

Abstract
The developing brain is uniquely susceptible to drug-induced increases in programmed cell death or apoptosis. Many compounds, including anticonvulsant drugs, anesthetic agents, and ethanol, when administered in a narrow postnatal window in rodents, result in increased pruning of neurons. Here, we report that dimethyl sulfoxide (DMSO) triggers widespread neurodegeneration in the immature (postnatal day, P7) rat brain, an effect consistent with a prior report in neonatal mice. We found that the synthetic cannabinoid receptor agonist WIN 55,212–2 (WIN) exerts a neuroprotective effect against DMSO-induced cell death. We extended these findings to determine if WIN is neuroprotective against another drug class known to increase developmental cell death, namely anti-seizure drugs. The anti-seizure drug phenobarbital (PB) remains the primary treatment for neonatal seizures, despite significantly increasing cell death in the developing rodent brain. WIN exerts anti-seizure effects in immature rodent seizure models, but increases the toxicity associated with neonatal ethanol exposure. We thus sought to determine if WIN would protect against or exacerbate PB-induced cell death. Unlike either the prior report with ethanol or our present findings with DMSO, WIN was without effect on PB- induced cell death; it neither increased nor decreased PB-induced apoptosis. WIN alone did not increase cell death over levels observed in vehicle-treated rats. These data suggest that WIN has a favorable safety profile in the developing brain, and could potentially serve as an adjunct therapy with phenobarbital (albeit one that does not attenuate PB-induced toxicity).
Keywords: apoptosis, cell death, degeneration, development, rat, toxicity, brain growth spurt, barbiturate, cannabinoid
Introduction
Programmed cell death, or apoptosis, is a tightly regulated process that is essential for network development and homeostasis in the developing brain; cells that make appropriate synaptic connections are maintained, whereas those that fail to wire appropriately are pruned (Elmore 2007). During development, apoptosis in the rodent brain exhibits high temporal specificity with peak programmed cell death detected between postnatal day (P) 2 and the end of the first postnatal week (Ferrer et al. 1992). After P7, the rate of apoptosis dramatically decreases into the second week and developmental cell death is undetectable by the end of the first month (Ferrer et al. 1992).
During this period of developmental vulnerability, a variety of compounds that suppress synaptic transmission can potentiate ongoing apoptosis. Exposure to several drug classes, including ethanol, (Ikonomidou et al. 2000; Olney et al. 2004; Hansen et al. 2008) anxiolytics, (Ikonomidou et al. 2000; Olney et al. 2004) anesthetics, (Ikonomidou et al. 1999; Jevtovic- Todorovic et al. 2003) and some (but not all) anticonvulsants (Bittigau et al. 2003; Bittigau et al. 2006; Forcelli et al. 2011) significantly increase cell death during the first postnatal week. Similarly, dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system (CNS) of mice with vulnerability to degeneration peaking at P7 (Hanslick et al. 2009). This profile is strikingly different from that observed in the adult brain, where these agents do not trigger apoptosis, and in some cases, actually exert neuroprotective effects.
For example, in adult animal models of traumatic brain injury, spinal cord injury, and stroke, DMSO has been reported to significantly reduce cell death and damage (Bardutzky et al. 2005; Giorgio et al. 2008; Jacob and de la Torre 2009). Developmentally restricted increases in cell death by otherwise neuroprotective agents is not a wholly new phenomenon. Plant-based and synthetic cannabinoids have demonstrated neuroprotective effects in several neurodegenerative disease models (Ramirez et al. 2005; Gowran et al. 2011; Sanchez and Garcia-Merino 2012; Concannon et al. 2015; Bisogno et al. 2016). Cannabinoid receptor agonists can potentiate developmental neurotoxicity induced by other compounds, but have no proapoptotic effect alone despite the early ontogeny of the cannabinoid system and reported neuroprotective effects in adult animals (Harkany et al. 2007; Hansen et al. 2008). For example, in P7 mice, administration of the mixed CB1/CB2 receptor synthetic agonist WIN 55,212–2 (WIN) did not increase cell death compared to vehicle controls (Hansen et al. 2008). However, WIN dose-dependently increased the apoptotic response induced by ethanol administration in these animals (Hansen et al. 2008). Given the increasing interest in cannabinoids as therapeutic agents for neonatal seizures, a more detailed toxicity profile is needed.
Provided the pattern of age-dependent toxicity of otherwise neuroprotective agents and the report of potentiated cell death by cannabinoid agonists, we aimed to (1) confirm that DMSO increases developmental cell death in P7 rats and (2) determine if DMSO- or PB- induced cell death would be altered by co-treatment with the cannabinoid agonist WIN.
Methods
Animals
Female Sprague-Dawley rats with male pups were obtained from Harlan/Envigo (Envigo, Frederick, MD, USA) and were delivered to the Division of Comparative Medicine at Georgetown University at postnatal day (P) 5. On P7, pups were treated, returned to the dam and allowed to survive for 24 hours before tissue collection. A total of 120 pups, derived from 12 litters were used for the present experiments.
For all experiments, treatments were counterbalanced within and across litters. Animals were maintained in temperature-controlled (21 °C) rooms with a 12 h light cycle and food (Lab Diet #5001) and water available ad libitum in the Georgetown University Division of Comparative Medicine. All experimental manipulations occurred during the light phase. Experimental procedures were performed in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care standards and were approved by the Georgetown University Animal Care and Use Committee.
Drug preparation
All drug treatments were administered intraperitoneally (ip) at a volume of 10 mL/kg. WIN 55,212–2 (Cayman Chemical, Ann Arbor, Ml, USA) was prepared in either 100% dimethyl sulfoxide (DMSO) ora vehicle (VEH) solution of ethanol: Kolliphor: 0.9% saline (2:1:17; Sigma- Aldrich, St. Louis, MO, USA) in concentrations of 0.1, 0.3, or 1.0 mg/ml (corresponding to doses of 1, 3, and 10 mg/kg, respectively; Hansen et al. 2008; Huizenga et al. 2017). We have previously reported that 1–3 mg/kg of WIN protects immature animals from seizures evoked by methyl-6,7-dimethoxy-4-ethyl-beta-carboline-3-carboxylate (DMCM) and pentylenetetrazole (PTZ; Huizenga et al. 2017). Phenobarbital (PB, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 0.9% saline at a concentration of 75 mg/ml (corresponding to a dose of 75 mg/kg). This dose has been demonstrated to exert anti-seizure efficacy against PTZ-induced seizures in P7 rat pups (Kubova and Mares 1991; Forcelli et al. 2013). Moreover, this dose of PB robustly increases cell death above developmental levels (Bittigau et al. 2006; Forcelli et al. 2011). Drug combination treatments were administered in a single injection.
Drug treatment
P7 pups were randomly assigned to receive a single dose of: DMSO, WIN (1, 3, or 10mg/kg in DMSO), saline, PB (75mg/kg in saline), vehicle (VEH; 2:1:17 ethanol, Kolliphor, saline), WIN (1, 3, or 10mg/kg in vehicle) or a combination of WIN (3 or 10mg/kg in vehicle) plus PB (75mg/kg).
Tissue Preparation
Twenty-four hours after drug treatment, pups were perfused transcardially with phosphate buffered saline (pH 7.4, 10mL; Thermo Fisher Scientific, Waltham, MA, USA) followed by 4% paraformaldehyde (10mL; Sigma-Aldrich, St. Louis, MO, USA). This survival interval was based on prior reports examining drug-related apoptosis in the developing brain (Bittigau et al. 2002; Kim et al. 2007). Brains were removed and post-fixed for 24 h before transferred to 30% sucrose for at least 72 h before cryosectioning. 40 μm thick coronal sections were mounted and stained for Fluoro-Jade B as described below.
Fluoro Jade B staining:
40 μm coronal sections were stained using the Fluoro-Jade B histological staining protocol (Histo-Chem Inc., Jefferson, AR, USA) to fluorescently label neurons undergoing degeneration. Slides were first immersed in a solution containing 1% sodium hydroxide in 80% ethanol for 5 min. Next, slides were transferred to 70% ethanol followed by distilled water for 2 min each. Then, slides were transferred to a 0.06% potassium permanganate solution in distilled water, for 5–10 min. After a 2 min rinse in distilled water, slides were immersed in the Fluoro-Jade B (EMD Millipore, Burlington, MA, USA) staining solution with a final dye concentration of 0.0004%, for 15 min. Last, the slides were rinsed 3 times for 1 min each before dried, cleared by at least 1 min of xylene immersion and coverslipped using Cytoseal 60 (Thermo Fisher Scientific, Waltham, MA, USA) mounting media.
Microscopy
Fluorescent photomicrographs were collected on a Nikon 80i microscope with a Qlmaging QlClick camera. Images at 10x magnification of three sequential sections at 200 pm intervals for each brain region were taken. Brain regions were defined using the neonatal rat brain atlas of Ramachandra and Subramanian (Ramachandra and Subramanian 2011) and selected based on prior reports of anti-seizure drug induced enhancements in cell death from our lab (Forcelli et al. 2011; Kaushal et al. 2016; Brown et al. 2016) and others (Bittigau et al. 2006). Quantification of neurodegeneration within each region was performed by manual counting of the Fluoro-Jade B positive cells within the anatomical boundaries for each brain regions. Cell counting was performed using IMAGEJ software (National Institutes of Health, Bethesda, MD, USA). For analysis, the mean number of degenerating (Fluoro-Jade B positive) cells for each region in each animal was calculated from the three sequential sections. Microscopy and analysis was performed by investigators blinded to treatment conditions.
Statistics
Statistical analyses were performed using GraphPad Prism 7.0c (GraphPad Software; La Jolla, CA, USA). Cell death analyses were analyzed using one-way ANOVA with multiple comparisons corrected for by Sidak and Holm-Sidak’s multiple comparisons test. Probability values < 0.05 were considered statistically significant. One statistical outlier (treated with the highest dose of WIN + DMSO) was identified using the ROUT algorithm (Q=0.1%) and was removed.
Results
Effect of WIN on DMSO-induced Cell Death
DMSO has been shown to induce neuronal toxicity, particularly during vulnerable periods of development (Hanslick et al. 2009). To determine the effect of CB receptor agonism on DMSO-induced cell death, we quantified Fluoro-Jade B positive cells after treatment with WIN dissolved in DMSO (1, 3, and 10 mg/kg). In line with a prior report that found DMSO increases developmental apoptosis (Hanslick et al. 2009), we found DMSO alone increases neuronal cell death across the developing brain as compared to saline treatment. This effect was striking, with 8 to 26-fold increases in cell death observed. Administration of WIN resulted in a dose-dependent decrease in DMSO-induced cell death in cortical brain regions (Fig. 1). There was a significant treatment effect in the cingulate cortex (ANOVA, F4,37=6.47, p=0.0005; Fig. 1A), motor cortex (ANOVA, F4,30=11.99, p<0.0001; Fig. 1B) and somatosensory cortex (ANOVA, F4,30 = 14.73, p<0.0001; Fig. 1C). Sidak’s multiple comparisons test revealed that DMSO significantly increased cell death compared to saline treated controls in the cingulate (p=0.0004), motor (p<0.0001), and somatosensory (p<0.0001) cortices. WIN, when given at a dose of 10 mg/kg, significantly decreased the DMSO-induced cell death in the cingulate (p=0.0084) and motor cortices (p=0.0005). In the somatosensory cortex, WIN reduced DMSO- induced cell death at both the 3 mg/kg (p=0.0157) and 10 mg/kg (p=0.0003) doses.
Figure 1. WIN dose-dependently decreases DMSO-induced cell death across cortical regions.
