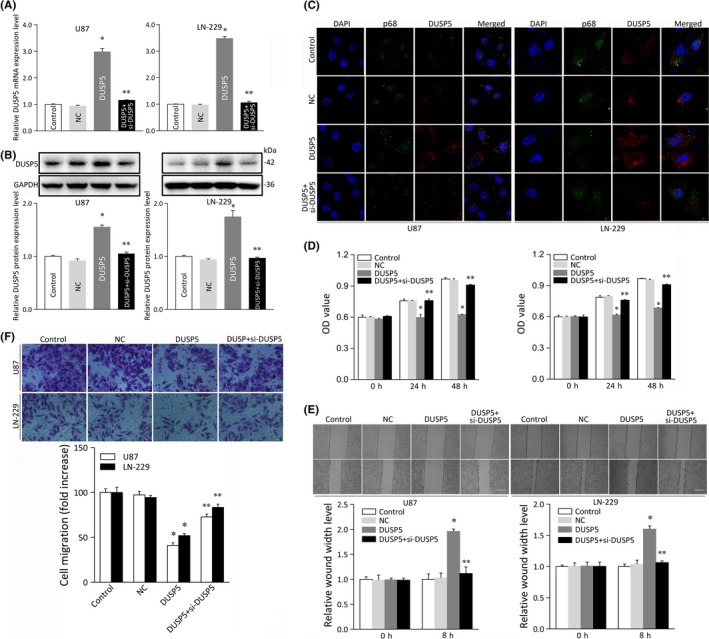
Analysis of cell proliferation, invasion, and migration in glioma cell lines overexpressing dual specificity phosphatase 5 (DUSP5). A, Relative mRNA expression of DUSP5 in U87 and LN‐229 glioma cell lines transfected with control plasmid vector (Control), empty plasmid vector (NC), DUSP5 plasmid vector (DUSP5), or DUSP5 siRNA (si‐DUSP5). B, Protein expression of DUSP5 in U87 and LN‐229 cells transfected with DUSP5 or cotransfected with DUSP5 and si‐DUSP5. Histogram showing the relative DUSP5 protein expression level with relative ratios shown as percentages of the DUSP5/GAPDH band. C, Immunofluorescence staining showing the cellular localization and transfection rates of DUSP5 or DUSP5 and si‐DUSP5. Scale bar = 50 μm. D, MTT assay of cell growth in U87 and LN‐229 cells transfected with DUSP5 or cotransfected with DUSP5 and si‐DUSP5. OD, optical density. E, Wound healing assay of cellular invasive capacity in U87 and LN‐229 cells overexpressing DUSP5 or in which DUSP5 overexpression was abolished. Upper panels, micrographs of invaded cells; lower panels, relative quantitative analysis of scratch width of invaded cells shown in upper panels. Scale bar, 50 μm. F, Transwell assay of U87 and LN‐229 cells transfected with DUSP5 or/and si‐DUSP5. Upper panels, micrographs of migrated cells; lower panels, relative quantitative analysis of migrated cells. Bars represent SD; *P < .05, **P < .05 vs adjacent control
In cell proliferation assays, proliferation was significantly lower in DUSP5‐overexpressing U87 and LN‐229 cells (mean ± SD, 0.63 ± 0.003 and 0.64 ± 0.02, respectively; P < .05) than in the control groups (0.97 ± 0.01 and 0.96 ± 0.01, respectively; and 0.97 ± 0.005 and 0.95 ± 0.004; respectively, Figure 5D). The effects of DUSP upregulation on cell invasion were next tested, and the results showed that the rate of cell invasion in monolayer cell surfaces was also significantly lower in U87 and LN‐229 cells transfected with a DUSP plasmid (1.96 ± 0.05 and 1.60 ± 0.05, respectively; P < .05) than in control‐transfected cells (1.00 ± 0.03 and 1.03 ± 0.04, respectively and; 1.00 ± 0.02 and 1.03 ± 0.06, respectively; Figure 5E). Similarly, migration assays indicated that the rate of migration was significantly lower in U87 and LN‐229 cells in which DUSP was upregulated (41.06 ± 3.04 and 52.00 ± 2.36, respectively; P < .05) than in those transfected with the control vector (100.00 ± 4.13 and 97.35 ± 3.97, respectively; and 100.00 ± 5.94 and 94.49 ± 2.36, respectively; Figure 5F). A normal DUSP5‐activated level of glioma cell motility was rescued in glioma cells cotransfected with both a DUSP5 vector and DUSP5 siRNA but not in those treated with the DUSP5 vector alone. These data confirmed that DUSP5 suppresses proliferation, invasion, and migration in U87 and LN‐229 cells and suggest that DUSP5 affects glioma chemotactic behavior in vitro.
