

Abstract
Understanding the mechanism of chemoresistance and disease progression in patients with prostate cancer is important for developing novel treatment strategies. In particular, developing resistance to cabazitaxel is a major challenge in patients with docetaxel‐resistant and castration‐resistant prostate cancer (CRPC) because cabazitaxel is often administered as a last resort. However, the mechanism by which cabazitaxel resistance develops is still unclear. C‐C motif chemokine ligands (CCL) were shown to contribute to the castration resistance of prostate cancer cells via an autocrine mechanism. Therefore, we focused on CCL as key factors of chemoresistance in prostate cancer cells. We previously established a cabazitaxel‐resistant cell line, DU145‐TxR/CxR, from a previously established paclitaxel‐resistant cell line, DU145‐TxR. cDNA microarray analysis revealed that the expression of CCL2 was upregulated in both DU145‐TxR and DU145‐TxR/CxR cells compared with DU145 cells. The secreted CCL2 protein level in DU145‐TxR and DU145‐TxR/CxR cells was also higher than in parental DU145 cells. The stimulation of DU145 cells with CCL2 increased the proliferation rate under treatments with cabazitaxel, and a CCR2 (a specific receptor of CCL2) antagonist suppressed the proliferation of DU145‐TxR and DU145‐TxR/CxR cells under treatments of cabazitaxel. The CCL2‐CCR2 axis decreased apoptosis through the inhibition of caspase‐3 and poly(ADP‐ribose) polymerase (PARP). CCL2 is apparently a key contributor to cabazitaxel resistance in prostate cancer cells. Inhibition of the CCL2‐CCR2 axis may be a potential therapeutic strategy against chemoresistant CRPC in combination with cabazitaxel.
Keywords: apoptosis, cabazitaxel, CCL2, chemoresistance, prostate cancer
1. INTRODUCTION
Prostate cancer is the most common malignant cancer in men and the second leading cause of cancer‐related deaths in the United States.1 Androgen deprivation therapy is the main treatment for metastatic prostate cancer.2 However, the majority of patients with prostate cancer eventually develop castration‐resistant prostate cancer (CRPC).3, 4, 5 Cabazitaxel is used as a subsequent treatment in these patients after developing resistance to docetaxel.6 Because cabazitaxel is often administered as a last resort in this setting, developing resistance to it is a major challenge in the treatment of patients with CRPC. However, the mechanism by which cabazitaxel resistance develops is still unclear. We previously established a cabazitaxel‐resistant cell line, DU145‐TxR/CxR, from our previously established paclitaxel‐resistant cell line, DU145‐TxR.7 Although the activation of the multidrug resistance gene 1 (MDR1) is believed to play an important role in the development of cabazitaxel resistance in prostate cancer cells, we observed no significant difference in the expression levels between DU145‐TxR and DU145‐TxR/CxR cells.7 Identification of the definitive mechanism of development of cabazitaxel resistance is a key imperative to overcome prostate cancer. C‐C motif chemokine ligands (CCL) were shown to contribute to the progression of several cancers, including prostate cancer.8 The expression of CCL5 in bone stromal cells was shown to contribute to prostate cancer cell migration in the setting of bone metastasis.9 Furthermore, CCL21 was shown to promote the migration of prostate cancer cells through the phosphorylation of protein kinase p38 in the setting of lymph node metastasis.10 Moreover, CCL2 was shown to induce castration resistance in prostate cancer cells through an autocrine mechanism.11 These findings suggest that CCL may be involved in the acquisition of chemoresistance among prostate cancer cells. In this study, we identified some candidate CCL that may induce cabazitaxel resistance in prostate cancer cells on the basis of cDNA microarray data and explored the mechanism of development of cabazitaxel resistance through the CCL2‐CCR2 axis.
2. MATERIALS AND METHODS
2.1. cDNA microarray analysis data
To unravel the mechanisms that contribute to the development of cabazitaxel resistance, we compared gene expression profiles among DU145, DU145‐TxR and DU145‐TxR/CxR cells using cDNA microarray analysis as previously reported.7 Briefly, 48 hours after plating of 4 × 105 DU145, DU145‐TxR and DU145‐TxR/CxR cells, total RNA was purified using an RNeasy Mini Kit (Qiagen, Germantown, MD, USA). RNA samples were sent to Takara (Otsu, Japan) and were analyzed using Agilent Technologies Human 8x60K ver. 2.0 (Santa Clara, CA, USA).
2.2. Reagents and antibodies
Recombinant human CCL2 (rhCCL2) was obtained from BioLegend (San Diego, CA, USA). The following antibodies were used in western blotting: rabbit anti‐human CCR2 antibody (ab155321) and rabbit anti‐caspase‐3 (ab32351) from Abcam (Cambridge, MA, USA); rabbit anti‐poly(ADP‐ribose) polymerase (PARP, D64E10), rabbit anti‐Bcl‐xL (54H6) and HRP‐conjugated anti‐rabbit IgG (7074) antibodies from Cell Signaling Technology (Danvers, MA, USA); and mouse anti‐glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH, NB300‐221) antibody from Novus Biologicals (Littleton, CO, USA). Selective CCR2 receptor antagonists for in vitro (ab120812) and in vivo (sc202525) were obtained from Abcam and Santa Cruz Biotechnology (Santa Cruz, CA, USA), respectively.