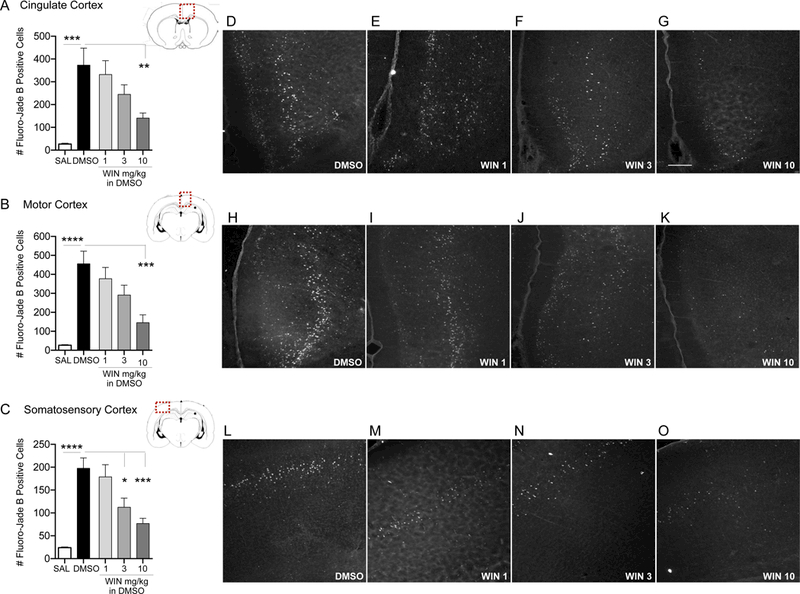
Quantification of cell death as indicated by Fluoro-Jade B positive cells in the cingulate cortex (A), motor cortex (B) and somatosensory cortex (C) from P7 rat pups treated with DMSO or WIN 55,212–2 (WIN; 1, 3, and 10 mg/kg i.p.). Values are expressed as mean +S.E.M of 3 sequential 40μm tissue samples per animal (n=6–10 animals). *p<0.05, ***p<0.005, ****p<0.0001; significantly different from SAL or DMSO-treated group. Photomicrographs of Fluoro-Jade B stained section in cingulate cortex (D-G), motor cortex (H-K), and somatosensory cortex (L-O) by treatment. Scale bar in Panel G = 200 μm.
In subcortical regions, we observed a similar pattern (Fig. 2): there was a significant main effect of drug treatment in striatum (ANOVA, F4,29=8.10, p=0.0002; Fig. 2A), lateral thalamus (ANOVA, F4,30=24.72, p<0.0001; Fig. 2B), and lateral septum (ANOVA, F4,35=6.39, p=0.0006; Fig. 2C). Sidak’s multiple comparisons test confirmed that DMSO significantly increased cell death in the striatum (p<0.0001), lateral thalamus (p<0.0001), and lateral septum (p=0.0002), as compared to saline treated controls. As in the cortical regions, the 10 mg/kg dose of WIN significantly decreased DMSO-induced cell death in the striatum (p=0.0049) and lateral thalamus (p=0.0005). WIN was without effect on DMSO-increased cell death in the lateral septum.
Figure 2. High-dose WIN decreases DMSO-induced cell death in the striatum and lateral thalamus.
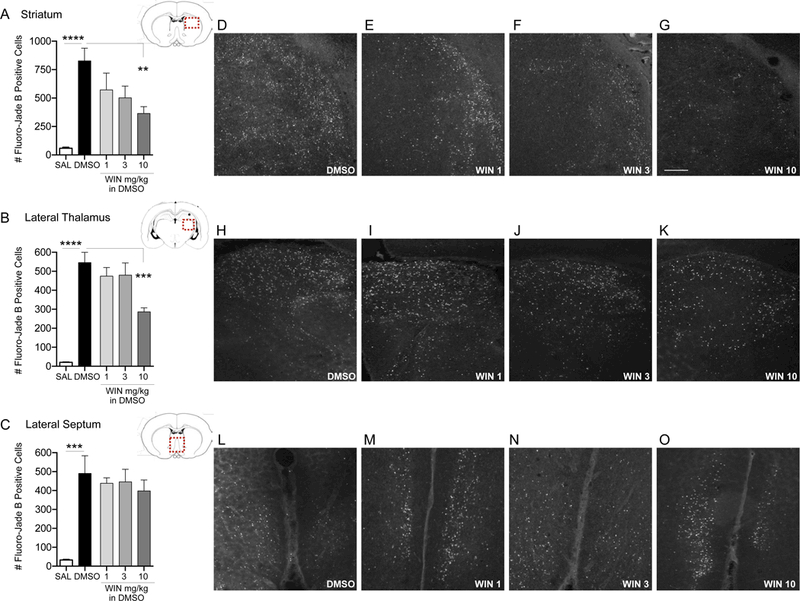
Quantification of cell death as indicated by Fluoro-Jade B positive cells in the striatum (A), lateral thalamus (B), and lateral septum (C) from P7 rat pups treated with DMSO or WIN 55,212–2 (WIN; 1, 3, and 10 mg/kg i.p.). Values are expressed as mean +S.E.M of 3 sequential 40μm tissue samples per animal (n=6–10 animals). **p<0.01, ***p<0.0005, ****p<0.0001 significantly different from SAL or DMSO-treated group. Photomicrographs of Fluoro-Jade B stained section in striatum (D-G), lateral thalamus (H-K), and lateral septum (L- O) by treatment. Scale bar in Panel G = 200 μm.
Effect of WIN alone, and in combination with PB, on Cell Death.
The neuroprotective effect of WIN against DMSO-induced cell death led us to next examine if: (1) WIN, when administered in an inert vehicle, impacts basal levels of cell death in the developing brain, and (2) if WIN would attenuate the cell death caused by a common anti-seizure medication, phenobarbital.