3.5. DUSP5 impairs p68‐induced proliferation, invasion, and migration and negatively regulates ERK signaling
To gain further insight into the potential role of the DUSP5 gene in the malignant behavior of human gliomas, we undertook proliferation, invasion, and migration assays in cells transfected with p68 siRNA and/or DUSP5 siRNA. The mRNA and protein levels of DUSP5 were assessed by qRT‐PCR and immunostaining in cells cotransfected with p68 siRNA and DUSP5 siRNA (Figure 6A,B). A chemotactic functional analysis revealed that, in U87 and LN‐229 cells, DUSP5 knockdown resulted in a higher rate of cell motility than was observed in the control‐transfected cells (Figure 5). Cotransfection with p68 siRNA and DUSP5 siRNA (mean ± SD, 0.83 ± 0.008 and 0.88 ± 0.005, respectively; P < .05; Figure 6C) rescued the proliferation‐inhibiting effect of p68 siRNA in U87 and LN‐229 cells, suggesting that DUSP5 acts in part by impairing p68‐induced proliferation in glioma cells.
Figure 6.
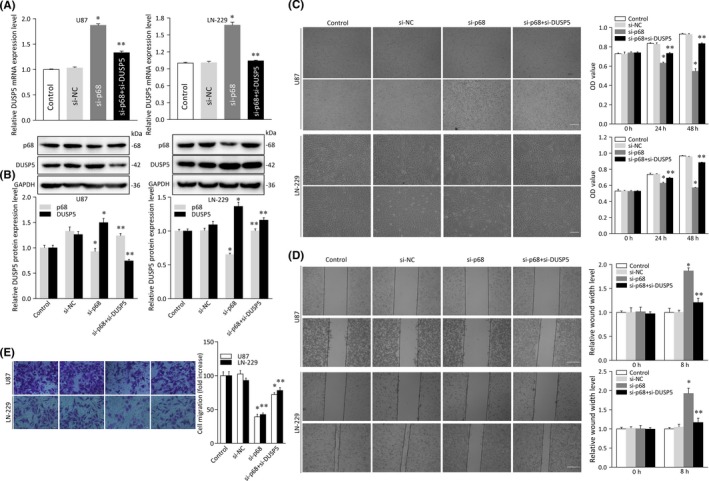
Analysis of cell proliferation, invasion, and migration in glioma cell lines cotransfected with p68 and dual specificity phosphatase 5 (DUSP5) siRNA. A, Relative mRNA expression of DUSP5 in U87 and LN‐229 glioma cell lines transfected with siRNA control (control), siRNA negative control (si‐NC), or p68 siRNA (si‐P68) and/or DUSP5 siRNA (si‐DUSP5). B, Protein expression of DUSP5 in U87 and LN‐229 cells cotransfected with si‐P68 and si‐DUSP5. Histogram of relative p68 or DUSP5 protein expression levels with relative ratios shown as a percentage of the p68/GAPDH or DUSP5/GAPDH band. C, MTT assay of cellular growth in U87 and LN‐229 cells cotransfected with the siRNAs mentioned above. OD, optical density. D, Wound healing assay of cellular invasive capacity in U87 and LN‐229 cells cotransfected with the siRNAs mentioned above. Left panels, micrographs of invaded cells; right panels, relative quantitative analysis of scratch width in invaded cells shown in left panels. Scale bar = 50 μm. E, Transwell assay of U87 and LN‐229 cells cotransfected with the siRNAs mentioned above. Left panels, micrographs of migrated cells; right panels, relative quantitative analysis of migrated cells shown in left panels. Experiments were repeated three times independently. Bars represent SD; *P < .05, **P < .05 vs adjacent control
Similar findings were observed in an invasion and migration assay. As shown in Figure 6D, the rate of scratch‐stimulated cell invasion on the bottom of the well was higher in U87 and LN‐229 cells treated with p68 siRNA (1.87 ± 0.06 and 1.93 ± 0.14, respectively; P < .05) than in control‐transfected cells (1.00 ± 0.02 and 1.01 ± 0.08, respectively; Figure 6D), whereas U87 and LN‐229 cells cotransfected with p68 siRNA and DUSP5 siRNA had lower rates of invasion (1.20 ± 0.09 and 1.17 ± 0.11, respectively; P < .05). We found a similar result in migration assays carried out in cells cotransfected with p68 siRNA and DUSP5 siRNA (Figure 6E). Collectively, these functional assays indicate that DUSP5 impairs p68‐induced motility in glioma cells.
