

Abstract
Macromolecular cancer drugs such as therapeutic antibodies and nanoparticles are well known to display slow extravasation and incomplete penetration into tumors, potentially protecting cancer cells from therapeutic effects. Conventional assays to track macromolecular drug delivery are poorly matched to the heterogeneous tumor microenvironment, but recent progress on optical tissue clearing and three-dimensional (3D) tumor imaging offers a path to quantitative assays with cellular resolution. Here, we apply Transparent Tissue Tomography (T3) as a tool to track perfusion and delivery in the tumor and to evaluate target binding and vascular permeability. Using T3, we mapped anti-programmed cell death protein-ligand 1 (PD-L1) antibody distribution in whole mouse tumors. By measuring 3D penetration distances of the antibody drug out from the blood vessel boundaries into the tumor parenchyma, we determined spatial pharmacokinetics of anti-PD-L1 antibody drugs in mouse tumors. With multiplex imaging of tumor components, we determined the distinct distribution of anti-PD-L1 antibody drug in the tumor microenvironment with different PD-L1 expression patterns. T3 imaging revealed CD31+ capillaries are more permeable to anti-PD-L1 antibody transport compared to the blood vessels composed of endothelium supported by vascular fibroblasts and smooth muscle cells. T3 analysis also confirmed that isotype IgG antibody penetrates more deeply into tumor parenchyma than anti-Her2 or anti-EGFR antibody, which were restrained by binding to their respective antigens on tumor cells. Thus, T3 offers simple and rapid access to three-dimensional, quantitative maps of macromolecular drug distribution in the tumor microenvironment, offering a new tool for development of macromolecular cancer therapeutics.
Keywords: Three-dimensional imaging, Macromolecular drug distribution, Transparent tissue tomography
Introduction
Macromolecular agents including antibodies, proteins, polymer-drug conjugates, and drug-loaded nanoparticles for chemotherapy, hormone therapy, targeted therapy and/or immunotherapy are critical tools in the cancer treatment armamentarium (1-4). Macromolecular drugs display distinct pharmacokinetic and tumor distribution profiles from small molecule drugs (5). The Enhanced Permeability and Retention (EPR) effect (6), based on the hypothesis that a disordered vasculature that favors macromolecule accumulation would be a common feature of tumors, has long provided a rationale for development of macromolecular oncology drugs (7-9). However, real-world performance has been disappointing, with many agents displaying very low tumor specificity (10-12). Further, what does reach the tumor may only reach perivascular cells, leaving much of the parenchyma untreated (13). Subsequent work has revealed multiple barriers that limit tumor delivery, leading to a range of pharmacological and physical approaches to enhancing extravasation and penetration (14-16). These studies have exposed a critical need for assays to track macromolecular drug delivery in three dimensions and with cellular resolution.
A major advantage of in vivo drug tracking methods such as positron-emission tomography (PET) with CT or magnetic resonance imaging (MRI) is access to real-time monitoring of appropriately tagged macromolecular drugs in large volumes, but these approaches are limited to millimeter resolution and offer limited anatomical detail (17, 18). While intravital fluorescence microscopy offers cellular resolution, imaging is often limited to a specific tumor region and just one or two features, such as the drug and microvasculature (19). Biopsy followed by fixation, embedding and sectioning enables analysis by multiplexed immunohistochemistry (IHC) or immunofluorescence (20), allowing simultaneous detection of the drug along with multiple features of the microenvironment at micrometer resolution (21, 22). However, the tortuous microvasculature makes estimating delivery from 2D thin sections unreliable while 3D reconstruction from serial sections may be impractical for multiple samples (23).
Recent advances in tissue optical clearing combined with multiplex immunofluorescent detection and new microscopy methods have dramatically improved capabilities to map cellular markers in “whole mount” samples such as intact organs and/or tissue fragments that are stained and imaged without sectioning (24, 25). Several tissue clearing methods have been successfully applied to tumor tissue, providing high resolution, 3D images of the microenvironment and demonstrating feasibility for tracking nanoparticle and macromolecular drug delivery (26, 27). At the same time, these pioneering efforts have exposed potential drawbacks of current approaches including slow processing speed, antigen loss, and destructive methodologies that may limit their application to drug distribution and pharmacokinetic studies (28).
To address these challenges, we have adapted Transparent Tissue Tomography (T3) (29), a simple and fast tissue clearing and multiplex 3D imaging method, to track macromolecular drug distribution in the tumor microenvironment. With T3, perfusion and extravasation of macromolecular drugs are readily measured and the agents can be localized with respect to their molecular target and in the context of cell types and other features in the microenvironment. To apply T3 to track protein drug delivery, we modeled immune checkpoint blockade with anti-programmed cell death protein-ligand 1 (PD-L1) monoclonal antibody, examining antibody distribution and PD-L1 targeting in 3D at cellular resolution in murine mammary tumors. T3 also enabled direct comparison of the tumor penetration of model therapeutic monoclonal antibodies targeting tumor antigens versus an isotype control. Thereby, we examined delivery of anti-Her2 to a Her2 transgenic murine mammary tumor and anti-EGFR antibodies to a lung cancer patient derived xenograft (PDX) tumor, comparing each to isotype controls. Similarly, we also validated T3 with fluorescent dextran as a model for polymer-drug conjugates and PEGylated liposomal doxorubicin (Doxil) as a model nanoparticle. This work establishes T3 as a tool for quantitative, 3D spatial analysis of macromolecular drug distribution in the tumor microenvironment.
Materials and Methods
Mouse tumor models
BALB/c female mice (6-8 week old) were purchased from Envigo (Indianapolis, IN, USA). Transgenic BALB/c males carrying the mutated rat Her2/neu oncogene driven by the MMTV promoter (BALB-NeuT (30)) were bred with wild type BALB/c females. Genotypes of offspring were determined by PCR of tail snips. BALB-NeuT female mice developed spontaneous mammary carcinoma in each mammary gland between 5 and 33 weeks of age. TUBO cells, derived from a spontaneous mammary tumor in a BALB-NeuT female mouse, and 4T1 cells (ATCC CRL-2539TM) were cultured in 5% CO2 and maintained in RPMI 1640 and DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, and 100 units/ml penicillin, respectively. Subcutaneous tumors were formed by injection of 5 × 105 TUBO or 4T1 cells into the right flank of female BALB/c mice and used for the indicated studies 14 days after injection. A patient derived xenograft (PDX) mouse model of non-small cell lung cancer (Model ID: TM00231) was purchased from The Jackson Laboratory and engrafted into immunodeficient NSG mice. All mice were maintained under specific pathogen-free conditions. The study has been approved by the Institutional Animal Care and Use Committee of the University of Chicago, and all experiments conformed to the relevant regulatory standards.
Antibodies
Commercially available anti-PD-L1, anti-ER-TR7, anti-CD8, anti-SMA, anti-CD31, anti-EGFR, IgG isotype antibodies were obtained from commercial sources (Supplementary Table S1). Anti-rat Her2 antibody was isolated from culture supernatant from hybridoma 7.16.4 (ATCC) at the Frank W. Fitch Monoclonal Antibody Facility at the University of Chicago. Primary antibodies diluted in phosphate buffered saline pH 8.0 (PBS, Corning) were conjugated to NHS-DyLight488, 550, 594, 633, or 680 fluorescent dyes (Thermo) as shown in Supplementary Table S1. Reactions were incubated overnight at 4° C with gentle agitation. Unreacted dye was removed by dialysis in 10K MWCO Slide-A-Lyzer cassettes (Thermo) against PBS pH 7.4 at 4° C for 3 days. Fluorescent antibody solutions were stored at 4° C.
Tumor macrosectioning
TUBO tumors were harvested at time points after tail vein injection of 0.2 mg of DyLight594-conjugated anti-PD-L1 antibody in 0.1 ml PBS pH 7.4. At 10 min after injection of the antibody drug, tumors were washed with cold PBS, fixed with 0%, 2%, or 4% paraformaldehyde in PBS for 10 min, 30 min, or 1 h at room temperature, and washed with PBS. Then, tumors were cast in 2% agarose gel (dissolved in distilled water, LE Quick Dissolve Agarose, GeneMate) in 24 well plates. The gel plugs containing tumors were marked for orientation and mounted on a vibrating microtome (VT1200S, Leica) equipped with a buffer tray. 400 μm thick sections were collected in order in cold PBS.
Immunofluorescence staining
400 μm tumor macrosections obtained from tumors treated with DyLight594-conjugated anti-PD-L1 were fixed in PFA at 2% for 10 min, 2% for 1 h or 4% for 1 h, stained with fluorescent anti-Her2 and anti-CD31 for 18 h at 4° C, clarified in D-fructose and examined by confocal microscopy to evaluate staining depth (Supplementary Fig. S4A). While 1 h of PFA preserved circulating anti-PD-L1 (Supplementary Fig. S4B), it prevented DyLight488-conjugated anti-Her2 and DyLight633-conjugated anti-CD31 penetration (Supplementary Fig. S4C, D), making injection of DyLight633-conjugated anti-CD31 necessary to map the microvasculature (Supplementary Fig. S4E). Light fixation failed to preserve circulating anti-PD-L1 but allowed anti-Her2 and anti-CD31 to homogenously stain the macrosections. To obtain complementary spatial information of macromolecular drug transport and distribution in macrosections, tumors were divided and tissue was treated with brief or long fixation in parallel.
Thus, for multiplex immunostaining, after 2% PFA fixation for 10 min, tumor macrosections were stained with antibody cocktails in staining buffer (SB, RPMI 1640 with 10 mg/ml BSA and 0.3% Triton X-100). For Supplementary Fig. S4, 50 μl DyLight488-anti-Her2 and 20 μl of DyLight633-anti-CD31 were combined in 0.5 ml SB. For Fig. 3 and 4, 70 μl DyLight488-anti-Her2, 20 μl DyLight550-anti-CD8, 20 μl of DyLight633-anti-CD31, and 80 μl DyLight680-anti-PD-L1 were combined in 0.5 ml SB. For Fig. 5 and Supplementary Fig. S5, 50 μl of DyLight488-anti-SMA, 20 μl of DyLight633-anti-CD31, and 80 μl of DyLight680-anti-ER-TR7 were combined in 0.5 ml SB. The macrosections were incubated with the antibody cocktails for 18 h at 4° C. After incubation, macrosections were washed three times for 10 min in PBS pH 7.4 at 4° C, fixed with 2% paraformaldehyde in PBS for 10 min at room temperature, and washed in PBS pH 7.4.
Figure 3.
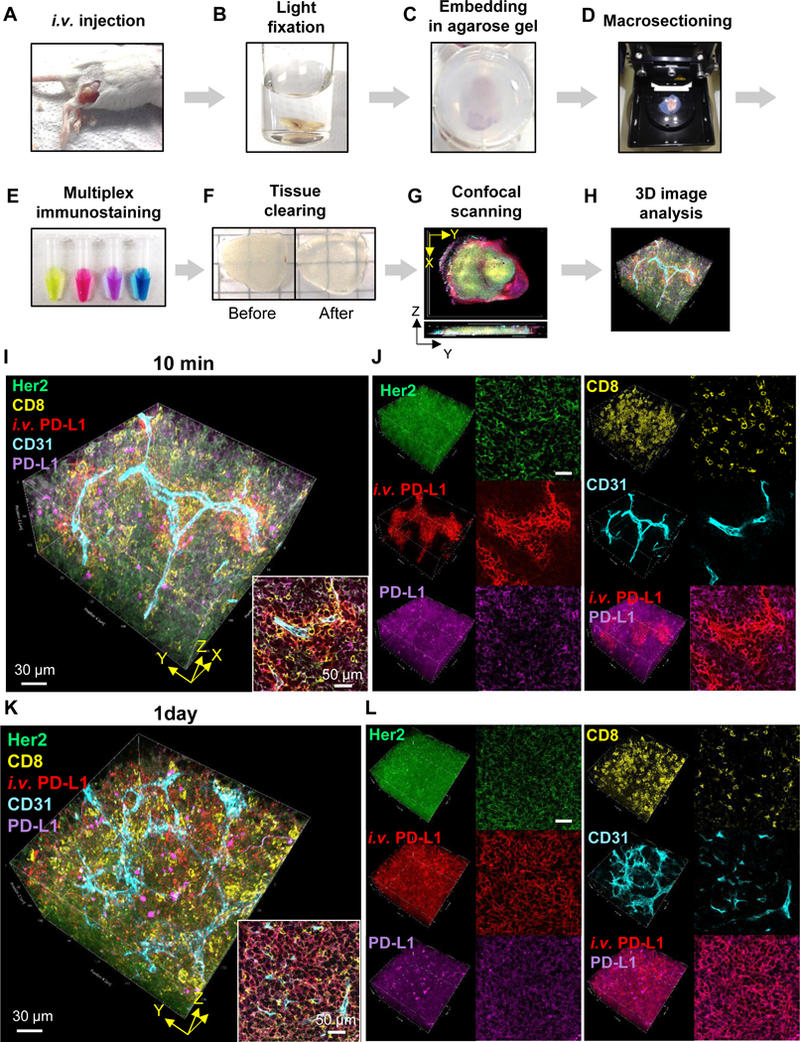
Transparent Tissue Tomography (T3) for mapping drug distribution in tumor microenvironment. A-H, Workflow of T3, A, Fluorescent macromolecular drug was intravenously injected into tumor bearing mice. B, The tumors were excised, fixed with 2% PFA solution for 10 min, C, embedded in 2% agarose gel, and D, sectioned at 400 μm thickness using a vibratome. E, The macrosections were stained with fluorescent primary antibodies. F, After washing and fixing, the macrosections were optically cleared by incubating with D-fructose solutions. G, Complete immunostaining for each macrosection was determined by optical cross-section (Z) scanning using a confocal microscope, and then each macrosection was imaged by XYZ scanning with multi-excitation and emission filters. H, 3D macrosection images were reconstructed, and macromolecular drug distribution in the tumor microenvironment was quantitatively analyzed using Fiji software. I-L, Spatial mapping of anti-PD-L1 antibody distribution kinetics in the tumor microenvironment. High resolution 3D images show distribution patterns of anti-PD-L1 antibody (red) in TUBO tumors at I, J, 10 min and K, L, 1 d post injection. The tumors were lightly fixed, sectioned, and immunostained for Her2+ cancer cells (green), CD8+ T lymphocytes (yellow), CD31+ blood vessels (cyan), and PD-L1+ cells (magenta). I, K, 3D rendering of anti-PD-L1 distribution in the tumor microenvironment. Scale bar: 30 μm. Right bottom inserts show 2D optical section images. Scale bar: 50 μm. J, L, 3D (left) and 2D (right) channel images for each cellular marker and antibody drug.
Figure 4.
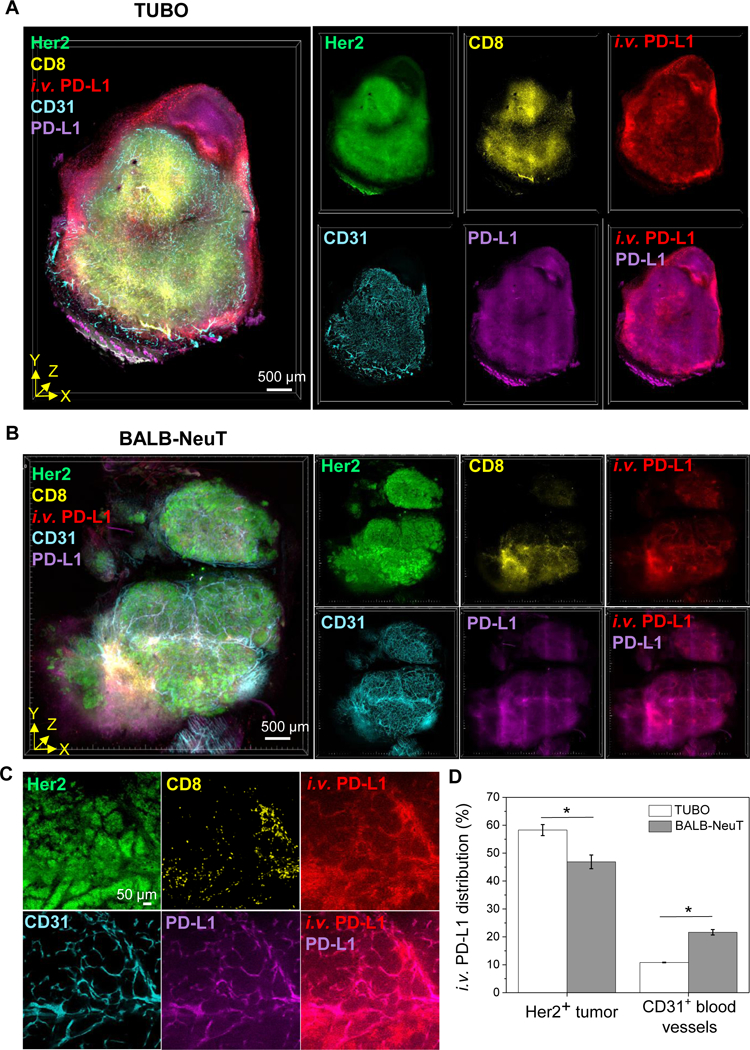
T3 analysis of anti-PD-L1 antibody distribution in tumor microenvironment with different PD-L1 expression patterns. A, B, 3D rendering of tumor perfusion of anti-PD-L1 antibody (red). At 1 day after fluorescent anti-PD-L1 antibody injection to TUBO (A) and BALB-NeuT (B) tumor mouse models, the tumors were excised, lightly fixed, sectioned, and immunostained for Her2 (green), CD8 (yellow), CD31 (cyan), and PD-L1 (magenta). Scale bar: 500 μm. C, 2D optical section images of BALB-NeuT tumor show PD-L1 expression in CD31+ vascular endothelium and colocalization of i.v. anti-PD-L1 with PD-L1+CD31+ microvessels. Scale bar: 50 μm. D, Relative quantification of i.v. anti-PD-L1 distribution (%) into Her2+ tumor and CD31+ vascular endothelium in the TUBO and BALB-NeuT tumor macrosections (n=3, mean ± SEM). *P<0.01.
Figure 5.
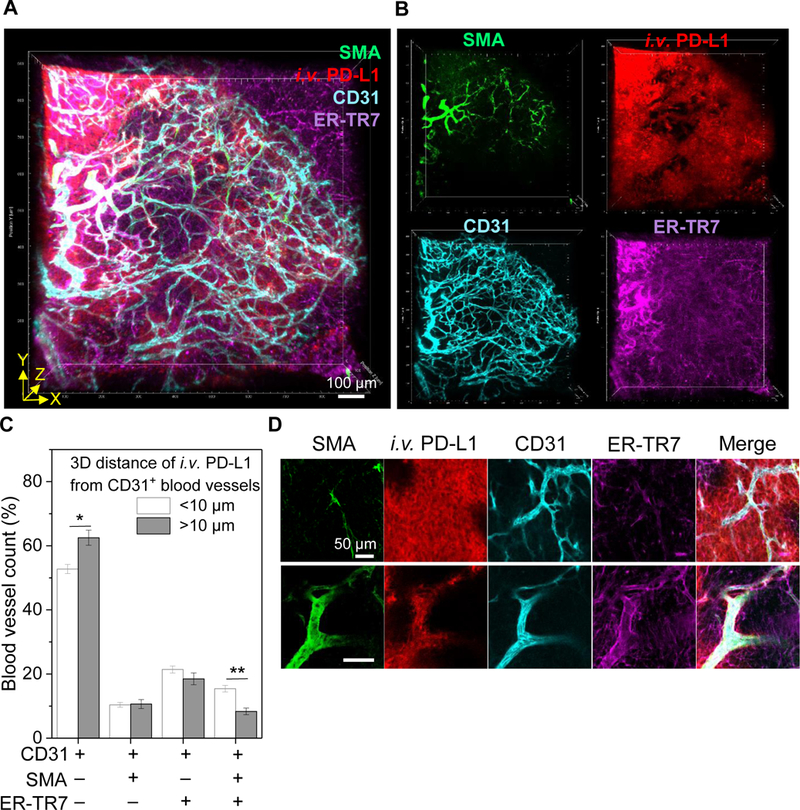
Vascular permeability to anti-PD-L1 antibody drug transport. A, 3D rendering of tumor perfusion of anti-PD-L1 antibody (red) through tumor microvessels immunostained for SMA+ smooth muscle cells, CD31+ endothelium, and ER-TR7+ fibroblasts. Tumors were harvested at 10 min post fluorescent anti-PD-L1 antibody injection, lightly fixed, sectioned, and immunostained. Scale bar: 100 μm. B, 3D channel images for each vascular cell marker and antibody drug. C, Relative quantification of permeability of different types of microvessels with regards to anti-PD-L1 transport to tumor (n=6, total blood vessels in three random 3D volume images in two macrosections, mean ± SEM). Mean 3D tumor penetration distance (10 μm) of anti-PD-L1 antibody at 10 min after injection was used as a guideline to determine high and low permeability of microvessels composed of different vascular cells. *P<0.05, **P<0.001. D, 2D optical section images show representative high and low permeability of CD31+-only and SMA+CD31+ER-TR7+ microvessels with regards to anti-PD-L1 antibody transport to tumors, respectively. Scale bar: 50 μm.
Optical clearing of macrosections
To prepare D-fructose solutions for optical clearing, 20, 50, and 80% (w/v solutions of D-fructose were prepared in 10 mM phosphate buffer pH 7.8 to a final volume of 10 ml. 30 μl of α-thioglycerol (Sigma Aldrich) was added to each D-fructose solution. Immunostained macrosections were incubated sequentially in 10 ml of 20, 50, and 80% D-fructose solutions for 1 h each at 25° C with gentle agitation in 20 ml glass vials. The immunostained macrosections and agarose were then mounted between coverslips in 80 % (w/v) D-fructose solutions.
Confocal microscopic imaging
Imaging was performed using a Leica TCS SP8 confocal laser scanning microscope, white light laser, Leica HCX PL APO 10X/0.4 NA dry objective (2.2 mm working distance) or Leica HC PL APO 40X/1.25 NA oil objective (0.24 mm working distance), and a SuperZ galvometric scanning stage. 3D scanning of macrosections was performed in a defined X/Y/Z (1.82/1.82/7.5 μm) volume with 10 frame averaging and bidirectional scanning using 488 nm excitation and 496-530 nm emission filter for DyLight488, 550 nm and 563-579 nm filters for DyLight550, 594 nm and 603-624 nm filter for DyLight594, 633 nm and 648-662 nm filter for DyLight633, and 670 nm excitation and 698-721 nm filter for DyLight680.
Comparison of 2D and 3D images
To obtain thin sections for conventional analysis, TUBO tumors were excised at 12 h post DyLight594-anti-PD-L1 antibody injection, and cut in half. One of the tumor halves was embedded in optimal cutting temperature compound (OCT, Tissue-Tek), frozen at −80° C, and sectioned at 10 μm thickness in a cryostat at −20° C. Sections were transferred to microscope slides, dried at room temperature and imaged by confocal microscopy. The other half of the tumor was analyzed using the T3 procedure for 3D imaging of 400 μm macrosection.
Spatial pharmacokinetics of anti-PD-L1 antibody
For analysis of antibody transport, 0.2 mg of DyLight594-anti-PD-L1 antibody in 0.1 ml PBS pH 7.4 was injected by tail vein (intravenous, i.v.) into mice 14 d after TUBO cell implantation and tumors were harvested at 10 min, 2 h, 12 h, 1 d, 2 d, and 4 d. 10 min before sacrificing the mice, 0.05 mg of DyLight633-anti-CD31 antibody in 0.1 ml PBS pH 7.4 was injected by tail vein. Tumors were fixed in 4% PFA for 1 h, sectioned, optically cleared, and imaged as described above.
Measuring concentration of anti-PD-L1 antibody in blood
0.2 mg of DyLight594-anti-PD-L1 antibody in 0.1 ml PBS were intravenously injected into BALB/c mice (n=4, average weight: 19 g). Blood samples (5 μl) were collected from the dorsal pedal vein at different time points. Blood samples were mixed with 10% K3-EDTA (0.5 μl) and centrifuged (RCF 4600 x g). The plasma (2 μl) was placed in a 1536 well microplate (Corning 3891), and the fluorescent intensity was measured using a plate reader (BioTeck, Synergy Neo2) at Ex 570 nm/Em 625 nm. Using a calibration curve (Supplementary Fig. S3), the data were profiled and fit to a two-compartment pharmacokinetic model for anti-PD-L1 profile (R2 = 0.998). The two-compartment model was y = A1 e(-x/t1) + A2 e(-x/t2), where the term y represents the fluorescent intensity of anti-PD-L1 antibody in plasma, 1/t1 and 1/t2 are rate constants for the distribution phase and elimination phase, A1 and A2 are intercepts on the y axis for each exponential segment of the curve. For terminal half-time, t1/2, term = 2 Ln[2] / [k + kpt + ktp − ((k + kpt + Ktp)2 − 4 k ktp)0.5]. For distribution half-time, t1/2, dist = Ln[2] [k + kpt + ktp − ((k + kpt + Ktp)2 − 4 k ktp)0.5] / [2 k ktp], where the terms k, kpt, and ktp represent elimination rate constant, plasma to tissue rate constant, and tissue to plasma rate constant, respectively (20).
T3 to compare tumor perfusion of anti-Her2 or anti-EGFR and isotype IgG antibodies
0.2 mg of DyLight594-anti-Her2 antibody and 0.2 mg of DyLight680-isotype IgG antibody in 0.2 ml PBS pH 7.4 were injected by tail vein (intravenous, i.v.) into a BALB-NeuT tumor-bearing mouse. 0.2 mg of DyLight594-anti-EGFR antibody and 0.2 mg of DyLight680-isotype IgG antibody in 0.2 ml PBS pH 7.4 were injected by tail vein into a lung cancer PDX tumor-bearing NSG mouse. BALB-NeuT and PDX tumors were harvested at 1 h after injection. 10 min before sacrificing the mice, 0.05 mg of DyLight633-anti-CD31 antibody in 0.1 ml PBS pH 7.4 was injected by tail vein. Tumors were fixed in 4% PFA for 1 h, sectioned, optically cleared, and imaged as described above.
T3 imaging of dextran and Doxil
Dextran amine (Invitrogen, MW 40K) was conjugated with a fluorescent dye. In brief, 5 mg of dextran amine was dissolved in 0.5 ml of PBS (pH 8.0), and 13 μl of DyLight594-NHS (at 10 mg/ml in dimethylformamide) was added to the dextran solution. The reaction was incubated with gentle agitation at room temperature for 3 h. Unreacted dye was removed by dialysis (MWCO 10K) against PBS pH 7.4 at room temperature for 3 d and the fluorescent dextran solution was stored at 4° C. DyLight594-dextran (1 mg in 0.1 ml PBS) or PEGylated liposomal doxorubicin (Doxil, 0.2 mg in 0.1 ml) were injected by tail vein in 4T1 tumor-bearing mice and tumors were harvested after 30 min and 4 h. Prior to harvesting tumors, 0.05 mg DyLight633-anti-CD31 antibody in 0.1 ml PBS was injected by tail vein and mice were sacrificed after 10 min. Excised tumors were fixed, macrosectioned, cleared, and imaged as described above.
Image processing
We applied the Fiji macros developed in our previous study (29) for automated 3D reconstruction of whole tumor images (Supplementary Table S2). In brief, macro LIFtile-restitcher aligned and stitched 3D mosaics for multi-channel “hyperstack” images (0-999 image tiles). To compensate for depth-related intensity losses, the mean intensity value (of Otsu-thresholded areas; maximum gain limited to 3X) of each optical slice in the macrosection images was normalized to the mean value of the entire macrosection for each channel using the macro HPRstackConstantMean, facilitating thresholding and segmentation as used in measurement of drug penetration distance from blood vessel to tumor area. Macro composite big aligner enabled automated alignment and registration of the stitched macrosection images. To remove gaps between macrosections in the 3D reconstructed tumor image, the macro closeZvoids merged the top and bottom of adjoining macrosections by summing signal from two adjoining optical slices. Next, macro hyprBKGDfix cleared background outside the stained tissue to facilitate 3D volume visualization. Finally, the 3D images were deconvolved using Huygens Pro software v. 4.3 (Scientific Volume Imaging), to correct for refractive index mismatch, and to enhance spatial signals and cell morphology discrimination. We constructed 3D tomographic visualizations of the tumor images using Imaris software v. 9.0 (Bitplane).
Data analysis
For hyperstack segmentation of anti-PD-L1 antibody, Her2+, and PD-L1+ cells, we visually determined the cutoff threshold for the antibody drug, Her2, and PD-L1 channel images, and converted into binary (8 bit) images. Macro vessel extractor was used for automated segmentation of CD31+, SMA+, and ER-TR7+ blood vessels from the CD31, SMA, and ER-TR7 channel images. To measure radius penetration distance of anti-PD-L1 antibody from CD31+ tumor vasculature, we applied the 3D distance map plugin to the tumor blood vessels. In brief, the ImageJ 3D-EDT plugin calculates the nearest distance of each foreground pixel to background in 3D space (31). The vascular space (binarized) is treated as background, so that each pixel in the non-vascular tissue is assigned a (32-bit) value corresponding to its distance to the closest vessel in 3D space by the plugin. The antibody drug signal is then identified by thresholding and this mask is measured by redirection to the 3D-EDT volume. In this way the 3D distance of each antibody drug voxel is read as the corresponding 3D distance from the vasculature. All other image analysis was conducted with basic analyze functions in Fiji software (http://fiji.sc/Fiji).
Statistical analysis
Statistical values are expressed as mean ± SD and SEM, and statistical comparisons between groups were made using one-way ANOVA and Student’s t test. P value of <0.05 was considered statistically significant.
Results
Transparent Tissue Tomography (T3) for macromolecular drug distribution in the tumor microenvironment
Using immune checkpoint blockade immunotherapy (32, 33) as a model for macromolecular drug therapy, we adapted the T3 method to enable tracking drug perfusion and penetration into tumors (Fig. 1A-G and Table 1). A rat IgG2b monoclonal anti-murine PD-L1 antibody (anti-B7/H1, clone 10F.9G2) was conjugated to an amine-reactive N-hydroxysuccinimide (NHS)-ester activated fluorescent dye, NHS-DyLight594, dialyzed to remove free dye and injected by tail vein. Prior to harvesting tumors, the luminal endothelial surface of the tumor vasculature was labeled by injecting anti-murine CD31 antibody conjugated to NHS-DyLight633 and, after 10 min, animals were sacrificed and their tumors were excised, fixed in 4% paraformaldehyde (PFA) for 1 h and cast in agarose. Tumors were cut into 400 μm thick macrosections by vibratome and optically cleared by sequential incubation in 20, 50, and 80% (w/v) D-fructose, allowing the full volumes to be imaged by confocal microscopy. To reconstruct whole tumors, the digital images of each macrosection were stitched, registered and annealed using open-source Fiji image processing and analysis software. The resulting two-color, 3D maps enabled visualizing and quantifying anti-PD-L1 perfusion and extravasation in whole tumors.
Figure 1.
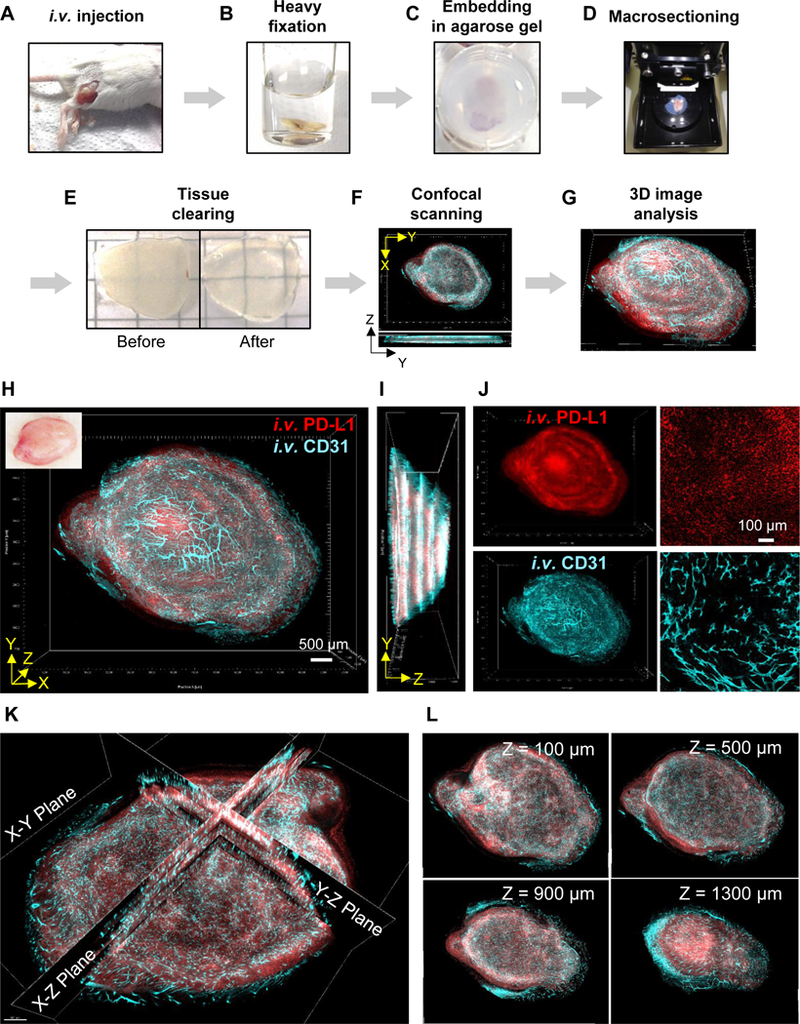
Transparent Tissue Tomography (T3) for tracking tumor perfusion and penetration of macromolecular drug. A-G, Workflow of T3, A, Fluorescent macromolecular drug was intravenously injected into tumor bearing mice. At 10 min before sacrificing the mice, fluorescent anti-CD31 antibody was additionally injected for in vivo labeling of vascular endothelium. B, The tumors were excised and fixed with 4% paraformaldehyde (PFA) solution for 1h, C, followed by embedding in 2% agarose gel. D, The embedded tumors were sectioned at 400 μm thickness using a vibratome. E, Collected tumor macrosections were optically cleared by incubating with D-fructose solutions. F, Each macrosection was imaged by XYZ scanning using a confocal microscope. G, Whole tumor images were reconstructed by concatenating macrosection images, and quantitative 3D spatial analysis of tumor perfusion and penetration of macromolecular drug was performed using Fiji software. H, 3D visualization of anti-PD-L1 antibody drug distribution in a whole mouse tumor. 3D rendering of a reconstructed tumor obtained by fusing images of four macrosections. TUBO tumors were excised at 2 days post intravenous (i.v.) injection of fluorescent anti-PD-L1 (red) with in vivo labeling of vascular endothelium (cyan) via i.v. injection fluorescent anti-CD31 antibody. Scale bar: 500 μm. Left top insert shows tumor prior to macrosectioning. I, Lateral view of reconstructed tumor. J, 3D (left) and 2D (right) channel images for antibody drug and tumor blood vessels. Scale bar: 100 μm. K, Tomographic visualization of the reconstructed tumor image with multiple orthogonal planes (X-Y, X-Z, Y-Z planes). L, Serial tomographic sections of X-Y planes at different Z-stack depths (100, 500, 900, 1300 μm).
Table 1.
Tracking tumor perfusion and penetration
| Process | Average Time | Notes |
|---|---|---|
| Drug Injection to mouse tumor model | - | -Intravenous injection of fluorescent macromolecular drug -Intravenous injection of fluorescent anti-CD31 antibody |
| Fixing, embedding, and macrosectioning | 2 h | -Fixing tumor in 4% paraformaldehyde (PFA) for 1 h -Embedding tumor in 2% agarose gel -Macro-sectioning at 400 μm thickness |
| Tissue clearing | 3 h | -Incubating sequentially for 1 h each in 20%, 50%, 80% D-fructose solutions (containing 0.3% (v/v) α-thioglycerol) at 25°C |
| Confocal imaging | 30 min /macrolayer (6×5×0.4 mm3) | -Using 10X objective (X/Y/Z grid resolutions=1.82/1.82/7.5 μm) -Using 2 different excitation and emission filters -Using 40X objective for high-resolution 3D imaging of interest area |
| Image processing and analysis | 1 day | -Using Fiji macros for 3D image reconstruction and analysis |
Thus, to evaluate antibody penetration in each tumor, we visualized the distribution of DyLight594 fluorescence to track anti-PD-L1 and compared it to DyLight633, marking the anti-CD31 bound to the perfused tumor vascular endothelium. T3 analysis yielded a 3D tumor model that could be examined by projections (Fig. 1H-J) or by extracting virtual sections (Fig. 1K, L), providing 2D representations of the data that facilitate examination of tumor penetration of anti-PD-L1 antibody in relation to the CD31+ microvasculature. As shown in Supplementary Video 1, scanning through different planes in the 3D model revealed significant heterogeneity at millimeter scales across tumors with respect to density and distribution of perfused vasculature and tissue levels of anti-PD-L1. Compared to imaging in cryosections (Supplementary Fig. S1A), 3D images obtained by T3 analysis of macrosections from the divided TUBO tumor (Supplementary Fig. S1B) allowed the correlation between tissue distribution of injected PD-L1 and the microvascular network to be far better appreciated.
Spatial pharmacokinetics of anti-PD-L1 antibody delivery to tumors
To probe distribution kinetics, we performed a time-course experiment where tumors were excised at 10 min, 2 h, 12 h, 1 d, 2 d or 4 d after i.v. injection of DyLight594-anti-PD-L1. Examining the whole macrosections, T3 analysis demonstrated an overall pattern of delivery favoring accumulation and persistence in the tumor periphery, with antibody concentrations appearing to peak between 12 h and 1 d and then decreasing close to baseline by 4 d (Fig. 2A and Supplementary Fig. S2). Examining antibody delivery at the 10 min time point at high magnification captured marked heterogeneity of anti-PD-L1 antibody extravasation at the level of individual microvascular elements (Fig. 2B, C and Supplementary Video 2).
Figure 2.

3D spatial pharmacokinetics of anti-PD-L1 antibody drug in tumors. A, 3D rendering of macrosections of TUBO tumors collected at 10 min, 2 h, 12 h, 1 d, 2 d, and 4 d after i.v. injection of fluorescent anti-PD-L1 antibody (red). Tumor blood vessels (cyan) in the macrosections were stained by i.v. injection of fluorescent anti-CD31 antibody. Scale bar: 500 μm. B, Heterogeneity of anti-PD-L1 antibody drug extravasation through blood vessels in the tumor collected at 10 min post injection. C, 3D vascular penetration of anti-PD-L1 antibody drug. High-resolution 3D images were obtained from the tumor collected at 10 min post injection. D, Concentration kinetics of anti-PD-L1 antibody in plasma (n=4, mean ± SD). Data were fitted to a two-compartment model (y=A1e(-x/t1) +A2e(-x/t2)). Insert represents two-compartment model (plasma (P), tissue (T), rate constants (kpt, ktp, k), half-time for distribution and termination (t1/2, dist and t1/2, term)). E, 3D tumor penetration distance profiles of anti-PD-L1 antibody away from CD31+ blood vessels at different time points after injection (n=3 for each time point, merged distance data derived from total i.v. PD-L1 voxels in macrosections). Insert illustrates 3D tumor penetration distance of antibody drug from blood vessels. F, Distribution kinetics of anti-PD-L1 antibody coverage of macrosection volume at the different time points (n=3 for each time point, mean ± SD).
Toward evaluating T3 as a tool for quantitative analysis of drug distribution, we first examined the kinetics of circulating anti-PD-L1 antibody after injection using a calibration curve (Supplementary Fig. S3). Fitting the DyLight594 dye fluorescence in blood samples to a two-compartment pharmacokinetic model (Fig. 2D) yielded a distribution half-life (t1/2, dist) of 46 min and a terminal half-life (t1/2, term) of 1 day. Then, 3D tumor penetration was examined by delineating all CD31+ blood vessels in each macrosection and determining radial penetration of the anti-PD-L1 antibody out from the blood vessel wall and into the tumor parenchyma via a 3D distance map (Fig. 2E). A mean penetration of 14 μm from blood vessel boundaries was observed at 1 d post injection. Finally, we measured distribution volume (%) of anti-PD-L1 antibody within the macrosections (Fig. 2F), revealing a maximal coverage of 35% at 1 d after injection. A simple interpretation of the data suggests that injected anti-PD-L1 antibody equilibrates between circulation and tumor by 1 d (34). Once PD-L1 enters the tumor, it dissipates slowly compared to the circulating fraction, raising the question whether these kinetics might reflect anti-PD-L1 binding its target.
Multiplex 3D imaging of anti-PD-L1 distribution in the tumor microenvironment
To map the delivered anti-PD-L1 with respect to other elements of the tumor microenvironment, we sought to analyze the tumors by T3 and multiplex immunostaining (Fig. 3A-H and Table 2). Examining antibody penetration of macrosections cut from tumors subjected to different fixation conditions indicated light fixation was optimal (Supplementary Fig. S4). Thus, at time points after injecting DyLight594-labeled anti-PD-L1, tumors were harvested and fixed for 10 min in 2% PFA, embedded in agar and macrosectioned. To enable multiplexed immunodetection, panels of primary antibodies were directly labeled by conjugation to different NHS fluorophores at specific dye:antibody molar ratios (Supplementary Table S1) and combined in a cocktail. The macrosections were immunostained to detect cell surface antigen antibodies against PD-L1 to detect the target, CD31 to identify endothelial cells, Her2 to identify cancer cells, and CD8 to identify cytotoxic T lymphocytes. Then, macrosections were optically cleared with D-fructose and imaged by confocal microscopy. By zooming in on specific regions, T3 revealed the distribution kinetics and patterns of anti-PD-L1 antibody drug in the tumor microenvironment, distinguishing PD-L1-expressing Her2+ cancer cells and CD8+ T lymphocytes at cellular resolution (Fig. 3I-L). As shown in Supplementary Video 3, by 10 min after injection, a front of concentrated anti-PD-L1 antibody was observed to have reached tumor cells in proximity to CD31+ blood vessels, apparently sufficient to saturate binding sites on PD-L1 antigen on cancer cell membranes and thereby preventing PD-L1 immunostaining (Fig. 3J, i.v. PD-L1/PD-L1 channel images). By 1 day post injection, anti-PD-L1 antibody had permeated the tumor to reach essentially all PD-L1-expresssing cancer cells (Fig. 3L). We determined target engagement of the anti-PD-L1 antibody drug by measuring spatial colocalization (%) with PD-L1-expressing Her2+ cancer cells in a whole macrosection, revealing that anti-PD-L1 antibody reached 48% of PD-L1+Her2+ expressing tumor volume at 1 day post injection (Fig. 4A).
Table 2.
Mapping drug distribution in tumor microenvironment
| Process | Average Time | Notes |
|---|---|---|
| Drug Injection to mouse tumor model | - | -Intravenous injection of fluorescent macromolecular drug |
| Fixing, embedding, and macrosectioning | 1 h | -Fixing tumor in 2% paraformaldehyde (PFA) for 10 min -Embedding tumor in 2% agarose gel -Macro-sectioning at 400 μm thickness -Fixing macrosections in 2% PFA for 5 min |
| Immunofluorescence staining | 18 h | -Incubating with 4 different fluorescent primary antibodies at 4°C |
| Washing and fixing | 40 min | -Washing with PBS x3 times, and fixing in 2% PFA for 10 min |
| Tissue clearing | 3 h | -Incubating sequentially for 1 h each in 20%, 50%, 80% D-fructose solutions (containing 0.3% (v/v) α-thioglycerol) at 25°C |
| Confocal imaging | 1 h/macrolayer (6×5×0.4 mm3) | -Using 10X objective (X/Y/Z grid resolutions=1.82/1.82/7.5 μm) -Using 5 different excitation and emission filters -Using 40X objective for high-resolution 3D imaging of interest area |
| Image processing and analysis | 1 day | -Using Fiji software and created macros for 3D image reconstruction and analysis |
T3 analysis of anti-PD-L1 distribution in the tumor microenvironment with different PD-L1 expression patterns.
We examined T3 for whether it can provide sufficient sensitivity to distinguish drug distribution patterns among different tumor microenvironments. Our previous report described the high expression of PD-L1 in CD31+ blood vessels in BALB-NeuT tumors (29). However, T3 imaging of tumors formed in BALB/c mice by subcutaneous injection of TUBO cells, a Her2+ cell line derived from a BALB-NeuT tumor, failed to demonstrate a similar pattern (Fig. 3L). Using the multiplex T3 protocol, we compared the distributions of DyLight594-labeled anti-PD-L1 in the microenvironment of BALB-NeuT and TUBO tumors 1 day after injection (Fig. 4A, B). High-resolution optical section images of BALB-NeuT macrosections displayed both the previously described PD-L1 expression in CD31+ vascular endothelium and a corresponding accumulation of i.v. injected anti-PD-L1 antibody in proximity to the PD-L1+CD31+ blood vessels (Fig. 4C). Comparing the relative distribution (%) of i.v. injected anti-PD-L1 antibody associated with the Her2+ tumor and CD31+ blood vessels in TUBO and BALB-NeuT macrosections (Fig. 4D) demonstrated a greater association of anti-PD-L1 antibody with CD31+ blood vessels in the BALB-NeuT tumor versus a higher level of binding to the Her2+ tissue in TUBO tumors. The different distributions appear to reflect distinct patterns of PD-L1 expression in the TUBO and BALB-NeuT tumor microenvironments.
T3 analysis of microvascular permeability
Despite the known diversity of types of microvasculature in tumors, the vascular barrier to macromolecular drug delivery is often modeled as a single parameter. Several lines of evidence suggest considerable heterogeneity in tumor vascular permeability (35, 36). The T3 multiplex imaging strategy enabled three-dimensional mapping of the full tumor vasculature and quantitative analysis of local perfusion and permeability with regards to anti-PD-L1 antibody transport. To investigate microvascular permeability, we visualized anti-PD-L1 antibody drug distribution at 10 min after injection by staining tumor macrosections to image vascular fibroblasts (ER-TR7), smooth muscle cells (SMA), and endothelial cells (CD31) (Fig. 5A, B and Supplementary Video 4). We then used a computational strategy to classify the microvasculature based on vascular cell composition and then to examine the dependence of anti-PD-L1 antibody distribution on vascular composites (Supplementary Fig. S5). The local radial penetration of anti-PD-L1 antibody from the CD31 endothelium at 10 min post injection compared to the average value of 10 μm (Fig. 2E) was used to evaluate local permeability. “Naked”, endothelium-only microvessels that stained with CD31 but lacked ER-TR7 or SMA showed greater permeability compared to the microvessels composed of multiple cell types (Fig. 5C). High-resolution longitudinal optical section images confirmed increased penetration along CD31-only sections of microvessels, suggesting a disproportionate role in overall anti-PD-L1 transport (Fig. 5D).
T3 to compare tumor penetration of antibody drugs
Immobilization and sequestration of monoclonal antibody drugs upon binding to their target antigens on tumor cell membranes is considered to be a constraint on tumor penetration and, thereby, the effects of therapeutic agents. Toward measuring this effect by T3, we compared the relative tumor perfusion of fluorescently labeled antibodies targeting tumor cell surface antigens and their isotype controls labeled with a different fluorophore. Thus, we modeled antibody therapy targeting the oncogenes Her2 and EGFR, which are recognized by the clinical agents Herceptin (trastuzumab) and Erbitux (cetuximab). To observe impact of antibody binding on delivery, we examined the relative penetration of the IgG2a anti-rat Her2 clone 7.16.4 versus IgG2a isotype control (DyLight594-anti-Her2/DyLight680-isotype IgG) into Her2+ mammary tumors formed in BALB-NeuT mice. Then, we compared penetration of an IgG2a anti-human EGFR clone 528 to IgG2a isotype control (DyLight594-anti-EGFR/DyLight680-isotype IgG) into an EGFR+ non-small cell lung cancer PDX tumor formed in an NSG mouse. We injected a mixture of the fluorescent antibodies i.v. and used T3 imaging to examine the distribution of specific and control antibodies in macrosections at 1 h (Fig. 6A, B). With CD31+ microvasculature visualization, examining antibody distribution at high magnification captured deeper penetration of isotype IgG antibody into tumor nests compared to the anti-Her2 or anti-EGFR antibodies (Fig. 6A bottom and 6B right, Supplementary Video 5 and 6). Using 3D distance mapping, the spatial tumor penetration of each antibody was determined by delineating all CD31+ blood vessels in the BALB-NeuT or lung cancer PDX tumor nest (shown in Fig. 6A bottom and 6B right) and determining radial penetration of the antibodies out from the blood vessel wall and into the tumor parenchyma (Fig. 6C). The mean penetrations of anti-Her2 and isotype IgG antibodies in the BALB-NeuT tumor nest were 38 μm and 63 μm from CD31+ blood vessel boundaries at 1 h after injection. The mean penetrations of anti-EGFR and isotype antibodies in lung cancer PDX tumor nest were 45 μm and 114 μm at 1 h after injection. The distribution volumes (%) of anti-Her2 and isotype IgG antibodies within the BALB-NeuT macrosection were 27% and 29%. The distribution volumes (%) of anti-EGFR and isotype IgG antibodies within the lung cancer PDX macrosection were 17% and 27%. These data support prior studies of therapeutic antibodies showing penetration into tumor parenchyma being limited by binding to target proteins on tumor membranes, potentially leaving watershed areas untreated.
Figure 6.
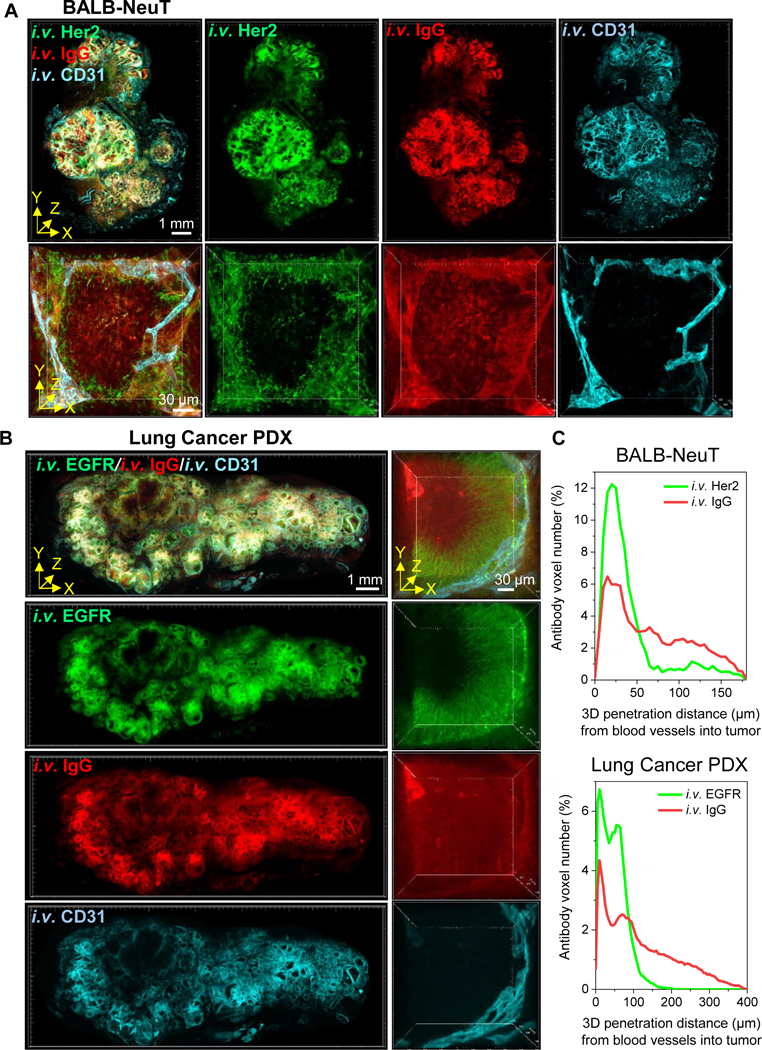
T3 analysis of tumor penetration of anti-Her2 or anti-EGFR and isotype IgG antibodies. Tumors collected 1 h after i.v. co-injection of fluorescently labeled specific and isotype control antibodies and 10 min after i.v. injection of fluorescent anti-CD31 antibody. A, 3D rendering of macrosection of BALB-NeuT tumor indicating distribution of anti-Her2 (green), IgG isotype (red) and blood vessels (cyan). Low magnification 3D image (upper, scale bar: 1 mm) demonstrates antibodies spreading through stroma with less delivery to tumor nests. High magnification 3D image (lower, scale bar: 30 μm) of a single tumor nest suggests anti-Her2 antibody remains proximal to vasculature while isotype control displays complete penetration. B, 3D rendering of macrosection of lung cancer PDX tumor indicating distribution of anti-EGFR (green), IgG isotype (red) and blood vessels (cyan). High magnification view (right) similarly displays limited penetration of anti-EGFR antibody compared to isotype control. C, 3D tumor penetration distance profiles of anti-Her2 and IgG antibodies in BALB-NeuT tumor (top) and anti-EGFR and IgG antibodies in lung cancer PDX tumor (bottom) determined as radial distance from CD31+ blood vessels at 1 h after injection. Merged distance data are derived from total antibody voxels detected in the 3D images of tumor nests represented in A (bottom) and B (right).
T3 imaging of polymers and nanoparticles distribution in the tumor microenvironment
Toward establishing T3 as a general tool to visualize distribution of macromolecular drugs in tumors, we examined delivery of a fluorescent polymer and a model nanoparticle. Dextran has been broadly used to develop polymer-drug conjugates and nano-drug formulations (37, 38), and PEGylated liposomal doxorubicin (Doxil) is an FDA approved nanoformulation for treating cancer patients (39, 40). With T3, we mapped 3D tumor distributions of fluorescent dextran and Doxil with CD31+ blood vessels at 30 min (dextran) and 4 h (Doxil) after injection in 4T1 tumor-bearing mice (Supplementary Fig. S6 and Supplementary Video 7 (Doxil)). T3 images revealed that dextran broadly transported from microvessels to tumor, while the fluorescence of DNA-bound doxorubicin was limited to the nuclei of perivascular cells.
Discussion
Recent advances in biology (41), immunology (42), and chemistry (43) have enabled dramatic progress in the development of biologics and other macromolecular drugs as targeted cancer therapeutics. Similar progress has been made in nanomedicines. However, without sufficient tools to track macromolecular drug distribution, it remains a challenge to overcome transport barriers, sequestration mechanisms and other interactions with the tumor microenvironment that may limit efficacy (44). For example, nanoscale drug formulations have been designed and developed based on their potential to leverage the enhanced permeability and retention (EPR) effect (9, 45). However, translation has lagged as it has become apparent that the EPR effect is negligible in many human tumors. Even in mice, the EPR effect is typically only sufficient to deliver a tiny fraction of circulating nanoparticles to tumors (10, 46). In turn, while monoclonal antibodies may extravasate more efficiently, their binding to target antigens and resulting sequestration make characterization of spatial distribution challenging (47). Methods such as spectroscopic imaging in tissue or microscopy in thin sections (48-50) are well-established as tools to track drug delivery but a technology gap has remained which would facilitate analysis of drug distribution in the heterogeneous tumor microenvironment.
Recent progress in tissue optical clearing and light microscopy have led to significant advances in 3D immunofluorescence imaging of tissues. In particular, CLARITY (51) and related methods (52) have provided powerful tools to establish connectivity in the central nervous system. Applications for cancer are emerging (53-55), but initial studies have revealed challenges, including rendering the tumor tissue sufficiently transparent while maintaining structural integrity and protein immunogenicity, obtaining satisfactory penetration of antibodies and conjugates, and performing multiplexed detection with high sensitivity and specificity at cellular resolution (56).
To overcome these challenges and advance technologies available for PK/PD studies, we have adapted the Transparent Tissue Tomography (T3) platform (29) to enable multiplex 3D imaging of macromolecular drugs in mouse tumor tissues and perform computational analysis of spatial distribution, interaction with target cells, and vascular penetration using open-source Fiji macros (57). Using in vivo CD31 vasculature labeling and heavy fixation after excising tumors soon after intravenous injection, it was possible to capture the macromolecular drugs still circulating in the blood stream and track their initial extravasation from blood vessels into tumor parenchyma. Lighter fixation facilitated tissue staining with fluorescent primary antibodies, allowing detection of multiple antigens along with tracking drug binding to its target.
Thereby, we used T3 to map the distribution of anti-PD-L1 antibody in whole mouse tumors and analyze the spatial pharmacokinetics of anti-PD-L1 antibody by determining 3D vascular penetration distance at different time points post injection. Potential limitations of applying the T3 method for spatial pharmacokinetics are that it depends on harvesting tumor tissues at a series of time points and concentration is measured by relative fluorescent intensity. However, T3 data can be readily linked to plasma drug concentration profiling and in vivo imaging to enhance both the quantitative and temporal resolution, which along with cell-level spatial distribution mapping, would provide a route to truly comprehensive analysis of drug delivery.
Because T3 is a multiplex method, it enables not only drug tracking but also analysis of features in the tumor and microenvironment that may influence delivery. Thus, T3 allowed cell-resolution analysis of the efficiency of targeting membrane-bound PD-L1 by anti-PD-L1 antibody. In turn, T3 also enabled analysis of the distribution pattern of anti-PD-L1 antibody with respect to tumor-scale heterogeneity of PD-L1 expression. Studying microvascular permeability, we found that CD31+ capillaries were leakier with regards to anti-PD-L1 antibody transport than microvessels surrounded by SMA+ smooth muscle cells and ER-TR7+ fibroblasts. Further, T3 made it straightforward to track the relative perfusion of two antibodies in a tumor, with T3 analysis confirming that co-injected non-specific IgG extravasates deeper into tumor parenchyma than targeted antibodies. T3 revealed the extent to which anti-Her2 and anti-EGFR antibodies were sequestered by binding to their target antigens at the periphery of tumor nests. The T3 strategy was also validated by tracking dextran to demonstrate transport through the interstitial space and to demonstrate intracellular delivery of doxorubicin to tumor cells by Doxil, demonstrating potential for broad applications for macromolecular drugs.
We report Transparent Tissue Tomography (T3) as a new tool for 3D spatial imaging and quantitative analysis of macromolecular drug transport to tumors. By assessing multiple spatial parameters simultaneously, T3 provides a unique 3D window into macromolecular drug distribution in the tumor microenvironment. Although demonstrated here as a tool to examine delivery of macromolecular drugs, T3 might also be adapted to tracking small molecule agents in the tumor microenvironment using covalent immobilization and detection with anti-drug antibodies (58). Toward determining the absolute local concentration of a drug in the tumor microenvironment, we are pursuing further development of methods to improve fluorescence detection and calibration that will enable direct measurement of drug levels by fluorescence intensity. Combined with T3’s multiplexing capabilities, this would enable determination of drug concentration-therapeutic effect relationships in pharmacokinetic/pharmacodynamic analysis at cellular resolution. To address another limitation of T3, we intend to automate sample staining, image acquisition and image processing, and implement machine learning strategies to enhance pattern recognition during data analysis, thus decreasing hands-on effort and speeding up sample-to-answer time while increasing the information gained from each sample. Thereby, we anticipate that T3 can be applied broadly to facilitate the development of new cancer drugs and strategies for targeted drug delivery to tumors.
Supplementary Material
Acknowledgments
This study was supported by National Cancer Institute (NCI) R01s CA164492 and CA199663 to S.J. Kron and by National Institute of Biomedical Imaging and Bioengineering (NIBIB) K99 EB022636 and Susan G. Komen Postdoctoral Fellowship PDF15333618 (made possible through funding from American Airlines) to S.S.-Y. Lee. Imaging, image processing, and data analysis were performed at the University of Chicago Integrated Light Microscopy Facility directed by V.P. Bindokas, supported by National Cancer Institute (NCI) P30 CA014599. TUBO cells were generously provided by the Emerling Lab at Sanford Burnham Prebys Medical Discovery Institute.
Financial Support: HHS | NIH | National Cancer Institute (NCI) - Ref.# R01 CA164492 and R01 CA199663 / Stephen Kron (skron@uchicago.edu), HHS | NIH | National Institute of Biomedical Imaging and Bioengineering (NIBIB) - Ref.# K99 EB022636 and Susan G. Komen for the Cure - Ref.# PDF 15333618 to Steve Seung-Young Lee (ssylee@uchicago.edu), HHS | NIH | National Cancer Institute (NCI) - Ref.# P30 CA014599 to Vytautas Bindokas (vbindoka@bsd.uchicago.edu)
Footnotes
Conflict of Interest Disclosure Statement
The authors are founders of Transnostics LLC to commercialize T3.
References
- 1.Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater 2017;2:17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov 2017;17:197–223. [DOI] [PubMed] [Google Scholar]
- 3.Serna N, Sánchez-García L, Unzueta U, Díaz R, Vázquez E, Mangues R, et al. Protein-based therapeutic killing for cancer therapies. Trends Biotechnol 2017;36:318–35. [DOI] [PubMed] [Google Scholar]
- 4.Jiang W, von Roemeling CA, Chen Y, Qie Y, Liu X, Chen J, et al. Designing nanomedicine for immuno-oncology. Nat Biomed Eng 2017;1:0029. [Google Scholar]
- 5.Thurber GM, Weissleder R. A systems approach for tumor pharmacokinetics. PLOS ONE 2011;6:e24696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986;46:6387–92. [PubMed] [Google Scholar]
- 7.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev 2013;65:71–9. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura H, Jun F, Maeda H. Development of next-generation macromolecular drugs based on the EPR effect: challenges and pitfalls. Expert Opin Drug Deliv 2015;12:53–64. [DOI] [PubMed] [Google Scholar]
- 9.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer 2016;17:20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater 2016;1:16014. [Google Scholar]
- 11.Nakamura Y, Mochida A, Choyke PL, Kobayashi H. Nanodrug delivery: is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjugate Chem 2016;27:2225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park K Facing the truth about nanotechnology in drug delivery. ACS Nano 2013;7:7442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer 2017;17:738–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Q, Ojha T, Kiessling F, Lammers T, Shi Y. Enhancing tumor penetration of nanomedicines. Biomacromolecules 2017;18:1449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, Hu Y, Pang Z. Modulating the tumor microenvironment to enhance tumor nanomedicine delivery. Front Pharmacol 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Kohane DS. External triggering and triggered targeting strategies for drug delivery. Nat Rev Mater 2017;2:17020. [Google Scholar]
- 17.Weissleder R, Schwaiger MC, Gambhir SS, Hricak H. Imaging approaches to optimize molecular therapies. Sci Transl Med 2016;8:355ps16–ps16. [DOI] [PubMed] [Google Scholar]
- 18.Beckmann N, Kaltashov IA, Windhorst AD. Editorial: in vivo imaging in pharmacological research. Front Pharmacol 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller MA, Weissleder R. Imaging the pharmacology of nanomaterials by intravital microscopy: toward understanding their biological behavior. Adv Drug Deliv Rev 2017;113:61–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SS-Y, Li J, Tai JN, Ratliff TL, Park K, Cheng J-X. Avasimibe encapsulated in human serum albumin blocks cholesterol esterification for selective cancer treatment. ACS Nano 2015;9:2420–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blom S, Paavolainen L, Bychkov D, Turkki R, Mäki-Teeri P, Hemmes A, et al. Systems pathology by multiplexed immunohistochemistry and whole-slide digital image analysis. Sci Rep 2017;7:15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remark R, Merghoub T, Grabe N, Litjens G, Damotte D, Wolchok JD, et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci Immunol 2016;1:aaf6925–aaf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C-W, Budiman Gosno E, Li Y-S. Fully automatic and robust 3D registration of serial-section microscopic images. Sci Rep 2015;5:15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feuchtinger A, Walch A, Dobosz M. Deep tissue imaging: a review from a preclinical cancer research perspective. Histochem Cell Biol 2016;146:781–806. [DOI] [PubMed] [Google Scholar]
- 25.Lagerweij T, Dusoswa SA, Negrean A, Hendrikx EML, de Vries HE, Kole J, et al. Optical clearing and fluorescence deep-tissue imaging for 3D quantitative analysis of the brain tumor microenvironment. Angiogenesis 2017;20:533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobosz M, Ntziachristos V, Scheuer W, Strobel S. Multispectral fluorescence ultramicroscopy: three-dimensional visualization and automatic quantification of tumor morphology, drug penetration, and antiangiogenic treatment response. Neoplasia 2014;16:1–W7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sindhwani S, Syed AM, Wilhelm S, Glancy DR, Chen YY, Dobosz M, et al. Three-dimensional optical mapping of nanoparticle distribution in intact tissues. ACS Nano 2016;10:5468–78. [DOI] [PubMed] [Google Scholar]
- 28.Sindhwani S, Syed AM, Wilhelm S, Chan WCW. Exploring passive clearing for 3D optical imaging of nanoparticles in intact tissues. Bioconjug Chem 2017;28:253–9. [DOI] [PubMed] [Google Scholar]
- 29.Lee SS-Y, Bindokas VP, Kron SJ. Multiplex three-dimensional optical mapping of tumor immune microenvironment. Sci Rep 2017;7:17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boggio K, Nicoletti G, Di Carlo E, Cavallo F, Landuzzi L, Melani C, et al. Interleukin 12–mediated prevention of spontaneous mammary adenocarcinomas in two lines of Her-2/neu transgenic mice. J Exp Med 1998;188:589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borgefors G On digital distance transforms in three dimensions. Comput Vis Image Underst 1996;64:368–76. [Google Scholar]
- 32.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shargel L, Yu ABC. Applied biopharmaceutics and pharmacokinetics. McGraw-Hill; 1999. [Google Scholar]
- 35.Ekdawi SN, Stewart JMP, Dunne M, Stapleton S, Mitsakakis N, Dou YN, et al. Spatial and temporal mapping of heterogeneity in liposome uptake and microvascular distribution in an orthotopic tumor xenograft model. J Control Release 2015;207:101–11. [DOI] [PubMed] [Google Scholar]
- 36.Mittapalli RK, Adkins CE, Bohn KA, Mohammad AS, Lockman JA, Lockman PR. Quantitative fluorescence microscopy measures vascular pore size in primary and metastatic brain tumors. Cancer Res 2017;77:238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varshosaz J Dextran conjugates in drug delivery. Expert Opin Drug Deliv 2012;9:509–23. [DOI] [PubMed] [Google Scholar]
- 38.Foerster F, Bamberger D, Schupp J, Weilbächer M, Kaps L, Strobl S, et al. Dextran-based therapeutic nanoparticles for hepatic drug delivery. Nanomedicine 2016;11:2663–77. [DOI] [PubMed] [Google Scholar]
- 39.Szebeni J, Fülöp T, Dézsi L, Metselaar B, Storm G. Liposomal doxorubicin: the good, the bad and the not-so-ugly. J Drug Target 2016;24:765–7. [DOI] [PubMed] [Google Scholar]
- 40.Rivankar S An overview of doxorubicin formulations in cancer therapy. J Cancer Res Ther 2014;10:853–8. [DOI] [PubMed] [Google Scholar]
- 41.Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci USA 2016;113:2868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuesta-Mateos C, Alcaraz-Serna A, Somovilla-Crespo B, Muñoz-Calleja C. Monoclonal antibody therapies for hematological malignancies: not just lineage-specific targets. Front Immunol 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi H, Turkbey B, Watanabe R, Choyke PL. Cancer drug delivery: considerations in the rational design of nanosized bioconjugates. Bioconjugate Chem 2014;25:2093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenblum D, Joshi N, Tao W, Karp JM, Dan P. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun 2018;9:1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aftab S, Shah A, Nadhman A, Kurbanoglu S, Aysıl Ozkan S, Dionysiou DD, et al. Nanomedicine: an effective tool in cancer therapy. Int J Pharm 2018;540:132–49. [DOI] [PubMed] [Google Scholar]
- 46.Hansen AE, Petersen AL, Henriksen JR, Boerresen B, Rasmussen P, Elema DR, et al. Positron emission tomography based elucidation of the enhanced permeability and retention effect in dogs with cancer using copper-64 liposomes. ACS Nano 2015;9:6985–95. [DOI] [PubMed] [Google Scholar]
- 47.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci 2008;29:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schluep T, Hwang J, Hildebrandt IJ, Czernin J, Choi CHJ, Alabi CA, et al. Pharmacokinetics and tumor dynamics of the nanoparticle IT-101 from PET imaging and tumor histological measurements. Proc Natl Acad Sci USA 2009;106:11394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoppin J, Orcutt KD, Hesterman JY, Silva MD, Cheng D, Lackas C, et al. Assessing antibody pharmacokinetics in mice with in vivo imaging. J Pharmacol Exp Ther 2011;337:350–8. [DOI] [PubMed] [Google Scholar]
- 50.Lee CM, Tannock IF. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 2010;10:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chung K, Wallace J, Kim S-Y, Kalyanasundaram S, Andalman AS, Davidson TJ, et al. Structural and molecular interrogation of intact biological systems. Nature 2013;497:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tainaka K, Kuno A, Kubota SI, Murakami T, Ueda HR. Chemical principles in tissue clearing and staining protocols for whole-body cell profiling. Annu Rev Cell Dev Biol 2016;32:713–41. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka N, Kanatani S, Tomer R, Sahlgren C, Kronqvist P, Kaczynska D, et al. Whole-tissue biopsy phenotyping of three-dimensional tumours reveals patterns of cancer heterogeneity. Nat Biomed Eng 2017;1:796–806. [DOI] [PubMed] [Google Scholar]
- 54.Cuccarese MF, Dubach JM, Pfirschke C, Engblom C, Garris C, Miller MA, et al. Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D organ imaging. Nat Commun 2017;8:14293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lloyd-Lewis B, Davis FM, Harris OB, Hitchcock JR, Lourenco FC, Pasche M, et al. Imaging the mammary gland and mammary tumours in 3D: optical tissue clearing and immunofluorescence methods. Breast Cancer Res 2016;18:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson DS, Lichtman JW. Clarifying tissue clearing. Cell 2015;162:246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lawson ADG. Antibody-enabled small-molecule drug discovery. Nat Rev Drug Discov 2012;11:519–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.