When prepared in an inert vehicle solution (VEH, see methods), WIN alone did not modify cell death from the levels seen in vehicle treated controls (Figs. 3–5). However, as expected, phenobarbital exposure caused a significant increase in the number of Fluoro-Jade B positive cells in each region analyzed. This increase in degenerating cells ranged from 2.5 to 10-fold above saline treated controls. This is a similar increase to that previously reported following PB exposure (Bittigau et al. 2006; Forcelli et al. 2011; Kaushal et al. 2016; Brown et al. 2016).
Figure 3. WIN modifies PB-induced enhancement of developmental cell death levels in cortical regions.
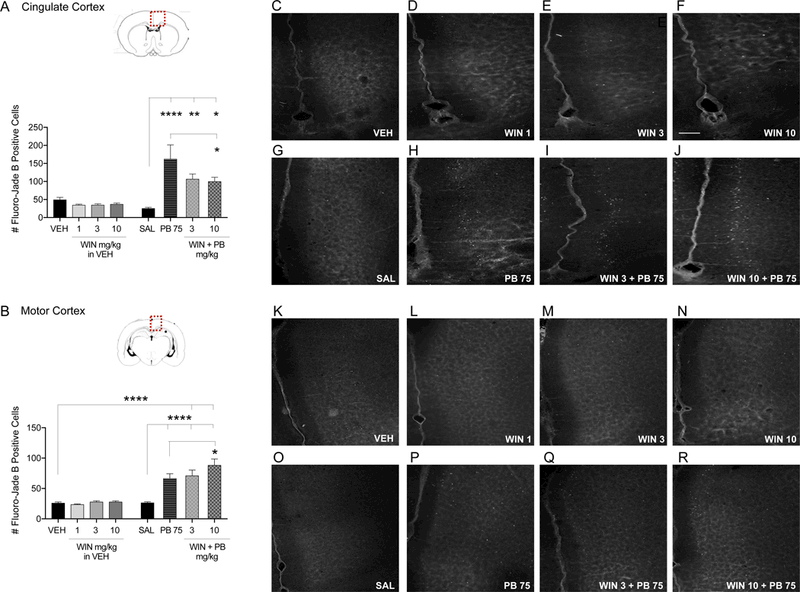
Quantification of cell death as indicated by Fluoro-Jade B positive cells in the cingulate cortex (A) and motor cortex (B) from P7 rat pups treated with VEH, WIN (1, 3, and 10 mg/kg prepared in VEH), SAL, PB 75 mg/kg, or combination of WIN 3 and 10 mg/kg plus PB 75 i.p. Values are expressed as mean +S.E.M of 3 sequential 40μm tissue samples averaged per animal (n=7–9 animals). *p<0.05, **p<0.01, ****p<0.001; significantly different from SAL, VEH, or PB 75 groups. Photomicrographs of Fluoro-Jade B stained section in cingulate cortex (C-J) and motor cortex (K-R) by treatment. Scale bar in Panel F = 200 μm.
Figure 5. WIN has no effect on PB-induced enhancement of developmental cell death in the lateral thalamus and septum.
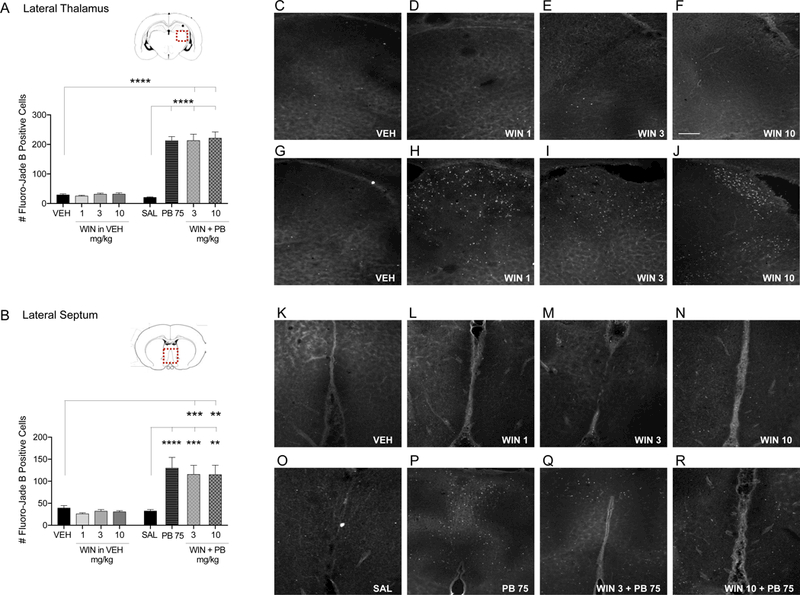
Quantification of cell death as indicated by Fluoro-Jade B positive cells in the lateral thalamus (A) and lateral septum (B) from P7 rat pups treated with VEH, WIN (1, 3, and 10 mg/kg prepared in VEH), SAL, PB 75 mg/kg, or combination of WIN 3 and 10 mg/kg plus PB 75 i.p. Values are expressed as mean +S.E.M of 3 sequential 40μm tissue samples averaged per animal (n=6–8 animals). **p<0.01, ***p<0.005, ****p<0.001; significantly different from VEH or SAL control groups. Photomicrographs of Fluoro-Jade B stained section in lateral thalamus (B-l) and lateral septum (K-R) by treatment. Scale bar in Panel F = 200 μm.
These effects were confirmed by one-way analysis of variance, which revealed significant treatment effects in each brain region analyzed (cingulate cortex: F7,55=9.67, p<0.0001; motor cortex: F7,48=20.01, p<0.0001, somatosensory cortex: F7,48=21.39, p<0.0001; striatum: F7,47=32.72, p<0.0001; lateral thalamus: F7,48=68.32, p<0.0001; lateral septum: F7,46=11–48, p<0.0001). These effects were driven by PB exposure, a finding consistent with prior reports. In each region, Holm-Sidak corrected pairwise comparisons revealed significant differences between the PB treated group and the saline treated groups for each region (cingulate cortex: p=0.0004, Fig. 3A; motor cortex: p<0.0001, Fig. 3B; somatosensory cortex: p<0.0001, Fig. 4A; striatum: p<0.0001, Fig. 4B; lateral thalamus: p<0.0001, Fig. 5A; lateral septum: p=0.0002, Fig. 5B).
Figure 4. WIN has no effect on PB-induced enhancement of developmental cell death in the somatosensory cortex and striatum.
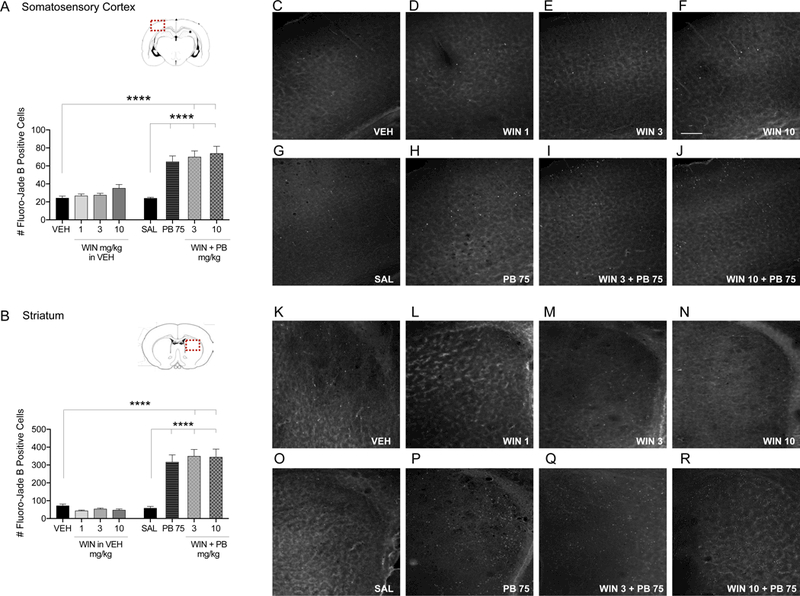
Quantification of cell death as indicated by Fluoro- Jade B positive cells in the somatosensory cortex (A) and striatum (B) from P7 rat pups treated with VEH, WIN (1, 3, and 10 mg/kg prepared in VEH), SAL, PB 75 mg/kg, or combination of WIN 3 and 10 mg/kg plus PB 75 i.p. Values are expressed as mean +S.E.M of 3 sequential 40μm tissue samples averaged per animal (n=6–8 animals). ****p<0.001; significantly different from VEH or SAL control groups. Photomicrographs of Fluoro-Jade B stained section in somatosensory cortex (C-J) and striatum (K-R) by treatment. Scale bar in Panel F = 200 μm.
Surprisingly, despite the robust effect of WIN against DMSO-induced cell death, the effects of WIN against PB-induced cell death were limited. Only within the cingulate cortex, and only at the 10 mg/kg dose, did WIN significantly reduce cell death compared to PB alone (Holm- Sidak, p=0.0445). The reduction in cell death observed with 3 mg/kg dose of WIN approached but did not reach the level of statistical significance (p=0.07). The protection observed with WIN was incomplete, as levels of cell death still exceeded, although not significantly, the vehicle treated group (WIN 3 + PB 75: p=0.0561; WIN 10 + PB 75: p=0.0843) and significantly exceeded the saline treated group (WIN 3 + PB 75: p=0.0067; WIN 10 + PB 75: p=0.0148).
In the motor cortex, a different pattern was observed. While PB significantly increased cell death above saline control levels (Holm-Sidak, p<0.0001), co-treatment with WIN (10 mg/kg) produced a small but significant exacerbation of injury (Holm-Sidak p=0.0447; Fig. 3B).
In the somatosensory cortex and striatum, PB alone (p<0.0001) and in combination with WIN (3 and 10 mg/kg) produced significant increases in cell death compared to vehicle (Holm- Sidak, p<0.0001) and saline controls (Holm-Sidak, p<0.0001; Fig. 4A,B). Co-treatment with WIN did not produce an effect that differed from that of PB alone. The same pattern was observed in lateral thalamus and lateral septum. PB alone (p<0.0001) and in combination with WIN (3 and 10 mg/kg) produced significant increases in cell death compared to vehicle (lateral thalamus: WIN 3 + PB 75: p<0.0001, WIN 10 + PB 75: p<0.0001; lateral septum: WIN 3 + PB 75: p=0.0010; WIN 10 + PB 75: p=0.0014) and saline treated controls (lateral thalamus: WIN 3 + PB 75: p<0.0001, WIN 10 + PB 75: p<0.0001; lateral septum: WIN 3 + PB 75: p=0.0009, WIN 10 + PB 75: p=0.0013; Fig. 5A,B).
Discussion
Here we evaluated the toxicity and neuroprotective potential of DMSO, phenobarbital, and the CB1/2 receptor agonist WIN 55,212–2 in immature (P7) rats. We report (1) robust induction of cell death throughout the developing brain following administration of DMSO, (2) a smaller magnitude induction of cell death after treatment with phenobarbital, (3) a benign profile of WIN 55,212–2, and (4) a divergent neuroprotective effect of WIN 55,212–2 against DMSO and PB-induced cell death.
This profile of enhanced neurodegeneration with DMSO is consistent with a prior report indicating DMSO increases apoptosis in developing mice (Hanslick et al. 2009). In that report, DMSO-induced apoptosis (as measured through activated caspase-3 immunohistochemistry and electron microscopy) was most prominent in the first two postnatal weeks (Hanslick et al. 2009). Toxicity was evident with doses of DMSO as low as 0.3 ml/kg, with maximal effects seen at 10 ml/kg doses (which is the dose we used in the present study). DMSO is an organic solvent often used as a vehicle for drug delivery in preclinical studies, and these data indicate that it should not be considered biologically inert.
While the mechanisms by which DMSO induces cell death in developing animals are unknown, the phenomena appear similar to that observed after early exposure to sedative, anxiolytic, anesthetic, and anticonvulsant drugs. First, as with these other drugs, DMSO-induced cell death occurs during a restricted developmental period, primarily during the first two postnatal weeks (Hanslick et al. 2009). Second, a central hypothesis behind drug induced apoptosis is the suppression of network activity. During development, neurons are suggested to require synaptic input to allow them to integrate into a functioning neuronal network. Network integration is a requirement for survival and suppression of activity consequently increases apoptosis (Heck et al. 2008). Furthermore, blockade of NMDA receptors triggers apoptotic neurodegeneration, supporting the idea that network activity suppresses an apoptotic response to encourage functional network development (Ikonomidou et al. 1999; Dikranian et al. 2001). Consistent with this hypothesis, DMSO suppresses NMDA- and AMPA-mediated currents in cultured hippocampal neurons (Lu and Mattson 2001). GABA, glutamate, and acetylcholine receptor activity is also reduced by DMSO (Sawada and Sato 1975; Hulsmann et al. 1999). suggesting that suppression of synaptic transmission may be a mechanism underlying its developmental toxicity.
We found that the CB1/2 receptor agonist WIN 55,212–2, which is commonly dissolved in DMSO, produced a dose-dependent neuroprotective effect against DMSO-induced cell death. This is interesting in light of a prior report of WIN enhancing developmental apoptosis triggered by ethanol (Hansen et al. 2008). Cannabinoid receptor agonists have been reported to display CB1 receptor mediated neuroprotective effects by lowering extracellular glutamate to inhibit excitotoxic release (Shen et al. 1996; Szabo et al. 2000; Gerdeman and Lovinger 2001; Robbe et al. 2001) and through HINT1-mediated modulation of NMDA receptor (Vicente-Sánchez et al. 2013; Sánchez-Blázquez et al. 2013). Along these lines WIN has been reported to protect against a variety of excitotoxic insults, including glutamate (Shen et al. 1996), 3-nitropropionic acid (3-NP; Maya-López et al. 2017), and quinolinic acid toxicity (Rangel-López et al. 2015). However, these effects are likely do not explain the neuroprotective effects of WIN in the present study, as rather than excitotoxic injury, PB and DMSO-induced damage likely results from a suppression of activity.
Together these data provide comprehensive evidence of extensive neuroprotective effects via CB receptor activation. In fact, the endocannabinoid system itself appears to play a role in the brain response to injury: both the CB1 receptor and its endogenous ligand anandamide, increase in early postnatal rats following injury or NMDA receptor blockade (Hansen et al. 2001; Marsicano et al. 2003; Mechoulam and Parker 2013). Although principally expressed in peripheral immune cells, CB2 receptors are also expressed in select neuronal populations and receptor activation can promote neuronal survival by modulating the release of immunomodulators from astrocytes and microglial cells to inhibit excitotoxicity, oxidative stress, and neuronal apoptosis (Mechoulam and Parker 2013; Bisogno et al. 2016). Therefore, WIN activation of both CB1 and CB2 receptors underlies the neuroprotective effects that we detected.
A considerable amount of research reveals a strong and consistent proapoptotic effect of the anticonvulsant drug, phenobarbital (PB) on the developing rodent brain (Olney et al.; Bittigau et al. 2006; Ikonomidou 2009; Forcelli et al. 2011; Kaushal et al. 2016; Brown et al. 2016). PB is a positive allosteric modulator of the GABAA receptor, which results in a suppression of neuronal activity. As described above, suppression of neurotransmission during synaptogenesis in the developing brain triggers enhanced apoptosis (Olney et al. 2004; Bittigau et al. 2006; Ikonomidou 2009). PB is of particular interest, as it is the current first-line treatment for neonatal seizures (World Health Organization et al. 2011), despite its toxicity in the developing brain, even at therapeutically relevant doses (Kubova and Mares 1991; Bittigau et al. 2006; Forcelli et al. 2011). This underscores the importance of identifying adjunct therapies that can mitigate the neurotoxicity of PB without sacrificing its antiseizure effects.
We recently described the antiseizure efficacy of CB1 receptor agonists in multiple models of seizures in developing animals (Huizenga et al. 2017). WIN, in particular, significantly reduced seizure severity in three different epilepsy models in P10 rats (Huizenga et al. 2017). The antiseizure effect of WIN in developing animals, together with its neuroprotective effect against DMSO-induced cell death, led us to examine if WIN, in combination with PB, would reduce PB-induced cell death. However, unlike the protective effect WIN exerted against DMSO-induced cell death, it produced a mixed effect against PB-induced cell death. In one brain region WIN increased PB-induced cell death, in another it decreased it, and in the remaining regions it was without effect. The mechanism behind this region-specificity are unknown, but unlikely related to CB receptor expression as the receptors are expressed early in development, and WIN was effective in all regions examined against DMSO-induced cell death.
The combined administration of WIN and PB did not exhibit a consistent enhancement in cell death similar to the widespread apoptosis seen with PB and ∆9-tetrahydrocannabinol (THC; (Hansen et al. 2008). One reason for this difference could be a result of the multimodal mechanism of action of THC compared to the CB receptor specificity of WIN. Despite both acting at the CB1 receptor, THC is only a partial agonist, and has been shown to transiently activate and desensitize the transient receptor potential (TRP) family of receptors (Felder et al. 1995; Pertwee 2008; De Petrocellis et al. 2011). Additionally, THC is reported to antagonize the action of endogenous ligands, which could effectively blunt any otherwise neuroprotective response from endocannabinoid production and signaling (Pertwee 2008). However, further studies are required to delineate the role of the complex cannabinoid signaling during development, in response to pathological conditions, and in conjunction with other compounds.
Here we have shown the cannabinoid receptor agonist WIN displays neuroprotective effects against toxicity induced by neonatal DMSO administration. This effect was selective and did not extend to toxicity induced by neonatal PB exposure. These data are relevant to both research and clinical practice. As WIN is sparingly soluble in aqueous solvents and is recommended by the manufacturer to be wholly or initially dissolved in organic solvents such as DMSO or ethanol (Caymen Chemical, Ann Arbor, Ml, USA). However, provided the biological activity of both DMSO and ethanol alone, especially in the developing brain, careful attention should be paid to the source of any detected experimental outcomes when using either as a drug solvent. Additionally, with a growing interest and use of CB targeted compounds for pediatric epilepsy (Friedman and Devinsky 2015), the neurodevelopmental toxicity of DMSO limits its clinical use as a solvent. While the precise mechanisms of WIN’s neuroprotective action remain unknown, these data underscore its potential as a neuroprotective agent against select insults during critical periods of brain development.
Acknowledgments
Funding: MNH was supported by TL1TR001431; PAF was supported by R01NS097762 and KL2TR001432.
Abbreviations:
- DMSO
dimethyl sulfoxide
- WIN
WIN 55,212–2
- PB
phenobarbital)
- P
postnatal day
References
- Bardutzky J, Meng X, Bouley J, Duong T, Ratan R, et al. (2005) Effects of intravenous dimethyl sulfoxide on ischemia evolution in a rat permanent occlusion model. J Cereb Blood Flow Metab 25:968–977. doi: 10.1038/sj.jcbfm.9600095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Oddi S, Piccoli A, Fazio Domenico, Maccarrone M (2016) Type-2 cannabinoid receptors in neurodegeneration. Pharmacol Res 111:721–730. doi: 10.1016/j.phrs.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D et al. (2002) Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proceedings of the National Academy of Sciences 99:15089–15094. doi: 10.1073/pnas.222550499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Ikonomidou C (2003) Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci 993:103–114; discussion 123–124 [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Ikonomidou C (2006) Antiepileptic Drugs and Apoptosis in the Developing Brain. Annals of the New York Academy of Sciences 993:103–114. doi: 10.1111/j.1749-6632.2003.tb07517.x [DOI] [PubMed] [Google Scholar]
- Brown L, Gutherz S, Kulick C, Soper C, Kondratyev A et al. (2016) Profile of retigabine-induced neuronal apoptosis in the developing rat brain. Epilepsia 57:660–670. doi: 10.1111/epi.13335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon R, Okine B, Finn D, Dowd E (2015) Differential upregulation of the cannabinoid CB2 receptor in neurotoxic and inflammation-driven rat models of Parkinson’s disease. Exp Neurol 269:133–141. doi: 10.1016/j.expneurol.2015.04.007 [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allara M, Bisogno T et al. (2011) Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol 163:1479–1494. doi: 10.1111/j.1476-5381.2010.01166.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikranian K, Ishimaru M, Tenkova T, Labruyere J, Qin YQ et al. (2001) Apoptosis in the in Vivo Mammalian Forebrain. Neurobiology of Disease 8:359–379. doi: 10.1006/nbdi.2001.0411 [DOI] [PubMed] [Google Scholar]
- Elmore S (2007) Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol 35:495–516. doi: 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder C, Joyce K, Briley EM, Mansouri J, Mackie K et al. (1995) Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48:443–450 [PubMed] [Google Scholar]
- Ferrer I, Soriano E, Del Rio J, Alcántara S, Auladell C et al. (1992) Cell death and removal in the cerebral cortex during development. Progress in Neurobiology 39:1–43. doi: 10.1016/0301-0082(92)90029-E [DOI] [PubMed] [Google Scholar]
- Forcelli P, Kim J, Kondratyev A, Gale K (2011) Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia 52:e207–211. doi: 10.1111/j.1528-1167.2011.03297.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli P, Soper C, Duckies A, Gale K, Kondratyev A (2013) Melatonin potentiates the anticonvulsant action of phenobarbital in neonatal rats. Epilepsy Research 107:217–223. doi: 10.1016/j.eplepsyres.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman D, Devinsky O (2015) Cannabinoids in the Treatment of Epilepsy. New England Journal of Medicine 373:1048–1058. doi: 10.1056/NEJMra1407304 [DOI] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger D (2001) CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol 85:468–471. doi: 10.1152/jn.2001.85.1.468 [DOI] [PubMed] [Google Scholar]
- Giorgio D,, Hou Y, Zhao X, Zhang B et al. (2008) Dimethyl sulfoxide provides neuroprotection in a traumatic brain injury model. Restorative Neurology and Neuroscience 26:501–507 [PubMed] [Google Scholar]
- Gowran A, Noonan J, Campbell VA (2011) The multiplicity of action of cannabinoids: implications for treating neurodegeneration. CNS Neurosci Ther 17:637–644. doi: 10.1111/j.1755-5949.2010.00195.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen H, Krutz B, Sifringer M, Stefovska V, Bittigau P et al. (2008) Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Ann Neurol 64:42–52. doi: 10.1002/ana.21287 [DOI] [PubMed] [Google Scholar]
- Hansen H, Schmid P, Bittigau P, Lastres-Becker I, Berrendero F et al. (2001) Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem 78:1415–1427 [DOI] [PubMed] [Google Scholar]
- Hanslick JL, Lau K, Noguchi K, Olney J, Zorumski C et al. (2009) Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol Dis 34:1–10. doi: 10.1016/j.nbd.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Guzmán M, Galve-Roperh I, Berghuis P, Devi L et al. (2007) The emerging functions of endocannabinoid signaling during CNS development. Trends in Pharmacological Sciences 28:83–92. doi: 10.1016/j.tips.2006.12.004 [DOI] [PubMed] [Google Scholar]
- Heck N, Golbs A, Riedemann T, Sun J, Lessmann V et al. (2008) Activity-Dependent Regulation of Neuronal Apoptosis in Neonatal Mouse Cerebral Cortex. Cereb Cortex 18:1335–1349. doi: 10.1093/cercor/bhm165 [DOI] [PubMed] [Google Scholar]
- Huizenga M, Wicker E, Beck V, Forcelli P (2017) Anticonvulsant effect of cannabinoid receptor agonists in models of seizures in developing rats. Epilepsia 58:1593–1602. doi: 10.1111/epi.13842 [DOI] [PubMed] [Google Scholar]
- Hülsmann S, Greiner C, Köhling R, Wölfer J, Moskopp D et al. (1999) Dimethyl sulfoxide increases latency of anoxic terminal negativity in hippocampal slices of guinea pig in vitro. Neurosci Lett 261:1–4 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C (2009) Triggers of apoptosis in the immature brain. Brain and Development 31:488–492. doi: 10.1016/j.braindev.2009.02.006 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru M, Wozniak D, Koch C et al. (2000) Ethanol-Induced Apoptotic Neurodegeneration and Fetal Alcohol Syndrome. Science 287:1056–1060. doi: 10.1126/science.287.5455.1056 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J et al. (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74 [DOI] [PubMed] [Google Scholar]
- Jacob S, de la Torre J (2009) Pharmacology of dimethyl sulfoxide in cardiac and CNS damage. Pharmacological Reports 61:225–235. doi: 10.1016/S1734-1140(09)70026-X [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff N, Dikranian K et al. (2003) Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23:876–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S, Tamer Z, Opoku F, Forcelli P (2016) Anticonvulsant drug-induced cell death in the developing white matter of the rodent brain. Epilepsia 57:727–734. doi: 10.1111/epi.13365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kondratyev A, Gale K (2007) Antiepileptic Drug-Induced Neuronal Cell Death in the Immature Brain: Effects of Carbamazepine, Topiramate, and Levetiracetam as Monotherapy versus Polytherapy. J Pharmacol Exp Ther 323:165–173. doi: 10.1124/jpet.107.126250 [DOI] [PubMed] [Google Scholar]
- Kubova H, Mares P (1991) Anticonvulsant effects of phenobarbital and primidone during ontogenesis in rats. Epilepsy Res 10:148–155 [DOI] [PubMed] [Google Scholar]
- Lu C, Mattson M (2001) Dimethyl Sulfoxide Suppresses NMDA- and AMPA-Induced Ion Currents and Calcium Influx and Protects against Excitotoxic Death in Hippocampal Neurons. Experimental Neurology 170:180–185. doi: 10.1006/exnr.2001.7686 [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M et al. (2003) CB1 Cannabinoid Receptors and On-Demand Defense Against Excitotoxicity. Science 302:84–88. doi: 10.1126/science.1088208 [DOI] [PubMed] [Google Scholar]
- Maya-López M, Colín-González A, Aguilera G, de Lima M, Colpo-Ceolin A et al. (2017) Neuroprotective effect of WIN55,212–2 against 3-nitropropionic acid-induced toxicity in the rat brain: involvement of CB1 and NMDA receptors. Am J Transl Res 9:261–274 [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA (2013) The endocannabinoid system and the brain. Annu Rev Psychol 64:21–47. doi: 10.1146/annurev-psych-113011-143739 [DOI] [PubMed] [Google Scholar]
- Olney J, Wozniak D, Jevtovic-Todorovic V, Farber N, Bittigau P et al. Drug-induced Apoptotic Neurodegeneration in the Developing Brain. Brain Pathology 12:488–498. doi: 10.1111/j.1750-3639.2002.tb00467.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney J, Young C, Wozniak D, Jevtovic-Todorovic V, Ikonomidou C (2004) Do pediatric drugs cause developing neurons to commit suicide? Trends in Pharmacological Sciences 25:135–139. doi: 10.1016/j.tips.2004.01.002 [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2008) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: A9-tetrahydrocannabinol, cannabidiol and A9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215. doi: 10.1038/sj.bjp.0707442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandra R, Subramanian T (2011) Atlas of the Neonatal Rat Brain. CRC Press, Boca Raton [Google Scholar]
- Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos M (2005) Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-López E, Colín-González A, Paz-Loyola A, Ρίηzόη E, Torres I et al. (2015) Cannabinoid receptor agonists reduce the short-term mitochondrial dysfunction and oxidative stress linked to excitotoxicity in the rat brain. Neuroscience 285:97–106. doi: 10.1016/j.neuroscience.2014.11.016 [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni O (2001) Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21:109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez AJ, García-Merino A (2012) Neuroprotective agents: cannabinoids. Clin Immunol 142:57–67. doi: 10.1016/j.clim.2011.02.010 [DOI] [PubMed] [Google Scholar]
- Sánchez-Blázquez P, Rodríguez-Muñoz M, Vicente-Sánchez A, Garzón J (2013) Cannabinoid receptors couple to NMDA receptors to reduce the production of NO and the mobilization of zinc induced by glutamate. Antioxid Redox Signal 19:1766–1782. doi: 10.1089/ars.2012.5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, Sato M (1975) The effect of dimethyl sulfoxide on the neuronal excitability and cholinergic transmission in Aplysia ganglion cells. Ann N Y Acad Sci 243:337–357 [DOI] [PubMed] [Google Scholar]
- Shen M, Piser T, Seybold V, Thayer S (1996) Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci 16:4322–4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Wallmichrath I, Mathonia P, Pfreundtner C (2000) Cannabinoids inhibit excitatory neurotransmission in the substantia nigra pars reticulata. Neuroscience 97:89–97 [DOI] [PubMed] [Google Scholar]
- Vicente-Sánchez A, Sánchez-Blázquez P, Rodríguez-Muñoz M, Garzón J (2013) HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Mol Brain 6:42. doi: 10.1186/1756-6606-6-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, Department of Mental Health and Substance Abuse, Agarwal R, et al. (2011) Guidelines on neonatal seizures