In these experiments, we attempted to identify which downstream signaling pathway is involved in the regulation of DUSP5. As shown in Figure S1, overexpressing DUSP5 inhibited the level of phosphorylation of ERK, whereas silencing DUSP5 increased the forced phosphorylation of ERK. To further evaluate the effects of DUSP5 on ERK signaling, we cotransfected cells with the DUSP5 vector and DUSP5 siRNA and confirmed that the effect on p‐ERK signaling that was induced by upregulating DUSP5 was rescued in comparison with the results obtained from overexpressing DUSP5 alone.
4. DISCUSSION
Gliomas are the most common type of primary brain tumors in adults, in which they account for approximately 80% of all malignant CNS tumors and are associated with substantial morbidity and mortality.22 Gliomas are classified into WHO grades I‐IV according to differences in morphological characteristics, with a higher grade usually indicating a poorer prognosis.23, 24 Glioblastomas (WHO grade IV) are the most malignant tumors in the spectrum of diffuse astrocytomas; they have the highest recurrence rate and the worst prognosis, in addition to a high risk of radioresistance and chemoresistance.25 Thus, it is important to investigate the detailed mechanisms underlying growth and invasion in gliomas. In our study, we show that p68 is highly expressed in both glioma cells and tissues and that silencing p68 suppressed proliferation, invasion, and migration in glioma cells. Using microarray gene expression profiling, we identified a novel target of p68, DUSP5. Following p68 knockdown, both the mRNA and the protein level of DUSP5 were increased, and p68 colocalized with DUSP5 in glioma cells. Moreover, DUSP5 expression was lower in high‐grade glioma tissues than in low‐grade glioma tissues. We also found that aberrant expression of DUSP5 efficiently suppressed growth and invasion in glioma cells and negatively regulated the downstream ERK pathway. Additionally, we downregulated DUSP5 using DUSP5 siRNA and p68 siRNA and found that the capacity of the cells to grow and invade was stronger in cotransfected cells than in cells treated with p68 knockdown alone. Taken together, these results indicate that p68 negatively regulates DUSP5 to promote proliferation, invasion, and migration in glioma cells. Our previous study showed that p68 combined with nuclear factor‐κB p50 to promote glioma proliferation. These studies emphasize the notion that p68 plays a chemotactic role in gliomagenesis, and we predict a novel therapeutic target, DUSP5, for glioma therapy.17
P68 is an oncogene that is overexpressed in colorectal tumors,15 in which downregulating p68 reduced proliferation and tumor formation in nude mice.26 P68 is overexpressed in prostate tumors, in which it promotes prostate tumorigenesis,13 and it is overexpressed in breast tumors, in which it regulates target gene transcription and promotes tumorigenesis.16 Our results verify that p68 is overexpressed in glioma tumors, and we show that p68 knockdown inhibited growth and invasiveness in glioma, suggesting that p68 plays an important role in glioma therapy. Additional analyses of the functions of the downstream signaling pathways of p68 in glioma will be required to further address this question. In our study, we show that a novel gene, DUSP5, functions downstream of p68 and interacts with p68 to regulate glioma growth. DUSP5 interacts specifically with p68; however, the exact binding site at which DUSP5 binds p68 was not identified.
The DUSP family comprises a group of tyrosine or serine/threonine phosphatases that are involved in cancer progression and resistance and could be regarded as a novel rational target.27 As a vital member of the DUSP family, DUSP5 localizes solely in cellular nuclei,28 possesses a functional nuclear localization signal, and has been proposed to act as a nuclear anchor for ERK.19 As a growth factor‐inducible phosphatase, DUSP5 is critical for both vascular development and disease.29 Previous studies showed that DUSP5 is a novel prognostic biomarker for advanced colorectal cancer patients30 and that it can prevent progression in prostate cancer patients.31 However, the precise role of DUSP5 in glioma remains unclear. The results of our study show that DUSP5 expression is associated with glioma differentiation and that DUSP5 expression was lower in high‐grade gliomas. Here, our findings also indicate that DUSP5 might act as a tumor suppressor during progression in glioma. To our knowledge, our study is the first to explore the molecular functions and roles underlying the clinicopathological effects of DUSP5 in glioma.
In this study, we reveal that DUSP5 functions as a tumor suppressor and inhibits tumor growth. As a member of the dual‐specificity phosphatase family that specifically inactivates ERK,32 DUSP5 is a component in one of three branches of the MAPK cascade that function in tumors. Abnormal MAPK signaling is closely related to the development and progression of tumors.32 Moreover, increasing evidence indicates that DUSP5 plays a key role in inhibiting tumor proliferation and growth.31 A recent study showed that DUSP5 suppressed skin cancer by regulating nuclear ERK activation.33 In gastric cancer, DUSP5 inhibited tumor cell growth and colony‐forming ability by regulating the cell cycle transition from G1 to S phase that was induced by the dephosphorylation of nuclear ERK1/2.34 Furthermore, DUSP5 plays a critical role in tumor metastasis progression by inhibiting vascular development. Bellou et al.'s study showed that DUSP5 localizes to endothelial cell nuclei and suppresses proliferation in endothelial cells by dephosphorylating vascular endothelial growth factor (VEGF)‐phosphorylated ERK1/2.35 Pramanik et al.'s study indicated that DUSP5 plays a key role in embryonic vascular development and could be mutated in human patients with vascular anomalies.29 DUSP5 also counteracted functions underlying the angioblast development‐promoting effects of a serine threonine kinase, Snrk‐1. In our study, we found that DUSP5 negatively regulated ERK expression and phosphorylation, indicating a novel molecular mechanism involving p68/DUSP5/ERK in glioma. In summary, p68/DUSP5 might participate in various functions during tumorigenesis by negatively targeting ERK signaling, which facilitates glial tumorigenesis. However, the underlying molecular mechanism by which it exerts these effects needs to be further explored.
Finally, the results of our study reveal that p68 might exert inhibitory effects on the regulation of chemotactic cell invasion and migration by negatively regulating DUSP5/ERK signaling. Improving our understanding of the mechanisms by which p68 negatively regulates DUSP5 will expand our knowledge of the molecular pathogenesis of glioma. Furthermore, the discoveries presented here represent a new facet of the complex interactions of the p68 gene and its tumor‐promoting functions and provide novel insights into the potential roles of a p68/DUSP5/ERK signaling pathway in carcinogenesis.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81372173 to R. Wang, 81502404 to WZ Du, and 81402062 to DY Han), the First Clinical College of Harbin Medical University Foundation (2013Lx03 to L. Teng), Fund of Science Research, Education Department of Heilongjiang Province (1254HQ006 to L. Teng), and Fund for returned researchers, Department of International Cooperation and Communication, Ministry of Education of the People’s Republic of China (No. 49 to L. Teng).
