

SUMMARY
The TET2 DNA dioxygenase regulates gene expression by catalyzing demethylation of 5-methylcytosine, thus epigenetically modulating the genome. TET2 does not contain a sequence-specific DNA-binding domain, and how it is recruited to specific genomic sites is not fully understood. Here we carried out a mammalian two-hybrid screen and identified multiple transcriptional regulators potentially interacting with TET2. The SMAD nuclear interacting protein 1 (SNIP1) physically interacts with TET2 and bridges TET2 to bind several transcription factors, including c-MYC. SNIP1 recruits TET2 to the promoters of c-MYC target genes, including those involved in DNA damage response and cell viability. TET2 protects cells from DNA damage-induced apoptosis dependending on SNIP1. Our observations uncover a mechanism for targeting TET2 to specific promoters through a ternary interaction with a co-activator and many sequence-specific DNA-binding factors. This study also reveals a TET2-SNIP1-c-MYC pathway in mediating DNA damage response, thereby connecting epigenetic control to maintenance of genome stability.
Graphical Abstract
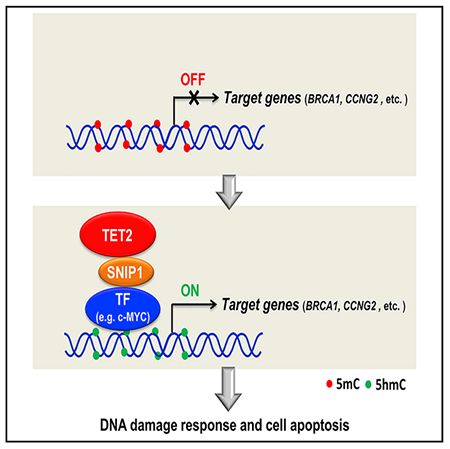
In Brief
Chen et al. show SNIP1 recruits TET2 to the promoters of c-MYC target genes, including those involved in DNA damage response and cell viability. This study uncovers a mechanism for targeting TET2 to specific promoters through a ternary interaction with a co-activator and sequence-specific DNA-binding factors and also reveals a TET2-SNIP1-c-MYC pathway in mediating DNA damage response, thereby connecting epigenetic control to maintenance of genome stability.
INTRODUCTION
The ten-eleven translocation (TET) family of proteins, which includes TET1, TET2, and TET3 in mammalian cells, catalyzes three sequential oxidation reactions: first converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), then to 5-for mylcytosine (5fC), and finally to 5-carboxylcytosine (5caC) (He et al., 2011; Ito et al., 2011; Tanida et al., 2012). A subsequent base-excision repair, by thymine-DNA glycosylase (TDG) or other yet unknown DNA repair enzymes, leads to eventual DNA demethylation (Kohli and Zhang, 2013). Pathologically, the TET2 gene is frequently mutated in human hematopoietic malignancies of both myeloid, in particular acute myeloid leukemia (AML; ~15%–20%), and lymphoid lineages, such as angioimmunoblastic T cell lymphoma (AITL; ~30%–40%) (Delhommeau et al., 2009; Quivoron et al., 2011; Tefferi et al., 2009). Genetic ablation of individual Tet gene has demonstrated broad functions of TET dioxygenases, including meiosis (Yamaguchi et al., 2012), zygotic development (Gu et al., 2011), induced pluripotent stem cell (iPSC) reprogramming (Costa et al., 2013; Doege et al., 2012; Piccolo et al., 2013), somatic cell differentiation (Moran-Crusio et al., 2011), immune response (Ichiyama et al., 2015; Yang et al., 2015; Zhang et al., 2015), cardiac protection (Fuster et al., 2017; Jaiswal et al., 2017), and tumor suppression (Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). How TET enzymes achieve such diverse functions is currently not well understood but is believed to be linked to the regulation of specific target genes.
All three TET proteins contain a conserved, cysteine-rich dioxygenase (CD) domain in their C-terminal region that binds to Fe(II) and α-ketoglutarate (α-KG) and catalyzes the oxidation reaction (Iyer et al., 2009; Tahiliani et al., 2009). The N-terminal region is more divergent among three TET proteins, and its function is unclear. Both TET1 and TET3 contain a CXXC-type zinc finger domain. However, TET2 lacks the CXXC DNA-binding domain and instead interacts with a CXXC domain protein, IDAX (Ko et al., 2013). The IDAX CXXC domain binds to DNA sequences containing unmethylated CpG dinucleotides in promoters but do not appear to recognize specific DNA sequences (Ko et al., 2013). How TET2, like other chromatin-modifying enzymes that in general do not have specific DNA-binding domains, is recruited to specific sites in the genome to modulate target gene expression is not fully understood.
Immunopurification coupled with mass spectrometry (IP-MS) has been previously used by a number of groups in attempt to identify TET-interacting proteins. By this approach, only very few proteins have been identified and functionally characterized, including O-linked β-N-acetylglucosamine transferase (OGT) (Chen et al., 2013; Deplus et al., 2013; Vella et al., 2013; Zhang et al., 2014). Guided by their mutual exclusive mutations in AML, we and others have previously demonstrated that DNA sequence-specific transcription factor Wilms tumor protein (WT1) physically interacts with TET2 (Rampal et al., 2014; Wang et al., 2015). These results provide early evidence supporting a possible mechanism, by interacting with a DNA sequence-specific transcription factor, for targeting TET2 to particular genes.
In this study, we hypothesized that TET2 is generally recruited to specific genes in part through interaction with transcriptional regulators that either contain sequence-specific DNA recognition domains or can interact with DNA-binding proteins. We carried out a mammalian two-hybrid screen and identified transcriptional regulators that interact with TET2. Functional characterizations of one newly identified TET2-interacting transcriptional co-activator, the SMAD nuclear interacting protein 1 (SNIP1), led to the discovery of a mechanism for targeting TET2 to specific promoters through a ternary interaction with SNIP1 and sequence-specific DNA-binding factors, including c-MYC. Previous studies have reported that TET2 is required for the generation of damage-associated 5hmC foci in HeLa cells (Kaferetal., 2016). Moreover, altered expression of many DNA damage repair genes and spontaneous progressive accumulation of γH2AX are observed in Tet2 and Tet3 double-knockout mouse myeloid cells (An et al., 2015). These studies imply a potential role of TET2 in regulating DNA damage response and ensuring genome integrity, but the underlying mechanisms remain unclear. Our present study reveals a molecular basis for the co-activator function of SNIP1 to recruit TET2 as well as a TET2-SNIP1-c-MYC pathway in DNA damage response.
RESULTS
Identification of SNIP1 as One of Multiple TET2-Interacting Transcription Regulators
To investigate the mechanism of TET2 recruitment to genomic sites, we carried out a mammalian two-hybrid screen of a human transcription factor library containing 1,126 known or putative DNA-binding proteins (Zhao et al., 2008) to search for potential TET2-interacting DNA-binding proteins (Figures 1A and 1B). To optimize the screening system, we fused one or two copies of VP16 transactivation domain (AD) to human full-length TET2, its catalytic domain (TET2CD), and non-catalytic domain (TET2n) in different ways. OGT, a known TET2-interacting protein as a positive control, was fused to Gal4 DNA-binding domain (DBD). Gal4(DBD)-OGT and the 9 × UAS-luciferase reporter, which was driven by the nine Gal4-binding elements, were co-transfected into HEK293T cells with or without various TET2 fusion (Figure 1C). After cell transfection, the luciferase reporter activity was measured as an indicator for protein-protein interaction. As shown, fusion of VP16 to the N-terminal domain of TET2 produced stronger reporter activation than the similar fusion to the C-terminal domain of TET2, and fusion of two copies of VP16 (2 × VP16) to TET2 produced stronger reporter activation than one copy of VP16. In addition, our data also demonstrated that the TET2CD, but not TET2N, interacted with OGT, which is in line with previous studies (Chen etal., 2013; Deplus et al., 2013).
Figure 1. Identification of SNIP1 as One of the TET2 Potential Interacting Transcription Regulators.
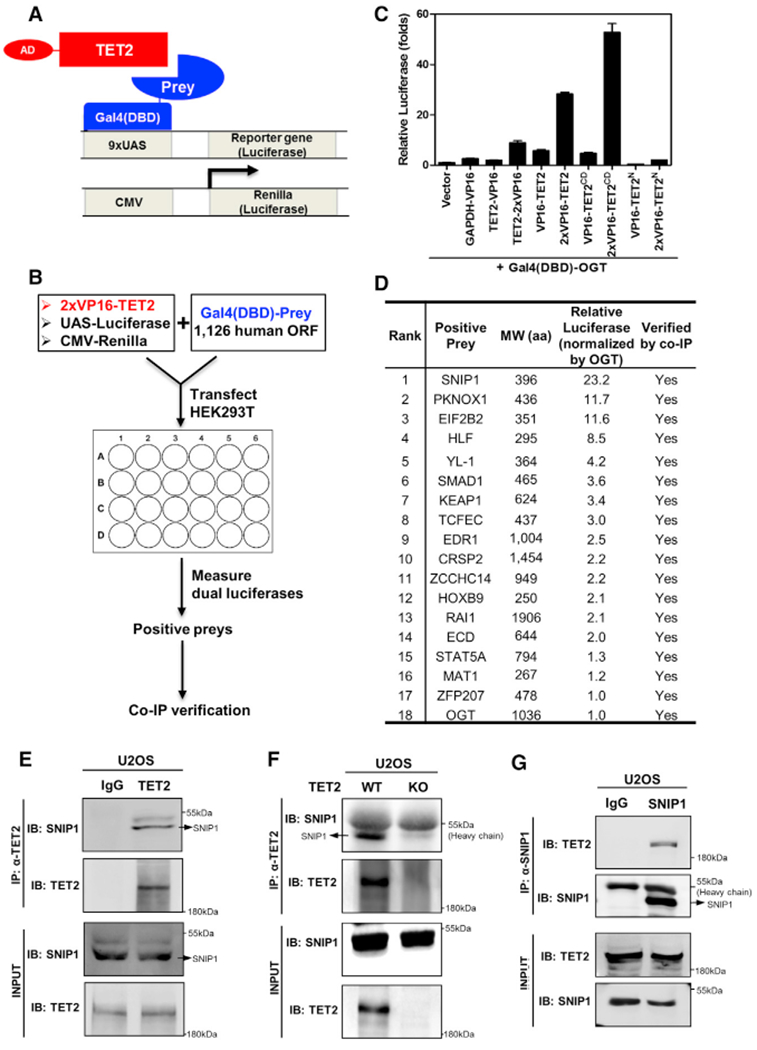
(A and B) Schematic representation of the mammalian two-hybrid system and dual-luciferase reporter system used to search for TET2-interacting proteins. Human full-length TET2 fused toVP16transactivation domain (AD), preys (1,126 human open reading frames [ORFs]) fused to Gal4 DNA-binding domain (DBD) (A), UAS-luciferase reporter plasmid, and CMV-Renilla control plasmid were co-transfected in HEK293T cells. At 30 hr after transfection, the luciferase reporter activity was measured as an indicator for protein-protein interaction (B). Moredetailed information is provided in STAR Methods.
(C) Optimization of the mammalian two-hybrid screening system. One or two copies of VP16 were fused to either the N or C terminus of human full-length or truncated TET2, and OGT as a positive control was fused to C terminus of Gal4(DBD). Gal4(DBD)-OGT and the 9 × UAS-luciferase reporter, which was driven by nine Gal4-binding elements, were co-transfected into HEK293 cells with various TET2 fusion as indicated. At 30 hr after transfection, the luciferase reporter activity was measured.
(D) The reporter-based screen identifies SNIP1 as the strongest positive. Candidates with relative luciferase activation higher than OGT were shown, and their protein interaction with TET2 was verified by co-immunoprecipitation (Figure S1).
(E and F) Endogenous TET2 interacts with endogenous SNIP1 in U2OS cells. TET2 protein in U2OS cells (E) or TET2 deficient U2OS cells (F) was purified by immunoprecipitation with an antibody against TET2, followed by western blot to detect endogenous SNIP1 using a SNIP1 antibody. Rabbit IgG was used as negative control.
(G) Endogenous SNIP1 interacts with endogenous TET2 in U2OS cells. SNIP1 protein in U2OS cells was purified by immunoprecipitation with an antibody against SNIP1, followed by western blot to detect endogenous TET2 using a TET2 antibody. Rabbit IgG was used as negative control.
After optimization, we transfected the 2 × VP16 fused to the N-terminal domain of TET2 together with 1,126 “prey” proteins fused to Gal4(DBD) as well as luciferase reporters to identify positive preys. The protein association of ectopically expressed positive prey proteins with TET2 was verified by co-immunoprecipitation (co-IP) and western blotting. Eventually, we discovered 17 transcription regulators that exhibited stronger luciferase activation than OGT when co-overexpressed with 2 × VP16-TET2 (Figures 1D and S1A; Table S1).
Among the transcription regulators identified to interact with TET2, SNIP1 is one of the strongest positives (Figure 1D). SNIP1 is a 396-amino acid Forkhead-associated (FHA) domain-containing protein, which was first identified as a SMAD nuclear interacting protein and can interact with p300/CBP histone acetyltransferase (Kim et al., 2000). Subsequent investigations have linked the function of SNIP1 as a co-activator for c-MYC (Fujii et al., 2006) in DNA damage response (Yu et al., 2008) and cell-fate decision of stem cells (Chng et al., 2010).
The interaction between Flag-TET2 and SNIP1 was confirmed by co-IP with two differentially tagged SNIP1 constructs (Figures S1B and S1C) and by reciprocal co-IP (Figure S1D). When overexpressed in HEK293T cells, we found that Myc-tagged SNIP1 specifically interacted with full-length TET2 but not full-length TET1 and TET3 (Figure S1E). Furthermore, Flag-SNIP1 was found to interact with endogenous TET2 in U2OS cells (Figure S1F). The association between endogenous TET2 and SNIP1 was observed using immunoprecipitation with TET2 antibody followed by western blotting with SNIP1 antibody in U2OS cells (Figure 1E). To confirm the specificity of the endogenous co-IP, we used a CRISPR/Cas9-mediated genome-editing technique to delete TET2 in U2OS cells (Figures S2A and S2B) and found that SNIP1 was immunoprecipitated by TET2 antibody in wild-type cells but not the TET2-knockout cells (Figure 1F). Reciprocal co-IP confirmed the association of endogenous SNIP1 with TET2 in U2OS cells (Figure 1G). Collectively, all these data demonstrate that SNIP1 physically interacts with TET2.
The N-Terminal Domains of SNIP1 and TET2 Are Responsible for Their Interaction
To gain insight into the TET2-SNIP1 interaction, we mapped the domain(s) of TET2 and SNIP1 responsible for their interaction. The N-terminal (residues 1–1127) and C-terminal (residues 1128–2002) domains of human TET2 were co-overexpressed with Gal4(DBD)-tagged SNIP1 in HEK293T cells. We observed that Gal4(DBD)-SNIP1 interacted with the N-terminal but not C-terminal domain of TET2 (Figure 2A). GST pull-down assay confirmed that the N terminus of TET2 directly interacted with SNIP1 (Figure 2B). Given that the N terminus of TET2 is mutated in solid tumors (Table S2), we generated constructs with patient-derived mutants of N-terminal TET2 from osteosarcoma, breast carcinoma, and lung adenocarcinoma. However, none of the ten selected mutations in the N terminus of TET2 affected the TET2-SNIP1 interaction (Figure S3).
Figure 2. The N Terminus of SNIP1 Interacts with the N Terminus of TET2.
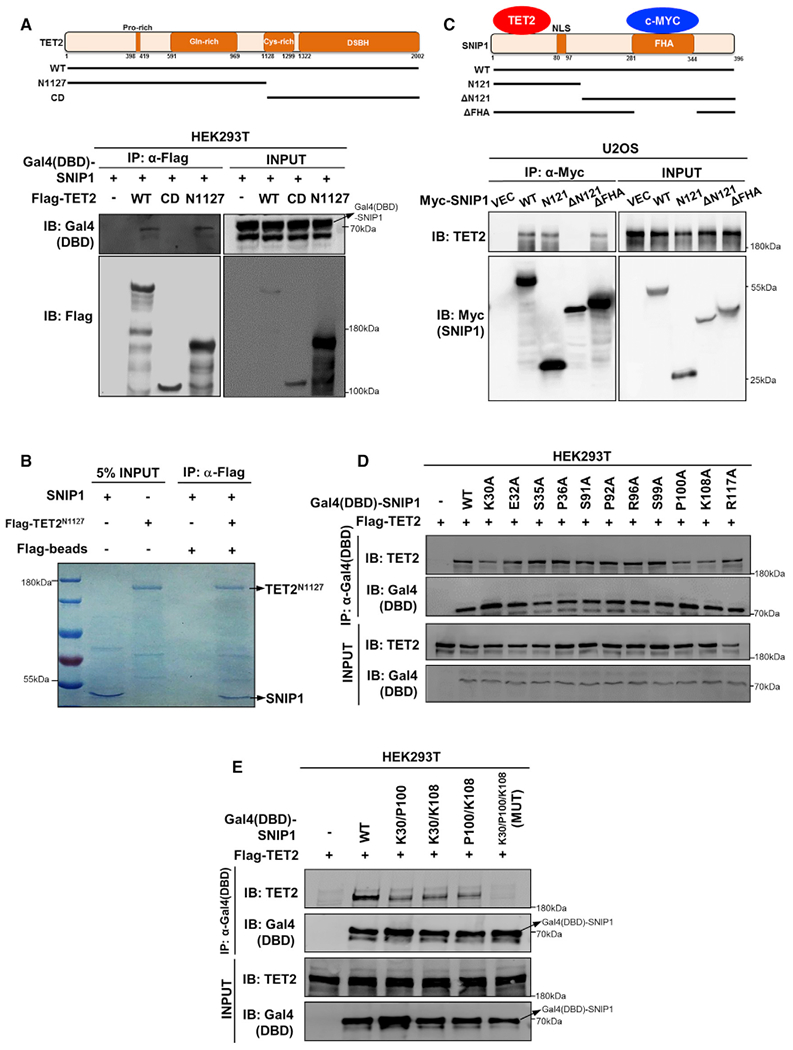
(A) SNIP1 interacts with the N-terminal domain of TET2. Schematic representation of human full-length TET2 is shown (top). Flag-tagged TET2 truncations were individually co-overexpressed with Gal4(DBD)-SNIP1 in HEK293T cells. Each truncation ofTET2 was purified by immunoprecipitation with Flag beads, followed by western blot to detect Gal4(DBD)-SNIP1 with the Gal4-DBD antibody.
(B) SNIP1 interacts with the N terminus of TET2 in vitro. Flag-tagged TET2 N terminus was overexpressed in HEK293T cells and purified by immunoprecipitation with Flag beads and subsequent Flag peptide competition. Meanwhile, GST-tagged SNIP1 was expressed in Escherichia coli and purified by immunoprecipitation with GST beads and enzyme digestion (to remove GST tag). The aforementioned purified proteins were incubated and then immunoprecipitated with Flag beads to detect the interaction of SNIP1 by Coommassie blue staining.
(C) TET2 interacts with the N-terminal domain of SNIP1. SNIP1 protein contains a nuclear localization signal domain (NLS) and a FHA domain (top). Myc-tagged truncations of SNIP1 were transiently overexpressed in U2OS cells. Each truncation of SNIP1 was purified by immunoprecipitation with a Myc antibody, followed by western blot to detect endogenous TET2.
(D and E) K30/P100/K108 residues in the N-terminal domain of SNIP1 are crucial forTET2 interaction. Gal4(DBD)-tagged wild-type or single point mutant SNIP1 was transiently co-overexpressed with Flag-TET2 in HEK293T cells. Each truncated protein of SNIP1 was purified by immunoprecipitation with the Gal4-DBD antibody, followed by western blot to detect the co-precipitated TET2 using a commercial TET2 antibody (D). Furthermore, K30/P100/K108 double- or triplemutant SNIP1 was transiently co-overexpressed with Flag-TET2 in HEK293T cells. Each wild-type or mutant SNIP1 protein was purified by immunoprecipitation with the Gal4-DBD antibody, followed by western blot to detect the co-precipitated TET2 using a commercial TET2 antibody (E). MUT, K30/P100/K108 triple-mutant SNIP1.
Next, we mapped the domain(s) in SNIP1 required for interaction with TET2. We found that ectopically expressed wild-type, N-terminal (residues 1–121; N121), and central FHA domain deletion (without residues 281–343; ΔFHA), but not C-terminal (residues 122–396; ΔN121), interacted with endogenous TET2 in U2OS cells (Figure 2C), indicating that the N-terminal domain of SNIP1 interacts with TET2. Next, the conserved amino acids in the N-terminal domain of SNIP1 were individually mutated (Figure S4). We found that single mutations of K30A, P100A, or K108A in SNIP1 partially reduced the interaction with TET2 (Figure 2D), while K30A/P100A/K108A triple mutations completely abolished the interaction with TET2 (Figure 2E). To the best of our knowledge, these results identify SNIP1 as the first factor interacting with the N-terminal domain of TET2.
SNIP1 Mediates TET2 Binding to Several Transcription Factors, Including c-MYC
Like TET2, SNIP1 does not contain a domain for sequence-specific DNA recognition either, raising an intriguing question of how a TET2-SNIP1 association regulates the expression of specific genes. SNIP1 was reported to act as a transcriptional co-activator and may cooperate with other DNA sequence-specific transcriptional factors to regulate target gene expression. Supporting this notion, SNIP1 can interact with c-MYC to regulate the expression of c-MYC targets (Fujii et al., 2006). This prompted us to test the interaction of TET2, SNIP1, and c-MYC. We found that TET2 interacted with c-MYC when all three proteins were ectopically expressed and, importantly, that SNIP1 was indispensable for the TET2-c-MYC interaction (Figure 3A). Furthermore, ectopically expressed TET2 failed to interact with c-MYC when co-expressed with the TET2-binding defective K30A/P100A/K108A mutant of SNIP1 (Figure 3B). The association of endogenous TET2 with SNIP1 and c-MYC was further confirmed in MCF-7 human breast adenocarcinoma cells and U2OS human osteosarcoma cells (Figure 3C). By using the lentiviral CRISPR/Cas9-mediated genome-editing system, we generated stable U2OS cells with SNIP1 depletion by three different single-guide RNAs (sgRNAs) (Figures 3D and S2C). In these SNIP1-knockdown cells, c-MYC protein level was not changed, but the association of endogenous TET2 and c-MYC was substantially reduced by more than 80% (Figure 3D). These data suggest a model whereby SNIP1 bridges the protein interaction between TET2 and c-MYC to form a TET2-SNIP1-c-MYC ternary complex.
Figure 3. SNIP1 Mediates the Interaction between TET2 and Several Transcription Factors, Including c-MYC.
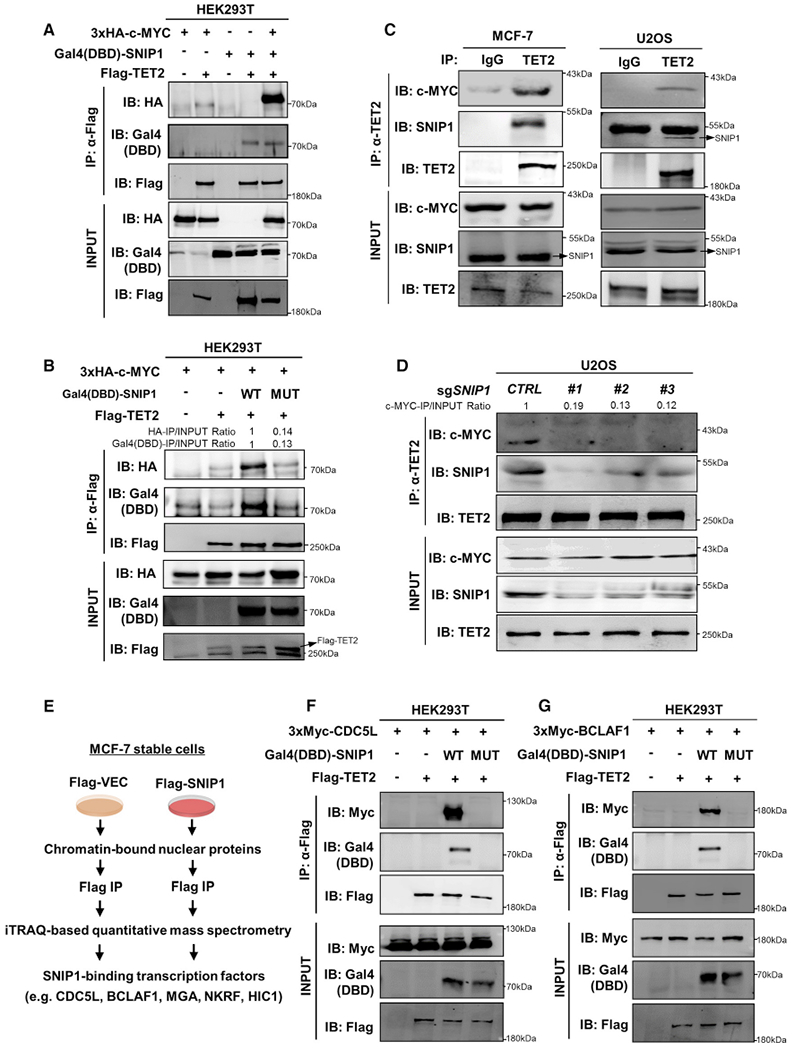
(A) SNIP1 bridges TET2 binding with c-MYC. Plasmids expressing the indicated proteins were transiently transfected in HEK293T cells. TET2 protein was purified by immunoprecipitation with Flag beads, followed by western blot to detect the co-precipitated Gal4(DBD)-SNIP1 and HA-c-MYC with antibodies against Gal4-DBD and HA, respectively.
(B) K30/P100/K108 residues in the N-terminal domain of SNIP1 are crucial for TET2-SNIP1-c-MYC complex formation. Plasmids expressing the indicated proteins were transiently transfected in HEK293T cells. TET2 protein was purified by immunoprecipitation with Flag beads, followed by western blotto detect the co-precipitated Gal4(DBD)-SNIP1 and HA-c-MYC with antibodies against Gal4-DBD and HA, respectively.
(C) Endogenous TET2 interacts with SNIP1 and c-MYC in MCF-7 and U2OS cells. TET2 protein in the indicated cells was immunoprecipitated with a TET2 antibody and followed by western blot to detect the co-precipitated endogenous SNIP1 and c-MYC. Rabbit IgG was used as a negative control.
(D) SNIP1 is essential for TET2 and c-MYC interaction. SNIP1 knockout U2OS cells were generated by three different sgRNAs (#1, #2, and #3). CTRL, control sgRNA. TET2 protein was immunoprecipitated and followed by western blot to detect the co-precipitated endogenous SNIP1 and c-MYC.
(E) Schematic representation of the mass spectrometry assay to identify SNIP1-binding transcription factors. In MCF-7 stable cells with knockdown of endogenous SNIP1 and put-back of Flag-tagged empty vector or SNIP1, chromatin-bound nuclear proteins were isolated, followed by immunoprecipitation with Flag beads. The immunoprecipitated proteins were then proceeded with isobaric tags for relative and absolute quantification by mass spectrometry.
(F and G) SNIP1 bridges TET2 binding with other transcription factors. Plasmids expressing the indicated proteins were transiently transfected in HEK293T cells. TET2 protein was purified by immunoprecipitation with Flag beads, followed by western blot to detect the co-precipitated Gal4(DBD)-SNIP1 and Myc-CDC5L (F)or Myc-BCLAF1 (G) with antibodies against Gal4-DBD and Myc tag, respectively.
To search for more SNIP1-interacting transcriptional factors, we generated MCF-7 stable cells with knockdown of endogenous SNIP1 and put-back of Flag-tagged empty vector or SNIP1. In these cells, chromatin-bound nuclear proteins were isolated, immunoprecipitated with Flag beads, and then subjected to iTRAQ-based quantitative mass spectrometry (Figure 3E). As expected, mass spectrometry analysis identified TET2 as one of the SNIP1-interacting proteins, and importantly, several transcription factors were recovered in the SNIP1 immunoprecipitates, including CDC5L (cell division cycle 5 like), BCLAF1 (BCL2 associated transcription factor 1), MGA (MAX dimerization protein), NKRF (NFKB repressing factor), and HIC1 (HIC ZBTB transcriptional repressor 1) (Table S3). The protein association of Flag-TET2 with Gal4 (DBD)-SNIP1 and Myc-tagged CDC5L or BCLAF1 was confirmed when all three proteins were co-overexpressed in HEK293T cells (Figures 3F and 3G). Moreover, ectopically expressed TET2 failed to interact with CDC5Lor BCLAF1 when co-expressed with the TET2-binding defective K30A/P100A/K108A mutant of SNIP1 (Figures 3F and 3G), further supporting the role of SNIP1 in bridging TET2 to transcription factors.
TET2 Is Recruited to and Regulates c-MYC Target Genes through SNIP1
To test the model that SNIP1 bridges TET2 to bind c-MYC and thereby facilitates TET2 to regulate c-MYC target genes, we conducted RNA sequencing analysis in SNIP1-knockdown U2OS cells (Figure S2C). Gene set enrichment analysis (GSEA) demonstrated that SNIP1 depletion significantly (p < 0.01) affected the expression of c-MYC targets as well as genes involved in apoptosis (Figure 4A). Among the genes that were significantly downregulated by SNIP1 depletion, we selected those that contain the c-MYC-binding E-box motifs (Seitz et al., 2011) (Figures S5 and S6B) and confirmed their downregulation in U2OS cells with SNIP1 knockdown or TET2 knockout (Figure 4B). Notably, SNIP1 knockdown and TET2 knockout did not have an additive effect on downregulating c-MYC target genes in U2OS cells, including SOCS3, CCNG2, BRCA1, and ST3GAL3 (Figure 4C). Furthermore, put-back of wild-type SNIP1, but not TET2-binding defective K30A/P100A/K108A mutant, could rescue the inhibitory effect of SNIP1 depletion on the expression of SOCS3, CCNG2, BRCA1, and ST3GAL3 (Figure 4D).
Figure 4. TET2 Is Recruited by SNIP1 to Regulate c-MYC Target Gene Expression.
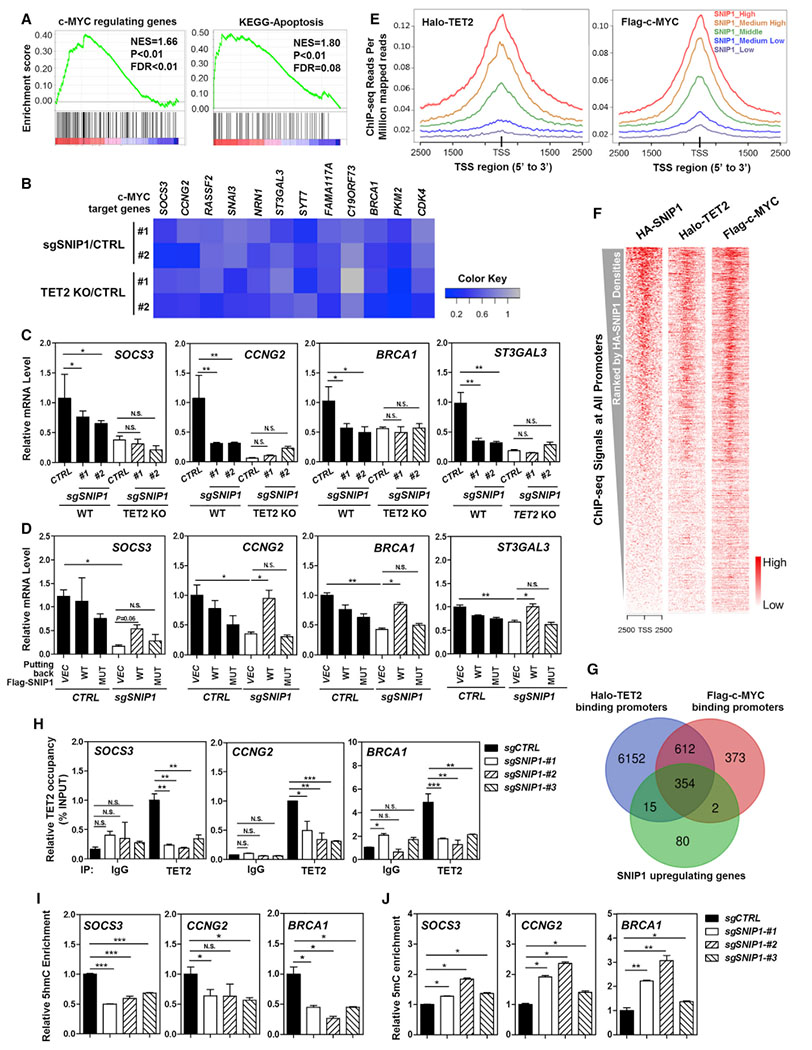
(A) RNA sequencing analysis reveals that SNIP1 knockdown causes significant alterations in mRNA expression of genes that are c-MYC targets or involved in cell apoptosis. More detailed information is provided in STAR Methods.
(B) Real-time qPCR confirms that c-MYC target genes are commonly downregulated by SNIP1 knockdown or TET2 knockout in U2OS cells. Data were normalized by mRNA expression in control cells. A gray-to-blue color scale indicates values from no change to downregulation, and row scaling is used to facilitate visualization.
(C) SNIP1 knockdown and TET2 knockout do not have an additive effect on downregulating c-MYC target genes. In SNIP1-knockdown and/or TET2-knockout U2OS cells, mRNA expression of the indicated c-MYC direct target genes was determined using real-time qRT-PCR, as described in STAR Methods.
(D) TET2 association is indispensable for SNIP1 to activate c-MYC target genes. Wild-type or TET2-binding defective K30A/P100A/K108A mutant SNIP1 was overexpressed in SNIP1-knockdown U2OS cells, and mRNA expression of the indicated c-MYC direct target genes was determined using real-time qRT-PCR.
(E–G) Enrichment of ectopically expressed TET2, SNIP1, and c-MYC in the genome of HEK293T cells. Previous ChIP-seq data (GSM897576 and GSM1493021) were re-analyzed using Bowtie 1.2.1.1 (Langmead et al., 2009) and MACS 2.1.1 (Zhang et al., 2008). ChIP-seq densities of Halo-TET2 and Flag-c-MYC were profiled according to the indicated levels of HA-SNIP1 ChIP-seq signals around TSS regions (E). Heatmap analysis of total 27,569 TSSs showed that HA-SNIP1, Halo-TET2, and Flag-c-MYC had similar binding patterns across all TSSs. All TSSs were ranked by HA-SNIP1 signal intensities (F). The overlap of TET2 and c-MYC-binding promoters and SNIP1 upregulating genes in U2OS identified by RNA-seq is displayed by Venn diagram (G) (p < 10−10).
(H) TET2 binds to c-MYC target gene promoters in a manner dependent on SNIP1. In SNIP1-knockdown and the control U2OS cells, the occupancy of endogenous TET2 on the promoters of indicated c-MYC target genes was determined using ChIP-qPCR as described in STAR Methods. Rabbit IgG was used as a negative control.
(I and J) SNIP1 knockdown decreases 5hmC and increases 5mC at the promoters of c-MYC target genes. In SNIP1-knockdown and the control U2OS cells, 5hmC and 5mC levels at the promoter regions of indicated c-MYC direct target genes were determined using hMeDIP-qPCR (I) and MeDIP-qPCR (J), respectively, as described in STAR Methods.
Shown are average values of triplicated experiments with SD. *p < 0.05, **p < 0.01, and ***p < 0.001 for the indicated comparisons. N.S., not significant.
To examine the co-occupancy of TET2, SNIP1, and c-MYC across the genome, we conducted chromatin immunoprecipitation followed by sequencing (ChIP-seq) of HA-tagged SNIP1 (GEO: GSE118811) and re-analyzed previous data of Halo-tagged TET2 or Flag-tagged c-MYC in HEK293T cells (Deplus et al., 2013; Thomas et al., 2015). We divided all TSS (transcription start site) regions into five groups according to HA-SNIP1 ChIP-seq intensities and found strong correlations between the binding intensities of Halo-TET2 as well as Flag-c-MYC with those of HA-SNIP1 (Figure 4E), which was further supported by heatmap analysis (Figure 4F).
Strikingly, 369 of 451 genes (81.8%) downregulated by SNIP1 depletion (Table S4) are bound by TET2 (Figure 4G), indicating that a significant portion of SNIP1 function is linked to TET2. Moreover, Halo-TET2 and Flag-c-MYC co-bound 966 promoter regions, accounting for 72.0% of c-MYC-binding promoters (Figure 4G). The binding events by ectopically expressed Halo-TET2, HA-SNIP1, and Flag-c-MYC at the same promoter regions of c-MYC target genes, including ST3GAL3, BRCA1, CCNG2, and PKM2 (Figure S6A), support the notion that SNIP1 bridges the interaction between TET2 and c-MYC and forms a TET2-SNIP1-c-MYC ternary complex to co-regulate target gene expression.
The binding events by endogenous TET2 at the promoters of c-MYC target genes, including SOCS3, CCNG2, BRCA1, ST3GAL3, and PKM2, were further validated by chromatin immunoprecipitation and qPCR (ChIP-qPCR) analysis in U2OS cells using a TET2 antibody (Figures 4H, S6C, and S6D). Importantly, TET2 bindings at these promoters were significantly decreased upon SNIP1 knockdown (Figures 4H and S6D). Consistently, hydroxymethylated DNA immunoprecipitation and qPCR (hMeDIP-qPCR) experiments showed that the 5hmC levels at the selected c-MYC target gene promoters were significantly reduced in the U2OS cells with SNIP1 knockdown (Figures 4I and S6E), which was accompanied by significant increases of 5mC levels at the same gene promoters (Figure 4J).
Collectively, our findings indicate that TET2 and SNIP1 act in the same pathway to regulate the expression of c-MYC target genes by promoting DNA demethylation at the promoters.
TET2 Plays a Crucial Role in DNA Damage Response and Cell Apoptosis
SNIP1 is implicated in the regulation of DNA damage response, cell cycle, and apoptosis (Roche et al., 2004, 2007; Yu et al., 2008). In accord, our RNA sequencing analysis revealed the enrichment of genes associated with cell apoptosis in SNIP1-knockdown U2OS cell pools (Figure 4A). Immunofluorescence staining demonstrated that SNIP1-knockdown cells displayed a significant increase of S139-phosphorylated histone variant H2AX (γH2AX), which normally forms surrounding DNA damage sites and serves as a platform for recruitment of DNA damage response factors (Lowndes and Toh, 2005), compared with control cells (p < 0.05; Figures S7A and S7B). Western blot analysis confirmed the increase of γH2AX in SNIP1-knockdown U2OS cells under basal culture condition (Figures S7D and S7E). Cisplatin (cis-diamminedichloroplatinum [II]; CDDP) is a commonly used chemotherapeutic agent that triggers DNA damage response and induces cell apoptosis (Cvitkovic and Misset, 1996; Koizumi et al., 2008). SNIP1 knockdown exacerbated γH2AX foci in U2OS cells upon cisplatin treatment (p < 0.05; Figures S7A, S7B, and S7D). The increased γH2AX was associated with a significant reduction (p < 0.001) of cell viability (both PI and annexin V negative) in SNIP1-knockdown U2OS cells (Figure S7C). SNIP1-knockdown cells also exhibited higher γH2AX level than the wild-type control cells after exposure to UV radiation (Figure S7E), further supporting a role of SNIP1 in DNA damage response and cell apoptosis.
Likewise, we observed that TET2-knockout U2OS cells exhibited a significant increase (p < 0.05) in spontaneous accumulation of γH2AX foci at basal condition (Figures 5A–5C), and a significant reduction (p < 0.05) in cell viability (Figure 5D). Upon cisplatin treatment, more γH2AX foci were detected in TET2-knockout cells than the wild-type control cells (p < 0.01; Figures 5A–5C). The increase of γH2AX correlated with a significant decrease (p < 0.001) of cell viability in TET2-knockout cells upon cisplatin treatment (Figure 5D). Additionally, TET2-knockout cells also exhibited higher γH2AX level than the wild-type control cells after exposure to UV radiation (Figure 5E). These findings suggest that like SNIP1, TET2 also plays a crucial role in controlling cellular susceptibility to cisplatin- or UV-induced DNA damage.
Figure 5. Catalytic Activity Is Indispensable for TET2 to Protect Cells from Cisplatin-Induced Double-Strand Breaks and Cell Apoptosis.
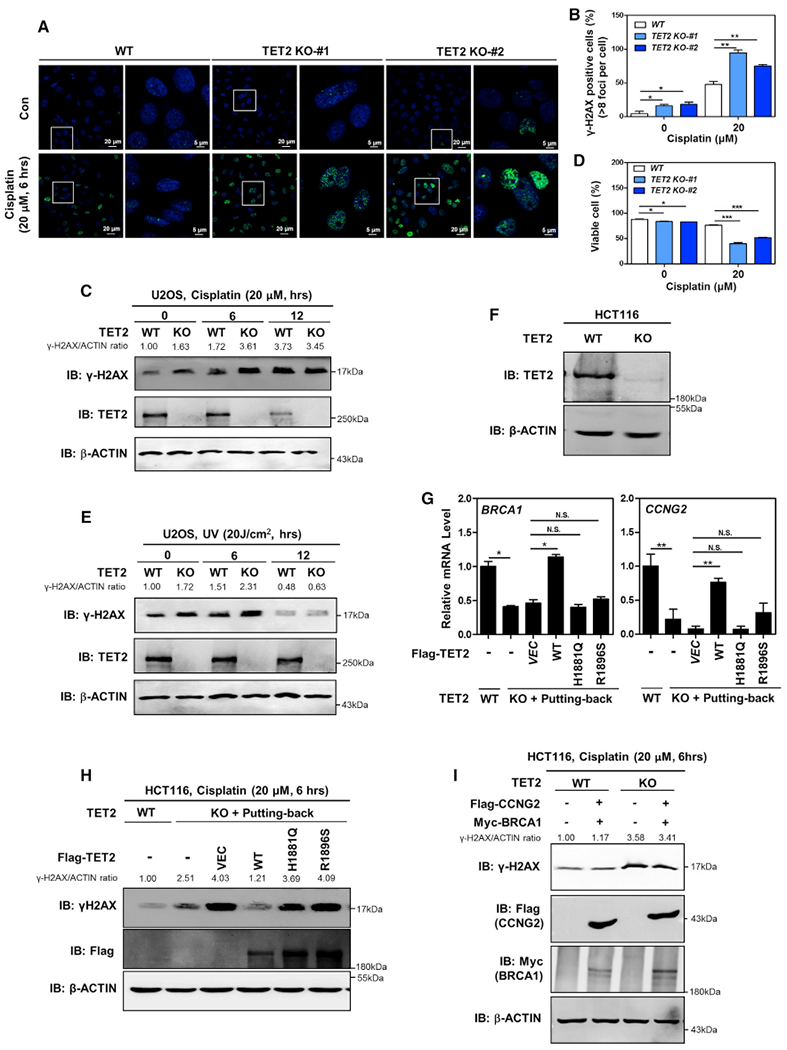
(A–D) TET2-knockout U2OS cells show an elevated basal γH2AX foci and a reduction in cell viability and higher susceptible to cisplatin-induced double-strand breaks and cell apoptosis. The indicated U2OS stable cells were treated with or without cisplatin (20 μM for 6 hr), the γH2AX level was determined using immunofluorescence (A), and the percentage of γH2AX-positive cells (more than eight foci per cell) was counted from randomly selected cells of three independent experiments (B). Additionally, γH2AX level was also determined using western blotting in these cells (C). Moreover, these cells were treated with or without cisplatin (20 μM for 24 hr), and the percentage of viable cells (both PI and annexin V negative) was determined using flow cytometry (D). More detailed information is provided in STAR Methods.
(E) TET2-knockout U2OS cells exhibit increased DNA double-strand breaks upon UV treatment. The indicated stable cells were treated UV radiation, and the protein level of γH2AX in cells was determined using western blotting, normalized by β-ACTIN.
(F) Verification of TET2 deletion in HCT116 cells by western blotting. By using the lentiviral CRISPR/Cas9-mediated genome-editing technique, TET2-knockout HCT116 cells were generated. TET2 protein was determined using western blot analysis.
(G) The catalytic activity is indispensable for TET2 to activate c-MYC target genes. In TET2-knockout HCT116 cells, wild-type TET2 or catalytic inactive mutants (H1881 and R1896) were transiently overexpressed. At 48 hr after transfection, mRNA expression of the indicated two c-MYC direct target genes was determined using real-time qRT-PCR.
(H) Catalytic activity is indispensable for TET2 to protect cells from cisplatin-induced double-strand breaks. In TET2-knockout HCT116 cells, wild-type TET2 or catalytic inactive mutants (H1881 and R1896) were transiently overexpressed. At 48 hr after transfection, the cells were treated with cisplatin (20 μM for 6 hr), and the γH2AX level was determined using western blotting, normalized by β-ACTIN.
(I) Put-back of CCNG2 and BRCA1 is not sufficient to rescue the effect of TET2 depletion on increasing γH2AX. In HCT116 cells without or with TET2 deletion, CCNG2 was stably overexpressed by retrovirus infection, and BRCA1 was transiently overexpressed. At 48 hr after transfection, the cells were treated with cisplatin (20 μM for 6 hr), and the γH2AX level was determined using western blotting, normalized by β-ACTIN.
Next, we set out to investigate whether TET2 catalytic activity is required for its downstream effect on regulating c-MYC target genes and DNA damage response. To this end, HCT116 cells with TET2 deletion were generated by using the lentiviral CRISPR/Cas9-mediated genome-editing system (Figure 5F). These cells were then transiently overexpressed with wild-type TET2 or catalytic inactive mutants, H1881 and R1896 (Hu et al., 2013; Ko et al., 2010). We found that put-back of wild-type TET2, but not these two patient-derived catalytic inactive mutants, could rescue the effect of TET2 deletion on downregulation of BRCA1 and CCNG2 (Figure 5G) and upregulation of γH2AX level upon cisplatin treatment (Figure 5H). These results thus indicate that catalytic activity is indispensable for TET2 to activate c-MYC target genes and mediate DNA damage response.
Furthermore, we found that overexpression of BRCA1 and CCNG2, two c-MYC targets involved in DNA damage repair processes, failed to rescue the effect of TET2 deletion on increasing γH2AX level (Figure 5I), suggesting that other TET2-SNIP1-c-MYC regulated genes may contribute to this phenotype.
Depletion of SNIP1 and TET2 Has No Additive Effect on Increasing DNA Double-Strand Breaks and Cell Apoptosis
Immunofluorescence staining revealed that SNIP1 knockdown and TET2 knockout did not additively increase the basal (Figures 6A–6C) or cisplatin-induced (Figures 6D and 6E) γH2AX foci in U2OS cells. Western blot analysis demonstrated that in TET2-knockout U2OS cells, SNIP1 knockdown did not further increase the levels of γH2AX and cleaved PARP (an apoptotic marker) after cisplatin treatment (Figures 6F and 6G). Flow cytometry analysis showed that SNIP1 knockdown and TET2 knockout had no additive effect on exacerbating cisplatin-induced apoptosis (Figure 6H). Together, these results suggest that SNIP1 and TET2 may act on the same pathway to regulate DNA damage response and cell apoptosis.
Figure 6. Depletion of SNIP1 and TET2 Has No Additive Effect on Increasing DNA Double-Strand Breaks and Cell Apoptosis.
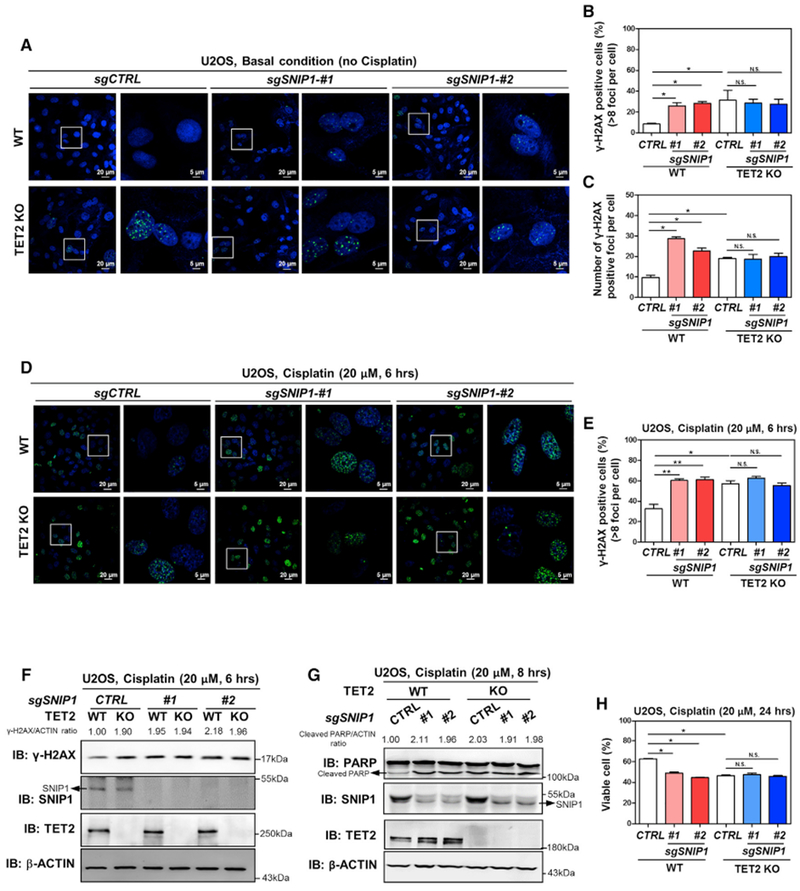
(A–C) SNIP1 knockdown and TET2 knockout have no additive effect on increasing internal DNA double-strand breaks. In the indicated U2OS stable cells, γH2AX foci was determined using immunofluorescence (A), and the percentage of γH2AX-positive cells (more than eight foci per cell) was counted from randomly selected cells of three independent experiments (B). Meanwhile, the number of γH2AX foci per cell was counted from randomly selected cells (C).
(D–F) SNIP1 knockdown and TET2 knockout have no additive effect on regulating cisplatin-induced DNA damage response. The indicated U2OS stable cells were treated with cisplatin (20 μM for 6 hr), γH2AX foci was determined using immunofluorescence (D), and the percentage of γH2AX-positive cells (more than eight foci per cell) was counted from randomly selected cells of three independent experiments (E). Additionally, γH2AX level was also determined using western blotting (F) and normalized by β-ACTIN in these U2OS stable cells.
(G and H) SNIP1 knockdown and TET2 knockout has no additive effect on regulating cisplatin-induced apoptosis. The indicated U2OS stable cells were treated with cisplatin (20 μM) as indicated, and the protein level of cleaved PARP was determined using western blotting (G), normalized by β-ACTIN. The percentage of viable cells (both PI and annexin V negative) was determined using flow cytometry (H).
Shown are average values of triplicated experiments with SD. *p < 0.05 and **p < 0.01 for the indicated comparisons. N.S., not significant.
SNIP1-TET2 Interaction Is Required for Their Function in Mediating DNA Damage Response
To provide further support for the function of SNIP1-TET2 interaction in mediating DNA damage response, we re-introduced wild-type or the TET2-binding defective K30/P100/K108A mutant SNIP1 into SNIP1-knockdown U2OS cells. Our data demonstrated that put-back of wild-type SNIP, but not the mutant, rescued the effects of SNIP1 depletion on increasing γH2AX foci (Figures 7A and 7B), the levels of γH2AX and cleaved PARP (Figures 7C and 7D), and cell apoptosis in response to cisplatin treatment (Figure 7E). Similarly, put-back of wild-type SNIP, but not TET2-binding defective mutant, rescued the effects of SNIP1 depletion on increasing the γH2AX level in stable MCF-7 cells (Figures S7F and S7G). These findings thus support the notion that SNIP1-TET2 interaction is critically important for SNIP1 to mediate DNA damage response and apoptosis in cultured cells.
Figure 7. SNIP1-TET2 Interaction Plays a Crucial Role in Regulating DNA Damage Response and Cell Apoptosis.
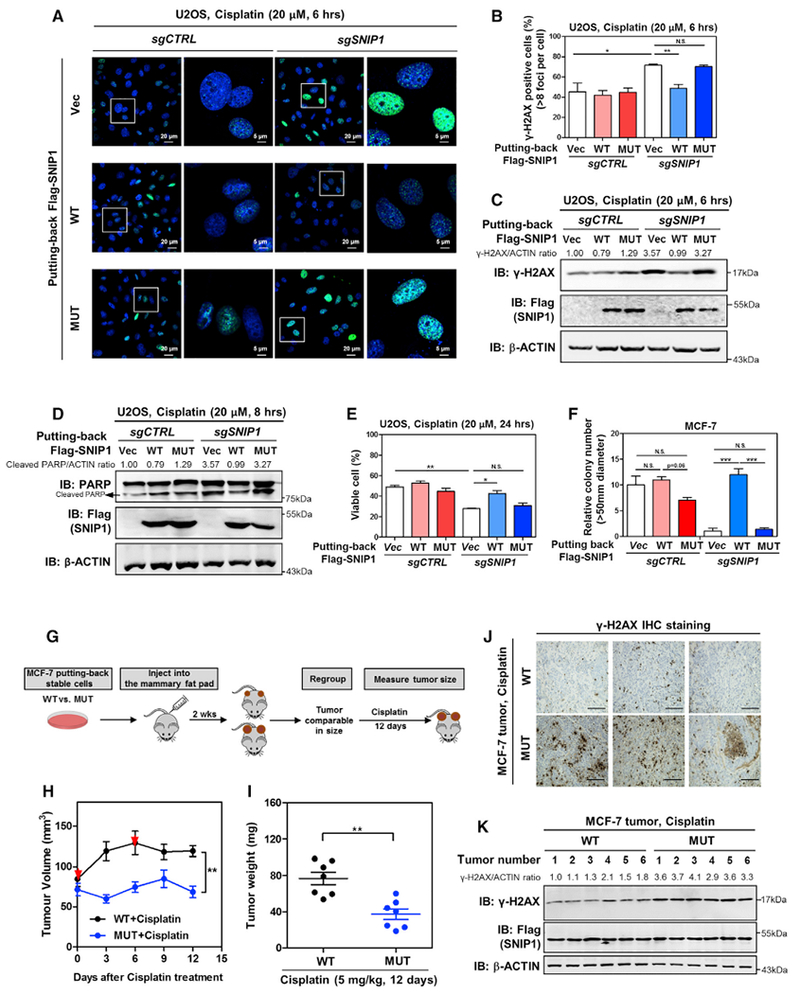
(A–C) TET2 association is required for SNIP1 to protect cells from cisplatin-induced DNA damage. The indicated U2OS stable cells were treated with cisplatin (20 μM for 6 hr), and the γH2AX level was determined by immunofluorescence (A), and the percentage of γH2AX-positive cells (more than eight foci per cell) was counted from randomly selected cells of three independent experiments (B). In addition, the protein level of γH2AX was also determined using western blotting (C), normalized by β-ACTIN.
(D and E) TET2 association is required for SNIP1 to protect cells from cisplatin-induced apoptosis. The indicated U2OS stable cells were treated with cisplatin (20 μM) as indicated, and the protein level of cleaved PARP was determined using western blotting (D), normalized by β-ACTIN. The percentage of viable cells (both PI and annexin V negative) was determined using flow cytometry (E).
(F) TET2 association is required for SNIP1 to promote anchorage-independent cell growth. The ability of indicated MCF-7 stable cells to exhibit anchorage-independent growth was determined by soft-agar colony formation assay as described in STAR Methods. Note that cells were maintained under basal condition (without cisplatin).
(G) Schematic representation of the orthotopic xenograft experiment. In brief, SNIP1-knockdown MCF-7 cells expressing wild-type or TET2-binding defective mutant SNIP1 were injected into the mammary pat of nude mice (female, 4–6 weeks old). At 2 weeks after injection, tumor-bearing nude mice with similar tumor volumes (73.69 ± 15.14 mm3 [n = 10] and 68.68 ± 14.04 mm3 [n = 10] for tumors expressing wild-type SNIP1 and TET2-binding defective mutant SNIP1, respectively) were selected and then subjected to intraperitoneal injection with cisplatin (5 mg/kg for every 6 days). The tumor volume was monitored for 12 days, and tumor weight was determined at sacrifice.
(H and I) SNIP1-TET2 interaction is vital for cisplatin sensitivity of breast cancer in vivo. The orthotopic xenograft experiment was performed as described in (G), and the tumor volume (H) and tumor weight (I) were measured at the indicated time points. Red arrows indicate cisplatin injection.
(J and K) SNIP1-TET2 interaction isvital for regulating cisplatin-induced DNA damage response in vivo. The level of γH2AX in tumor samples was detected using IHC staining (J). Representative IHC images (original magnification, 200×; scale bar, 50 μm) are shown. Meanwhile, the γH2AX level in tumor samples was also determined using western blotting (K), normalized by β-ACTIN.
Shown are average values of triplicated results with SD. *p < 0.05, **p < 0.01, and ***p < 0.001 for the indicated comparisons. N.S., not significant.
It is well established that the ability of anchorage-independent cell growth correlates with the tumorigenic potentials of cancer cells in vivo. We found that knockdown of SNIP1 suppressed the anchorage-independent growth of MCF-7 breast cancer cells (Figure 7F). Re-introduction of wild-type SNIP1, but not the TET2-binding defective mutant, rescued the ability of anchorage-independent cell growth in SNIP1-knockdown MCF-7 cells, suggesting that TET2 association is essential for the role of SNIP1 in promoting anchorage-independent cell growth.
Finally, we compared cisplatin sensitivity of tumors expressing wild-type and TET2-binding defective mutant SNIP1. For this purpose, we performed orthotopic injection of breast cancer cells into the mouse mammary fat pads using SNIP1-knockdown MCF-7 cells re-expressing either wild-type or TET2-binding defective mutant SNIP1 (Figure 7G). Mice with similar tumor volumes were regrouped and subjected to intraperitoneal injection with cisplatin (5 mg/kg per injection) at day 0 and day 6. Tumor volume was monitored every 3 days for 12 days, and tumor weight was determined at sacrifice (Figures 7H and 7I). Our data showed that tumors from cells expressing the TET2-binding defective mutant SNIP1 were significantly more sensitive to cisplatin treatment (p < 0.01). In agreement with our finding in cultured cells (Figures S7F and S7G), the γH2AX level was increased by >2-fold in tumors expressing TET2-binding defective mutant SNIP1 than those expressing wild-type SNIP1 after cisplatin treatment (Figures 7J and 7K). Thus, our results indicate that SNIP1-TET2 interaction is vital for cisplatin sensitivity of MCF-7 breast cancer cells in vivo.
DISCUSSION
There is increasing evidence that general chromatin-modifying enzymes may not bind to specific DNA sequences by themselves (Smith and Shilatifard, 2010). Instead, they must be recruited to specific targets in the genome by other factors, presumably sequence-specific DNA-binding proteins, to regulate specific genes and cellular processes. In this study, we performed an unbiased screening and identified multiple transcription regulators that interact with TET2, including the coactivator SNIP1. It should be noted that SNIP1 itself does not directly bind DNA but rather interacts with other DNA-binding proteins, such as c-MYC. Our study thus reveals a mechanism by which SNIP1 bridges the interaction of TET2 and c-MYC, thereby recruiting TET2 to specific sequences in the genome to induce DNA demethylation and target gene expression. Besides c-MYC, we show that SNIP1 also interacts with additional transcription factors, such as CDC5L and BCLAF1. We therefore propose that TET2 is recruited by the co-activator SNIP1 and cooperates with multiple DNA sequence-specific transcriptional factors to regulate genes and cellular processes. Future studies are needed to determine how SNIP1 recruits TET2 to selective transcription factors in response to both extracellular and intracellular stimuli/conditions.
Previous studies have reported that full-length TET2 exhibits higher enzymatic activity than its C-terminal catalytic domain (He et al., 2011; Hu et al., 2013). Moreover, lysine acetylation enhances TET2 enzymatic activity with two key regulatory residues, K110/111, in the N terminus of TET2 (Zhang et al., 2017b). These findings suggest that the N terminus of TET2 may contain positive regulatory mechanisms. In this study, we show that the N terminus plays an important role in TET2 regulation at least in part by forming a ternary complex with SNIP1 and DNA sequence-specific transcriptional factors to induce downstream targets. Notably, the N-terminal domain of TET2 is frequently mutated in hematopoietic neoplasm and lymphoid neoplasm and to a less degree in solid tumors (https://cancer.sanger.ac.uk/cosmic). The issue of whether tumor-derived mutations or changes in posttranslational modifications (e.g., acetylation) in the N terminus of TET2 would disturb its interaction with SNIP1 remains to be addressed and may connect the TET2-SNIP1 interaction to cancer biology.
Increasing evidence suggests that TET proteins regulate DNA damage response and maintain genome integrity. For instance, Tet2/3 double-knockout myeloid cells show DNA damage and impaired DNA repair (An et al., 2015). Moreover, 5hmC was found to be localized to sites of DNA damage and repair, and TET deficiency eliminates DNA damage-induced 5hmC accumulation and increases spontaneous accumulation of γH2AX foci in cells (Kafer et al., 2016). A recent study reported that upon oxidative stress-induced DNA damage, TET2 forms “yin-yang” complexes with DNMTs and is targeted to chromatin, actively removing abnormal DNA methylation in promoter CpG islands and enhancers by converting unwanted 5mC to 5hmC (Zhang et al., 2017b). In this study, we show that TET2-knockout U2OS cells exhibit spontaneous accumulation of γH2AX foci and high sensitivity of U2OS, HCT116, and MCF-7 cells to cisplatin- and/or UV-induced DNA damage, reaffirming the crucial role of TET2 in regulating DNA damage response and maintaining genome integrity. Furthermore, our present study uncovers a biochemical mechanism for how TET2 is recruited to certain sites of the chromatin and regulates target genes involved in DNA damage response. Mechanistically, TET2 is recruited by SNIP1 to the promoters of c-MYC target genes. The binding of TET2 to c-MYC target genes depends largely on the presence of SNIP1 protein. The binding of TET2 to SNIP1-c-MYC-target genes is functional, as seen by the increased 5hmC near the E-box sites and mRNA levels of c-MYC target genes, including BRCA1, CCNG2, SOCS3, and ST3GAL3. Among these c-MYC targets, BRCA1 is well known to participate in DNA double-strand break repair by homologous recombination (Silver and Livingston, 2012). Cyclin G2 can be recruited to sites of DNA repair to promote dephosphorylation of γH2AX (Naito et al., 2013). SOCS3 is considered as a critical attenuator of pro-apoptotic pathways in mammary development (Sutherland et al., 2006). ST3GAL3 modulates cisplatin-induced apoptosis via affecting caspase-8 and caspase-3 (Zhang et al., 2017a). We thus propose a model that TET2 modulates DNA damage response at least in part by stimulating expression of c-MYC target genes. The physiological significance of SNIP1-TET2 interaction is further supported by the observations that triple mutations at K30/P100/K108 in the N-terminal domain of SNIP1 disrupt TET2 association, leading to the loss of TET2’s ability to trans-activate c-MYC target genes and to regulate anchorage-independent cell growth and cisplatin sensitivity of MCF-7 breast cancer cells.
In conclusion, our present study uncovers a TET2-SNIP1-c-MYC pathway in regulating gene expression and provides insights into the biochemical mechanism for how TET2 regulates DNA damage response and ensures genome stability. The biological significance of the TET2-SNIP1 interaction needs further exploration, and with the newly defined TET2-SNIP1-c-MYC axis, the importance of DNA sequence-specific recruitment for TET2 functions is reinforced.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TET2 | Abclonal | A1526 |
| SNIP1 | Abcam | ab19611 |
| GAL4 | RK5C1 | SC-510 |
| GAL4 | RK5C1 | RK5C1 |
| HA | Santa Cruz | sc-7392 |
| Flag | Shanghai Genomics Technology | 4110-20 |
| c-MYC | Abcam | ab32 |
| β-actin | genscript | A00702 |
| Myc tag | Santa Cruz | sc-40 |
| PARP | Cell Signaling Technology | 9532S |
| γH2AX | Cell Signaling Technology | 9718s |
| TET2 for immunoprecipitation | YouKe Biotechnology | Immunogen:1-232 aa of TET2 |
| 5hmC | Active Motif | 39769 |
| Flag-beads | SIGMA | A2220 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cisplain | Sigma | Cat#P4394-25MG |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Deposited Data | This paper | GSE118811 |
| Raw imaging data | This paper | https://doi.org/10.17632/kf7jknsvn4.1 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL-247 |
| MCF-7 | ATCC | HTB-22D |
| HEK293T | (Wang et al., 2015) | N/A |
| U2OS | ATCC | HTB 96 |
| Experimental Models: strains | ||
| BALB/c-nude | Beijing Vital River Laboratory Animal Technology |
401 |
| Recombinant DNA | ||
| pcDNA3-Flag-TET2 WT | This paper | N/A |
| pcDNA3-Gal4-SNIP WT/MUT | This paper | N/A |
| pcDNA3-HA-c-MYC WT | This paper | N/A |
| pcDNA3-Flag-TET2 H1881Q | This paper | N/A |
| pcDNA3-Flag-TET2 R1896S | This paper | N/A |
| plentiCRISPR v2 | (Sanjana et al., 2014) | N/A |
| pCDH-Flag-SNIP WT/MUT | This paper | N/A |
| pCDH-GFP-SNIP WT/MUT | This paper | N/A |
| pcDNA3-VP16-TET2-WT | This paper | N/A |
| pcDNA3-Gal4(DBD)-TFs | (Zhao et al., 2008) | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kun-Liang Guan (kuguan@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal studies were performed according to the investigator’s protocol approved by the Ethics Committee of the Institutes of Biomedical Sciences (IBS), Fudan University. Charles River Japan(CRJ)crossed BALB/cABom-nu and BALB/cAnNCrj-nu to get CAnN.Cg-Foxn1nu/Crl (BALB/c-nude) mice. All mice used in this study were female and were maintained in the animal center of Fudan University. Mice were group-housed in individually ventilated cages and maintained under specific pathogen-free conditions. mice were used between 12 and 14 weeks of age.
Cell lines
HEK293T and MCF-7 cells were cultured by Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, Shanghai, China) added with 5% fetal bovine serum (GBICO), penicillin, and streptomycin; U2OS cells was cultured by Roswell Park Memorial Institute (RPMI) medium (Invitrogen, Shanghai, China) in a supplement with 10% fetal bovine serum (GBICO), penicillin, and streptomycin; HCT116 cells were maintained in McCoy’s 5A Medium (GIBCO), supplemented with 10% fetal bovine serum in the presence of penicillin, streptomycin.
METHOD DETAILS
Plasmids
For co-immunoprecipitation assay, cDNAs encoding full-length human SNIP1, TET2, c-MYC were cloned into Flag, HA, Myc or Gal4(DBD)-tagged vectors (pcDNA-Flag, pcDNA3-HA, pcDNA-Gal4(DBD), pcDNA3-Myc, pMCB-Flag), and the truncated proteins of SNIP1 and TET2 were sub-cloned into pcDNA-Flag or pcDNA3-Myc vector; For putting-back experiments, Flag-SNIP1 was sub-cloned into pCDH-GFPor pCDH-puro vector; For depleting SNIP1 or TET2, sgSNIPI and sgTET2 were cloned into Plenti-Crispr or pX458 vector. For overexpression, CCNG2 was cloned into PQCXIH (hygromycin) vector, and BRCA1 was cloned into pcDNA3-Myc vector. All constructs have been verified by DNA sequencing.
Transfection and immunoprecipitation
Plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen) or polyJet (SignaGen) following the manufacturer’s instruction. Cells were washed with cold phosphate buffered saline (PBS) once, and lysed in lysis buffer (50mM Tris-HCl and 0.1% Np-40, pH 7.4). After centrifugation, the supernatant was used to immunoprecipitation with indicated antibodies.
Mammalian two-hybrid screen
Human full-length TET2 fused to VP16 transactivation domain (AD), Preys (1,126 human ORFs) fused to Gal4 DBD, UAS-Luciferase reporter plasmid, and CMV-Renilla control plasmid were co-transfected in HEK293T cells. At 30 hours after transfection, the luciferase reporter activity was measured by a commercial kit (Promega E1910) using a Turner BioSystems Luminometer Reader (Promega).
When the AD-TET2 exists, the luciferase value is labeled as L1, and the Renilla value is labeled as R1. When the AD-TET2 does not exist, the luciferase value is labeled as L2, and the Renilla value is labeled as R2. The blank luciferase background is low and thus can be ignored. The relative luciferase activation is calculated as following: (L1/R1) / (L2/R2).
Generation of stable cell lines
For generation of stable cell pools with SNIP1 knockdown, three different sgRNAs against SNIP1 in Plenti-Crispr vector were used. Lentivirus was harvested after 24 hours post transfection, and mixed with 8 μg/mL polybrene. U2OS or MCF-7 cells were infected and selected in 1 μg/ml puromycin (Amresco, OH, USA) for 5 days.
For generation of stable monoclonal cells with TET2 knockout, sgRNA against TET2 in Px458 vector were transfected in U2OS cells. At 24 hours post transfection, flow cytometry (Beckman) was applied to sort GFP-positive monoclonal cells in 96-well plates, following verification of TET2 deletion by DNA sequencing and western blotting.
For generation of stable cells with TET2 knockout and SNIP1 knockdown, TET2 knockout monoclonal cells were infected with lentivirus carrying sgRNA against SNIP1, and then selected in 1 μg/ml puromycin for 5 days.
For generation of putting-back stable cells, wild-type or mutant SNIP1 was sub-cloned into pCDH-puro vector to produce lentivirus. SNIP1 knockdown cell pools in U2OS were infected and then subjected to functional assays (without puromycin selection). Meanwhile, wild-type or mutant SNIP1 was sub-cloned into pCDH-GFP vector to produce lentivirus. SNIP1 knockdown cell pools in MCF-7 were infected. At 48 hours post infection, flow cytometry (Beckman) was applied to sort GFP-positive cells, and the transfection efficiency was verified by western blotting.
For generation of stable HCT116 cells with TET2 deletion, cells were infected with lentivirus carrying sgRNA against TET2, and then selected in 1 μg/ml puromycin for 5 days.
For rescue experiments in HCT116 stable cells, Flag-tagged CCNG2 was sub-cloned into PQCXIH vector to produce retrovirus. Infected cells were then selected in 100 μg/mL hygromycin for 7 days. Myc-tagged BRCA1 were transiently overexpressed by using polyJet (SignaGen).
Protein interaction assay in vitro
Flag-tagged N terminus of TET2 was overexpressed in HEK293T cells, and was purified by immunoprecipitation with Flag beads, followed by Flag peptide competition to elute. Meanwhile, GST-tagged SNIP1 was transformed in Escherichia coli BL21, and was induced to express with 1 mg/mL IPTG at 16°C. GST-SNIP1 protein was purified by immunoprecipitation with GST beads, followed by enzyme digestion to remove GST tag. Purified N-terminal domain of TET2 and SNIP1 were co-incubated with Flag-beads at 4°C for 3 hours. Afterward, the protein sample was immunoprecipitated with Flag beads to detect the interaction of SNIP1 by Commassie blue staining.
Chromatin-bound nuclear proteins isolation
To isolate chromatin-bound nuclear proteins, cell pellet was harvested after centrifugation and proceeded following the manufacturer’s instruction (Thermo 78840). The enriched chromatin-bound nuclear proteins were immunoprecipitated with Flag-beads.
iTRAQ-based quantitative mass spectrometry
To search for SNIP1-interacting transcription factors, multiplexed isobaric tag for relative and absolute quantitation (iTRAQ)-based quantitative proteomic analysis was performed as described previously (Zhou et al., 2013). In brief, proteins were precipitated by adding six volumes of cold (−20°C) acetone and resolubilized in digestion buffer containing 8 M urea and 0.1 M NH4HCO3. Total protein levels were measured by BCA. 10 mM Dithiothreitol (DTT) was added and incubated for 30 minutes at 60°C, followed by addition of methyl methanethiosulfonate (MMTS) (ThermoFisher Scientific, Waltham, MA) to 20 mM. After 30 min incubation in the dark at room temperature, excess MMTS was quenched by addition of 20 mM DTT. Reduced and alkylated proteins were diluted in 0.1M ammonium bicarbonate, followed by addition of trypsin, with overnight digestion 37°C and end-over-end rotation. Digested peptides were labeled with 4-plex iTRAQ reagents (AB Sciex, Framingham, MA). For each reaction, peptides were resuspended in 500 mM triethylammonium bicarbonate and mixed with the appropriate iTRAQ reagent in ethanol. Labeling was allowed to proceed at room temperature for one hour. Samples were then combined and dried by vacuum centrifugation.
Gene expression profiling by RNA sequencing
The total RNA extracts (1 μg per sample) from stable cell pools with SNIP1 knockdown and the control cells were treated with VAHTS mRNA Capture Beads (Vazyme) to enrich polyA+ RNA. RNA-seq libraries were prepared using VAHTS mRNA-seq V2 Library Prep Kit for Illumina (Vazyme) following the manufacturer’s instruction. Briefly, polyA+ RNA samples were fragmented and then used for first- and second-strand cDNA synthesis with random hexamer primers. The cDNA fragments were treated with DNA End Repair Kit to repair the ends, then modified with Klenow to add an A at the 3′ end of the DNA fragments, and finally ligated to adapters. Purified dsDNA was subjected to 12 cycles of PCR amplification, and the libraries were sequenced by the Illumina sequencing platform on a 150 bp paired-end run. Sequencing reads from RNA-seq data were aligned using the spliced read aligner HISAT2, which was supplied with the Ensembl human genome assembly (Genome Reference Consortium GRCh38) as the reference genome. Gene expression levels were calculated by the FPKM (fragments per kilobase of transcript per million mapped reads). Gene Set Enrichment Analysis (GSEA) pre-ranked was run on the ranked list using the KEGG dataset as the gene set.
RNA isolation and qRT-PCR analysis
Total RNA was extracted from cultured cells by Trizol reagent (Invitrogen) following the manufacturer’s instruction. RNA was reversely transcribed with oligo-dT primers. Diluted cDNA was then used for real-time PCR with gene-specific primers in the presence of SYBR Premix ExTaq (TaKaRa) by 7500 real-time PCR system (Applied Biosystems). β-ACTIN was used as a housekeeping control. Primer sequences were listed in the Table S5.
Chromatin Immunoprecipitation (ChIP)-seq assays
The DNA fragments achieved above were further proceeded by KAPA Hyper Prep Kit KAPA (Biosystems, kk8502) for library construction following the manufacturer’s instruction. The workflow combines enzymatic steps and employs minimal bead-based cleanups, thereby reducing sample handling. After library construction, samples were sequenced in BasePair Biotechnology (Su Zhou, China) by Hiseq × 10 (illumina).
Chromatin Immunoprecipitation (ChIP)-qPCR assays
ChIP-qPCR assays were performed as described previously (Lan et al., 2007). Briefly, cells were cross-linked with 1% paraformaldehyde. After sonication at 4°C for 20 min (Bioruptor, low mode), chromatin was immunoprecipitated at 4°C for 3 hours with the antibody against TET2 (Shanghai Youke) or rabbit IgG. Antibody-chromatin complexes were pulled-down using protein A-Sepharose (RepliGen), and then washed and eluted by elution buffer. After cross-link reversal and proteinase K (TaKaRa) treatment, immunoprecipitated DNA was extracted with PCR Purification Kit (QIAGEN). The DNA fragments were further analyzed by real-time quantitative PCR using the primers as listed in Table S5.
(h)MeDIP-qPCR analysis
The (h)MeDIP assay was performed as previously described (Ito et al., 2010). Briefly, 2 μg genomic DNA was extracted from cells by the phenol-chloroform method and was then denatured and immunoprecipitated with the anti-5(h)mC or rabbit IgG antibody (Millipore) and protein G-Sepharose (Invitrogen). Beads were washed for three times and treated with proteinase K for 4 hours. DNA was extracted with PCR Purification Kit (QIAGEN) and the isolated DNA was analyzed by real-time quantitative PCR using the primers as listed in Table S5.
Immunofluorescence assay
Cells were washed with cold PBS and fixed with 4% PMSF (Sangon) for 15 min at room temperature. Then, cells were treated with 0.3% Triton X-100 for cell perforation at room temperature for 15 min, and were incubated with blocking buffer (3% BSA in PBS) for 1 hour, followed by incubation at 4°C overnight with the primary antibody against γH2AX, and Alex Fluor 594 (Green) conjugated secondary antibody (Invitrogen) at room temperature for 1 hour. Cell nucleus was stained with DAPI (Invitrogen). Images were captured using Leica fluorescence optical microscope.
Cell viability assay
Cells was seeded in 6-well plates at the density of 2×106 cells per well, and were treated with Cisplatin (Sigma) as indicated. Subsequently, cells were collected by trypsinization, and were stained by FITC Apoptosis Detection Kit (BD) following the manufacturer’s instruction. The stained cells were detected by BD Accuri C6 to calculate the percentage of viable cells (both PI and Annexin V-negative).
Soft-agar colony formation assay
1.5 mL medium containing 0.7% agar was poured into 6-well plates as the bottom layer. After solidification, a top layer medium containing 0.35% agar and MCF-7 stable cells (6×103) were seeded. The plates were placed in the incubator and the medium was changed every 2 days for 30 days. Then the colonies were fixed by 4% paraformaldehyde in PBS and stained with 0.01% crystal violet solution. The colonies larger than 0.1 mm were counted per well as described previously (Chernicky et al., 2000).
Xenograft studies
Nude mice (nu/nu, female, 4 to 6-week-old) were performed orthotopic injection of MCF-7 cells stable cells (2 × 106) mixed with Matrigel (BD Biosciences, Bedford, MA, USA) at a 1:3 vol ratio in 100 μL. Mice were primed with 17β-estradiol (0.18 mg per pellet, Innovative Research of American) subcutaneously on the neck. After 2 weeks, nude mice with similar tumor volumes were regrouped and intraperitoneal injection of Cisplatin was conducted in tumor-bearing mice at the dose of 5 mg/kg for every 6 days. The diameters of tumors were measured every 3 days during a total period of 12 days. Upon sacrifice, the tumors were dissected for further analysis. The procedures were approved by the Ethics Committee of the Institutes of Biomedical Sciences (IBS), Fudan University
Immunohistochemistry (IHC) analysis
Tumor samples were mounted in paraffin, and then were cut into 5 μm thin sections. Sections were normally deparaffinized and incubated with 3% H2O2 in PBS for 30 min to eliminate the endogenous peroxidase activity. After microwave repair for 10 min, the sections were incubated with blocking buffer (5% goat serum and 0.3% Triton X-100 in PBS) at 25°C for 1-2 hours, followed by incubation at 4°C overnight with the primary antibody against γH2AX (CST, dilution at 1:200). Horseradish peroxidase (HRP)-conjugated secondary antibody (MXB) was then applied and incubated at 37°C for 1 hour. Sections were developed with DAB kit (Vector Laboratories) and stopped with water according to the manufacturer’s instructions. Images were captured using OLYMPUS digital camera (DP71), original magnification, 200 ×; a single focal plane, scale bar, 50 μm are shown
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with a two-tailed unpaired Student’s t test. All data shown represent the results obtained from triplicated independent experiments with standard errors of the mean (mean ± SD). The values of p < 0.05 were considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the ChIP-seq data reported in this paper are GEO: GSE118811. We also deposited unprocessed gel data at Mendeley data (https://doi.org/10.17632/kf7jknsvn4.1)
Supplementary Material
Highlights.
SNIP1 bridges TET2 to interact with multiple transcription factors, including c-MYC
TET2 protects cells from DNA damage-induced apoptosis in a SNIP1-dependent manner
The TET2-SNIP1-c-MYC axis supports the DNA sequence-specific recruitment of TET2
ACKNOWLEDGMENTS
We thank members of the Fudan MCB laboratory for discussions and support throughout this study. We are grateful for the kind offer of Myc-tagged human TET3 from Dr. Jiemin Wong (East China Normal University) and the BRCA1 overexpression construct from Dr. Jun Huang (Zhejiang University). This work was supported by the National Key R&D Program of China (2016YFA0501800 to D.Y. and Y.X.), a National Natural Science Foundation of China (NSFC) grant (81522033 to D.Y., 31670836 to F.Z.), the Shanghai Committee of Science and Technology, China (16JC1404000 to D.Y.), and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institute of Higher Learning (TP2015003 to F.Z.). This work was also supported by NIH grants (GM067113 and CA063834 to Y.X. CA196878 and GM51586 to K.-L.G.).
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.028.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- An J, González-Avalos E, Chawla A, Jeong M, López-Moyado IF, Li W, Goodell MA, Chavez L, Ko M, and Rao A (2015). Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun 6, 10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen Y, Bian C, Fujiki R, and Yu X (2013). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernicky CL, Yi L, Tan H, Gan SU, and Ilan J (2000). Treatment of human breast cancer cells with antisense RNA to the type I insulin-like growth factor receptor inhibits cell growth, suppresses tumorigenesis, alters the metastatic potential, and prolongs survival in vivo. Cancer Gene Ther. 7, 384–395. [DOI] [PubMed] [Google Scholar]
- Chng Z, Teo A, Pedersen RA, and Vallier L (2010). SIP1 mediates cell-fate decisions between neuroectoderm and mesendoderm in human pluripotent stem cells. Cell Stem Cell 6, 59–70. [DOI] [PubMed] [Google Scholar]
- Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, Fidalgo M, Saunders A, Lawrence M, Dietmann S, et al. (2013). NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvitkovic E, and Misset JL (1996). Chemotherapy for ovarian cancer. N. Engl. J. Med 334, 1269. [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. (2009). Mutation in TET2 in myeloid cancers. N. Engl. J. Med 360, 2289–2301. [DOI] [PubMed] [Google Scholar]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, Fujita R, Guarnieri P, Bhagat G, Vanti WB, Shih A, et al. (2012). Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 488, 652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Lyakh LA, Bracken CP, Fukuoka J, Hayakawa M, Tsukiyama T, Soll SJ, Harris M, Rocha S, Roche KC, et al. (2006). SNIP1 is a candidate modifier of the transcriptional activity of c-Myc on E box-dependent target genes. Mol. Cell 24, 771–783. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T-P, Guo F, Yang H, Wu H-P, Xu G-F, Liu W, Xie Z-G, Shi L, He X, Jin SG, et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610. [DOI] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, and Xu Y (2013). Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155, 1545–1555. [DOI] [PubMed] [Google Scholar]
- Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, et al. (2015). The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 42, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, and Zhang Y (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Tahiliani M, Rao A, and Aravind L (2009). Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 8, 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med 377, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, and Carlton PM (2016). 5-Hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep 14, 1283–1292. [DOI] [PubMed] [Google Scholar]
- Kim RH, Wang D, Tsang M, Martin J, Huff C, de Caestecker MP, Parks WT, Meng X, Lechleider RJ, Wang T, and Roberts AB (2000). A novel smad nuclear interacting protein, SNIP1, suppresses p300-dependent TGF-beta signal transduction. Genes Dev. 14, 1605–1616. [PMC free article] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. (2010). Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, An J, Bandukwala HS, Chavez L, Aijö T, Pastor WA, Segal MF, Li H, Koh KP, Lähdesmäki H, et al. (2013). Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, and Zhang Y (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W, et al. (2008). S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 9, 215–221. [DOI] [PubMed] [Google Scholar]
- Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, and Shi Y (2007). Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, and Xu M (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes NF, and Toh GW (2005). DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol 15, R99–R102. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Yabuta N, Sato J, Ohno S, Sakata M, Kasama T, Ikawa M, and Nojima H (2013). Recruitment of cyclin G2 to promyelocytic leukemia nuclear bodies promotes dephosphorylation of γH2AX following treatment with ionizing radiation. Cell Cycle 12, 1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo FM, Bagci H, Brown KE, Landeira D, Soza-Ried J, Feytout A, Mooijman D, Hajkova P, Leitch HG, and Tada T (2013). Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol. Cell 49, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. (2011). TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38. [DOI] [PubMed] [Google Scholar]
- Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, Li Y, Ahn J, Abdel-Wahab O, Shih A, et al. (2014). DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 9, 1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KC, Wiechens N, Owen-Hughes T, and Perkins ND (2004). The FHA domain protein SNIP1 is a regulator of the cell cycle and cyclin D1 expression. Oncogene 23, 8185–8195. [DOI] [PubMed] [Google Scholar]
- Roche KC, Rocha S, Bracken CP, and Perkins ND (2007). Regulation of ATR-dependent pathways by the FHA domain containing protein SNIP1. Oncogene 26, 4523–4530. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz V, Butzhammer P, Hirsch B,Hecht J,Gütgemann I, Ehlers A, Lenze D, Oker E, Sommerfeld A, von der Wall E, et al. (2011). Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS ONE 6, e26837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DP, and Livingston DM (2012). Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, and Shilatifard A (2010). The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 40, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Vaillant F, Alexander WS, Wintermantel TM, Forrest NC, Holroyd SL, McManus EJ, Schutz G, Watson CJ, Chodosh LA, et al. (2006). c-Myc as a mediator of accelerated apoptosis and involution in mammary glands lacking Socs3. EMBO J. 25, 5805–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida S, Mizoshita T, Ozeki K, Tsukamoto H, Kamiya T, Kataoka H, Sakamuro D, and Joh T (2012). Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: potential of BIN1 to act as a potent predictor of cisplatin sensitivity in gastric cancer treatment. Int. J. Surg. Oncol 2012, 862879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A, et al. (2009). TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 23, 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, et al. (2015). Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 58, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K, and Pasini D (2013). Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 49, 645–656. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, Wang P, Yang H, Ma S, Lin H, et al. (2015). WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 57, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, Diep D, Zhang K, and Zhang Y (2012). Tet1 controls meiosis by regulating meiotic gene expression. Nature 492, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, Chen C, Liu S, Liu D, Chen Y, et al. (2015). Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 43, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Bi L, Zheng B, Ji L, Chevalier D, Agarwal M, Ramachandran V, Li W, Lagrange T, Walker JC, and Chen X (2008). The FHA domain proteins DAWDLE in arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc. Natl. Acad. Sci. U S A 105, 10073–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Modelbased analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, and Wong J (2014). Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked b-N-acetylglucosamine transferase (OGT). J. Biol. Chem. 289, 5986–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. (2015). Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang Z, Wang L, Han J, Li F, Shen C, Li H, Huang L, Zhao X, Yue D, et al. (2017a). Pseudomonas aeruginosa-mannose sensitive hemagglutinin injection treated cytokine-induced killer cells combined with chemotherapy in the treatment of malignancies. Int. Immunopharmacol 51, 57–65. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Wang Z, Xie W, Cai Y, Xia L, Easwaran H, Luo J, Yen RC, Li Y, and Baylin SB (2017b). Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol. Cell 65, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Lu Y, Ficarro SB, Adelmant G, Jiang W, Luckey CJ, and Marto JA (2013). Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.