2.3. Cell culture and proliferation assay
The human prostate cancer cell line DU145 obtained from the ATCC (Manassas, VA, USA) was cultured in DMEM (D5796; Sigma‐Aldrich, St. Louis, MO, USA) containing 5% FBS and 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) in a humidified incubator at 37°C with 5% CO2. Androgen‐independent DU145‐TxR (paclitaxel‐resistant) and DU145‐TxR/CxR (paclitaxel‐cabazitaxel‐resistant) cell lines were established in our laboratory as previously reported.7, 12 Briefly, DU145‐TxR cells were established after long‐term subculture of parental DU145 cells in DMEM supplemented with a low concentration of paclitaxel. DU145‐TxR/CxR cells were established after long‐term subculture of DU145‐TxR cells in DMEM supplemented with a low concentration of cabazitaxel.7 DU145‐TxR cells have already been shown to exhibit cross‐resistance to docetaxel.13 A cell proliferation assay was performed by plating 1 × 105 cells on 6‐well plates. After 8 hours, the cells were treated with predetermined concentrations of cabazitaxel, rhCCL2 or CCR2 antagonist for 24, 48 and 72 hours. At the end of the culture period, the cells were trypsinized, harvested and counted using a hemocytometer.
2.4. RNA extraction and RT‐PCR analysis
Twenty‐four hours after the plating of 1 × 105 DU145, DU145‐TxR and DU145‐TxR‐CxR cells on 6‐well plates, total RNA was purified with the RNeasy Mini Kit, following the manufacturer's instructions. The RNA concentration was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific). First‐strand cDNA was prepared from an RNA template (500 ng) using the iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA, USA). Reverse transcription was performed at 25°C for 5 minutes, 42°C for 30 minutes and then 85°C for 5 minutes. PCR amplification for GAPDH (98°C for 10 seconds, 60°C for 30 seconds and 72°C for 60 seconds), CCR2 and CCL2 (98°C for 10 seconds, 60°C for 30 seconds and 72°C for 60 seconds) was performed using template cDNA and the TaKaRa Ex Taq Hot Start Version PCR kit (Takara Bio, Kusatsu, Japan). Each of the amplified PCR products was identified using electrophoresis on a 1.5% agarose gel. Primer sequences were as follows: GAPDH forward: 5′‐TCC ACC ACC CTG TTG GTG TA‐3′, GAPDH reverse: 5′‐GAC CAC AGT CCA TGC CAT CA‐3′; CCR2 forward: 5′‐CTG TCC ACA TCT CGT TCT CGG TTT A‐3′, CCR2 reverse: 5′‐CCC AAA GAC CCA CTC ATT TGC AGC‐3′.
2.5. Western blotting
Prostate cancer cells were seeded in 6‐well plates and allowed to achieve 60%‐70% confluence, followed by incubation with rhCCL2, CCR2 antagonist or cabazitaxel. Cell lysates were prepared with M‐PER (Mammalian Protein Extraction Reagent, Thermo Fisher Scientific). The soluble lysate (20 μg) was mixed with lithium dodecyl sulfate sample buffer and a sample reducing agent (Thermo Fisher Scientific) and then the components were separated using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes, which were blocked with 1% gelatin in .05% Tween in Tris‐buffered saline for 1 hour at room temperature. The membranes were then incubated overnight at 4°C with primary anti‐CCR2, anti‐caspase‐3, anti‐PARP, anti‐Bcl‐xL or anti‐GAPDH antibodies following the manufacturers’ instructions. The membranes were then washed 3 times before incubation with HRP‐conjugated anti‐rabbit secondary antibodies for 1 hour at room temperature. Protein bands were detected using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
2.6. ELISA for CCL2
Human CCL2 secretion in a serum‐free conditioned medium from DU145, DU145‐TxR and DU145‐TxR‐CxR cells was quantified using a Quantikine Human ELISA Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. Absorbance was measured at 450 nm and was corrected at 540 nm on a microplate reader.
2.7. Apoptosis assay
DNA fragmentation was analyzed using a DeadEnd Fluorometric terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Briefly, DU145, DU145‐TxR and DU145‐TxR/CxR cells (2 × 104 cells/mL) were exposed to 10 μg/mL CCR2 antagonist or 3 nmol/L cabazitaxel for 24 hours. The cells were fixed in 4% formaldehyde and treated with a TUNEL buffer containing fluorescein‐12‐dUTP for 1 hour at 37°C in the dark. Finally, 4′,6‐diamidino‐2‐phenylindole was added and the positive cells were counted.
2.8. Xenograft study in mice
Intact male SCID mice (aged 6‐7 weeks) were obtained from CLEA Japan (Tokyo, Japan). After a 2‐week acclimatization period, 2 × 106 DU145 and DU145‐TxR/CxR cells were implanted with 50% Matrigel (Corning, Corning, NY, USA) subcutaneously in SCID mice. When tumors became detectable, a CCR2 antagonist and/or cabazitaxel were administrated intraperitoneally. As the first set, 2 groups were planned to confirm cabazitaxel's activity using DU145 cells (control group and cabazitaxel group). Each group included 5 mice. Next, 4 groups implanted with DU145‐TxR/CxR cells were planned: control, CCR2 antagonist alone, cabazitaxel alone, and CCR2 antagonist plus cabazitaxel. Each group included 6 mice. The control group was injected with 20 μL of DMSO. A CCR2 antagonist was injected every other day at a dose of 50 μg/kg, and cabazitaxel was injected weekly at a dose of 7 mg/kg diluted with 20 μL of DMSO. The tumor size and the body weight were measured every other day using a caliper and a scale, respectively. On day 21, mice were killed and the tumors were extracted. The animal protocol was approved by the Institutional Animal Care and Use Committee of the Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan.
2.9. Statistical analysis
Statistical analyses were performed using the commercially available software GraphPad Prism (GraphPad Software, San Diego, CA, USA). Student's t test was used to assess between‐group differences. Significance was defined as *P < .05, **P < .01 and ***P < .001.
3. RESULTS
3.1. CCL2 expression in DU145‐TxR and DU125‐TxR/CxR cells increased remarkably among the 24 CCL genes
We extracted the available 24 CCL genes from cDNA microarray analysis data. Compared to DU145 cells, the expressions levels of most CCL genes were not increased. However, the expression levels of CCL2 and CCL28 in DU145‐TxR cells increased 70‐fold and 13‐fold, respectively, and the expression levels of CCL1, CCL2 and CCL28 in DU145‐TxR/CxR cells increased 5‐fold, 43‐fold and 11‐fold, respectively. We focused on CCL2 as a key factor for the development of cabazitaxel resistance among DU145 cells, because the upregulation of CCL2 in DU145‐TxR and DU145‐TxR/CxR cells was prominent compared to other CCL (Table 1).
Table 1.
Expression change of CCL genes in DU145‐TxR and DU145‐TxR/CxR compared to DU145 cells by cDNA microarray
| Chemokine | DU145 | DU145‐TxR | Fold changea | DU145‐TxR/CxR | Fold changea | |||
|---|---|---|---|---|---|---|---|---|
| Normalized | Raw | Normalized | Raw | Normalized | Raw | |||
| CCL1 | .0275 | 10.82 | .0248 | 10.38 | .90 | .1500 | 57.88 | 5.46 |
| CCL2 | .0386 | 15.21 | 1.7300 | 725.06 | 69.81 | 1.1028 | 425.43 | 43.24 |
| CCL3 | .9645 | 379.91 | 1.0854 | 454.88 | 1.13 | .9257 | 357.13 | .96 |
| CCL4 | .0494 | 19.47 | .0380 | 15.92 | .83 | .0200 | 7.70 | .57 |
| CCL5 | .1677 | 66.07 | .1263 | 52.94 | .75 | .0553 | 21.35 | .33 |
| CCL6 | Not available | |||||||
| CCL7 | .0261 | 1.28 | .0243 | 10.17 | .93 | .0365 | 14.09 | 1.40 |
| CCL8 | .0257 | 10.12 | .0241 | 10.11 | .94 | .0224 | 8.65 | .87 |
| CCL9/10 | Not available | |||||||
| CCL11 | .0232 | 9.15 | .0188 | 7.86 | .81 | .0247 | 9.55 | 1.07 |
| CCL12 | Not available | |||||||
| CCL13 | .0210 | 8.26 | .0195 | 8.18 | .93 | .0177 | 6.84 | .84 |
| CCL14 | .0223 | 8.79 | .0207 | 8.68 | .93 | .0235 | 9.06 | 1.05 |
| CCL15 | .0862 | 33.97 | .0997 | 41.79 | 1.16 | .0942 | 36.34 | 1.09 |
| CCL16 | 1.0943 | 431.03 | 1.0640 | 445.93 | .95 | 1.0265 | 396.01 | .90 |
| CCL17 | .0420 | 16.53 | .0335 | 14.05 | .80 | .0548 | 21.13 | 1.31 |
| CCL18 | .0234 | 9.21 | .0217 | 9.07 | .93 | .0196 | 7.57 | .84 |
| CCL19 | .1270 | 50.04 | .0604 | 25.33 | 1.31 | .0493 | 19.03 | 1.13 |
| CCL20 | 3.9027 | 1537.24 | .3152 | 132.10 | .08 | .0848 | 32.71 | .02 |
| CCL21 | .0238 | 9.39 | .0221 | 9.24 | .92 | .0200 | 7.72 | .84 |
| CCL22 | .0231 | 9.11 | .0221 | 9.25 | .95 | .0201 | 7.77 | .87 |
| CCL23 | .0249 | 9.82 | .0233 | 9.78 | .94 | .0411 | 15.86 | 1.65 |
| CCL24 | 13.1511 | 5180.09 | 12.8215 | 5373.62 | .97 | 12.1863 | 4701.23 | .93 |
| CCL25 | .0223 | 8.77 | .0207 | 8.69 | .93 | .0201 | 7.75 | .90 |
| CCL26 | .0848 | 33.42 | .0392 | 16.44 | .46 | .0219 | 8.45 | .26 |
| CCL27 | .2616 | 103.05 | .2903 | 121.65 | 1.22 | .2578 | 99.44 | 1.09 |
| CCL28 | .0897 | 35.31 | 1.1894 | 498.49 | 13.27 | .9595 | 370.16 | 10.70 |
Compared to DU145.
3.2. DU145‐TxR and DU145‐TxR/CxR cells secreted CCL2 protein abundantly
The difference among DU145, DU145‐TxR and DU145‐TxR/CxR cells was examined. The proliferation assay showed that the proliferation of DU145 cells was significantly higher than that of both DU145‐TxR and DU145‐TxR/CxR cells and that there was no significant difference between DU145‐TxR and DU145‐TxR/CxR cells in this respect (Figure 1A). To confirm cabazitaxel resistance in DU145‐TxR and DU145‐TxR/CxR cells, cells were treated with cabazitaxel at different concentrations (0, 1, 3, 10 and 30 nmol/L) for 48 hours. The concentration that inhibited proliferation by 50% (IC50) of DU145, DU145‐TxR and DU145‐TxR/CxR cells for 48 hours was approximately .8, 7 and 28 nmol/L, respectively (Figure 1B). We regarded 3 and 10 nmol/L as the optimal concentrations for further experiments to compare these cell lines. Although the rate of proliferation of DU145 cells was higher than that of DU145‐TxR and DU145‐TxR/CxR cells (Figure 1A), the proliferation of DU145 cells 72 hours after treatment with 3 nmol/L cabazitaxel was significantly lower than that of DU145‐TxR and DU145‐TxR/CxR cells (Figure 1C). To explore the mechanism of development of cabazitaxel resistance, ELISA of CCL2 was performed. The CCL2 protein level in the medium of DU145‐TxR cells was significantly higher than that in the medium of DU145 cells. Furthermore, the CCL2 protein level in the medium of DU145‐TxR/CxR cells was significantly higher than that in the medium of DU145‐TxR cells, regardless of cabazitaxel treatment (Figure 1D). To verify that CCL2 can act on prostate cancer cells via an autocrine mechanism, we examined the expression of CCR2, a specific receptor of CCL2. The results of RT‐PCR and western blotting showed high expressions of CCR2 gene and protein in all prostate cancer cells (Figure 1E). Cabazitaxel treatment did not change the expression level of CCR2 in any of the prostate cancer cells (Figure 1F). Collectively, these findings suggest that the CCL2‐CCR2 axis may play a key role in the development of cabazitaxel resistance in DU145 cells.
Figure 1.
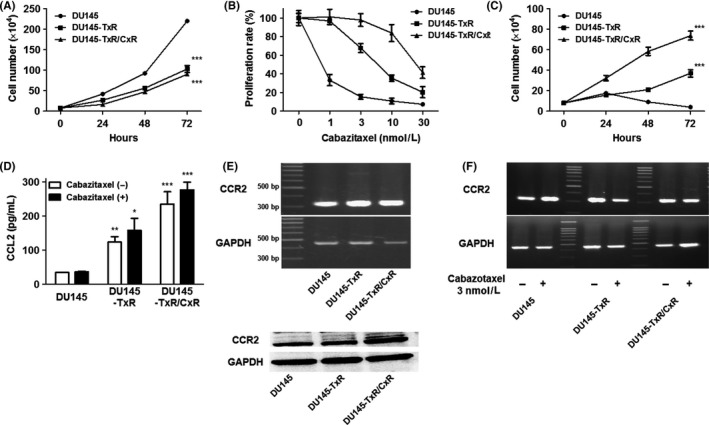
Proliferation assay and expression of CCL2 and CCR2. A‐C, DU145, DU145‐TxR and DU145‐TxR/CxR cells (1 × 105) were plated on 6‐well plates. Cells were treated with different concentrations of cabazitaxel after being plated for 8 h and were counted at 48 h (B). Cells were treated with 3 nmol/L cabazitaxel after being plated for 8 h and were counted at 24, 48 and 72 h (C). D, The concentration of human CCL2 secretion was measured in serum‐free conditioned media from DU145, DU145‐TxR and DU145‐TxR‐CxR cells with or without 3 nmol/L cabazitaxel using a Quantikine Human ELISA kit. E, The expression level of CCR2 in DU145, DU145‐TxR and DU145‐TxR‐CxR cells was examined using RT‐PCR and western blotting. F, The expression level of CCR2 was further examined with or without 3 nmol/L cabazitaxel using RT‐PCR. Data are shown as means ± SEM (n = 3)
3.3. CCL2 increased the proliferation rate under treatments with cabazitaxel
A TUNEL assay of DU145, DU145‐TxR and DU145‐TxR/CxR cells was performed with or without 3 nmol/L cabazitaxel treatment for 24 hours. The cabazitaxel‐induced apoptosis rate of DU145‐TxR (47%) and DU145‐TxR/CxR (8%) cells was significantly lower than that of DU145 cells (72%) (Figure 2A). To explore the effect of CCL2 on cell proliferation, rhCCL2 was added to DU145 cells under cabazitaxel treatment. RhCCL2 significantly increased the cell proliferation under cabazitaxel treatment in a dose‐dependent manner (Figure 2B). To inhibit the functional role of CCL2 in the proliferation of DU145‐TxR and DU145‐TxR‐CxR cells, a CCR2 antagonist was added to DU145‐TxR and DU145‐TxR/CxR cells under cabazitaxel treatments. The CCR2 antagonist clearly restored the sensitivity of DU145‐TxR and DU145‐TxR/CxR cells to cabazitaxel in a dose‐dependent manner (Figure 2C,D). The effect of the CCR2 antagonist on DU145‐TxR/CxR cells was more prominent than that on DU145‐TxR cells, which indicated that the secreted level of CCL2 had a direct impact on cabazitaxel resistance.
Figure 2.
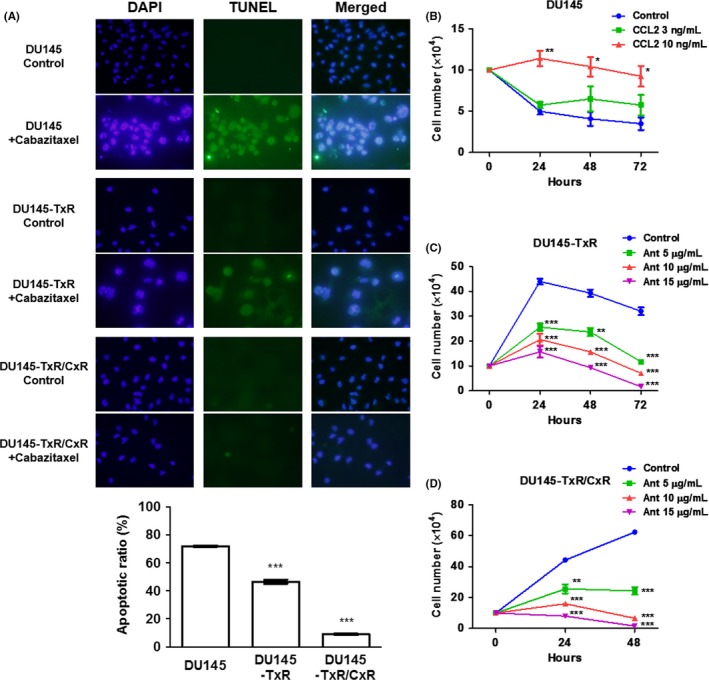
Cabazitaxel induced apoptosis and the CCL2‐CCR2 axis inhibited the sensitivity to cabazitaxel. A, DU145, DU145‐TxR and DU145‐TxR/CxR cells (2 × 104 cells/mL) were exposed to 3 nmol/L cabazitaxel for 24 h, and a TUNEL assay was performed. B‐D, DU145, DU145‐TxR and DU145‐TxR/CxR cells (1 × 105) were plated on 6‐well plates. Cells were treated with 3 nmol/L cabazitaxel and different concentrations of human recombinant CCL2 (B) and with 3 nmol/L cabazitaxel and different concentrations of CCR2 antagonist (Ant) (C,D) after being plated for 8 h and were counted at 24, 48 and 72 h (B,C) and at 24 and 48 h (D). Data are shown as means ± SEM (n = 3)
3.4. A CCR2 antagonist increased the apoptosis of DU145‐TxR and DU145‐TxR/CxR cells under cabazitaxel treatment
A TUNEL assay of DU145‐TxR and DU145‐TxR/CxR cells was performed with or without a CCR2 antagonist under cabazitaxel treatments. Although the CCR2 antagonist alone did not increase the apoptosis of DU145‐TxR cells, the combination of cabazitaxel and CCR2 antagonist significantly increased apoptosis compared to cabazitaxel alone (Figure 3A). Similarly, the CCR2 antagonist alone or cabazitaxel alone hardly increased the apoptosis of DU145‐TxR/CxR cells, whereas a combination of a CCR2 antagonist and cabazitaxel dramatically increased apoptosis compared to cabazitaxel alone (Figure 3B).
Figure 3.
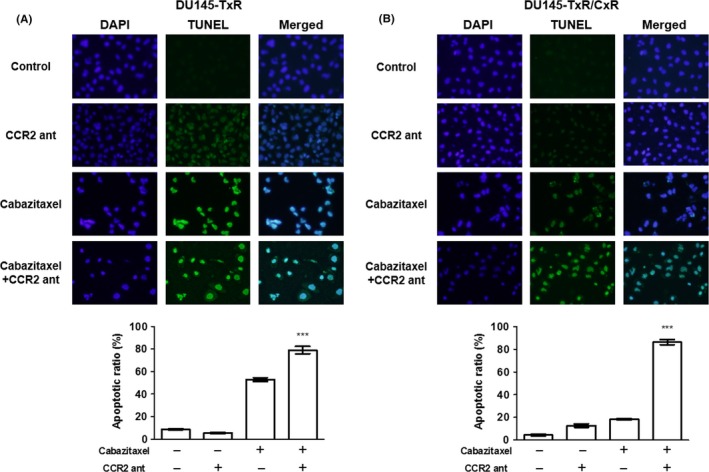
The CCR2 antagonist restored the sensitivity of DU145‐TxR and DU145‐TxR/CxR cells to cabazitaxel. (A) DU145‐TxR and (B) DU145‐TxR/CxR cells (2 × 104 cells/mL) were exposed to 10 μg/mL CCR2 antagonist or 3 nmol/L cabazitaxel or a combination of both for 24 h and a TUNEL assay was performed. Data are shown as means ± SEM (n = 3)
3.5. Cabazitaxel induced apoptosis through the induction of cleaved caspase‐3 and cleaved PARP
To explore the mechanism of the effect of cabazitaxel, apoptosis‐associated protein expression levels were detected by western blotting. The levels of cleaved caspase‐3 and cleaved PARP in DU145, DU145‐TxR and DU145‐TxR/CxR cells were increased in a dose‐dependent manner, and the respective intensities showed a strong association with the number of apoptotic cells (Figure 4A,B,C). In contrast, the expression level of Bcl‐xL (an antiapoptotic protein) in DU145 and DU145‐TxR cells was decreased under cabazitaxel treatment in a dose‐dependent manner (Figure 4A,B). The expression of Bcl‐xL in DU145‐TxR/CxR cells was hardly decreased (Figure 4C).
Figure 4.
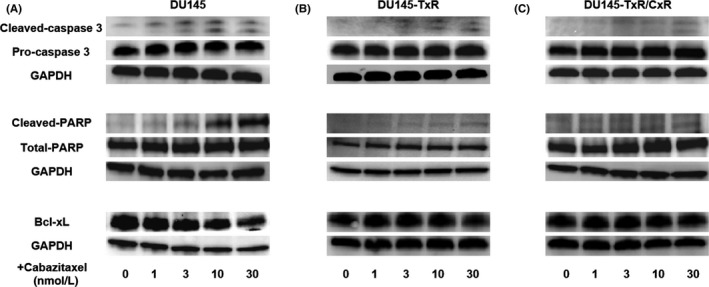
Cabazitaxel induced apoptosis through the induction of cleaved caspase‐3 and cleaved PARP. A‐C, The levels of apoptosis‐related proteins, caspase‐3, PARP and Bcl‐xL, in DU145 (A), DU145‐TxR (B) and DU145‐TxR/CxR (C) cells were determined using western blotting. Cells were seeded on 6‐well plates and allowed to attain 60%‐70% confluence, followed by incubation with different concentrations of cabazitaxel
3.6. Cleaved caspase‐3 and cleaved PARP were inhibited by the activation of the CCL2‐CCR2 axis
To confirm that the CCL2‐CCR2 axis plays a key role in cabazitaxel resistance, an rhCCL2 or CCR2 antagonist was added to prostate cancer cells under cabazitaxel treatment. RhCCL2 reduced cleaved caspase‐3 and cleaved PARP in DU145 cells under cabazitaxel treatment (Figure 5A). In contrast, the CCR2 antagonist increased cleaved caspase‐3 and cleaved PARP in DU145‐TxR and DU145‐TxR/CxR cells under cabazitaxel treatments (Figure 5B,C). These data suggest that the CCL2‐CCR2 axis induced cabazitaxel resistance in DU145‐TxR and DU145‐TxR/CxR cells through the antiapoptotic function, with a reduction in cleaved caspase‐3 and cleaved PARP.
Figure 5.
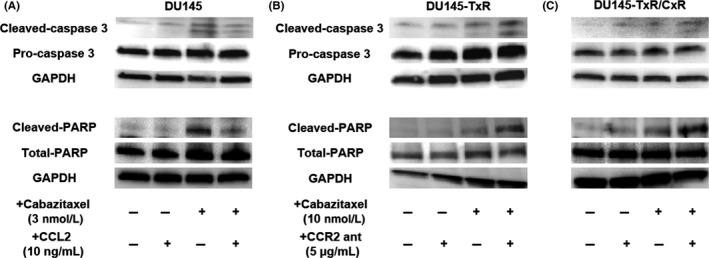
Cleaved caspase‐3 and cleaved poly(ADP‐ribose) polymerase (PARP) were inhibited by the activation of the CCL2‐CCR2 axis. A‐C, The levels of apoptosis‐related proteins, caspase‐3 and PARP, in DU145, DU145‐TxR and DU145‐TxR/CxR cells were examined using western blotting. DU145 cells were seeded on 6‐well plates and treated with 3 nmol/L cabazitaxel or 10 ng/mL CCL2 or a combination of both (A). DU145‐TxR (B) and DU145‐TxR/CxR (C) cells were seeded on 6‐well plates and treated with 10 nmol/L cabazitaxel or 5 μg/mL CCR2 antagonist or a combination of both
3.7. A CCR2 antagonist recovered the sensitivity to cabazitaxel in vivo
To confirm that the blockade of the CCL2‐CCR2 axis in DU145‐TxR/CxR cells by the CCR2 antagonist restores sensitivity to cabazitaxel in vivo, we implanted these cells into SCID mice subcutaneously and treated them with a CCR2 antagonist alone, cabazitaxel alone, or both a CCR2 antagonist and cabazitaxel. As a reference, SCID mice implanted with DU145 cells were treated with or without cabazitaxel. Cabazitaxel clearly inhibited tumor growth in mice injected with DU145 cells, with no associated loss of body weight (Figure 6A,B). Tumor growth in mice injected with DU145‐TxR/CxR cells was not significantly inhibited by cabazitaxel alone or CCR2 antagonist alone (P = .998 and .054, respectively) compared to the control, but was significantly inhibited by the combination of cabazitaxel and CCR2 antagonist compared to the control, cabazitaxel alone, and CCR2 antagonist alone groups (P = .018, .004 and .046, respectively; Figure 6C,D). Loss of body weight was not observed in any of the treatment groups (Figure 6E).
Figure 6.
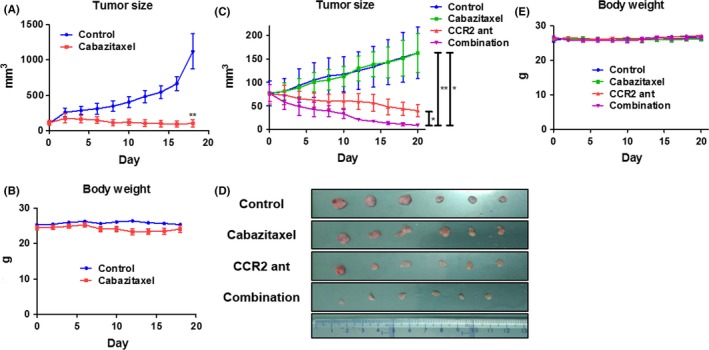
The CCR2 antagonist restored the sensitivity to cabazitaxel in vivo. A, After 2 wk of acclimatization, 2 × 106 DU145 cells were implanted subcutaneously in SCID mice. The control group was intraperitoneally injected with 20 μL of DMSO, and the cabazitaxel group was intraperitoneally injected with cabazitaxel weekly (days 0, 7 and 14) at a dose of 7 mg/kg diluted with 20 μL of DMSO (n = 5). The tumor size was measured every other day using a caliper. B, Body weight was measured every other day using a scale. C, After 2 wk of acclimatization, 2 × 106 DU145‐TxR/CxR cells were implanted subcutaneously in SCID mice. The following groups were compared: control, cabazitaxel alone, CCR2 antagonist alone, and a combination of cabazitaxel and CCR2 antagonist. The control group was injected with 20 μL of DMSO. Cabazitaxel was injected weekly (days 0, 7 and 14) at a dose of 7 mg/kg, and the CCR2 antagonist was injected every other day at a dose of 50 μg/kg (n = 6). The left, middle and right bars on the right side of the graph illustrate the comparison between the combination group and the CCR2 antagonist group, the cabazitaxel group and the control group, respectively. The tumor size was measured every other day using a caliper. D, On day 21, the mice were killed and the tumors extracted. E, Body weight was measured every other day using a scale. Data are shown as means ± SEM
4. DISCUSSION
In recent studies, the mechanism of development of docetaxel resistance in prostate cancer cells has been reported14; however, the mechanism of cabazitaxel resistance in prostate cancer cells is yet to be clearly elucidated. Within the tumor microenvironment, chemokines and their receptors play a key role in proliferation and metastasis.15, 16 Results of cDNA microarray analysis showed the upregulation of CCL2 and CCL28 in DU145‐TxR cells and the upregulation of CCL1, CCL2 and CCL28 in DU145‐TxR/CxR cells. These findings indicated that CCL1 alone was the strongest candidate for cabazitaxel resistance. Because the upregulation of the CCL1 mRNA level (5‐fold) was much lower than that of CCL2 (>40‐fold upregulation) and the upregulation of the CCL28 mRNA level was also relatively lower than that of CCL2, we first focused on CCL2 prior to CCL1 and CCL28. Indeed, data pertaining to the mRNA level and protein level of CCL2 were not consistent. The secreted CCL2 protein level in DU145‐TxR/CxR cells was much higher than that in DU145‐TxR cells, indicating that the CCL2 protein may be involved in cabazitaxel resistance in prostate cancer cells. CCL2 is abundantly expressed in the microenvironment of many cancers, including prostate cancer.17 Previous studies have shown that prostate cancer cells with high metastatic potential or CRPC cells have higher mRNA and protein expressions of CCL2 than earlier‐stage prostate cancer cells.18, 19 Interestingly, CCL2 was reported to be a key molecule for docetaxel resistance in prostate cancer cells.20 It is conceivable that a stronger expression of CCL2 contributes to not only docetaxel resistance but also cabazitaxel resistance in prostate cancer cells.
Other CCL may be involved in cabazitaxel resistance as the downstream of CCL2. CCL2 was shown to attract macrophages, and these macrophages transform to tumor‐associated macrophages in the tumor microenvironment and contribute to cancer progression.21 Tumor‐associated macrophages stimulate prostate cancer cells to secrete CCL17 and CCL22, which increase their migration ability via an autocrine mechanism.22 The expression level of CCR4 (a common receptor of CCL17 and CCL22) was shown to be associated with the expression level of CCR2 in prostate cancer tissues. In addition, prostate cancer tissues of patients with locally advanced or metastatic cancer show a high expression of CCR4.22 In addition, CCL17 and CCL22 were shown to activate regulatory T‐lymphocytes that inhibit the anticancer cell activity of cytotoxic T‐lymphocytes.23, 24 Hence, further studies with an immunological approach are required to further explore the complex role of CCL in cabazitaxel resistance of prostate cancer cells.
CCL2 also activates signal transducer and activator of transcription 3 (STAT3) and AKT in prostate cancer cells.11, 25, 26 In the process of developing castration resistance, the inhibition of androgen receptor signaling induces CCL2 secretion from prostate cancer cells, which, in turn, promotes the phosphorylation of STAT3 in an autocrine manner, resulting in an increase of the migratory and invasive ability of prostate cancer cells.11 Similarly, in the process of developing bicalutamide resistance, CCL2 promotes the phosphorylation of AKT via an autocrine mechanism, which results in an increase in the migratory and invasive ability of prostate cancer cells.25 Indeed, the activation of AKT contributes to docetaxel resistance via epithelial‐mesenchymal transition,27 and these pathways may contribute to cabazitaxel resistance of prostate cancer cells through the activation of the migratory and invasive ability.
Furthermore, because we previously demonstrated that MDR1 promotes cabazitaxel resistance of prostate cancer cells, CCL2 and MDR1 are probably directly or indirectly linked to each other. Although there are no studies proving a direct linkage between CCL2 and MDR1, STAT3 activation was shown to induce cisplatin resistance of lung cancer cells via the upregulation of MDR1.28 Moreover, the inhibition of STAT3 and AKT, as the upstream of MDR1, was shown to attenuate paclitaxel resistance of lung cancer cells and adriamycin resistance of breast and colon cancer cells.29 Paclitaxel disrupts the interaction of STAT3 with tubulin, and CCL2 may restore this interaction via activation of STAT3.30 Investigating the mechanisms related to STAT3 and AKT pathways may help unravel the mechanism of cabazitaxel resistance of prostate cancer cells.
In conclusion, although this study has a limitation that a single cell line was used for experiments, our data suggest that CCL2 is a contributor to cabazitaxel resistance of prostate cancer cells. The inhibition of the CCL2‐CCR2 axis may be a potential candidate for treatment of prostate cancer in combination with cabazitaxel.
CONFLICT OF INTEREST
The authors have no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI (Grant No. 16K10998 to K. Izumi) and Takeda Science Foundation.
Natsagdorj A, Izumi K, Hiratsuka K, et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2019;110:279–288. 10.1111/cas.13876
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists’ Collaborative Group. Lancet. 2000;355:1491‐1498. [PubMed] [Google Scholar]
- 3. Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sartor O, Gillessen S. Treatment sequencing in metastatic castrate‐resistant prostate cancer. Asian J Androl. 2014;16:426‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Izumi K, Namiki M. Optimal treatment for castration‐resistant prostate cancer. Asian J Androl. 2014;16:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration‐resistant prostate cancer progressing after docetaxel treatment: a randomised open‐label trial. Lancet. 2010;376:1147‐1154. [DOI] [PubMed] [Google Scholar]
- 7. Machioka K, Izumi K, Kadono Y, et al. Establishment and characterization of two cabazitaxel‐resistant prostate cancer cell lines. Oncotarget. 2018;9:16185‐16196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. 2014;2:1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Urata S, Izumi K, Hiratsuka K, et al. C‐C motif ligand 5 promotes migration of prostate cancer cells in the prostate cancer bone metastasis microenvironment. Cancer Sci. 2018;109:724‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maolake A, Izumi K, Natsagdorj A, et al. Tumor necrosis factor‐alpha induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci. 2018;109:1524‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Izumi K, Fang LY, Mizokami A, et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2‐induced STAT3 activation. EMBO Mol Med. 2013;5:1383‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Y, Mizokami A, Izumi K, et al. CTEN/tensin 4 expression induces sensitivity to paclitaxel in prostate cancer. Prostate. 2010;70:48‐60. [DOI] [PubMed] [Google Scholar]
- 13. Takeda M, Mizokami A, Mamiya K, et al. The establishment of two paclitaxel‐resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate. 2007;67:955‐967. [DOI] [PubMed] [Google Scholar]
- 14. Galletti G, Leach BI, Lam L, Tagawa ST. Mechanisms of resistance to systemic therapy in metastatic castration‐resistant prostate cancer. Cancer Treat Rev. 2017;57:16‐27. [DOI] [PubMed] [Google Scholar]
- 15. Tanaka T, Bai Z, Srinoulprasert Y, et al. Chemokines in tumor progression and metastasis. Cancer Sci. 2005;96:317‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Izumi K, Mizokami A, Lin HP, et al. Serum chemokine (CC motif) ligand 2 level as a diagnostic, predictive, and prognostic biomarker for prostate cancer. Oncotarget. 2016;7:8389‐8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kudo‐Saito C, Shirako H, Ohike M, et al. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin Exp Metastasis. 2013;30:393‐405. [DOI] [PubMed] [Google Scholar]
- 18. Lu Y, Cai Z, Galson DL, et al. Monocyte chemotactic protein‐1 (MCP‐1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate. 2006;66:1311‐1318. [DOI] [PubMed] [Google Scholar]
- 19. Loberg RD, Day LL, Harwood J, et al. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia. 2006;8:578‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qian DZ, Rademacher BL, Pittsenbarger J, et al. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel‐induced cytotoxicity. Prostate. 2010;70:433‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mizutani K, Sud S, McGregor NA, et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia. 2009;11:1235‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maolake A, Izumi K, Shigehara K, et al. Tumor‐associated macrophages promote prostate cancer migration through activation of the CCL22‐CCR4 axis. Oncotarget. 2017;8:9739‐9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizukami Y, Kono K, Kawaguchi Y, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122:2286‐2293. [DOI] [PubMed] [Google Scholar]
- 24. Maruyama T, Kono K, Izawa S, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to infiltration of regulatory T cells in esophageal squamous cell carcinoma. Dis Esophagus. 2010;23:422‐429. [DOI] [PubMed] [Google Scholar]
- 25. Lin TH, Izumi K, Lee SO, et al. Anti‐androgen receptor ASC‐J9 versus anti‐androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3‐CCL2 signaling. Cell Death Dis. 2013;4:e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Izumi K, Chang C. Targeting inflammatory cytokines‐androgen receptor (AR) signaling with ASC‐J9((R)) to better battle prostate cancer progression. Oncoimmunology. 2013;2:e26853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen H, Li H, Chen Q. INPP4B reverses docetaxel resistance and epithelial‐to‐mesenchymal transition via the PI3K/Akt signaling pathway in prostate cancer. Biochem Biophys Res Commun. 2016;477:467‐472. [DOI] [PubMed] [Google Scholar]
- 28. Fang Z, Chen W, Yuan Z, et al. LncRNA‐MALAT1 contributes to the cisplatin‐resistance of lung cancer by upregulating MRP1 and MDR1 via STAT3 activation. Biomed Pharmacother. 2018;101:536‐542. [DOI] [PubMed] [Google Scholar]
- 29. Yun M, Lee D, Park MN, et al. Cinnamaldehyde derivative (CB‐PIC) sensitizes chemo‐resistant cancer cells to drug‐induced apoptosis via suppression of MDR1 and its upstream STAT3 and AKT signalling. Cell Physiol Biochem. 2015;35:1821‐1830. [DOI] [PubMed] [Google Scholar]
- 30. Walker SR, Chaudhury M, Nelson EA, Frank DA. Microtubule‐targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol. 2010;78:903‐908. [DOI] [PubMed] [Google Scholar]