CONFLICT OF INTEREST
The authors disclose that they have no potential conflicts of interest.
Supporting information
{kind=link}
Wang R, Bao H‐B, Du W‐Z, et al. P68 RNA helicase promotes invasion of glioma cells through negatively regulating DUSP5 . Cancer Sci. 2019;110:107–117. 10.1111/cas.13858
Rui Wang, Hong‐Bo Bao, and Wen‐Zhong Du contributed equally to this work.
REFERENCES
- 1. Masui K, Kato Y, Sawada T, Mischel PS, Shibata N. Molecular and genetic determinants of glioma cell invasion. Int J Mol Sci. 2017;18:E2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta S, Lo Cascio C. Developmentally regulated signaling pathways in glioma invasion. Cell Mol Life Sci. 2018;75:385‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armento A, Ehlers J, Schotterl S, Naumann U. Molecular Mechanisms of Glioma Cell Motility Brisbane, AU: Codon Publications; 2017. [PubMed] [Google Scholar]
- 4. Crawford L, Leppard K, Lane D, Harlow E. Cellular proteins reactive with monoclonal antibodies directed against simian virus 40 T‐antigen. J Virol. 1982;42:612‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lane DP, Hoeffler WK. SV40 large T shares an antigenic determinant with a cellular protein of molecular weight 68,000. Nature. 1980;288:167‐170. [DOI] [PubMed] [Google Scholar]
- 6. Lin C, Yang L, Yang JJ, Huang Y, Liu ZR. ATPase/helicase activities of p68 RNA helicase are required for pre‐mRNA splicing but not for assembly of the spliceosome. Mol Cell Biol. 2005;25:7484‐7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zonta E, Bittencourt D, Samaan S, Germann S, Dutertre M, Auboeuf D. The RNA helicase DDX5/p68 is a key factor promoting c‐fos expression at different levels from transcription to mRNA export. Nucleic Acids Res. 2013;41:554‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukuda T, Yamagata K, Fujiyama S, et al. DEAD‐box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604‐611. [DOI] [PubMed] [Google Scholar]
- 9. Geissler V, Altmeyer S, Stein B, Uhlmann‐Schiffler H, Stahl H. The RNA helicase Ddx5/p68 binds to hUpf3 and enhances NMD of Ddx17/p72 and Smg5 mRNA. Nucleic Acids Res. 2013;41:7875‐7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore HC, Johnston M, Nicol SM, et al. An evolutionarily conserved, alternatively spliced, intron in the p68/DDX5 DEAD‐box RNA helicase gene encodes a novel miRNA. RNA. 2011;17:555‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicol SM, Bray SE, Black HD, et al. The RNA helicase p68 (DDX5) is selectively required for the induction of p53‐dependent p21 expression and cell‐cycle arrest after DNA damage. Oncogene. 2013;32:3461‐3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guturi KK, Sarkar M, Bhowmik A, Das N, Ghosh MK. DEAD‐box protein p68 is regulated by beta‐catenin/transcription factor 4 to maintain a positive feedback loop in control of breast cancer progression. Breast Cancer Res. 2014;16:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark EL, Coulson A, Dalgliesh C, et al. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008;68:7938‐7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang L, Lin C, Liu ZR. Phosphorylations of DEAD box p68 RNA helicase are associated with cancer development and cell proliferation. Mol Cancer Res. 2005;3:355‐363. [DOI] [PubMed] [Google Scholar]
- 15. Causevic M, Hislop RG, Kernohan NM, et al. Overexpression and poly‐ubiquitylation of the DEAD‐box RNA helicase p68 in colorectal tumours. Oncogene. 2001;20:7734‐7743. [DOI] [PubMed] [Google Scholar]
- 16. Wang D, Huang J, Hu Z. RNA helicase DDX5 regulates microRNA expression and contributes to cytoskeletal reorganization in basal breast cancer cells. Mol Cell Proteomics. 2012;11:M111 011932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang R, Jiao Z, Li R, Yue H, Chen L. p68 RNA helicase promotes glioma cell proliferation in vitro and in vivo via direct regulation of NF‐kappaB transcription factor p50. Neuro Oncol. 2012;14:1116‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caunt CJ, Keyse SM. Dual‐specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mandl M, Slack DN, Keyse SM. Specific inactivation and nuclear anchoring of extracellular signal‐regulated kinase 2 by the inducible dual‐specificity protein phosphatase DUSP5. Mol Cell Biol. 2005;25:1830‐1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang JH, Joo JC, Kim DJ, et al. Cordycepin promotes apoptosis by modulating the ERK‐JNK signaling pathway via DUSP5 in renal cancer cells. Am J Cancer Res. 2016;6:1758‐1771. [PMC free article] [PubMed] [Google Scholar]
- 21. Teng L, Nakada M, Furuyama N, et al. Ligand‐dependent EphB1 signaling suppresses glioma invasion and correlates with patient survival. Neuro Oncol. 2013;15:1710‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16:896‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayes J, Thygesen H, Droop A, et al. Prognostic microRNAs in high‐grade glioma reveal a link to oligodendrocyte precursor differentiation. Oncoscience. 2015;2:252‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high‐grade gliomas: response assessment in neuro‐oncology working group. J Clin Oncol. 2010;28:1963‐1972. [DOI] [PubMed] [Google Scholar]
- 26. Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572‐7578. [DOI] [PubMed] [Google Scholar]
- 27. Buffet C, Catelli MG, Hecale‐Perlemoine K, et al. Dual specificity phosphatase 5, a specific negative regulator of ERK signaling, is induced by serum response factor and Elk‐1 transcription factor. PLoS ONE. 2015;10:e0145484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3:REVIEWS3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pramanik K, Chun CZ, Garnaas MK, et al. Dusp‐5 and Snrk‐1 coordinately function during vascular development and disease. Blood. 2009;113:1184‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan X, Liu L, Li H, et al. Dual specificity phosphatase 5 is a novel prognostic indicator for patients with advanced colorectal cancer. Am J Cancer Res. 2016;6:2323‐2333. [PMC free article] [PubMed] [Google Scholar]
- 31. Cai C, Chen JY, Han ZD, et al. Down‐regulation of dual‐specificity phosphatase 5 predicts poor prognosis of patients with prostate cancer. Int J Clin Exp Med. 2015;8:4186‐4194. [PMC free article] [PubMed] [Google Scholar]
- 32. Keyse SM. Dual‐specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253‐261. [DOI] [PubMed] [Google Scholar]
- 33. Rushworth LK, Kidger AM, Delavaine L, et al. Dual‐specificity phosphatase 5 regulates nuclear ERK activity and suppresses skin cancer by inhibiting mutant Harvey‐Ras (HRasQ61L)‐driven SerpinB2 expression. Proc Natl Acad Sci USA. 2014;111:18267‐18272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin SH, Park SY, Kang GH. Down‐regulation of dual‐specificity phosphatase 5 in gastric cancer by promoter CpG island hypermethylation and its potential role in carcinogenesis. Am J Pathol. 2013;182:1275‐1285. [DOI] [PubMed] [Google Scholar]
- 35. Bellou S, Hink MA, Bagli E, et al. VEGF autoregulates its proliferative and migratory ERK1/2 and p38 cascades by enhancing the expression of DUSP1 and DUSP5 phosphatases in endothelial cells. Am J Physiol Cell Physiol. 2009;297:C1477‐C1489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials