

Abstract
Despite recent advances in cancer treatment, pancreatic cancer is a highly malignant tumor type with a dismal prognosis and it is characterized by dense desmoplasia in the cancer tissue. Cancer‐associated fibroblasts (CAF) are responsible for this fibrotic stroma and promote cancer progression. We previously reported that a novel natural compound conophylline (CnP) extracted from the leaves of a tropical plant reduced liver and pancreatic fibrosis by suppression of stellate cells. However, there have been no studies to investigate the effects of CnP on CAF, which is the aim of this work. Here, we showed that CAF stimulated indicators of pancreatic cancer malignancy, such as proliferation, invasiveness, and chemoresistance. We also showed that CnP suppressed CAF activity and proliferation, and inhibited the stimulating effects of CAF on pancreatic cancer cells. Moreover, CnP strongly decreased the various cytokines involved in cancer progression, such as interleukin (IL)‐6, IL‐8, C‐C motif chemokine ligand 2 (CCL2), and C‐X‐C motif chemokine ligand 12 (CXCL12), secreted by CAF. In vivo, CAF promoted tumor proliferation and desmoplastic formation in a mouse xenograft model, CnP reduced desmoplasia of tumors composed of pancreatic cancer cells + CAF, and combination therapy of CnP with gemcitabine remarkably inhibited tumor proliferation. Our findings suggest that CnP is a promising therapeutic strategy of combination therapy with anticancer drugs to overcome refractory pancreatic cancers.
Keywords: conophylline, cytokine, fibroblast, microenvironment, stellate cell
1. INTRODUCTION
Despite recent advances in cancer treatment, the prognosis of pancreatic cancer is extremely poor as a result of its rapid progression, early metastasis, and limited response to chemotherapy and radiotherapy.1, 2 Therefore, novel therapeutic strategies are needed to improve the prognosis of pancreatic cancer. Pancreatic cancer is characterized by dense fibrotic stroma, called desmoplasia, and pancreatic stellate cells (PSC) are responsible for its generation. Pancreatic stellate cells are activated by various stimuli from tumor‐stromal interactions, and transform into myofibroblast‐like cells, which express α‐smooth muscle actin (α‐SMA) as cancer‐associated fibroblasts (CAF),3 and these activated PSC are considered to be one of the main precursors of CAF.4 CAF are reported to promote malignant behavior of pancreatic cancer cells, such as proliferation, invasion, metastasis, and resistance to chemotherapy or radiotherapy through various cytokines, growth factors, and exosomes that contain microRNA.5, 6, 7, 8 These findings suggest that targeting CAF and desmoplasia would be a novel strategy to treat pancreatic cancer.
Conophylline (CnP) is a vinca alkaloid extracted from the leaves of the tropical plant, Ervatamia microphylla, and has been shown to promote β‐cell differentiation in pancreatic precursor cells.9 Recently, we reported that CnP suppressed thioacetamide‐induced liver fibrosis through the inhibition of hepatic stellate cell activation.10 Moreover, CnP was found to inhibit PSC and improve islet fibrosis in rat models of type‐2 diabetes.11 In these reports, CnP suppressed α‐SMA expression, a marker of stellate cell activation, and type I collagen secretion of PSC and hepatic stellate cells. Therefore, CnP has potential to be developed into an antifibrotic drug. Given the similarity of activated stellate cells and CAF, we hypothesized that CnP can inhibit the activity of CAF, which also express α‐SMA, in cancer tissue, and thereby attenuate the cancer‐promoting effects of CAF. However, to date, there have been no studies to investigate the effects of CnP on CAF derived from pancreatic cancer tissues.
Therefore, the aim of the present study was to clarify whether CnP could inhibit CAF and suppress desmoplasia in pancreatic cancer. We investigated the effects of CnP on CAF, and the effects of this CAF inhibition on measures of pancreatic cancer malignancy, such as proliferation, invasion, and chemoresistance. We also assessed the effects of CnP and combination therapy with gemcitabine using an in vivo xenograft model co‐implanted with CAF and pancreatic cancer cells.
2. MATERIALS AND METHODS
2.1. Cell cultures and cell isolation
The human PSC line, hPSC5, established from pancreatic cancer tissue, and human pancreatic cancer cell lines, SUIT‐2 and SW1990, were used in this study. hPSC5 cells were provided by RIKEN BRC through the National BioResource Project of MEXT, Japan, and SUIT‐2 and SW1990 cells were obtained from JCRB Cell Bank (Osaka, Japan) and ATCC (Manassas, VA, USA), respectively. hPSC5 cells were originally isolated by the outgrowth method after collagenase digestion of the resected pancreatic tissue of a patient undergoing surgery for pancreatic cancer. By this method, the isolated PSC are already activated by culture in serum‐containing medium and they express typical activated stellate cells and CAF markers including α‐SMA, vimentin, type I collagen, and fibronectin.12, 13 Indeed, the microarray analysis showed that hPSC5 cells express high levels of α‐SMA, vimentin, fibronectin and collagens (data not shown). In addition, hPSC5 cells express the stromal markers secreted protein, acidic and rich in cysteine (SPARC)14 and interleukin (IL)‐6, the latter plays a critical role in the interaction between CAF and pancreatic cancer cells.15 These results indicate that hPSC5 cells showed activated phenotypes, resembling CAF. Additionally, we established primary CAF (CAF1 and CAF2) from surgically resected pancreatic cancer tissues in Gunma University using the method described by Lau et al.16 We confirmed that CAF showed myofibroblast‐like morphology and were positive for α‐SMA by western blotting (data not shown). Cell line establishment from surgically resected pancreatic cancer tissues was approved by the Institutional Review Board of Gunma University (Approval number: 2016‐118). These cells were cultured in DMEM (Wako, Osaka, Japan) supplemented with 10% FBS and 1% penicillin‐streptomycin (Thermo Fisher Scientific, Kanagawa, Japan) and maintained at 37°C in a humidified 5% CO2 incubator. Growth speed of established CAF was slow compared with hPSC5. We used established CAF between passage numbers 3 and 6.
2.2. Preparation of CnP
The CnP used in this study was isolated and purified from the leaves of E. microphylla and Tabernaemontana divaricata, and prepared as described previously.10, 17 Briefly, pure CnP was dissolved in methanol and used in vitro; the final concentration of CnP was 0.05‐1.0 μg/mL, with a final methanol concentration of 0.1% (v/v). Crude CnP (containing 22 mg/g pure CnP) was used in vivo, diluted to 2 mg/mL in 0.5% (v/v) Tween‐80 solution.
