

Abstract
The success of targeted drug therapies for treating cancer patients has attracted broad attention both in the academic community and social society. However, rapidly developed acquired resistance is becoming a newly recognized major challenge to the continuing efficiency of these therapies. Metformin is a well‐known natural compound with low toxicity derived from the plant French lilac. Our previous work has highlighted research progress of the combination of clinically applied chemotherapies and metformin by different mechanisms. We have also launched a study to combine metformin with the small molecule targeted drug gefitinib to treat bladder cancer using intravesical administration. Thus, in this minireview, we summarize recent achievements combining metformin with various targeted therapies. This work directs the potential clinical future by selecting available cancer patients and providing precise medicine by the combination of metformin and targeted drugs to overcome resistance and enhance therapeutic efficacies.
Keywords: combination, metformin, small molecular inhibitor, synergistic effect, targeted therapy
Abbreviations
- AMPK
adenosine monophosphate activated protein kinase
- BAX
BCL‐2‐associated X
- BIM
Bcl‐2 interacting mediator of cell death
- CI
confidence interval
- EGFR
epidermal growth factor receptor
- HCC
hepatocellular carcinoma
- HER2
human epidermal growth factor receptor 2
- IGF‐1R
insulin‐like growth factor‐1 receptor
- NSCLC
non‐small‐cell lung cancer
- OS
overall survival
- PD‐L1
programmed death‐ligand 1
- PFS
progression‐free survival
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
1. INTRODUCTION
Cancer is one of the most deadly diseases that has been a considerable challenge in clinical treatment. The conventional chemotherapies provide limited benefits through inflicting DNA damage, with cancer cells and highly proliferative tissues particularly vulnerable to this damage.1 However, due to formidable side‐effects of chemotherapeutic drugs, such as myelosuppression and digestive tract side‐effects, cancer research scientists have focused their efforts on seeking better curative options. With the deeper understanding of cancer cell molecular biology, targeted therapies, including mAbs or small molecular inhibitors directed against genes or proteins that are crucial to cancer progression, novel treatment approaches have been developed, leading to therapeutic revolution. Well‐designed Abs or small molecules are able to specifically attack cancer cells through blocking signal transduction, which has been verified for cancer cell growth.2, 3 Targeted therapies have achieved dramatic success in treating cancer patients. However, rapidly developed resistance (acquired resistance) is a common phenomenon in the use of targeted drugs.4 Thus, finding approaches to overcome acquired resistance or avoid intrinsic resistance is becoming an unmet need to improve anticancer efficacies in targeted therapies. The combination of targeted drugs with other known agents is one efficient approach to overcome resistance and enhance efficacy.
Metformin, a widely prescribed drug for treating type II diabetes, is one of the most extensively recognized metabolic modulators, which has shown an important anticancer properties.5 Its general molecular mechanisms in inhibiting tumor growth are briefly summarized in Figure 1.5, 6, 7 Activation of AMPK and inhibition of mitochondria complex I are two major mechanisms of action.5, 6 It showed significant synergy with chemotherapeutic drugs in both in vitro and in vivo models through broad‐spectrum action mechanisms, as we previously described.8 More interestingly, both preclinical and clinical evidence has profoundly indicated the improvement of targeted therapeutic efficacies by combining metformin with targeted drugs, including our recent report.9 In this review, we summarize recent progress of strategies for treating cancer patients by combining metformin with targeted therapies, detailing the underlying mechanisms and discussing the future clinical transformation potential.
Figure 1.
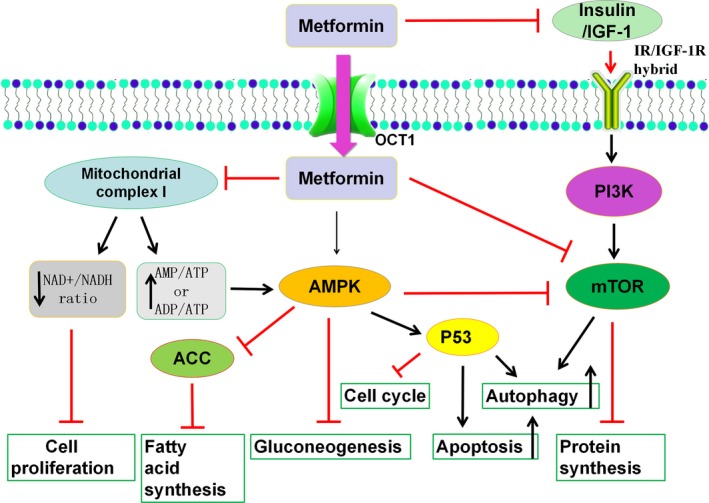
General molecular mechanisms of metformin on inhibiting cancer cell growth. Metformin inhibits mitochondria complex I, activates the adenosine monophosphate activated protein kinase (AMPK) signaling pathway, and/or inhibits the insulin signaling pathway. ACC, acetyl‐coa carboxylase; EMT, epithelial‐mesenchymal transition; IGF‐1, insulin‐like growth factor‐1; IGF‐1R, insulin‐like growth factor‐1 receptor; IR, insulin receptor; OCT1, organic cation transporter 1
2. COMBINATION OF METFORMIN WITH SMALL MOLECULAR INHIBITORS
Small molecular inhibitors are one of the major types of targeted drugs, which are usually organic compounds isolated from natural products or synthesized at an industrial scale. These small molecules exert their anticancer action through blocking signal transduction. The main portion of targeted drugs approved by the FDA for cancer treatment is made up of kinase inhibitors, with 38 approved to date.10 As shown in Figure 2, the number of FDA‐approved targeted inhibitors increased annually from 1995 to 2017.
Figure 2.
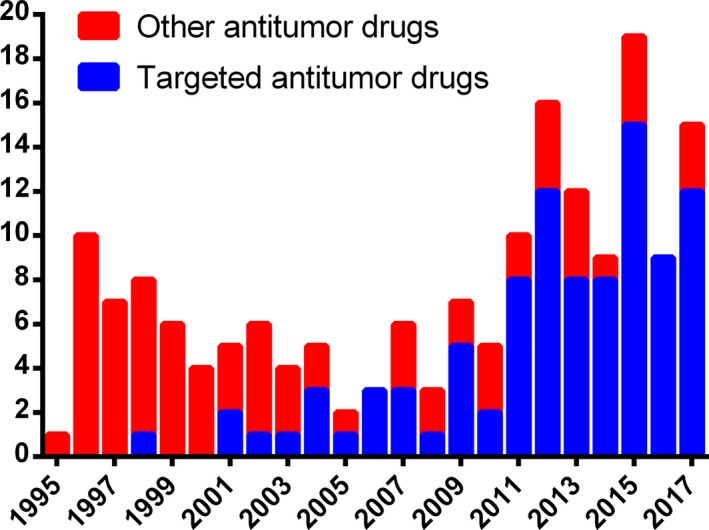
Trend of FDA‐approved targeted inhibitors between 1995 and 2017. The number of FDA‐approved targeted antitumor inhibitors increases dramatically, compared with other antitumor drugs
2.1. Combination of metformin with gefitinib
Gefitinib, as the first small molecular inhibitor targeting EGFR, was approved through an accelerated process by the FDA in May 2003 as monotherapy for the treatment of patients with locally advanced or metastatic NSCLC after failure of both platinum‐based and docetaxel chemotherapies.11 It blocks autophosphorylation of ligand‐induced receptor by binding to tyrosine kinase domain and disrupting tyrosine kinase activity, thereby abrogating intracellular downstream signaling.12 Approximately 90% of NSCLC patients with activating mutations (namely L858R) have high response rates to gefitinib and show complete responses with dramatic tumor shrink.13 Despite impressive clinical successes, it has been found that patients rapidly develop acquired resistance following long‐term gefitinib therapy. Developed resistance mechanisms include mutation, activation of the AKT/mTOR pathway, and upregulation of IGF‐1R.14 Thus, the approach combining gefitinib with metformin through inhibiting either the Akt/mTOR or insulin‐associated pathways to improve its efficacy has a strong rationale that warrants investigation.
The significant synergism of metformin with gefitinib in lung cancer has been reported. Adding metformin to gefitinib reduced proliferation and the anchorage‐independent colony‐forming ability of a panel of NSCLC cell lines.15 Chen et al16 found that metformin clearly inhibited antiapoptotic protein expression, increased BIM and BAX, leading to significantly improved sensitivity of gefitinib. It has been reported that metformin can revert resistance to gefitinib.16, 17 In xenografts with gefitinib‐resistant cancer cells, metformin blocked tumor growth and suppressed tumor relapse effectively. A study from Ko et al showed that metformin augmented the cytotoxic effect and growth inhibition of gefitinib in human squamous lung cancer cells through decreasing MSH2 expression, which plays a central role in promoting genetic stability by correcting DNA replication errors.18 Metformin also suppressed epithelial‐mesenchymal transition and the interleukin‐6/STAT3 pathway, abrogating the acquired resistance of gefitinib.17 Overcoming EGFR‐tyrosine kinase inhibitor primary resistance has also been reported in NSCLC by suppressing the IGF‐1R signaling pathway.19 Interstitial lung disease is a serious side‐effect of gefitinib treatment. Metformin attenuates gefitinib‐induced exacerbation of pulmonary fibrosis by inhibition of the transforming growth factor‐β signaling pathway both in vitro and in vivo NSCLC models.20
Studies aiming to evaluate the effect of metformin in combination with gefitinib on the prognosis of NSCLC patients with type 2 diabetes mellitus have been carried out in 6 hospitals in China between January 2006 and January 2014. Metformin significantly prolonged the median PFS and median OS compared with patients who received other hypoglycemic agents (19.0 months vs 8.0 months, P = 0.005; 32.0 months vs 23.0 months, P = 0.002). The objective response rate and disease control rate were also significantly higher (70.5% vs 45.7%, P = 0.017; 97.7% vs 80.4%, P = 0.009), indicating that metformin improved survival and delayed onset of acquired resistance to gefitinib.21 Based on these promising clinical data, a new multicenter double‐blind phase II study of metformin with gefitinib as first‐line therapy for locally advanced NSCLC is underway.22
Our group has studied the synergy between metformin with gefitinib in bladder cancer.9 We found that the combination of metformin and gefitinib induced a stronger antiproliferative and anticolony‐forming effect compared to either metformin or gefitinib alone.9 Apoptosis was significantly increased. Gefitinib suppressed EGFR signaling and inhibited phosphorylation of ERK and Akt, which was amplified when metformin was added.9 Interestingly, gefitinib induced activation of the AMPK signaling pathway, which was enhanced in metformin treatment as well. In vivo intravesical treatment of metformin and gefitinib on syngeneic orthotopic bladder cancer in mice confirmed the significant inhibitory effect on bladder tumor growth.9 These 2 drugs could be an excellent combination for the treatment of bladder cancer through intravesical instillation.9
Figure 3 summarizes the concise mechanisms underlying the combination of gefitinib and metformin, investigated so far.9, 23, 24
Figure 3.
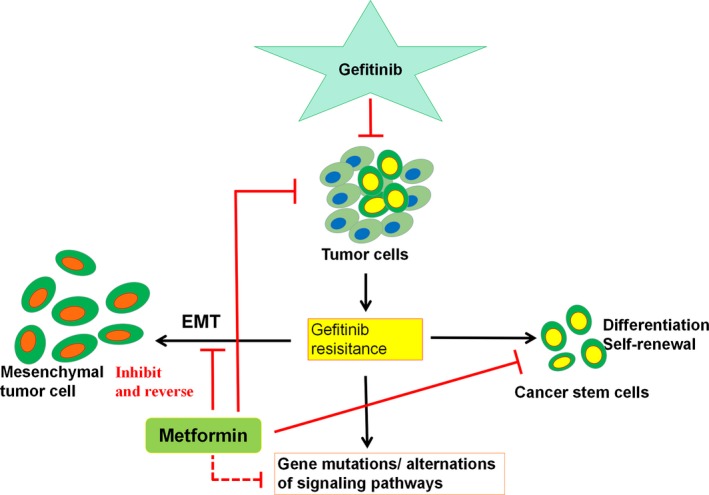
Gefitinib induces tumor cell death and develops resistance. Metformin either targets tumor cells directly or reverses and/or inhibits epithelial‐mesenchymal transition (EMT), differentiation, self‐renewal, and gene mutation/alteration of signaling pathways, which are the consequence of gefitinib‐induced resistance, thus sensitizing the antitumor effect or reducing the drug resistance to gefitinib
2.2. Combination of metformin with sorafenib
Sorafenib is a multiple target kinase inhibitor that targets the angiogenic receptor tyrosine kinases, VEGFR2 and platelet‐derived growth factor receptor‐β.25 Sorafenib obtained FDA approval for the treatment of advanced HCC in November 2007 and led to great success.26 However, low tumor response and serious side‐effects have been widely reported. Therefore, it is crucial to develop strategies to improve the efficacy of sorafenib. Combination of sorafenib with other drugs has attracted considerable attention.27 It has been shown that metformin sensitized cancer cells to certain clinical antitumor drugs, including sorafenib. These two drugs synergistically inhibited cancer cell proliferation and decreased sphere formation, especially in resistant cancer cells and cancer stem cells.28 Furthermore, adding metformin enabled a 25% dose reduction of sorafenib without loss of its tumor inhibitory efficacy.29 Metformin also enhanced the antimetastatic effects of sorafenib and reduced lung metastasis in HCC. Metformin suppressed the migration and invasion of HCC cells through downregulation of the ERK/JNK‐mediated nuclear factor‐κB‐dependent pathway, resulting in the reduction of uridylyl phosphate adenosine and MMP‐9 expression.30 Metformin also upregulated expression of Tat‐interacting protein 30, a protein that plays an important role in low‐dose sorafenib‐induced prometastasis.31 The combination of metformin and sorafenib suppresses proliferation and induces autophagy of HCC by targeting the mTOR pathway.32 In HCC xenograft tumors, combining metformin with sorafenib significantly minimized postoperative recurrence and lung metastasis through suppressing the mTOR pathway and CD37 and Ki67 expression, respectively.33, 34
Although both in vitro and vivo data have confirmed that metformin can enhance the antitumor effect of sorafenib, the results from clinical trials are not convincing. Unexpectedly, Casadei Gardini et al35 did observe that metformin‐treated patients experienced increased rather than decreased tumor aggressiveness and resistance to sorafenib. This might have been due to the small number of patients in the study. Thus, they recruited a larger number of patients and the results showed a lower response to sorafenib in those who developed HCC whilst undergoing chronic therapy with metformin, confirming their previous work. A possible explanation for these two clinical facts is that pretreatment of metformin decreased the levels of MAPK and inactivated PI3K pathways, which are the targets of sorafenib, inducing the resistance to sorafenib.36 These contradictory facts from clinical and experimental results might be due to the data collected from experimental studies were not imitating diabetic conditions while data collected from clinical studies were from diabetic patients. Thus, another type of clinical trial comparing sorafenib alone and sorafenib plus metformin under non‐diabetic conditions might be more helpful to explore whether there are any benefits of metformin on HCC patients using sorafenib. Another reason is that it is not easy to achieve the effective concentrations of metformin in patients’ bodies through conventional administration routes.37
2.3. Combination of metformin with everolimus
Mammalian target of rapamycin is a serine/threonine kinase that belongs to the family of PI3K‐related protein kinases.38 It acts as a key regulator of many cellular processes such as growth, protein synthesis, and cell‐cycle progression. It is also involved in several pathological conditions, including cancer, and has become a target for cancer treatments.39 Everolimus, an oral mTOR inhibitor, was approved to treat advanced pancreatic neuroendocrine tumors in 2011.40 Although everolimus showed valuable pharmacokinetic properties, its combination with other drugs as a potentially synergistic agent was encouraged.41 It has been reported that combining metformin with everolimus showed synergistic effects in in vitro and in vivo cancer models.42, 43
In breast cancer cells, cotreatment with metformin and everolimus intensified the inhibition of cell proliferation and colony formation.44 Metformin and everolimus significantly suppressed obesity‐induced tumor growth (0.5‐fold and 0.3‐fold).45 It was also reported that the effect of everolimus might be associated with insulin resistance, manifesting in impaired glucose tolerance or hyperglycemia, whereas metformin could restore it.46
Amazingly, a retrospective analysis of 445 patients with advanced pancreatic neuroendocrine tumors showed that the median PFS of patients treated with everolimus plus metformin was significantly longer than PFS for patients with diabetes receiving other treatments (median PFS, 20.8 months; hazard ratio, 0.49; 95% CI, 0.34‐0.69; P < .0001), indicating that metformin sensitized everolimus efficacy in patients with pancreatic neuroendocrine tumors.47 Unfortunately, a phase Ib study of everolimus combined with metformin for patients with advanced cancer showed that patients poorly tolerated the treatment and there are pharmacokinetic interactions between everolimus and metformin that might have implications for diabetic patients who are treated with these drugs.48
3. COMBINATION OF METFORMIN WITH ANTIBODIES
Antibodies, produced by genetic engineering, block cancer cell growth selectively through binding to specific antigen. Due to their specificities, Abs have showed excellent activities with minimal toxicities. Their excellent clinical benefits have resulted in a strong market for Ab drugs. Many Abs approved by the FDA are currently used in clinical cancer therapies, including trastuzumab, bevacizumab, rituximab, and cetuximab. Various studies listed here have reported that the combination of these Abs with metformin shows promising improvements on treatment efficacy.
3.1. Combination of metformin with trastuzumab
Trastuzumab was first approved for the treatment of HER2‐positive breast cancer in the USA in 1998.49 A member of the EGFR family, HER2 consists of 3 components: the intracellular tyrosine kinase domain, transmembrane lipophilic segment, and extracellular binding domain. Trastuzumab binds selectively and with high affinity to the extracellular domain, preventing HER2 cleavage and leading to its inactivation.50 Up to 30% of breast cancers overexpress HER2 and HER2 expression is positively associated with significantly worse outcomes in patient survival than HER2‐negative breast cancer.51 Over the last few decades, trastuzumab has been shown to prolong survival and improve outcomes of HER2‐positive breast cancer patients. However, trastuzumab is also associated with an increased risk of cardiotoxicity52 and prolonged exposure to trastuzumab leads to resistance.
To overcome these side‐effects of trastuzumab, the combination of trastuzumab with metformin has been explored. Vazquez‐Martin et al53 showed that metformin suppressed self‐renewal and proliferation of trastuzumab‐resistant tumor‐initiating breast cancer stem cells. Metformin significantly inhibited proliferation and clonogenicity of trastuzumab‐resistant HER2‐overexpressing breast cancer cells. The mechanisms of action involved disrupting HER2/IGF‐1R complexes that are present only in resistant sublines.54 In xenografts established from the pleural metastasis of a patient who was clinically resistant to trastuzumab, the combination of metformin with trastuzumab decreased tumor volume sharply by more than 4‐fold,55 implying that incorporation of metformin into trastuzumab‐based regimens could provide a valuable strategy for treatment of HER2‐positive breast cancer patients. Metformin also acts as a cardioprotective drug to attenuate the cardiac‐damaging effects of trastuzumab in the clinic and it has been confirmed that it does not interfere with the anticancer activity of trastuzumab.56 The phase III EMILIA trial showed trastuzumab emtansine, an Ab conjugate, significantly prolonged the PFS and OS in metastatic breast cancer patients. Metformin promotes trastuzumab emtansine drug efficacy through inducing caveolin‐1 expression in breast cancer.57 As for some HER2‐positive breast carcinomas patients after trastuzumab treatment, lapatinib was usually used to inhibit cancer progression. Metformin could be a clinically useful candidate of delaying or treating lapatinib resistance by inactivating mTOR and decreasing p70S6K1 activity.58
3.2. Combination of metformin with bevacizumab
Bevacizumab is a recombinant humanized mAb that targets all isoforms of VEGF‐A, preventing the binding of VEGF‐A to the endothelial cell surface receptors VEGFR‐1 (Flt‐1) and VEGFR‐2 (KDR/Flk‐1).59 Inhibition of VEGF‐A results in the regression of tumor vascularization and inhibition of new tumor vasculature formation, thereby inhibiting tumor growth.60 In 2004, bevacizumab was first approved by the FDA to treat metastatic colorectal cancer in combination with standard chemotherapy.61
Currently, bevacizumab is widely used to treat various cancers, including advanced nonsquamous NSCLC, metastatic breast cancer, advanced renal cell carcinoma, and advanced epithelial ovarian cancer.62 A case report showed that bevacizumab combined with metformin in recurrent type I endometrial cancer improved patient performance status: computed tomography scans showed reduced radiologic density of the lung and mediastinal lesions of liver disease, suggesting increased tumor necrosis.63 Moreover, combining bevacizumab with metformin was also found to be effective in ovarian cancer treatment, Metformin could specifically target cancer stem cells and synergistically inhibits angiogenesis with bevacizumab.64
A randomized phase II study of metformin plus bevacizumab‐based chemotherapy in advanced or metastatic nonsquamous NSCLC patients has been carried out. The 1‐year PFS on arm A (carboplatin, paclitaxel, and bevacizumab with metformin) was 47% (95% CI, 25%‐88%), which exceeded the historical control 1‐year PFS of 15%. Median overall survival of patients treated on arm A was 15.9 months (95% CI, 8.4‐not available) and 13.9 months (95% CI, 12.7‐not available) on arm B (carboplatin, paclitaxel, and bevacizumab without metformin).65 Disappointingly, the results indicated that there were no significant differences in toxicity between the two study arms.
4. DISCUSSION
Cancer is a disease with a growing mortality worldwide, with an increase to 19.3 million new cancer cases per year predicted in 2025.63 Targeted therapies have achieved notable success in some cancers; however, significant problems including resistance and high toxicity remain to be solved. To address these limits, combining targeted drugs with known drugs has drawn considerable attention. Metformin, a small molecule discovered from a natural plant, has been used as first‐line therapy for type II diabetes patients. Metformin has been newly recognized as a low‐toxicity “broad‐spectrum” therapeutic approach that could simultaneously target various key pathways and mechanisms in cancer treatment.66 Thus, combining targeted therapies with metformin is a potential strategy to enhance the efficacy of targeted drugs. Recently, Cha et al reported that metformin amplified the antitumor capacity of PD‐L1, a newly approved targeted Ab drug. It was shown that AMPK activated by metformin decreases PD‐L1 levels, which, in turn, increased CTL activities against cancer cells. The clinical data showed that metformin‐treated patients have reduced PD‐L1 levels with increased phospho‐AMPK, confirming the bench findings.67 In this minireview, we updated the combination of metformin with clinically used targeted drugs including small molecule inhibitors and Abs, summed up in Figure 4.6, 8, 68 Most studies drawing on both in vitro and in vivo data have shown promising synergy of metformin with these targeted drugs. However, results from clinical trials are contradictory. The main reason might be due to the difference of the dose, duration, and timing of metformin used between clinical trials and experimental studies.37 Another reason is that the concentration of metformin for anticancer activity is much higher than that for antidiabetic activity. Thus, novel administration routes, rather than the conventional method, to increase the concentration of this drug could be needed. The intravesical method we developed has shown much better benefits than oral administration.69 Another new direction for developing the clinical application of metformin is to characterize the precise targets of metformin, which could be helpful in personalized medicine and the clinical use of metformin. In summary, although the combination of metformin with clinically available targeted drugs shows a powerful synergistic efficiency in various cancer settings, experimental studies with a more sophisticated design and clinical trials are needed for future clinical application of metformin, one of the most important small natural products to treat cancer patients. There are limited studies exploring the molecular mechanisms behind the inhibition of mitochondria complex I in the combination of metformin with targeted drugs. Furthermore, it is necessary to evaluate the adverse effects of combinations compared with the single use of metformin, particularly acidosis. These studies could be future works warranting further efforts.
Figure 4.
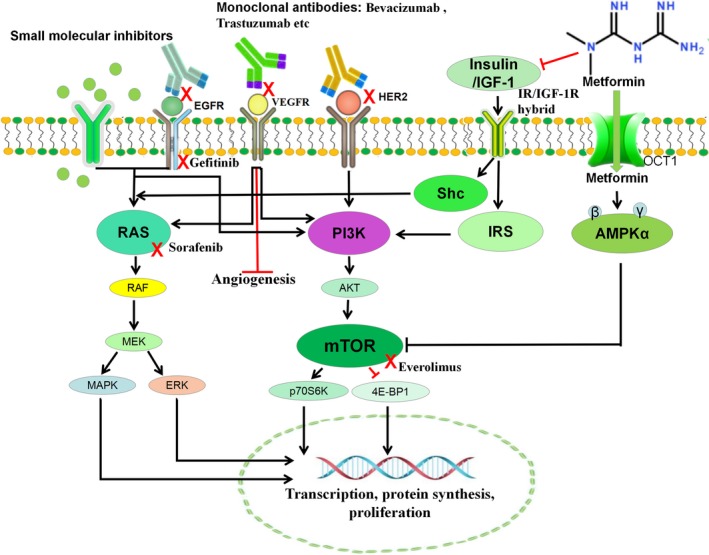
Mechanisms of metformin combined with Abs or small molecular inhibitors in cancer treatment. Metformin activates the adenosine monophosphate activated protein kinase (AMPK) pathway, eventually causing inhibition of the mTOR pathway. Metformin also indirectly reduces Akt activation, through insulin‐mediated insulin receptor (IR) and the insulin‐like growth factor‐1 receptor (IGF‐1R)/IR hybrid which activates the PI3/Akt/mTOR and RAS/RAF/MAPK/ERK pathways. These pathways are also inactivated by Abs or small molecule inhibitors. Thus, metformin shows synergy with targeted drugs to reduce protein synthesis and cellular proliferation. EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; IGF‐1, insulin‐like growth factor‐1; IRS, insulin/insulin‐like growth factor‐1 receptor substrate; Shc, Src homology 2/alpha‐collagen related protein; VEGFR, vascular endothelial growth factor receptor
CONFLICT OF INTEREST
No conflict of interest to declare.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81874212), Hunan Provincial Natural Science Foundation (2016JJ2187), Key Project of Hunan Province 2016 (2016JC2036), and Start‐up Funds of the Key Laboratory of Study and Discovery of Targeted Small Molecules of Hunan Province (2017TP1020) to X.Y., and the National Natural Science Foundation of China (81703008), Hunan Provincial Natural Science Foundation (2018JJ3831), and Institutional Fund of Xiangya Hospital Central South University (No 2016Q02) to M. P.
Deng J, Peng M, Wang Z, et al. Novel application of metformin combined with targeted drugs on anticancer treatment. Cancer Sci. 2019;110:23–30. 10.1111/cas.13849
Jun Deng and Mei Peng contributed equally to this study.
Contributor Information
Mei Peng, Email: meipeng@csu.edu.cn.
Xiaoping Yang, Email: xiaoping.yang@hunnu.edu.cn.
REFERENCES
- 1. Li Y, Wang Y, Zhou Y, et al. Cooperative effect of chidamide and chemotherapeutic drugs induce apoptosis by DNA damage accumulation and repair defects in acute myeloid leukemia stem and progenitor cells. Clin Epigenetics. 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Francoso A, Simioni PU. Immunotherapy for the treatment of colorectal tumors: focus on approved and in‐clinical‐trial monoclonal antibodies. Drug Des Devel Ther. 2017;11:177‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu W, Zhou M, Li Z, et al. A selective small molecule DNA2 inhibitor for sensitization of human cancer cells to chemotherapy. EBioMedicine. 2016;6:73‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Breslin S, Lowry MC, O'Driscoll L. Neratinib resistance and cross‐resistance to other HER2‐targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br J Cancer. 2017;116(5):620‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quinn BJ, Kitagawa H, Memmott RM, et al. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24(9):469‐480. [DOI] [PubMed] [Google Scholar]
- 6. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17‐29. [DOI] [PubMed] [Google Scholar]
- 7. Gui DY, Sullivan LB, Luengo A, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016;24(5):716‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng M, Darko KO, Tao T, et al. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat Rev. 2017;54:24‐33. [DOI] [PubMed] [Google Scholar]
- 9. Peng M, Huang Y, Tao T, et al. Metformin and gefitinib cooperate to inhibit bladder cancer growth via both AMPK and EGFR pathways joining at Akt and Erk. Sci Rep. 2016;6:28611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17(5):353‐377. [DOI] [PubMed] [Google Scholar]
- 11. Cohen MH, Williams GA, Sridhara R, et al. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8(4):303‐306. [DOI] [PubMed] [Google Scholar]
- 12. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786‐792. [DOI] [PubMed] [Google Scholar]
- 13. Juchum M, Günther M, Laufer SA. Fighting cancer drug resistance: opportunities and challenges for mutation‐specific EGFR inhibitors. Drug Resist Updat. 2015;20:12‐28. [DOI] [PubMed] [Google Scholar]
- 14. Wheeler Deric L, Dunn Emily F, Harari Paul M. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7(9):493‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgillo F, Sasso FC, Della Corte CM, et al. Synergistic effects of metformin treatment in combination with gefitinib, a selective EGFR tyrosine kinase inhibitor, in LKB1 wild‐type NSCLC cell lines. Clin Cancer Res. 2013;19(13):3508‐3519. [DOI] [PubMed] [Google Scholar]
- 16. Chen H, Wang Y, Lin C, et al. Vorinostat and metformin sensitize EGFR‐TKI resistant NSCLC cells via BIM‐dependent apoptosis induction. Oncotarget. 2017;8(55):93825‐93838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L, Han R, Xiao H, et al. Metformin sensitizes EGFR‐TKI‐resistant human lung cancer cells in vitro and in vivo through inhibition of IL‐6 signaling and EMT reversal. Clin Cancer Res. 2014;20(10):2714‐2726. [DOI] [PubMed] [Google Scholar]
- 18. Ko JC, Chiu HC, Wo TY, et al. Inhibition of p38 MAPK‐dependent MutS homologue‐2 (MSH2) expression by metformin enhances gefitinib‐induced cytotoxicity in human squamous lung cancer cells. Lung Cancer. 2013;82(3):397‐406. [DOI] [PubMed] [Google Scholar]
- 19. Pan YH, Jiao L, Lin CY, et al. Combined treatment with metformin and gefitinib overcomes primary resistance to EGFR‐TKIs with EGFR mutation via targeting IGF‐1R signaling pathway. Biologics. 2018;12:75‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li L, Huang W, Li K, et al. Metformin attenuates gefitinib‐induced exacerbation of pulmonary fibrosis by inhibition of TGF‐β signaling pathway. Oncotarget. 2015;6(41):43605‐43619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H, Yao W, Chu Q, et al. Synergistic effects of metformin in combination with EGFR‐TKI in the treatment of patients with advanced non‐small cell lung cancer and type 2 diabetes. Cancer Lett. 2015;369(1):97‐102. [DOI] [PubMed] [Google Scholar]
- 22. Li KL, Li L, Zhang P, et al. A multicenter double‐blind phase II study of metformin with gefitinib as first‐line therapy of locally advanced non‐small‐cell lung cancer. Clin Lung Cancer. 2017;18(3):340‐343. [DOI] [PubMed] [Google Scholar]
- 23. Cai Z, Cao Y, Luo Y, et al. Signalling mechanism(s) of epithelial‐mesenchymal transition and cancer stem cells in tumour therapeutic resistance. Clin Chim Acta. 2018;483:156‐163. [DOI] [PubMed] [Google Scholar]
- 24. Schrank Z, Chhabra G, Lin L. Current molecular‐targeted therapies in NSCLC and their mechanism of resistance. Cancers. 2018;10(7):E224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blumenschein GR Jr, Gatzemeier U, Fossella F, et al. Multicenter, uncontrolled trial of single‐agent sorafenib in patients with relapsed or refractory, advanced non‐small‐cell lung cancer. J Clin Oncol. 2009;27(26):4274‐4280. [DOI] [PubMed] [Google Scholar]
- 26. Guan YS, He Q. Sorafenib: activity and clinical application in patients with hepatocellular carcinoma. Expert Opin Pharmacother. 2011;12(2):303‐313. [DOI] [PubMed] [Google Scholar]
- 27. Sun W, Powell M, O'Dwyer PJ, et al. Phase II study of sorafenib in combination with docetaxel and cisplatin in the treatment of metastatic or advanced gastric and gastroesophageal junction adenocarcinoma: ECOG 5203. J Clin Oncol. 2010;28(18):2947‐2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groenendijk FH, Mellema WW, van der Burg E, et al. Sorafenib synergizes with metformin in NSCLC through AMPK pathway activation. Int J Cancer. 2015;136(6):1434‐1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen G, Nicula D, Renko K, et al. Synergistic anti‐proliferative effect of metformin and sorafenib on growth of anaplastic thyroid cancer cells and their stem cells. Oncol Rep. 2015;33(4):1994‐2000. [DOI] [PubMed] [Google Scholar]
- 30. Hsieh SC, Tsai JP, Yang SF, et al. Metformin inhibits the invasion of human hepatocellular carcinoma cells and enhances the chemosensitivity to sorafenib through a downregulation of the ERK/JNK‐mediated NF‐κB‐dependent pathway that reduces uPA and MMP‐9 expression. Amino Acids. 2014;46(12):2809‐2822. [DOI] [PubMed] [Google Scholar]
- 31. Guo Z, Cao M, You A, et al. Metformin inhibits the prometastatic effect of sorafenib in hepatocellular carcinoma by upregulating the expression of TIP30. Cancer Sci. 2016;107(4):507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ling S, Song L, Fan N, et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int J Oncol. 2017;50(1):297‐309. [DOI] [PubMed] [Google Scholar]
- 33. Saito T, Chiba T, Yuki K, et al. Metformin, a diabetes drug, eliminates tumor‐initiating hepatocellular carcinoma cells. PLoS ONE. 2013;8(7):e70010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. You A, Cao M, Guo Z, et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J Hematol Oncol. 2016;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Casadei Gardini A, Marisi G, Scarpi E, et al. Effects of metformin on clinical outcome in diabetic patients with advanced HCC receiving sorafenib. Expert Opin Pharmacother. 2015;16(18):2719‐2725. [DOI] [PubMed] [Google Scholar]
- 36. Casadei Gardini A, Faloppi L, De Matteis S, et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: validation study and biological rationale. Eur J Cancer. 2017;86:106‐114. [DOI] [PubMed] [Google Scholar]
- 37. Menendez JA, Quirantes‐Piné R, Rodríguez‐Gallego E, et al. Oncobiguanides: Paracelsus’ law and nonconventional routes for administering diabetobiguanides for cancer treatment. Oncotarget. 2014;5(9):2344‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dienstmann R, Rodon J, Serra V, et al. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13(5):1021‐1031. [DOI] [PubMed] [Google Scholar]
- 39. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729‐734. [DOI] [PubMed] [Google Scholar]
- 40. Saif MW. Pancreatic neoplasm in 2011: an update. J Ophthalmol. 2011;12(4):316‐321. [PubMed] [Google Scholar]
- 41. Calimeri T, Ferreri AJM. m‐TOR inhibitors and their potential role in haematological malignancies. Br J Haematol. 2017;177(5):684‐702. [DOI] [PubMed] [Google Scholar]
- 42. Wang Y, Wei J, Li L, et al. Combined use of metformin and everolimus is synergistic in the treatment of breast cancer cells. Oncol Res. 2014;22(4):193‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu H, Scholz C, Zang C, et al. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res. 2012;32(5):1627‐1637. [PubMed] [Google Scholar]
- 44. Ariaans G, Jalving M, Vries EG, et al. Anti‐tumor effects of everolimus and metformin are complementary and glucose‐dependent in breast cancer cells. BMC Cancer. 2017;17(1):232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fuentes‐Mattei E, Velazquez‐Torres G, Phan L, et al. Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor‐positive breast cancer. J Natl Cancer Inst. 2014;106(7):dju158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Opdam FL, Huitema AD, Beijnen JH, et al. Hyperglycaemia during treatment with everolimus. Ned Tijdschr Geneeskd. 2014;158:A7544. [PubMed] [Google Scholar]
- 47. Pusceddu S, Vernieri C, Di Maio M, et al. Metformin use is associated with longer progression‐free survival of patients with diabetes and pancreatic neuroendocrine tumors receiving everolimus and/or somatostatin analogues. Gastroenterology. 2018;155(2):479‐489.e7. [DOI] [PubMed] [Google Scholar]
- 48. Molenaar RJ, van de Venne T, Weterman MJ, et al. A phase Ib study of everolimus combined with metformin for patients with advanced cancer. Invest New Drugs. 2018;36(1):53‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thor A. HER2‐a discussion of testing approaches in the USA. Ann Oncol. 2001;12(Suppl 1):S101‐S107. [DOI] [PubMed] [Google Scholar]
- 50. Untch M, Rezai M, Loibl S, et al. Neoadjuvant treatment with trastuzumab in HER2‐positive breast cancer: results from the GeparQuattro study. J Clin Oncol. 2010;28(12):2024‐2031. [DOI] [PubMed] [Google Scholar]
- 51. Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science. 1987;235(4785):177‐182. [DOI] [PubMed] [Google Scholar]
- 52. Zeglinski M, Ludke A, Jassal DS, et al. Trastuzumab‐induced cardiac dysfunction: a “dual‐hit”. Exp Clin Cardiol. 2011;16(3):70‐74. [PMC free article] [PubMed] [Google Scholar]
- 53. Vazquez‐Martin A, Oliveras‐Ferraros C, Del Barco S, et al. The anti‐diabetic drug metformin suppresses self‐renewal and proliferation of trastuzumab‐resistant tumor‐initiating breast cancer stem cells. Breast Cancer Res Treat. 2011;126(2):355‐364. [DOI] [PubMed] [Google Scholar]
- 54. Liu B, Fan Z, Edgerton SM, et al. Potent anti‐proliferative effects of metformin on trastuzumab‐resistant breast cancer cells via inhibition of erbB2/IGF‐1 receptor interactions. Cell Cycle. 2011;10(17):2959‐2966. [DOI] [PubMed] [Google Scholar]
- 55. Cufi S, Corominas‐Faja B, Vazquez‐Martin A, et al. Metformin‐induced preferential killing of breast cancer initiating CD44+ CD24−/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012;3(4):395‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smith TA, Phyu SM, Akabuogu EU. Effects of administered cardioprotective drugs on treatment response of breast cancer cells. Anticancer Res. 2016;36(1):87‐93. [PubMed] [Google Scholar]
- 57. Chung YC, Chang CM, Wei WC, et al. Metformin‐induced caveolin‐1 expression promotes T‐DM1 drug efficacy in breast cancer cells. Sci Rep. 2018;8(1):3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vazquez‐Martin A, Oliveras‐Ferraros C, del Barco S, et al. mTOR inhibitors and the anti‐diabetic biguanide metformin: new insights into the molecular management of breast cancer resistance to the HER2 tyrosine kinase inhibitor lapatinib (Tykerb). Clin Transl Oncol. 2009;11(7):455‐459. [DOI] [PubMed] [Google Scholar]
- 59. Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti‐VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391‐400. [DOI] [PubMed] [Google Scholar]
- 60. Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res. 2005;65(3):671‐680. [PubMed] [Google Scholar]
- 61. Yamazaki K, Nagase M, Tamagawa H, et al. Randomized phase III study of bevacizumab plus FOLFIRI and bevacizumab plus mFOLFOX6 as first‐line treatment for patients with metastatic colorectal cancer (WJOG4407G). Ann Oncol. 2016;27(8):1539‐1546. [DOI] [PubMed] [Google Scholar]
- 62. Mortimer J, Zonder HB, Pal SK. Lessons learned from the bevacizumab experience. Cancer Control. 2012;19(4):309‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Indraccolo S, Randon G, Zulato E, et al. Metformin: a modulator of bevacizumab activity in cancer? A case report. Cancer Biol Ther. 2015;16(2):210‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Markowska A, Sajdak S, Markowska J, et al. Angiogenesis and cancer stem cells: new perspectives on therapy of ovarian cancer. Eur J Med Chem. 2017;142:87‐94. [DOI] [PubMed] [Google Scholar]
- 65. Marrone KA, Zhou X, Forde PM, et al. A randomized phase II study of metformin plus paclitaxel/carboplatin/bevacizumab in patients with chemotherapy‐naïve advanced or metastatic nonsquamous non‐small cell lung cancer. Oncologist. 2018;23(7):859‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Block KI, Gyllenhaal C, Lowe L, et al. Designing a broad‐spectrum integrative approach for cancer prevention and treatment. Semin Cancer Biol. 2015;35(Suppl):S276‐S304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cha JH, Yang WH, Xia W, et al. Metformin promotes antitumor immunity via endoplasmic‐ reticulum‐associated degradation of PD‐L1. Mol Cell. 2018;71(4):606‐62.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chong CR, Jänne PA. The quest to overcome resistance to EGFR‐targeted therapies in cancer. Nat Med. 2013;19(11):1389‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Peng M, Su Q, Zeng Q, et al. High efficacy of intravesical treatment of metformin on bladder cancer in preclinical model. Oncotarget. 2016;7(8):9102‐9117. [DOI] [PMC free article] [PubMed] [Google Scholar]