2.3. Protein extraction and western blotting
Total protein was extracted from cells using RIPA buffer (Wako) according to the manufacture's protocol. Western blotting was carried out as described previously.18, 19 The primary antibodies used in this study were anti‐α‐SMA mouse monoclonal antibody (A5247; 1:1000; Sigma‐Aldrich, St Louis, MO, USA), anti‐collagen I mouse monoclonal antibody (sc‐59772; 1:1000; Santa Cruz Biotechnology, Dallas, TX, USA) and transforming growth factor beta (TGF‐β) rabbit antibody (#3711; 1:1000; Cell Signaling Technology, Danvers, MA, USA). Anti‐β‐actin mouse monoclonal antibody (A5316; 1:1000; Sigma‐Aldrich) was used for loading control. Protein bands on the membrane were detected using ECL Prime Western Blotting Detection Reagent and an ImageQuant LAS 4000 imager (GE Healthcare, Buckinghamshire, UK).
2.4. Preparation of conditioned media
After CAF were cultured until 70%‐80% confluence in DMEM with 10% FBS, the medium was changed to serum‐free DMEM and the cells were cultured for 48 hours. Conditioned medium (CM) was collected as CAF‐CM, centrifuged for 10 minutes at 1500 g, and aliquots were stored at −80°C until use. To evaluate the effect of CnP, the CAF were cultured with serum‐free DMEM containing 0.3 μg/mL CnP for 48 hours, and collected as CnP‐treated CAF‐CM. Both types of CM were used for the following experiments.
2.5. Cell proliferation assay
Cell proliferation assay was carried out using CCK‐8 (Dojindo Laboratories, Kumamoto, Japan). CAF and pancreatic cancer cells were seeded at a density of 3000 cells/well in 96‐well plates and cultured in DMEM with 10% FBS. After an overnight incubation, the medium was changed to serum‐free medium with CnP between 0 and 1.0 μg/mL. To assess cell proliferation after stimulation with CM, the medium of pancreatic cancer cells was also replaced with CAF‐CM or CnP‐treated CAF‐CM, after overnight incubation. Cell proliferation was evaluated after culture for 48 hours. Absorbance of each well was measured using a spectrophotometer (Bio‐Rad, Hercules, CA, USA) at 450 nm with the reference wavelength set at 650 nm.
2.6. Invasion assay
Cell invasion assay was carried out using 24‐well Corning BioCoat Matrigel Invasion Chambers (Corning, Corning, NY, USA). SUIT‐2 and SW1990 cells (1 × 105 and 7.5 × 104 cells/well, respectively) were seeded in the upper chamber with 500 μL serum‐free medium, and the lower chamber was filled with 750 μL medium plus 0.3 μg/mL CnP, CAF‐CM, or CnP‐treated CAF‐CM. After incubation for 48 hours, the cells were fixed and stained with Diff‐Quik (Sysmex Corporation, Kobe, Japan). After staining, the cells that had invaded through the pores to the lower surface of the membrane were counted by microscopy. A total of 10 randomly selected fields were evaluated.
2.7. Gemcitabine sensitivity assay
SUIT‐2 and SW1990 cells were seeded at a density of 1 × 104 cells/well in 96‐well plates in 100 μL DMEM with 10% FBS. After 24 hours, the medium was changed to serum‐free medium, CAF‐CM, or CnP‐treated CAF‐CM, and the cells were treated with various concentrations of gemcitabine (0, 1, 10, or 100 nmol/L) for 48 hours. Cell viability was evaluated using CCK‐8 assays (Dojindo Laboratories), as described above. Gemcitabine was purchased from Selleck Chemicals (Houston, TX, USA).
2.8. Ultracentrifugation
Ultracentrifugation was carried out as previously described.20 Briefly, CAF‐CM was ultracentrifuged at 110 000 g for 70 minutes at 4°C. The supernatant was collected after the first centrifugation. The pellets were washed with 11 mL PBS, ultracentrifuged again, and then resuspended in serum‐free DMEM.
2.9. Cytokine array and ELISA
Cytokine profiles were compared between CAF‐CM and CnP‐treated CAF‐CM using the Proteome Profiler Human XL Cytokine Array Kit (ARY022B; R&D Systems, Minneapolis, MN, USA), according to the manufacturer's protocol. Detection and quantification of the array spots were carried out using the ImageQuant LAS 4000 imager (GE Healthcare). Concentration of cytokines in CAF‐CM and CnP‐treated CAF‐CM was measured by ELISA. IL‐6 (ab46027), IL‐8 (ab46032) and C‐C motif chemokine ligand 2 (CCL2) (ab100586) ELISA kits were purchased from Abcam (Cambridge, MA, USA) and used according to the manufacturer's protocol.
2.10. RNA extraction and RT‐qPCR
Cancer‐associated fibroblasts were incubated for 48 hours with and without 0.3 μg/mL CnP treatment. Total RNA was extracted using miRNeasy Mini Kit (Qiagen, Hilden, Germany) and quantified using an ND‐1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). RT‐qPCR was carried out as described previously.21 The following primers were used: IL‐6 forward, 5′‐TGCAATAACCACCCCTGACC‐3′; reverse,5′‐CCCAGTGGACAGGTTTCTGA‐3′;IL‐8 forward,5′‐TCCAAACCTTTCCACCCC‐3′;reverse,5′‐CACAACCCTCTGCACCCA‐3′;CCL2forward, 5′‐GAAGAATCACCAGCAGCAAGT‐3′;reverse, 5′‐TCCTGAACCCACTTCTGCTT‐3′; C‐X‐C motif chemokine ligand 12 (CXCL12) forward, 5′‐GTGGTCGTGCTGGTCCTC‐3′; reverse, 5′‐CACACTTGTCTGTTGTTGTTCTTC‐3′; and 18s rRNA forward, 5′‐GATGGTAGTCGCCGTGCC‐3′; reverse, 5′‐GCCTGCTGCCTTCCTTGG‐3′. For all RT‐qPCR analyses, 18s rRNA was used to normalize RNA input. mRNA levels of CAF with CnP treatment (0.3 μg/mL) is expressed relative to that of CAF without CnP treatment.
2.11. In vivo experiments
First, to investigate whether hPSC5 cells are actually activated in an in vivo xenograft model, hPSC5 was coinjected with SUIT‐2 cells at varying stroma‐to‐tumor ratios (1:1, 1:3 and 1:5). After 1 week, we evaluated the expression of activation marker α‐SMA in tumor by immunohistochemistry.
Moreover, to analyze the effects of CnP in vivo, we used a mouse xenograft model. SUIT‐2 cell suspensions (3 × 106 cells) in 200 μL PBS and SUIT‐2 (3 × 106 cells) + hPSC5 (1 × 106 cells) cell suspensions in 200 μL PBS were s.c. injected bilaterally into the flanks of 7‐week‐old female NOD‐SCID mice (CLEA Japan, Inc., Tokyo, Japan). We compared the proliferation rate between SUIT‐2 cells alone and SUIT‐2 + hPSC5 cells. We also analyzed the effects of CnP alone or in combination with gemcitabine in the SUIT‐2 + hPSC5 group. One week after implantation, we randomly divided mice into four groups: control; CnP; gemcitabine; and CnP plus gemcitabine groups. For CnP treatment, we gave 1.0 μg/g CnP by s.c. injection every other day. For gemcitabine treatment, we injected gemcitabine at a dose of 50 mg/kg i.p. twice a week for 28 days. Each group contained three mice (six xenografts). Tumor diameters and body weights were measured every other day and tumor volume was calculated using the formula: S × S × L/2, where S is the short diameter of the tumor, and L is the long diameter of the tumor. After excision, the xenografted tumors were microscopically evaluated by H&E staining, Sirius red staining, and immunohistochemistry. To evaluate adverse events of treatment, we sampled the blood of the mice, and serum biochemical tests were conducted by the Oriental Yeast Co. (Tokyo, Japan). All mouse experiments were carried out in compliance with the guidelines of the Institute for Laboratory Animal Research at Gunma University, Maebashi, Japan.
2.12. Immunohistochemistry and Sirius red staining
Immunohistochemistry was carried out on tumor samples as previously described.22 Primary antibodies were as follows: mouse monoclonal anti‐α‐SMA antibodies (A5247; 1:200; Sigma‐Aldrich) and anti‐Ki‐67 antibodies (M7240; 1:150; Dako; Agilent Technologies, Santa Clara, CA, USA). Sirius red staining was carried out using a Picro‐Sirius Red Stain Kit (ScyTek Laboratories, Inc., West Logan, UT, USA), according to the manufacturer's protocol. α‐SMA‐positive cells, Ki‐67 positive cells, and Sirius red‐stained areas were measured using ImageJ 1.51 image analysis software (NIH, Bethesda, MD, USA).
2.13. Statistical analysis
Data for continuous variables are expressed as means ± standard deviation (SD). Differences among groups were evaluated by ANOVA test. Results with P values <.05 were considered statistically significant. All statistical analyses were conducted using the JMP 13.0.0 software package (SAS Institute Inc., Cary, NC, USA).
3. RESULTS
3.1. Cancer‐associated fibroblasts promote cancer proliferation and invasion
To confirm the effect of CAF on cancer progression, we evaluated cancer proliferation and invasion using CM from CAF. CM from hPSC5 cells promoted cancer cell proliferation, and similar results were obtained using CM from the primary fibroblasts, CAF1 and CAF2 (Figure 1A). Moreover, although the invasion‐enhanced effect of CAF1 and CAF2 was small compared with that of hPSC5, the CM from hPSC5, CAF1, and CAF2 cells promoted the invasive ability of SUIT‐2 cancer cells (Figure 1B). Because of slow growth, the amount of secretion factors of CAF1 and CAF2 was considered to probably be small compared with that of hPSC5. These results suggest that CAF support cancer progression, and that the suppression of CAF is a potential therapeutic target for pancreatic cancer treatment.
Figure 1.
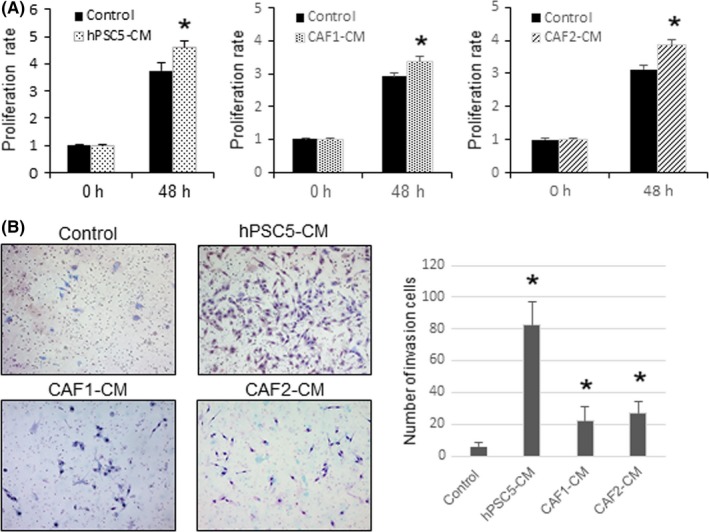
Effects of cancer‐associated fibroblasts conditioned medium (CAF‐CM) on pancreatic cancer cells (SUIT‐2 cells). A, Conditioned media from hPSC5, CAF1, and CAF2 enhance the proliferation of SUIT‐2 cells. B, Conditioned media from hPSC5, CAF1, and CAF2 enhance the invasive ability of SUIT‐2 cells. Representative photomicrographs are shown to the left and quantitative plots are shown to the right. *P < .05 (vs control). hPSC5, human pancreatic stellate cell line
3.2. Conophylline suppresses the activity of CAF
To examine the effect of CnP on CAF and pancreatic cancer cells, we evaluated the effect of CnP on hPSC5 and pancreatic cancer cell proliferation. Therefore, hPSC5 and SUIT‐2 were treated with various CnP concentrations for 48 hours. CnP significantly suppressed hPSC5 proliferation in a concentration‐dependent method compared with that of the control (Figure 2A). Furthermore, although there was an inhibitory effect on SUIT‐2 cells at high CnP concentrations greater than 1.0 μg/mL, there was no significant effect on the proliferation of SUIT‐2 at the lower CnP concentrations that inhibited hPSC5 proliferation (Figure 2B). Next, we investigated the effect of CnP on protein expression in hPSC5 cells. CnP remarkably suppressed the expression of collagen I. Likewise, the expression of α‐SMA was reduced by CnP in a concentration‐dependent method (Figure 2C). These results indicate the possibility of inhibiting CAF activity with CnP treatment.
Figure 2.
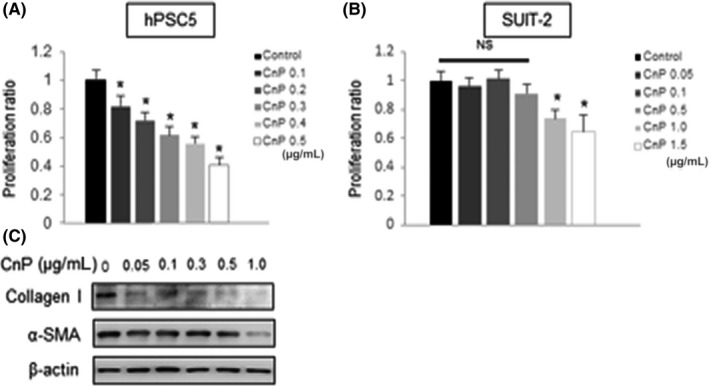
Effects of conophylline (CnP) on cancer‐associated fibroblasts (CAF) and SUIT‐2 cells. A, CnP inhibits the proliferation of hPSC5 concentration‐dependently. B, CnP inhibits the proliferation of SUIT‐2 cells only at concentrations >1.0 μg/mL. C, Collagen I and α‐smooth muscle actin (SMA) expression in hPSC5 cells treated by CnP is evaluated by western blotting. β‐Actin was used as the internal control. Representative blots are shown. *P < .05 (vs control). hPSC5, human pancreatic stellate cell line
3.3. Conophylline inhibits the stimulatory effects of CAF on pancreatic cancer cells
To determine the effects of CnP‐mediated inhibition of CAF on pancreatic cancer cells, we investigated pancreatic cancer cell proliferation, invasion, and chemosensitivity using CM from CnP‐treated fibroblasts. The proliferation‐enhancing effects of hPSC5‐CM were eliminated by CnP treatment of the hPSC5 cells and, further, similar results were obtained using primary CAF (Figure 3A). Similarly, CnP treatment of fibroblasts decreased the ability of their CM to enhance pancreatic cancer cell invasiveness (Figure 3B, Figure S1B). We confirmed that there was no significant direct effect of low concentrations of CnP on the invasiveness of pancreatic cancer cells (Figure S1A). Moreover, CAF‐CM decreased the sensitivity of pancreatic cancer cells to gemcitabine; however, this enhanced gemcitabine resistance was attenuated by CnP treatment of CAF (Figure 3C). Moreover, similar results were also obtained in SW1990 (Figure S2). These results suggest that CnP suppresses secretion of factors from CAF that are involved in cancer progression.
Figure 3.
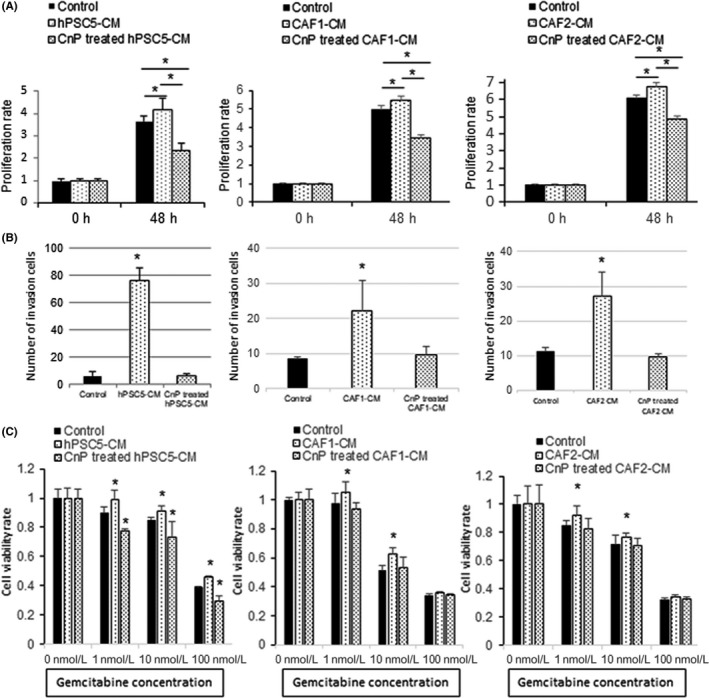
Conophylline (CnP) inhibits the promoting effects of cancer‐associated fibroblasts (CAF) on pancreatic cancer cells. A, Proliferation‐enhancing effects of CAF‐conditioned medium (CM) on SUIT‐2 are eliminated by CnP‐treated CAF‐CM. B, Invasiveness‐enhancing effects of CAF‐CM on SUIT‐2 are decreased by CnP‐treated CAF‐CM. C, Enhanced gemcitabine resistance of SUIT‐2 cells by CAF‐CM is improved by using CnP‐treated CAF‐CM. Left, hPSC5; middle, CAF1; right, CAF2, *P < .05 (vs control). hPSC5, human pancreatic stellate cell line
3.4. Conophylline decreases the cytokines produced by CAF that are involved in cancer progression
To assess the factors by which CnP suppresses CAF, we evaluated soluble factors, such as cytokines, and particulate factors, such as exosomes containing microRNA, by using ultracentrifugation. Supernatants after ultracentrifugation showed the same characteristics as the original solutions. In contrast, the resuspended pellets did not show significant effects on the pancreatic cancer cells (Figure 4A). These results suggest that CnP exerts its activity through soluble factors such as cytokines produced by CAF.
Figure 4.
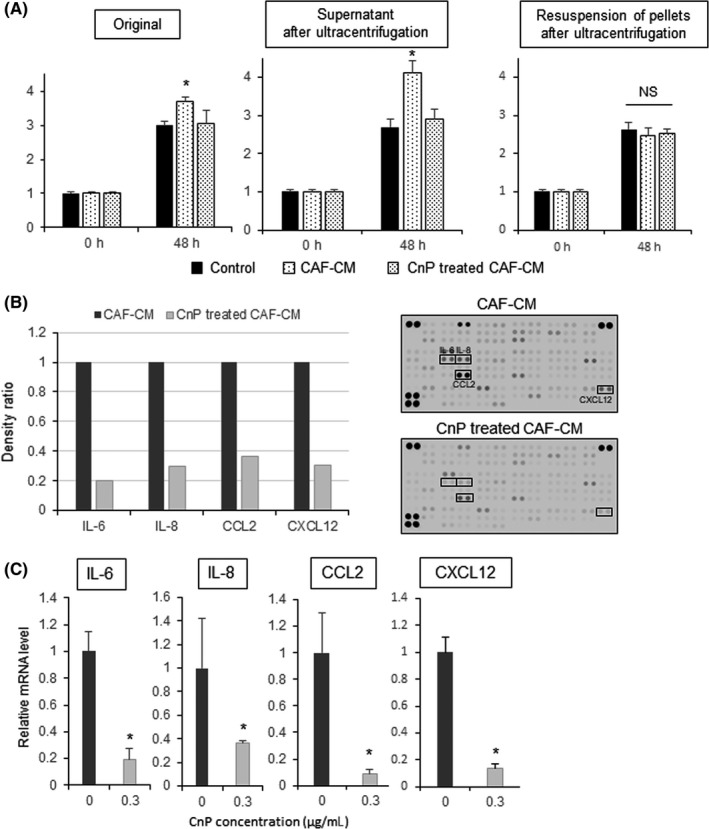
Conophylline (CnP) decreases the cytokines produced by cancer‐associated fibroblasts (CAF). A, After ultracentrifugation, supernatants showed the same phenotype as the original solutions. The resuspended pellets did not show this effect. B, Cytokine array comparing CAF‐conditioned medium (CM) and CnP‐treated CAF‐CM. Interleukin (IL)‐6, IL‐8, C‐C motif chemokine ligand 2 (CCL2), and C‐X‐C motif chemokine ligand 12 (CXCL12) were remarkably reduced by CnP treatment. Array images are shown to the right of the plotted data. C, mRNA levels of IL‐6, IL‐8, CCL2, and CXCL12. mRNA expression levels were quantified by RT‐qPCR and were normalized to 18S rRNA levels. *P < .05 (vs control)
Next, we analyzed a cytokine array to investigate the differences between CAF‐CM and CnP‐treated CAF‐CM. Although various cytokines were suppressed by CnP treatment, IL‐6, IL‐8, CCL2, and CXCL12 were decreased remarkably (Figure 4B). Moreover, we confirmed that these cytokines were also suppressed at the mRNA level by RT‐qPCR (Figure 4C). In fact, the concentration of IL‐6, IL‐8 and CCL2 decreased with CnP treatment (Figure S3). These results indicate that CnP inhibits tumor‐stromal interactions by suppressing the synthesis and secretion of paracrine factors from CAF, particularly IL‐6, IL‐8, CCL2, and CXCL12.
3.5. Cancer‐associated fibroblasts promote tumor proliferation and desmoplastic formation in an in vivo xenograft model
To indicate whether hPSC5 cells are activated when they are coinjected with a human pancreatic cancer cell line, we examined whether the expression of activation marker α‐SMA in hPSC5 cells was enhanced depending on the number of pancreatic cancer cells in an in vivo xenograft model. α‐SMA‐positive staining area are increased depending on the number of pancreatic cancer cells despite no change in the number of hPSC5 cells (Figure S4). This indicates that hPSC5 is activated in the presence of pancreatic cancer cells, and functioned similarly to CAF. Moreover, to examine the effect of CAF in vivo, we s.c. injected SUIT‐2 cells alone or SUIT‐2 + CAF (hPSC5) cells into the flanks of mice (Figure 5A). In the SUIT‐2 + CAF group, tumor growth was much greater compared with the tumor growth of SUIT‐2 cells alone (Figure 5A,B). Moreover, the tumors consisting of SUIT‐2 + CAF showed marked desmoplasia with higher Sirius red‐positive and α‐SMA‐positive staining compared with that in the tumors consisting of SUIT‐2 cells alone (Figure 5C‐E). Additionally, the proportion of Ki‐67‐positive pancreatic cancer cells was significantly higher in the SUIT‐2 + CAF group compared with that in the SUIT‐2 group (Figure 5C,F). These findings indicate that the stimulatory effects of CAF on pancreatic cancer cells are maintained even in vivo, and they play an important role in desmoplasia.
Figure 5.
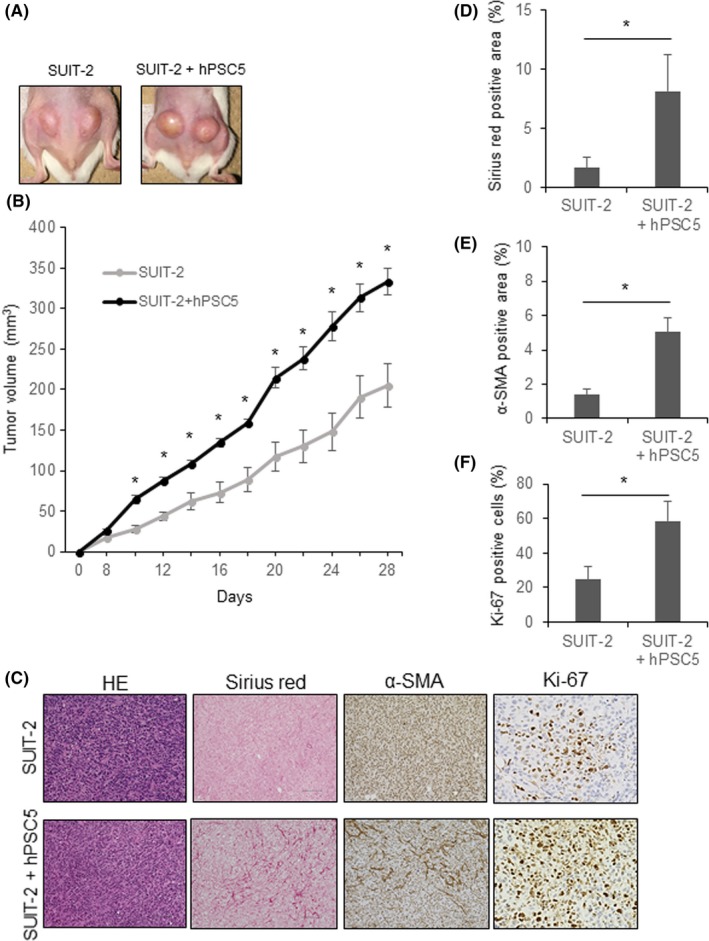
Cancer‐associated fibroblasts (CAF) promote tumor proliferation and desmoplastic formation in a mouse xenograft model. A, Representative photographs of tumors consisting of SUIT‐2 cells (LEFT), and SUIT‐2 + hPSC5 cells combined (right). B, Tumor growth curve of SUIT‐2 cells and SUIT‐2 + hPSC5 cells. Tumor growth of SUIT‐2 + hPSC5 cells was much greater compared with that of SUIT‐2 cells alone. C, Representative photomicrographs of histological evaluations of tumor tissue; original magnification (HE, Sirius red and α‐smooth muscle actin [SMA], ×200; scale bar, 100 μm; original magnification [Ki‐67], ×400). D, Sirius red‐positive stained area of tumors consisting of SUIT‐2 + hPSC5 cells was significantly higher than that of SUIT‐2 cells alone. E, α‐SMA‐positive stained area of tumors consisting of SUIT‐2 + hPSC5 cells was significantly higher than that of SUIT‐2 cells alone. F, Proportion of Ki‐67‐positive cells was significantly higher in tumors consisting of SUIT‐2 + hPSC5 cells compared with that of SUIT‐2 cells alone. *P < .05. hPSC5, human pancreatic stellate cell line
3.6. Conophylline reduces desmoplasia of tumors consisting of SUIT‐2 + CAF, and combination therapy with gemcitabine markedly inhibits tumor proliferation
To evaluate the possibility of using CnP alone or in combination with gemcitabine as a therapeutic tool, we investigated the effect of CnP, gemcitabine, and CnP plus gemcitabine on tumor proliferation in vivo. CnP itself inhibited tumor proliferation slightly (Figure 6A,B). Desmoplastic changes and proliferation‐promoting effects that were enhanced by CAF were attenuated by CnP treatment (Figure 6C‐F). Furthermore, although gemcitabine alone inhibited tumor proliferation, combination of CnP and gemcitabine therapy inhibited tumor growth to the greatest degree, and tumor shrinkage was only observed over time with this combination therapy (Figure 6A,B). Additionally, we evaluated adverse events associated with treatment. There were no significant differences among the groups regarding body weight loss and organ toxicity (Figure S5). These results indicate that CnP suppresses desmoplasia of pancreatic cancer, and combination therapy with gemcitabine has synergic effects during pancreatic cancer treatment.
Figure 6.
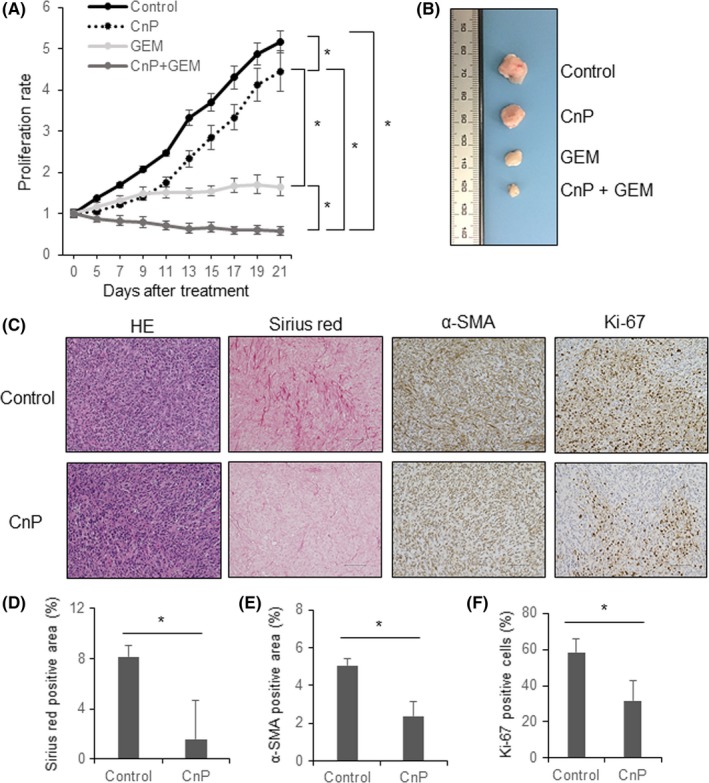
Conophylline (CnP) reduces desmoplasia of tumors consisting of SUIT‐2 + hPSC5 cells, and combination therapy with gemcitabine markedly inhibits tumor proliferation. A, Tumor growth curve showing that combination therapy with CnP plus gemcitabine suppresses the growth of tumors consisting of SUIT‐2 + hPSC5 cells to the greatest extent. B, Representative photographs of tumors from the four treatment groups (control, CnP, gemcitabine [GEM], CnP + GEM). C, Representative photomicrographs of histological examinations comparing control and CnP treatment (original magnification, ×200; scale bar, 100 μm); D‐F, CnP treatment significantly reduces Sirius red‐positive staining (D), α‐smooth muscle actin (SMA)‐positive staining (E), and percentage of Ki‐67‐positive cells (F) in tumors consisting of SUIT‐2 + hPSC5 cells. *P < .05. hPSC5, human pancreatic stellate cell line
4. DISCUSSION
Herein, we have shown that CAF promote pancreatic cancer malignancy in vitro and in vivo, and that CnP suppresses the ability of CAF to enhance pancreatic cancer cell activity. We also showed that various cytokines, such as IL‐6, IL‐8, CCL2, and CXCL12, produced by CAF are strongly suppressed by CnP treatment. Moreover, in an in vivo experiment, CnP suppressed the desmoplastic change in pancreatic cancer tissue derived from CAF, and combination therapy with gemcitabine inhibited tumor growth to the greatest extent, suggesting that suppression of CAF by CnP treatment may be a useful therapeutic tool to treat pancreatic cancer.
Recently, tumor‐stromal interactions in cancer tissues have been recognized as playing important roles in tumor progression, and have been highlighted in several reviews.8, 23, 24, 25 In particular, in dense fibrotic stromal tumors such as in pancreatic cancers, CAF play crucial roles in the tumor microenvironment and cancer progression. Considering this, anticancer therapy targeting CAF or the inhibitors of the cytokines secreted by CAF has been actively investigated.26 Antifibrotic drugs are thought to be candidates for targeting CAF because of their ability to suppress fibroblast activity. Actually, recent studies have shown that pirfenidone and the multi‐kinase inhibitor, nintedanib, which are drugs to treat idiopathic pulmonary fibrosis, can inhibit cancer progression through suppression of CAF in pancreatic cancer,27 breast cancer,28 and lung adenocarcinoma.29 It was reported that pirfenidone and nintedanib suppressed TGF‐β signaling and production of extracellular matrix components in tumor‐stromal interactions. Thus, targeting CAF themselves is a promising therapeutic strategy for cancer treatment.
In the present study, we showed that CnP suppressed the activities of CAF, and significantly inhibited the synthesis and production of various cytokines, especially IL‐6, IL‐8, CCL2, and CXCL12. These factors play important roles in the tumor microenvironment in pancreatic cancer.4, 15, 30 IL‐6 secreted by CAF was reported to induce the invasiveness of pancreatic cancer cells through STAT3 signal activation,31 and the inhibition of IL‐6 signaling significantly reduced tumor proliferation in a pancreatic cancer xenograft model.32 Additionally, IL‐8 derived from CAF was reported to promote pancreatic cancer invasion and metastasis.33 Moreover, CCL2 and CXCL12 were reported to be associated with cancer progression and resistance to radiotherapy, as well as resistance to gemcitabine chemotherapy in pancreatic cancer cells.34, 35, 36, 37, 38 In the current study, similar to these previous reports, CAF‐CM, which abundantly contained these cytokines, promoted pancreatic cancer progression and enhanced chemoresistance to gemcitabine. However, CnP‐treated CAF‐CM eliminated the ability of CAF to enhance the malignant potential and chemoresistance of pancreatic cancer cells. Therefore, the antitumor effects of CnP were considered to be derived from the inhibition of stromal activity by suppressing the synthesis and production of various cytokines, such as IL‐6, IL‐8, CCL2, and CXCL12.
We previously reported that CnP suppressed liver fibrosis induced by thioacetamide through the suppression of hepatic stellate cells.10 Similarly, in the present study, CnP suppressed the desmoplastic changes in tumors that consisted of pancreatic cancer cells and CAF in a mouse xenograft model. CnP significantly decreased the Sirius red‐positive area and α‐SMA‐positive cells within the tumors. These results suggest that the effects of CnP in vivo include suppression of CAF proliferation and suppression of the production of stromal components. In fact, CnP suppressed CAF proliferation and collagen expression concentration‐dependently in vitro. These effects of CnP may contribute to improved drug delivery within tumors. Previously, nab‐paclitaxel, which is a new agent for pancreatic cancer chemotherapy in the clinic, a sonic hedgehog inhibitor, and a vitamin D analog were reported to reduce stromal volumes and increase intratumoral gemcitabine concentrations.39, 40, 41 Thus, the reduction in stromal volume in tumors is expected to improve chemosensitivity. In our study, although CnP alone only partially inhibited tumor growth, CnP plus gemcitabine combination therapy was the most effective treatment, probably due to improved drug delivery within tumors. It is important to combine anticancer drugs such as gemcitabine based on the suppression of fibrotic changes. Considering these findings, combination therapy with CnP, which targets CAF desmoplastic activity, and gemcitabine, which targets the cancer cells, has synergistic antitumor effects. Thus, combination therapy with CnP and gemcitabine is a promising and potentially ideal treatment strategy for pancreatic cancer with desmoplasia.
Although the detailed mechanisms of CnP remain unclear, a previous report showed that CnP inhibits cAMP‐responsive element binding protein (CREB) activation.42 CREB is a crucial transcription factor, which regulates a wide range of biological processes to orchestrate proper cell differentiation and cell growth.43, 44 Moreover, CREB has previously been shown to regulate inflammatory responses by directly regulating gene transcription of proinflammatory genes, such as IL‐6 and TNF‐α,45 and CREB silencing has been shown to reduce the levels of IL‐6, IL‐8, and CCL2 in malignant mesothelioma cells.46 In contrast, another report showed that CREB promoted hepatic fibrosis through the transactivation of TGF‐β expression in rats.47 Thus, there is a possibility that CREB is a key factor involved in the activity of CnP. We confirmed that TGF‐β expression of CAF was decreased by CnP treatment (Figure S6). Thus, although CnP might affect TGF‐β signaling, further studies are clearly needed to identify the mechanism of CnP activity.
It is important to validate the results of in vivo xenograft experiments with established CAF; however, the growth speed of established CAF was very slow, and we did not immortalize CAF. Therefore, we could not obtain a sufficient number of established CAF for use in in vivo experiments. There are some studies where immortalized CAF have been used in in vivo experiments with cancer cells.5 However, when immortalized CAF is used in vivo experiments with cancer cells, it is unclear whether it is appropriate as an actual microenvironment of tumor. The origins and homology of CAF are still controversial.48 However, some studies have reported that a major source of CAF in pancreatic cancer is PSC.49, 50 Therefore, it is difficult to distinguish CAF from PSC. In general, CAF show myofibroblast‐like shaped cells and express α‐SMA. hPSC5 cells (PSC) used in the present study also showed similar characteristics; they were established from pancreatic cancer tissue. In this milieu, we used hPSC5 as CAF, which is a limitation of the present study.
In conclusion, to our knowledge for the first time, we showed that CnP suppressed CAF activity and the production of cancer‐promoting cytokines produced by CAF. There are two aspects to the effects of CnP. First, CnP inhibits tumor‐stromal interactions in pancreatic cancer directly by the suppression of cancer‐promoting cytokines derived from CAF. Second, CnP suppresses desmoplastic changes in tumors, which may improve drug delivery within the tumors. Therefore, combination therapy with CnP and anticancer drugs, such as gemcitabine, may be used as a therapeutic strategy to overcome refractory pancreatic cancers.
CONFLICTS OF INTEREST
Kazuo Umezawa received a fund donation from Shenzhen Wanhe Pharmaceutical Co., Ltd. The other authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
The present study was supported by Grants‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS; grant numbers JP17K16529). The present study was also supported by Yokoyama Foundation of Clinical Pharmacology and Japanese Society for Gastroenterological Carcinogenesis.
Ishii N, Araki K, Yokobori T, et al. Conophylline suppresses pancreatic cancer desmoplasia and cancer‐promoting cytokines produced by cancer‐associated fibroblasts. Cancer Sci. 2019;110:334–344. 10.1111/cas.13847
REFERENCES
- 1. Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22:9694‐9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605‐1617. [DOI] [PubMed] [Google Scholar]
- 3. Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144:1210‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pan B, Liao Q, Niu Z, Zhou L, Zhao Y. Cancer‐associated fibroblasts in pancreatic adenocarcinoma. Future Oncol. 2015;11:2603‐2610. [DOI] [PubMed] [Google Scholar]
- 5. Hwang RF, Moore T, Arumugam T, et al. Cancer‐associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikenaga N, Ohuchida K, Mizumoto K, et al. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology. 2010;139:1041‐1051, 51.e1–8. [DOI] [PubMed] [Google Scholar]
- 7. Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer‐associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2016;36:1770‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer‐associated fibroblasts in pancreatic cancer. J Hematol Oncol. 2017;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kojima I, Umezawa K. Conophylline: a novel differentiation inducer for pancreatic beta cells. Int J Biochem Cell Biol. 2006;38:923‐930. [DOI] [PubMed] [Google Scholar]
- 10. Kubo N, Saito R, Hamano K, et al. Conophylline suppresses hepatic stellate cells and attenuates thioacetamide‐induced liver fibrosis in rats. Liver Int. 2014;34:1057‐1067. [DOI] [PubMed] [Google Scholar]
- 11. Saito R, Yamada S, Yamamoto Y, et al. Conophylline suppresses pancreatic stellate cells and improves islet fibrosis in Goto‐Kakizaki rats. Endocrinology. 2012;153:621‐630. [DOI] [PubMed] [Google Scholar]
- 12. Bachem MG, Schünemann M, Ramadani M, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907‐921. [DOI] [PubMed] [Google Scholar]
- 13. Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin‐33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:821‐832. [DOI] [PubMed] [Google Scholar]
- 14. Mantoni TS, Schendel RR, Rödel F, et al. Stromal SPARC expression and patient survival after chemoradiation for non‐resectable pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1806‐1815. [DOI] [PubMed] [Google Scholar]
- 15. Hamada S, Masamune A, Yoshida N, Takikawa T, Shimosegawa T. IL‐6/STAT3 plays a regulatory role in the interaction between pancreatic stellate cells and cancer cells. Dig Dis Sci. 2016;61:1561‐1571. [DOI] [PubMed] [Google Scholar]
- 16. Lau EY, Lo J, Cheng BY, et al. Cancer‐associated fibroblasts regulate tumor‐initiating cell plasticity in hepatocellular carcinoma through c‐Met/FRA1/HEY1 signaling. Cell Rep. 2016;15:1175‐1189. [DOI] [PubMed] [Google Scholar]
- 17. Umezawa K, Ohse T, Yamamoto T, Koyano T, Takahashi Y. Isolation of a new Vinca alkaloid from the leaves of Ervatamia microphylla as an inhibitor of ras functions. Anticancer Res. 1994;14:2413‐2417. [PubMed] [Google Scholar]
- 18. Tsukagoshi M, Araki K, Yokobori T, et al. Overexpression of karyopherin‐a2 in cholangiocarcinoma correlates with poor prognosis and gemcitabine sensitivity via nuclear translocation of DNA repair proteins. Oncotarget. 2017;8:42159‐42172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishii N, Araki K, Yokobori T, et al. Reduced FBXW7 expression in pancreatic cancer correlates with poor prognosis and chemotherapeutic resistance via accumulation of MCL1. Oncotarget. 2017;8:112636‐112646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442‐17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Igarashi T, Araki K, Yokobori T, et al. Association of RAB5 overexpression in pancreatic cancer with cancer progression and poor prognosis via E‐cadherin suppression. Oncotarget. 2017;8:12290‐12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishii N, Araki K, Yokobori T, et al. Poor prognosis in cholangiocarcinoma patients with low FBXW7 expression is improved by chemotherapy. Oncol Lett. 2017;13:3653‐3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392‐401. [DOI] [PubMed] [Google Scholar]
- 24. Kubo N, Araki K, Kuwano H, Shirabe K. Cancer‐associated fibroblasts in hepatocellular carcinoma. World J Gastroenterol. 2016;22:6841‐6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Junttila MR, de Sauvage FJ. Influence of tumour micro‐environment heterogeneity on therapeutic response. Nature. 2013;501:346‐354. [DOI] [PubMed] [Google Scholar]
- 26. De Vlieghere E, Verset L, Demetter P, Bracke M, De Wever O. Cancer‐associated fibroblasts as target and tool in cancer therapeutics and diagnostics. Virchows Arch. 2015;467:367‐382. [DOI] [PubMed] [Google Scholar]
- 27. Kozono S, Ohuchida K, Eguchi D, et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013;73:2345‐2356. [DOI] [PubMed] [Google Scholar]
- 28. Takai K, Le A, Weaver VM, Werb Z. Targeting the cancer‐associated fibroblasts as a treatment in triple‐negative breast cancer. Oncotarget. 2016;7:82889‐82901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gabasa M, Ikemori R, Hilberg F, Reguart N, Alcaraz J. Nintedanib selectively inhibits the activation and tumour‐promoting effects of fibroblasts from lung adenocarcinoma patients. Br J Cancer. 2017;117:1128‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tjomsland V, Niklasson L, Sandstrom P, et al. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin Dev Immunol. 2011;2011:212810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagathihalli NS, Castellanos JA, VanSaun MN, et al. Pancreatic stellate cell secreted IL‐6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget. 2016;7:65982‐65992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goumas FA, Holmer R, Egberts JH, et al. Inhibition of IL‐6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int J Cancer. 2015;137:1035‐1046. [DOI] [PubMed] [Google Scholar]
- 33. Wang T, Notta F, Navab R, et al. Senescent carcinoma‐associated fibroblasts upregulate IL8 to enhance prometastatic phenotypes. Mol Cancer Res. 2017;15:3‐14. [DOI] [PubMed] [Google Scholar]
- 34. Monti P, Leone BE, Marchesi F, et al. The CC chemokine MCP‐1/CCL2 in pancreatic cancer progression: regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003;63:7451‐7461. [PubMed] [Google Scholar]
- 35. Kalbasi A, Komar C, Tooker GM, et al. Tumor‐derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2017;23:137‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang H, Wu H, Guan J, et al. Paracrine SDF‐1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL‐6 autocrine loop in pancreatic cancer cells. Oncotarget. 2015;6:3085‐3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo JC, Li J, Zhou L, et al. CXCL12‐CXCR7 axis contributes to the invasive phenotype of pancreatic cancer. Oncotarget. 2016;7:62006‐62018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen B, Zheng MQ, Lu JW, Jiang Q, Wang TH, Huang XE. CXCL12‐CXCR4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pac J Cancer Prev. 2013;14:5403‐5408. [DOI] [PubMed] [Google Scholar]
- 39. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab‐paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548‐4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor‐mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koide N, Kondo Y, Odkhuu E, et al. Inhibition of receptor activator of nuclear factor‐kappaB ligand‐ or lipopolysaccharide‐induced osteoclast formation by conophylline through downregulation of CREB. Immunol Lett. 2014;161:31‐37. [DOI] [PubMed] [Google Scholar]
- 43. Steven A, Seliger B. Control of CREB expression in tumors: from molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget. 2016;7:35454‐35465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413‐6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mayr B, Montminy M. Transcriptional regulation by the phosphorylation‐dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599‐609. [DOI] [PubMed] [Google Scholar]
- 46. Westbom CM, Shukla A, MacPherson MB, et al. CREB‐induced inflammation is important for malignant mesothelioma growth. Am J Pathol. 2014;184:2816‐2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang P, Deng L, Zhuang C, Cheng C, Xu K. p‐CREB‐1 promotes hepatic fibrosis through the transactivation of transforming growth factor‐beta1 expression in rats. Int J Mol Med. 2016;38:521‐528. [DOI] [PubMed] [Google Scholar]
- 48. Haviv I, Polyak K, Qiu W, Hu M, Campbell I. Origin of carcinoma associated fibroblasts. Cell Cycle. 2009;8:589‐595. [DOI] [PubMed] [Google Scholar]
- 49. Öhlund D, Handly‐Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moir JA, Mann J, White SA. The role of pancreatic stellate cells in pancreatic cancer. Surg Oncol. 2015;24:232‐238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials