

Abstract
FGF2 is an essential growth factor implicated in osteoarthritis (OA), and deletion of full-length FGF2 (Fgf2ALLKO) leads to murine OA. However, the FGF2 gene encodes both high-molecular-weight (HMW) and low-molecular-weight (LMW) isoforms, and the effects of selectively ablating individual isoforms, as opposed to total FGF2, has not been investigated in the context of OA. We undertook this study to examine whether mice lacking HMW FGF2 (Fgf2HMWKO) or LMW FGF2 (Fgf2LMWKO) develop OA and to further characterize the observed OA phenotype in Fgf2ALLKO mice. Fgf2HMWKO mice never developed OA, but 6- and 9-month-old Fgf2LMWKO and Fgf2ALLKO mice displayed signs of OA, including eroded articular cartilage, altered subchondral bone and trabecular architecture, and increased OA marker enzyme levels. Even with mechanical induction of OA, Fgf2HMWKO mice were protected against OA, while Fgf2LMWKO and Fgf2ALLKO displayed OA-like changes of the subchondral bone. Before exhibiting OA symptoms, Fgf2LMWKO or Fgf2ALLKO joints displayed differential expression of genes encoding key regulatory proteins, including IL-1β, insulin-like growth factor 1, bone morphogenetic protein 4, hypoxia-inducible factor 1, B-cell lymphoma 2, Bcl2- associated X protein, a disintegrin and metalloproteinase with thrombospondin motifs 5, ETS domain–containing protein, and sex-determining region Y box 9. Moreover, Fgf2LMWKO OA cartilage exhibited increased FGF2, FGF23, and FGFR1 expression, while Fgf2HMWKO cartilage had increased levels of FGFR3, which promotes anabolism in cartilage. These results demonstrate that loss of LMW FGF2 results in catabolic activity in joint cartilage, whereas absence of HMW FGF2 with only the presence of LMW FGF2 offers protection from OA.
Keywords: osteoarthritis, cartilage, fibroblast growth factor (FGF), animal models, ADAMTS5
INTRODUCTION
Osteoarthritis (OA) is the most common form of joint disease and a leading cause of chronic disability worldwide, characterized by overall joint degeneration, primarily of the knee (1). Specific hallmarks of OA include articular cartilage degeneration (2), osteophyte formation (3), and dynamic modification of trabecular architecture of the underlying bone characterized by thinning subchondral bone in early stages of OA disease due to an increase in bone resorption with subsequent sclerosis of bone in later stages (4). In the articular cartilage, excessive catabolic activity results in matrix degradation via upregulation of metalloprotease enzymes, including matrix metalloproteinase 13 (MMP-13) and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS-5) (5–7). The pathogenic mechanisms that underlie these morphological phenotypes remain largely unknown. There is an urgent need for additional studies in order to identify therapeutic targets to prevent and treat OA disease, as there are currently no effective treatment options beyond total joint replacement (8).
Recently, the involvement of members of the fibroblast growth factor (FGF) family, particularly FGF2, has received considerable attention in the regulation of cartilage and joint homeostasis (9,10). There is conflicting evidence which demonstrates both catabolic and anabolic potential of FGF2 in the joint. Specifically, exogenous FGF2 simulates MMP-13 and ADAMTS-5 production in articular cartilage explants in vitro (9,11) and the level of FGF2 in OA synovial fluid is nearly twice that of healthy knees (10), suggesting catabolic functions. Conversely, Fgf2 knockout (Fgf2ALLKO) mice develop spontaneous and surgically-induced OA more frequently and severely than their wild type (WT) littermates (11), suggesting an anabolic role for FGF2 in the joint. Additional studies are clearly necessary to further define the role that FGF2 plays in joint homeostasis and potentially in OA etiology.
Importantly, the FGF2 gene encodes multiple protein isoforms including the high molecular weight (HMW) isoforms and the low molecular weight (LMW) isoform. The LMW isoform is translated from the traditional AUG start codon, while the HMW isoforms are initiated upstream of the LMW start site from individual CUG codons (12). In humans there are three HMW FGF2 isoforms (22, 22.5, 24kDa), and in rodents there are two HMW FGF2 isoforms (21, 22kDa). The HMW FGF2 isoforms contain a nuclear localization sequence and remain in the nucleus to function as an intracrine factor (13). In humans and rodents there is one LMW FGF2 isoform, which is transported out of the cell to function as an autocrine/paracrine growth factor (14). Although deletion of full-length FGF2 leads to OA, the implications of separately ablating the individual isoforms in joint homeostasis and potentially in OA pathogenesis has not been addressed.
In this study we investigated if selectively deleting the HMW or LMW isoforms would affect the OA phenotype. Mice with a deficiency of HMW FGF2, Fgf2HMWKO mice, were previously generated and characterized (13) and of importance by dual beam xray absorptiometry (DEXA), microcomputed analysis (μCT) and quantitative histomorphometry were reported to have increased femur bone mineral density (BMD) and increased bone formation compared to WT littermate mice (14). In a similar manner mice lacking LMW FGF2, Fgf2LMWKO mice, were generated and previously characterized (15) and found to have decreased femur BMD and decreased bone formation compared to WT littermate mice (16). We have recently shown that mice that overexpress the HMW FGF2 isoforms develop degenerative joint disease (17), so we hypothesized that Fgf2LMWKO mice (which only express the high molecular weight FGF2 isoform), but not Fgf2HMWKO mice, would develop OA. The aim of our study was to examine Fgf2HMWKO and Fgf2LMWKO mice for any evidence of spontaneous and mechanically-induced OA, while confirming and further characterizing the OA phenotype in Fgf2ALLKO mice. The results of this novel study are of particular importance given the contrasting data of the involvement of FGF2 in OA, and for the first time show the differing effects of the distinct FGF2 isoforms in the development of the disease.
Materials and Methods
Animals
Fgf2ALLKO mice were previously developed on a Black Swiss 129Sv background (18) and genotyping was performed using primers as previously described (18,19). Fgf2HMWKO mice were formerly generated at the GEMM Core at the University of Arizona by gene-targeted introduction of polymorphisms, using a Tag and Exchange strategy, to remove the alternative CUG codon start sites required to translate the HMW isoforms of FGF2 (13) and genotyping was performed using primers previously described (13). Fgf2LMWKO mice were generated in a similar manner to remove the ATG translational start site (15), and genotyping was performed using primers previously described (15). All mice were developed on a Black Swiss 129Sv background and deletion of the gene or selective isoform is constitutive. Heterozygote mice for each genotype were bred and housed in the transgenic facility in the Center for Comparative Medicine at UConn Health and all experiments were performed using homozygote mice with wild-type littermate controls. Unless otherwise indicated, all data presented involved male mice. The UConn Health Institute of Animal Care and Use Committee approved all animal procedures.
Radiological analyses and Micro-computed tomography
Digital x-ray images of murine knee joints taken in the sagittal and frontal planes were obtained with a SYSTEM MX 20 from Faxitron X-ray Corporation (Faxitron X-ray Corp., Wheeling, IL) and taken under constant conditions (26 kV at 6-s exposure). Imaging of knee architecture was performed using ex vivo micro-computed tomography (μCT40, ScanCo Medical AG, Bassersdorf, Switzerland).
Histologic Assessment
Mice were sacrificed utilizing the approved CO2 method for euthanasia and whole knee joints were removed, fixed in 4% paraformaldehyde for 6 days, and decalcified with 14% EDTA solution at 4°C. Samples were processed for paraffin embedding in a frontal orientation and 7-μm alternate sections were obtained. Following de-paraffinization and rehydration of the sections, Safranin-O staining of glycosaminoglycans was performed using 0.1% aqueous Safranin-O and counterstained with Weigert’s Iron Hematoxylin and 0.02% aqueous Fast Green. Additional tissue sections were stained with hematoxylin and eosin (H&E). All histological images were captured using Nikon TS100 microscope interfaced with SPOT software. Using Safranin-O stained sections, the severity of cartilage destruction was assessed histologically by a single observer blinded with regard to the mouse group using the recommended scoring system from Glasson et al. (20) (0= normal; 0.5= loss of proteoglycan without structural changes, 1= small fibrillation without cartilage loss, 2= vertical clefts down to layer directly beneath superficial layer/some loss of surface lamina, 3= vertical clefts/erosion down to calcified cartilage layer which extends <25% of articular surface, 4= vertical clefts/erosion down to calcified cartilage layer which extends 25–50% of articular surface, 5= vertical clefts/erosion down to calcified cartilage layer which extends 50–75% of articular surface, 6= vertical clefts/erosion down to calcified cartilage layer which extends >75% of articular surface). Scoring was performed on all articular surfaces within each section using 4 separate representative sections from each knee, closest to the center of the joint, from 5 different samples from each genotype. The mean score of the medial tibial plateau (n=5/group) or mean overall (n=4–5/group) score was reported.
Histomorphometric measurements were made in the same blinded, nonbiased manner, using the OsteoMeasure image analysis system (R & M Biometrics, Nashville, TN) equipped with a Nikon E400 microscope (Nikon Inc., Melville, NY). The region of subchondral trabecular bone of the epiphysis in the femur and tibia, was previously established (21), and analyzed for the following histomorphometric parameters: bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular spacing (Tb.Sp), and trabecular number (Tb.N), using 5 representative samples closest to the center of the joint from each different genotype.
Immunohistochemistry
Immunohistochemical staining was performed on 7-μm paraffin embedded sections using the ImmunoCruz™ ABC Staining System (Santa Cruz Biotechnology, CA, USA). After sections were de-paraffinized and rehydrated, slides underwent antigen retrieval by incubation at 95°C with 10mM sodium citrate buffer for 10 minutes and then cooled to room temperature for 30 minutes. Endogenous peroxidase activity was then blocked by incubating sections with 3% hydrogen peroxide in water for 15 minutes. Following blocking sections with 10% serum for one hour at room temperature, the slides were incubated with primary antibodies in blocking buffer overnight at 4°C. A negative control slide was used each time, which was instead incubated with blocking serum overnight. The following primary antibodies were used: rabbit anti-MMP13, 1:100 (Abcam, ab39012), rabbit anti-ADAMTS5, 1:100 (Abcam, ab41037), rabbit anti-FGF2, 1:50 (Santa Cruz, sc-79), anti-FGF-23, 1:100 (R & D Systems, MN, USA, MAB26291), rabbit anti-FGFR-1 (Flg C-15)1:50 (Santa Cruz, sc-121), rabbit anti-pFGFR-1, 1:50 (Abnova, PAB0471), rabbit anti-pFGFR-3 (Tyr 724), 1:100 (Santa Cruz, sc-33041), and rabbit anti-phosphop44/42 MAPK (Cell Signaling, 4376S). After washing with TBS containing 0.1% Tween 20, the appropriate 1:200 biotinylated secondary antibody was applied at room temperature for 30 minutes. Finally, slides were washed and developed with DAB Peroxidase Substrate kit (Vector Laboratories, Burlingame, CA, USA), and counterstained with Harris hematoxylin.
Mechanical Induction of OA
Under isoflurane-induced anesthesia, mice were subjected to a single session of external compressive loading of the right tibiae, while left legs served as contralateral controls. The electromagnetic loading machine (ElectroForce 3100; Bose Co., Eden Prairie, MN, USA) applied a peak load level of 9.5N at a frequency of 4Hz for 1,200 cycles (5 min) and mice were sacrificed 2 weeks after mechanical compression and intact knees were fixed in 4% paraformaldehyde. This mechanical loading protocol was previously demonstrated to induce OA-like cartilage and bone alterations 2 weeks after loading (22).
RNA isolation and real time PCR
Total RNA was isolated from whole knee joints, to include all structures of the joint above the growth plate, following skin and bulk muscle removal from 2 month old male mice from all genotypes, in a method previously described (11). Following snap-freezing by liquid nitrogen, TRIzol reagent (Invitrogen) was used to extract RNA from the tissue. For real-time quantitative RT-PCR (qRT-PCR) analysis, RNA was reverse-transcribed to cDNA using RNA to cDNA EcoDry Premix kit (Clontech Inc., A Takara Bio Company). A Bio-Rad MyiQ™ instrument (BIO-RAD Laboratories Inc. Hercules, CA) using iTaq™ Universal SYBR® Green Supermix (Bio-Rad, CA, USA) was utilized for qPCR. The relative change in mRNA level was normalized to the mRNA level of beta-actin (β-actin), a housekeeping gene, which served as an internal reference for each sample. Table 1 lists the primers, synthesized by IDT (Integrated DNA Technologies, Inc., CA), for the genes of interest.
Table 1.
Primer used in qRT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| Actb | 5’-atggctggggtgttgaaggt-3’ | 5’-atctggcaccacaccttctacaa-3’ |
| Elk1 | 5’-ttggaggcctgtctggaggctgaa-3’ | 5’-agctcttccgatttcaggtttggg-3’ |
| Adamts5 | 5’-cctgcccacccaatggtaaa-3’ | 5’-ccacatagtagcctgtgccc-3’ |
| Col10a1 | 5’-gggaccccaaggacctaaag-3’ | 5’-gcccaactagacctatctcacct-3’ |
| Vegf | 5’-gcacatagagagaatgagcttcc-3’ | 5’-ctccgctctgaacaaggct-3’ |
| Fgf2 | 5’-gtcacggaaatactccagttggt-3’ | 5’-cccgttttggatccgagttt-3’ |
| Fgf18 | 5’ -aaggagtgcgtgttcattgag-3’ | 5’ -agcccacataccaaccagagt-3’ |
| Fgf23 | 5’-acttgtcgcagaagcatc-3’ | 5’-gtgggcgaacagtgtagaa-3’ |
| Fgfr1 | 5’-gactgctggagttaatacca-3’ | 5’-ctggtctctcttccagggct-3’ |
| Fgfr3 | 5’-gttctctctttgtagactgc-3’ | 5’-agtacctggcagcacca-3’ |
| Sox9 | 5’-agtacccgcatctgcacaac-3’ | 5’-acgaagggtctcttctcgct-3’ |
| Bmp4 | 5’-gccggagggccaagcgtagccctaag-3’ | 5’-ctgcctgatctcagcggcacccacatc-3’ |
| Igf1 | 5’-gtgagccaaagacacaccca-3’ | 5’-acctctgattttccgagttgc-3’ |
| Il1b | 5’-gcaactgttcctgaactcaact-3’ | 5’-atcttttggggtccgtcaact-3’ |
| Hif1a | 5’-caagatctcggcgaagcaa-3’ | 5’-ggtgagcctcataacagaagcttt-3’ |
| Bcl2 | 5’-gagagcgtcaacagggagatg-3’ | 5’-ccagcctccgttatcctgga-3’ |
| Bax | 5’-tgaagacaggggcctttttg-3’ | 5’-aattcgccggagacactcg-3’ |
Statistical analysis
Data is presented as mean ± standard deviation (SD). Student’s t-test was used to analyze differences between groups and considered significant at p values less than 0.05, unless otherwise stated.
RESULTS
Radiographic phenotype of joints of male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates
Digital radiographic examination revealed no abnormalities of the subchondral bone of the knee in all 2 months old samples of Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and WT littermate controls (Fig. 1). At 6 months of age all (10 of 10) Fgf2ALLKO and (9 of 9) Fgf2LMWKO knee joints displayed evidence of joint destruction, flattening of tibia, and osteophyte formation on the posterior tibial plateau compared to the WT controls. These OA-like alterations were also evident in 9 months old Fgf2ALLKO (4 of 4) and Fgf2LMWKO (8 of 8), while Fgf2HMWKO knees were phenotypically similar to WT at all ages and showed no radiographic indications of OA (Fig. 1). X-rays of knees in female mice displayed the same result as males of all genotypes (data not shown), indicating that gender does not influence the OA phenotype in these mice.
Figure 1. Time course of radiographs of male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates at 2, 6, and 9 months of age.
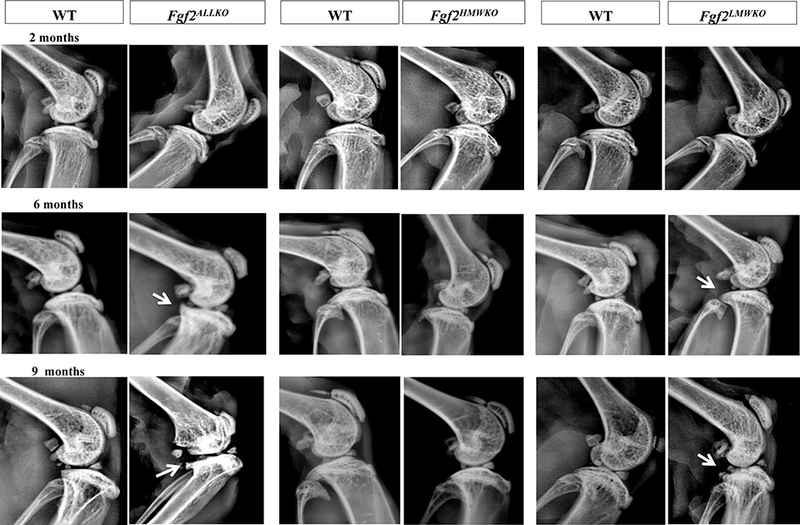
Sagittal digital x-ray images of all genotypes appeared to have the same subchondral bone phenotype at 2 months old. Osteophyte formation (arrows) was visible in the posterior tibial plateau of Fgf2ALLKO and Fgf2LMWKO knees at 6 and 9 months old. N=4–10/age group/genotype.
Histological phenotype of knees of male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates
Articular cartilage integrity was determined histologically by Safranin-O staining in 2, 6, and 9 months old Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and WT littermate controls. At 2 months of age there was no difference in proteoglycan content, cartilage thickness, or Safranin-O intensity between Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and WT knees. By 6 and 9 months of age, both Fgf2ALLKO and Fgf2LMWKO joints showed spontaneous fibrillation of cartilage (arrowheads), areas of complete cartilage loss (arrows), and evidence of osteophyte formation (Fig. 2A). Safranin-O stained Fgf2HMWKO cartilage appeared to have similar cartilage integrity to the WT at all ages (Fig. 2A). The severity of cartilage destruction and extent of chondral damage was graded in 6 months old samples from at least 20 Safranin-O sections from the joint, per group (see Experiment Procedures) and the average score of the medial tibial plateau was reported. The score of Fgf2ALLKO was significantly higher than WT, p=.002 and Fgf2LMWKO was significantly higher than the WT littermate, p=.004. The histologic score showed no significant difference between Fgf2HMWKO and WT, p=.126 (Fig. 2B). Also, H & E-stained 6 months old Fgf2LMWKO joints showed inflammation with larger cells in the medial tendon and synovium and enlarged meniscus (circled area), which was not present in WT littermates (Fig. 2C). Fgf2HMWKO knee joints did not show evidence of inflammation based on H & E-staining, as they appeared phenotypically similar to that of the WT littermate (Fig. 2C). Fgf2ALLKO H & E-stained sections showed cartilage loss, but less evidence of inflammation (Suppl. Fig. 1A).
Figure 2. Analysis of cartilage integrity of male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates.
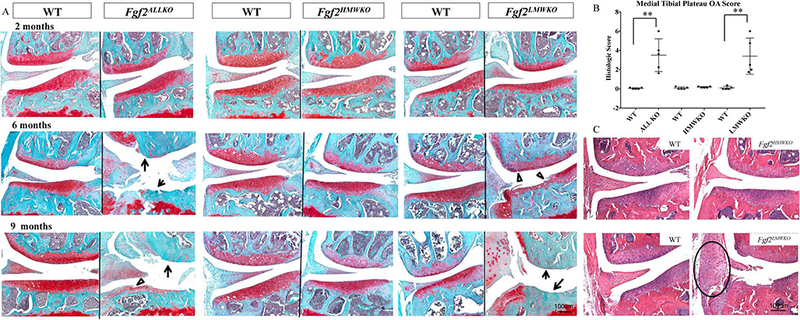
A, Safranin-O stained representative images of medial area of knees show similar histological appearances between all genotypes and their wild-type controls at 2 months of age. At 6 and 9 months of age Fgf2ALLKO and Fgf2LMWKO joints display fibrillation of articular cartilage (open arrowheads) and complete loss of cartilage (arrows) n=4–6/group. B, Mean medial tibial plateau OA score of Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and WT at 6 months of age. Values are means ± SD, *p <0.05, **P <0.01, n=5/group. C, H & E stained representative images of 6 month old Fgf2LMWKO displays inflammation of the tendon and synovium (circle). N=3/group. Magnification=10X.
Micro-computed tomography and histomorphometric analyses of male Fgf2HMWKO, Fgf2LMWKO, and wild-type littermate knees
Reconstructed 3D images of 6 months old Fgf2LMWKO joints display bony outgrowths (closed arrowheads), indentations, and wearing away of the tibial bone surface (open arrowhead), while the Fgf2HMWKO renderings show a smooth and flat shape of the subchondral surface which was consistent with both types of WT littermates (Fig. 3A). MicroCT-scanned 2D images show tibial osteophyte formation and thinning of subchondral femoral bone (dashed arrow) of Fgf2LMWKO compared to the WT (arrow). The Fgf2LMWKO images also display alterations in the trabecular architecture in the femoral and tibial epiphyses, particularly evident in the sagittal view, which includes loss of trabeculae and increased spacing between trabeculae. These OA characteristics were not observed in the Fgf2HMWKO or WT littermates (Fig. 3A). Although the long bones of Fgf2HMWKO mice were previously found to have increased bone formation (14), the histomorphometry of the actual subchondral bone revealed there was no significant difference between bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular spacing (Tb.Sp), and trabecular number (Tb.N) in both of the epiphyses of the femur and tibia between Fgf2HMWKO or WT littermates (Fig. 3B). However, the BV/TV of the femoral epiphyses was 50% less (p< .001) in the Fgf2LMWKO compared to the WT and 47% less (p< .001) in the tibia. Tb.Th was significantly lower in Fgf2LMWKO compared to the WT in the femur (p=.014) and tibia (p=.004). The Tb.Sp of the Fgf2LMWKO epiphysis was significantly increased in the femur and tibia by 96% (p< .001) and 61% (p< .001) respectively, compared to that of the WT. Also, there was a 39% reduction (p< .001) in Tb.N. of the femoral epiphysis and a 33% reduction (p=.001) in the tibia of the Fgf2LMWKO compared to the WT (Fig.3C). The findings were similarly significant in 6 month old Fgf2ALLKO epiphyses (Suppl. Fig. 1B). By histomorphometry, at 2 months of age the BV/TV, Tb.Th, Tb.Sp, and Tb.N of the epiphysis of both the femur and tibia was not significantly different between all genotypes and the littermate WT controls (Suppl. Fig. 2).
Figure 3. MicroCT imaging and histomorphometric analysis of subchondral bone of male Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates at 6 months old.
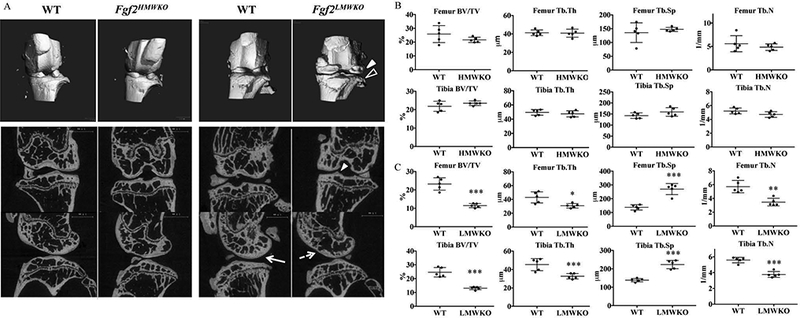
A, Three-dimensional (3D) renderings reveal pitting and erosion (open arrowhead) and osteophyte formation (closed arrowhead) of Fgf2LMWKO knees, while Fgf2HMWKO and all WT knees maintain a smooth contour. High-resolution micro-computed tomography (μCT) 2D images of representative sections of all Fgf2LMWKO samples show osteophyte formation of tibia (closed arrowhead) and thinning of femoral subchondral bone (dashed arrow) compared to the thicker femoral subchondral bone (arrow) of the WT littermate. Fgf2LMWKO epiphyses display decreased trabecular thickness and number and increased trabecular spacing compared to WT and Fgf2HMWKO epiphyses. N=3–7/group. B, Histomorphometric parameters of Fgf2HMWKO and WT femoral and tibial epiphyses are similar. C, Histomorphometric parameters of Fgf2LMWKO femoral and tibial epiphyses reveal decreases in BV/TV, Tb.Th, and Tb.N and an increase in Tb.Sp compared to WT. BV/TV, trabecular bone volume; Tb.Th, trabecular thickness Tb.Sp, trabecular separation; Tb.N, trabecular number. Values are the means ± SD. *P <0.05, **P <0.01, ***P <0.001 N=5/group.
Expression of OA markers in male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates
Immunohistochemistry was used to examine the presence of OA marker protein expression within the joint tissues. At 6 months of age, MMP-13 (a collagenase) was greatly enhanced within the articular cartilage of the Fgf2ALLKO and Fgf2LMWKO compared to the WT, whereas, Fgf2HMWKO cartilage had comparable MMP-13 staining to that of the WT littermate (Fig. 4A). ADAMTS-5 (the major aggrecan degrading enzyme in murine cartilage) was also increased throughout the tissues of the joints of 6-month-old Fgf2ALLKO and Fgf2LMWKO, particularly in the enlarged medial meniscus and tendon area and developing osteophyte region of Fgf2LMWKO. Examining the same area of the Fgf2HMWKO knee, immunohistochemistry revealed similar or slightly less ADAMTS-5 expression compared to WT (Fig. 4B).
Figure 4. Matrix metalloproteinase 13 (MMP-13), a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS-5) expression in knees of 6 month old male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates.
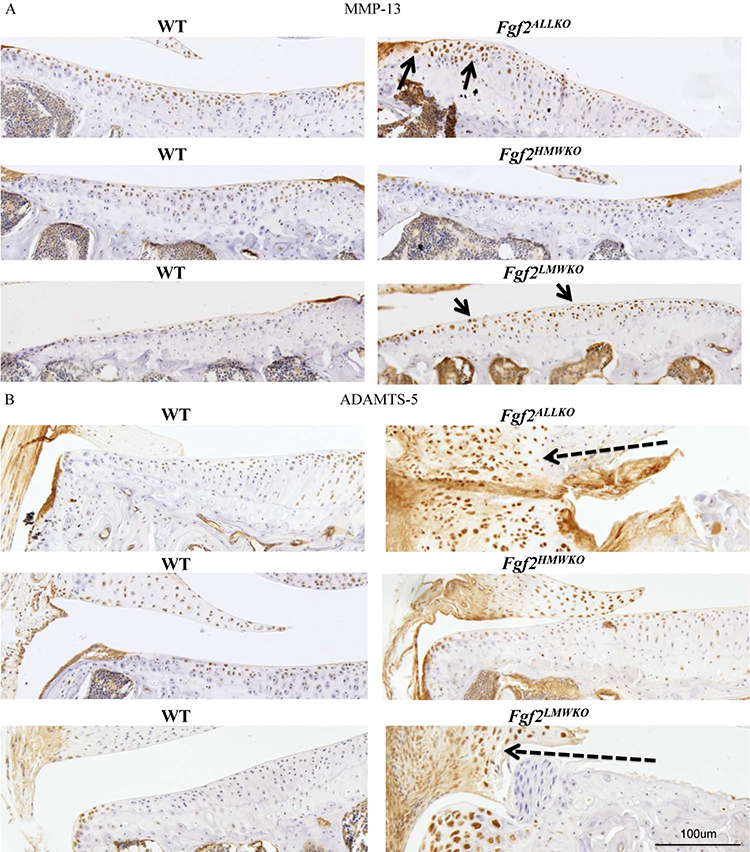
A, Representative immunohistochemical staining shows increased protein expression of MMP-13 along tibial articular cartilage of Fgf2ALLKO and Fgf2LMWKO (arrows) compared to WT littermates, while Fgf2HMWKO and WT have similar MMP-13 expression. B, Representative images show increased expression of ADAMTS-5 in the enlarged tendon and meniscus area of the medial tibia in both Fgf2ALLKO and Fgf2LMWKO (dashed arrows) compared to WT littermates, whereas Fgf2HMWKO and WT have similar ADAMTS-5 expression of the same area. Magnification=10X, n=3/group.
Examination of age induced OA in female Fgf2HMWKO, and mechanically induced OA in male Fgf2HMWKO, Fgf2LMWKO, and wild type littermates
In order to determine if Fgf2HMWKO mice are protected from age-related OA, we examined knees of 2-year-old female Fgf2HMWKO mice (n=6). The x-ray images of WT joints appeared to have some evidence of sclerotic bone formation, while the Fgf2HMWKO joints did not (Fig.5A). Safranin-O staining showed a loss of proteoglycan content, a decreased superficial zone layer, and slight fraying of cartilage in WT sections, while this was not observed in Fgf2HMWKO cartilage. The total joint OA score was significantly increased in WT compared to Fgf2HMWKO, p=.020 (Fig. 5A-B). Immunohistochemical staining of the OA marker, ADAMTS-5, showed little expression in tibial articular chondrocytes of Fgf2HMWKO mice compared to the many labeled chondrocytes of the WT (Fig. 5C). It would be expected that mice aged to 2 years would display signs of OA.
Figure 5. Fgf2HMWKO are protected from spontaneous (females) and mechanically-induced (males) OA.
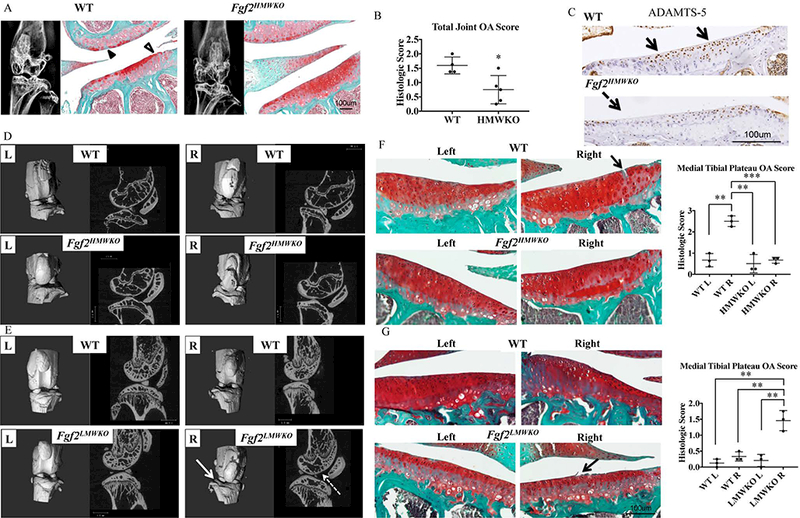
A, 2 year old Fgf2HMWKO and WT have similar subchondral bone phenotypes as determined by x-ray. Safranin-O staining of lateral side of joint shows loss of proteoglycan content (closed arrowhead) and fibrillation of cartilage (open arrowhead), compared to the more robust proteoglycan content of Fgf2HMWKO samples. N=4–6/group. B, Mean total joint OA score of Fgf2HMWKO is significantly lower than WT. Values are means ± SD, n=4–5/group. C, Representative immunohistochemical staining shows decreased protein expression of ADAMTS-5 along tibial articular cartilage of Fgf2HMWKO (dashed arrow) compared to expression in WT littermates (arrows). D-E, 3D reconstructed images and μCT images of sagittal sections of right tibial-loaded knees (2 weeks after loading) of 21 month old Fgf2HMWKO mice appear to have a similar appearance to the left contralateral control and both left and right knees of WT. Right tibial loaded knees of Fgf2LMWKO mice show erosion of the tibial surface (arrow) compared to the left contralateral control and all samples in D. Sagittal images display decreased femoral subchondral bone (dashed arrow) and increased trabecular spacing and decreased trabecular number in epiphyses compared to the contralateral control n=3–4/group. F, Safranin-O staining shows fissures (arrow) in cartilage of right tibial-loaded knees (2 weeks after loading) of 21 months old WT littermates of Fgf2HMWKO mice. OA score of right medial tibia of WT littermates of Fgf2HMWKO mice was significantly higher than all others. G, Right tibial loaded knees of 12 week old Fgf2LMWKO mice (2 weeks after loading) show fraying (arrow) and a significantly higher OA score than controls. Values are means ± SD, p <0.05**p <0.01, ***p <0.001.
To further verify that Fgf2HMWKO mice are protected from OA, we challenged them mechanically to assess induction of joint damage in 21 month old Fgf2HMWKO and WT mice by subjecting the right leg to a single session of cyclic tibial loading, previously shown to induce OA-changes in 2 weeks after loading (21). The 3D and 2D microCT images of Fgf2HMWKO right knees showed no indications of OA in the subchondral bone and had a similar phenotype to that of the left contralateral control and the right and left legs of the WT (Fig. 5D). In order to determine if challenging Fgf2LMWKO knees will accelerate the OA phenotype, right legs of approximately 12 week old Fgf2LMWKO mice underwent the same tibial loading procedure. Two weeks after loading, 3D microCT images of the right leg show an uneven tibial surface and evidence of erosion compared to the smooth surface observed in the left contralateral control and in both knees of the WT littermates. The sagittal view shows thinning of subchondral bone, loss of trabeculae and increased trabecular spacing in the epiphyses compared to the left control (Fig. 5E). To confirm the validity of the mechanically-induced OA model the same protocol was carried out on Fgf2ALLKO knees, which have previously shown to have accelerated OA following surgical induction (11), and 2 weeks after loading the microCT images revealed similar OA-like changes in the right leg that were observed in Fgf2LMWKO mice (Suppl. Fig. 1C).
Histologically, 21 months old WT joints that underwent tibial loading showed signs of cartilage damage, whereas, the contralateral controls and Fgf2HMWKO joints (both loaded and non-loaded) did not exhibit evidence of OA, and is reflected in the scores of the medial tibial plateau (Fig. 5F). Safranin-O staining of right loaded knees of 12 weeks old Fgf2LMWKO mice revealed fibrillation of tibial cartilage and decreased size of the superficial zone. The histologic score of loaded Fgf2LMWKO knees was significantly higher than the controls (Fig. 5G).
Expression of OA-like genes in male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates
Total RNA was isolated from knee joints of 2 month old Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and their WT littermates and used for measuring mRNA levels of genes that have been implicated in OA, including cytokines, growth factors, and components involved in OA-like changes in subchondral bone or hypertrophy of chondrocytes. Although many genes were examined only results in which there were significant differences between genotypes are presented. qPCR analysis showed significant increases in the mRNA from both Fgf2ALLKO and Fgf2LMWKO knees for interleukin 1 beta (Il-1β), insulin like growth factor 1(Igf1), bone morphogenetic protein 4 (Bmp4), hypoxia inducible factor 1(Hif1α), and the ratio of B-cell lymphoma 2 (Bcl2) and Bcl2-Associated X, apoptosis regulator (Bax), while Fgf2 was significantly increased in Fgf2LMWKO knees and decreased in Fgf2ALLKO knees (Table 2). Since FGF18 is a known player in cartilage homeostasis (23) we also examined its expression. As shown in in Table 2, Fgf18 mRNA was significantly increased in knees of Fgf2ALLKO compared with WT littermates. However there was no significant differences in Fgf18 mRNA expression in Fgf2HMWKO, Fgf2LMWKO, and their respective wild-type littermates. We also observed significant increases in the mRNA from only Fgf2ALLKO knees for Adamts5, ETS domain-containing protein-1 (Elk-1), Sex determining region Y box 9 (Sox9), and FGFR1 (Table 2). qPCR showed increases in the mRNA from only Fgf2LMWKO knees for vascular endothelial growth factor (Vegf), type X collagen (ColX) and FGFR3 (Table 2). Since recent studies demonstrated that another FGF ligand specifically FGF23 promoted the terminal differentiation of the mouse chondrogenic cell line ATDC5 (24) and mice that overexpressed the HMW FGF2 isoforms, developed degenerative joint disease associated with increased FGF23 (17) we assessed Fgf23 mRNA expression. The expression of Fgf23 was too low to be detected in Fgf2ALLKO joints, however Fgf23 mRNA expression was significantly increases in Fgf2LMWKO knees but was not significantly modulated in Fgf2HMWKO joints compared to WT (Table 2).
Table 2.
Gene expression of joints of 2 months old male WT, Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO.
| Genes | WT | ALLKO | HMWKO | LMWKO |
|---|---|---|---|---|
| Il1β | 1 | 1.26±0.08* | 1.79±0.22 | 1.35±0.13* |
| Igf1 | 1 | 2.15±0.25*** | 0.84±0.14 | 1.27±0.13* |
| Bmp4 | 1 | 4.09±0.53*** | 1.53±0.32 | 1.75±0.31* |
| Hif1α | 1 | 1.43±0.17* | 0.98±0.08 | 1.65±0.16*** |
| Bcl2/Bax | 1 | 2.23±0.21** | 0.84±0.13 | 1.50±0.11*** |
| Fgf2 | 1 | 0.17±0.06*** | 1.22±0.22 | 1.36±0.16* |
| Fgf18 | 1 | 2.27±0.25*** | 0.75±0.06 | 0.98±0.17 |
| Adamts5 | 1 | 1.59±0.15** | 1.05±0.05 | 1.01±0.11 |
| Elk1 | 1 | 1.26±0.10* | 1.15±0.06 | 0.92±0.04 |
| Sox9 | 1 | 1.33±0.08* | 0.98±0.06 | 1.31±0.18 |
| Fgfr1 | 1 | 1.26±0.08* | 1.05±0.10 | 0.97±0.05 |
| Vegf | 1 | 1.50±0.21 | 0.87±0.06 | 1.90±0.30* |
| ColX | 1 | 0.93±0.13 | 0.78±0.11 | 1.42±0.22 |
| Fgfr3 | 1 | 1.37±0.21 | 2.04±0.53 | 1.39±0.21* |
| Fgf23 | 1 | N/A | 1.93±0.61 | 2.22±0.51* |
Values are the means ± SD
P <0.05
P <0.01
P <0.001, n=6–8/group.
Comparison was made between Fgf2ALLKO and their WT littermates, Fgf2HMWKO and their WT littermates, Fgf2LMWKO and their WT littermates. However, for presentation purpose, WT of each genotype was set to 1.
Expression of FGF ligands and receptors in knees of male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates
Immunohistochemistry confirmed that Fgf2ALLKO joints did not have FGF2 expression, as shown by the absence of any labeled cells in the articular cartilage compared to the WT littermate at 2 and 6 months of age (Fig. 6A&B). Fgf2HMWKO cartilage had similar FGF2 expression to the WT at 2 months of age (Fig.6A), but appeared to have decreased FGF2 expression by 6 months of age (Fig. 6B). However, Fgf2LMWKO articular cartilage showed enhanced FGF2 expression at 2 and 6 months of age (Fig.6A&B) compared to the WT littermate. Localization of the protein confirmed that FGF2 was detected in the cytoplasm and the nuclei of chondrocytes in Fgf2HMWKO joints, similarly to the WT, whereas FGF2 was only detected in the nuclei of Fgf2LMWKO chondrocytes and not present in Fgf2ALLKO tissue (Fig. 6C). Immunostaining revealed increased FGF23 protein expression in Fgf2LMWKO articular cartilage at 2 months of age compared to WT, while cartilage of Fgf2ALLKO and Fgf2HMWKO had similar FGF23 expression to that of the WT (Fig. 7A). By 6 months of age immunostaining displayed enhanced FGF23 expression in Fgf2ALLKO along the articular surface compared to WT and Fgf2LMWKO maintained high FGF23 expression compared to WT. Fgf2HMWKO cartilage still had similar FGF23 expression to the WT littermate (Fig. 7B). FGF2 and FGF23 signal through FGFR1 and FGFR3, which are known to be involved in OA etiology. At 6 months of age, articular chondrocytes had increased total FGFR1 expression in Fgf2ALLKO knees and Fgf2LMWKO joint tissues had increased total FGFR1 expression, notably in developing osteophyte regions and the inflamed tendon areas compared to the WT. Fgf2HMWKO joints had similar total FGFR1 expression to that observed in WT (Fig. 8A). Phosphorylated FGFR1 expression was present in Fgf2ALLKO cartilage and appreciably expressed in Fgf2LMWKO chondrocytes (Fig. 8B). Conversely, at 6 months of age Fgf2ALLKO and Fgf2LMWKO knees had a marked decrease in phosphorylated FGFR3 expression as observed in articular cartilage in the femur (Fig. 8C). Also, phospho-FGFR3 protein expression was enhanced in knees of 6 months old Fgf2HMWKO compared to WT cartilage (Fig. 8C). Expression of phospho-ERK1/2, a downstream component of FGF2/FGF23/FGFR signaling, was only present in Fgf2LMWKO cartilage (Fig. 8D).
Figure 6. FGF2 protein expression in 2 and 6 month old male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates.
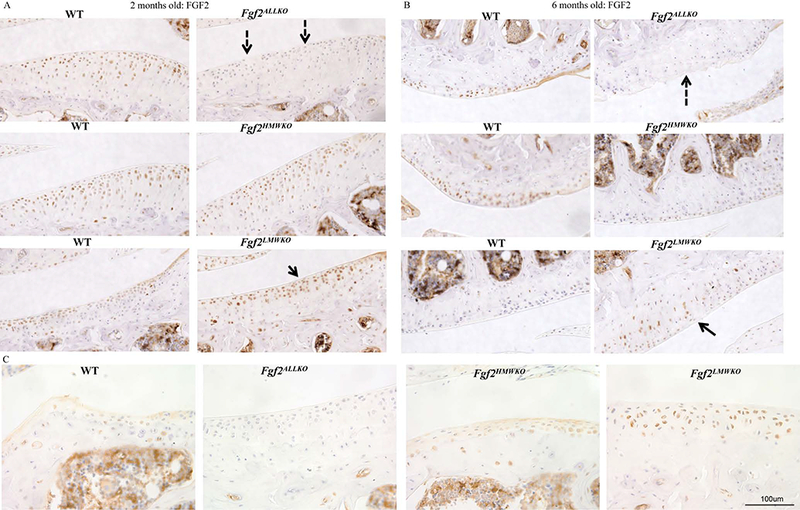
A, Immunohistochemical staining of tibial articular cartilage shows no FGF2 expression in 2 month old Fgf2ALLKO tissue (dashed arrows) compared to WT. Fgf2LMWKO cartilage had increased FGF2 expression (arrow) compared to WT. B, 6 month old femoral articular cartilage shows no FGF2 expression in Fgf2ALLKO cartilage (dashed arrow) compared to WT. FGF2 expression was increased in Fgf2LMWKO cartilage (arrow) compared to WT cartilage. Magnification=10X. C, Higher magnification of 6 months old joints shows localization of FGF2 isoforms within the lateral tibial plateau. In WT and Fgf2HMWKO cartilage, FGF2 is expressed in both the nucleus and cytoplasm of chondrocytes. FGF2 is only expressed in the nuclei Fgf2LMWKO cartilage. No FGF2 expression was detected in Fgf2ALLKO tissue. Magnification =40X, n=3/group.
Figure 7. FGF23 protein expression in 2 and 6 months old male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates.
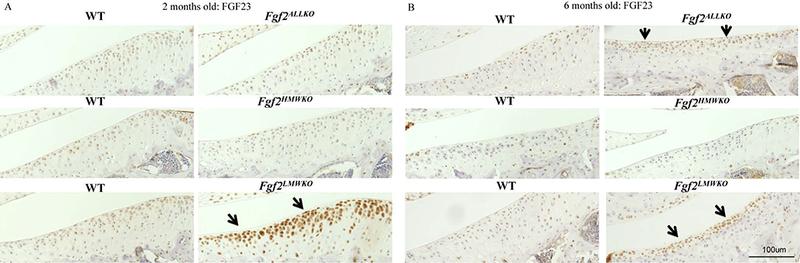
A, Representative immunohistochemical staining shows enhanced protein expression of FGF23 in tibial articular cartilage of Fgf2LMWKO (arrows) compared to WT littermates, while Fgf2ALLKO and Fgf2HMWKO had similar FGF23 expression to their WT littermates at 2 months of age. B, Both Fgf2LMWKO and Fgf2ALLKO had increased FGF23 staining in articular cartilage compared to WT, and Fgf2HMWKO had similar FGF23 expression to their WT at 6 months of age. Magnification=10X, n=3/group.
Figure 8. FGFR1, FGFR3, and ERK protein expression in 6 months old male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates.
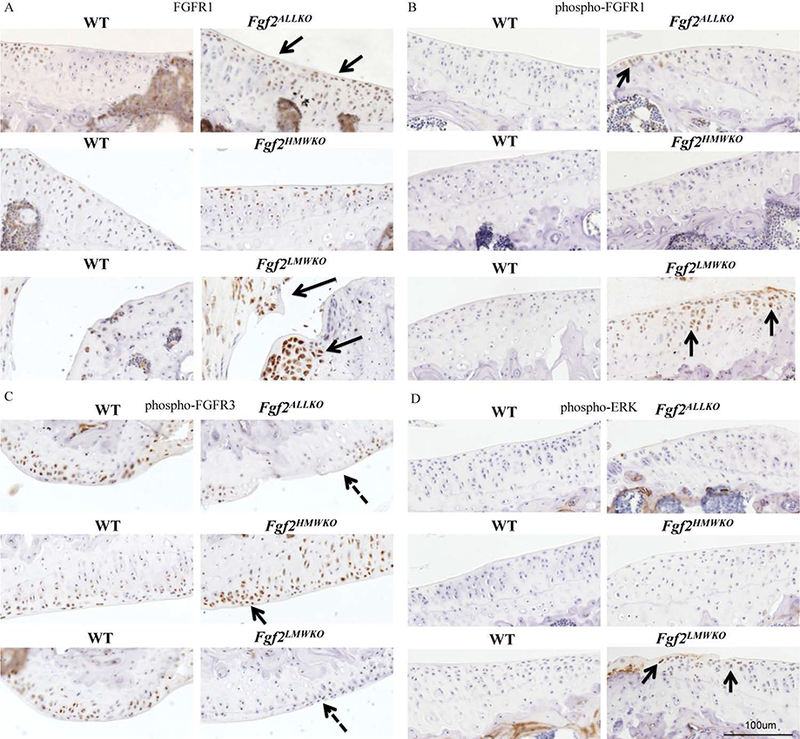
A, Immunohistochemical staining shows total FGFR1 expression was more robust in tibial articular cartilage of Fgf2ALLKO (arrows). FGFR1 was also increased in the joints of Fgf2LMWKO, particularly in the enlarged meniscus and developing osteophytes areas (arrows). Fgf2HMWKO had similar FGFR1 expression to the WT. B, phospho-FGFR1 was present in tibial cartilage of Fgf2ALLKO (arrow). Fgf2LMWKO cartilage had increased phopho-FGFR1 expression (arrows). C, phospho-FGFR3 expression was decreased in Fgf2ALLKO and Fgf2LMWKO femoral articular cartilage (dashed arrows) compared to WT, while Fgf2HMWKO had increased FGFR3 staining (arrow). D, phospho-ERK expression was only detected in Fgf2LMWKO cartilage (arrows). Magnification=10X, n=3/group.
DISCUSSION
Though there have been several publications reporting the contribution of FGF2 in the progression of OA, including a previous study which showed that deletion of full-length FGF2 in mice leads to enhanced OA (11), we describe for the first time that the varying FGF2 isoforms have differing roles in degenerative joint disease. In this study we demonstrated that mice lacking only the LMW FGF2 isoform (Fgf2LMWKO) develop signs of spontaneous and mechanically-induced OA, including cartilage destruction, inflammation, modifications of the subchondral bone, and upregulation of degradative enzymes. Fgf2HMWKO mice appeared to be protected from OA, as they did not display these characteristics, despite age or mechanical induction. Moreover, Fgf2ALLKO and Fgf2LMWKO knees already had enhanced expression of genes involved in multiple signaling pathways known to promote OA at just 2 months of age, which preceded the OA phenotype. Also, essential FGF family members including FGF23 and FGFR1 were upregulated in Fgf2ALLKO and Fgf2LMWKO joints, while FGFR3 was increased in Fgf2HMWKO cartilage.
Despite there being numerous contradictory findings about the involvement of FGF2 in cartilage homeostasis and degradation (9–11), this study supports the idea that FGF2 can have both an anabolic and catabolic effect dependent upon the presence or absence of the isoform. Fgf2LMWKO mice displayed classic hallmarks of OA that began at 6 months of age, which persisted to 9 months of age and were not restricted to simply one component of the joint. Histological examination revealed frayed cartilage, areas of complete cartilage loss, and evidence of endochondral ossification of developing osteophytes, which was most apparent on the medial side. As indicated by the OA score, these destructive alterations of cartilage were not present in joints of Fgf2HMWKO mice. Following mechanical induction the 21 months old WT littermates of Fgf2HMWKO mice displayed histologic evidence of OA, as their age would make them more susceptible when challenged, whereas, the Fgf2HMWKO mice appeared protected. Also, articular cartilage of Fgf2LMWKO and Fgf2ALLKO mice had increased expression of degradative enzymes, MMP-13 and ADAMTS-5, while Fgf2HMWKO joints did not. Additionally, subchondral bone was thinner, contained osteophytes, had a decrease in trabecular number and thickness and an increase in trabecular spacing in only Fgf2LMWKO and Fgf2ALLKO mice, indicating that this particular OA phenotype encompasses all joint tissues.
In order to determine if there are modifications of gene expression of pathways involved in OA induction before the disease phenotype occurs in Fgf2LMWKO and Fgf2ALLKO mice, RNA was isolated from whole knee joints at 2 months of age. It has been found that progression of OA may be caused by activation of hypertrophic differentiation of articular chondrocytes (25) and various signaling pathways are implicated which include cytokines such as Il-1β, growth factors such as IGF-1 and BMP4, and transcription factor Hif1α (26). Since joints from both Fgf2LMWKO and Fgf2ALLKO mice were found to have significant increased expression in genes of those pathways, it suggests that in addition to the observed modifications of the underlying bone, that hypertrophic differentiation of articular chondrocytes is driving the OA phenotype. Typically, the Bcl2/Bax ratio is decreased in mRNA taken from OA cartilage, as the Bcl2 protein protects chondrocytes from cell death, while Bax accelerates apoptosis (27). However, it was found that Bcl2 expression declines with age, while Bax expression remains constant, suggesting that the shift of this ratio to favor apoptosis would only occur in cartilage with age (28). Thus, the young age in which we examined the Bcl2/Bax ratio in Fgf2LMWKO and Fgf2ALLKO knees may be why we did not observe a decrease.
Although Fgf2ALLKO and Fgf2LMWKO shared a similar OA phenotype and modulation of related genes, there were some differences that were observed, indicating that joint degeneration might not be caused by the equivalent mechanism, as the Fgf2LMWKO mice still possess the HMW FGF2 isoforms. Through H & E staining we observed inflammation of the tendon and synovium in Fgf2LMWKO knees, whereas this particular aspect was absent in Fgf2ALLKO knees, which was consistent with the previous study that examined OA in Fgf2ALLKO mice and reported no evidence of synovitis (11). Also, the qPCR analysis showed some distinct differences in gene expression of mRNA taken from 2 month old joints of Fgf2ALLKO and Fgf2LMWKO mice. Fgf2ALLKO joints already had significant increases in the catabolic enzyme Adamts5 and the transcription factor, Elk-1, which induces MMP-13 expression (29) and Sox9, which although is a chondroprogenitor marker, has been shown to be increased in human chondrocytes with early-stage OA (30). Fgf2LMWKO mRNA did not have this early high expression of these genes, but instead had increased Vegf and ColX expression, signifying chondrocyte hypertrophy, another hallmark of OA cartilage.
As mentioned earlier there are numerous conflicting studies on the role that FGF2 plays in OA and this is mainly due in part to its binding affinity for FGFR1 and FGFR3, which have contrasting roles in cartilage homeostasis. While FGFR1 is increased in human OA chondrocytes, drives cartilage destruction (31), and its inhibition attenuates OA in murine cartilage (32), FGFR3 is decreased in human OA chondrocytes (31), promotes anabolic activity in cartilage (33), and its absence accelerates murine OA (34,35). Regardless of the essential absence of mRNA and protein expression of FGF2 in Fgf2ALLKO joints, there was a significant increase in FGFR1 mRNA at 2 months of age and protein expression at 6 months of age. This early increase of FGFR1 expression could be due to enhanced expression of other FGF ligands, as many other than FGF2, bind to FGFR1, including FGF1 which has been shown stimulate catabolic effects in chondrocytes and was increased in OA tissues of rat and humans (36,37). Once the Fgf2ALLKO mice displayed signs of OA, the cartilage had increased expression of FGF23, which can bind to FGFR1 and is known to drive MMP13 expression in osteoarthritic chondrocytes (38). Also, the expression of activated FGFR1 was not as great as the expression of total FGFR1, implying that the OA phenotype may be attributed to modulation of a different pathway.
Interestingly, Fgf2LMWKO had increased Fgf2 mRNA and protein expression at 2 months and increased protein expression still at 6 months of age, and while FGFR3 expression was enhanced at 2 months of age, phospho-FGFR3 was not detected at 6 months, yet total and phospho-FGFR1 expression was greatly increased. This initial increase in FGFR3 may be an early repair response, as the RNA was taken at only 2 months of age and signaling through FGFR3 can promote anabolic activities in cartilage (33). Furthermore, FGF23 expression was increased at 2 and 6 months of age in Fgf2LMWKO joints, which can also signal through FGFR1 to push the OA phenotype via regulation of the MEK/ERK cascade (38) and we observed enhanced phospho-ERK expression in only Fgf2LMWKO only. Intriguingly, Fgf2LMWKO knees showed upregulation of the same genes as mice that overexpressed the HMW FGF2 isoforms, including FGF23, which also developed degenerative joint disease (17), suggesting that lack of LMW FGF2 may be causing a compensatory effect, which would increase HMW FGF2, which is driving the OA phenotype and would explain the significant increase in FGF2 protein and mRNA. Mice that overexpress the LMW FGF2 isoform do not develop OA (17) and Fgf2HMWKO mice were protected from OA despite age and mechanical induction and had an increase in FGFR3 expression at 6 months of age. Similarly, this suggests that it is the HMW FGF2 isoform responsible for the OA phenotype and that the presence (or possible overcompensation) of LMW FGF2 confers a chondroprotective effect.
We demonstrated that chondrocytes of Fgf2LMWKO expressed FGF2 solely and robustly in the nucleus, confirming that Fgf2LMWKO mice only express HMW FGF2 since this isoform remains in the nucleus (13). Fgf2HMWKO mice expressed FGF2 in the nucleus and cytoplasm, having similar expression to WT. This data is consistent with previous investigators who showed that HMW FGF2 has nuclear localization and LMW FGF2 is expressed in cytoplasm and nucleus (12) and others who demonstrated that cardiomyocytes from Fgf2LMWKO and Fgf2HMWKO mice have varying localization of FGF2 via immunohistochemistry (39) (as there are no commercially available antibodies to detect the isoforms). Notably, this study highlights the importance of the possible roles that each FGF2 variant may have in cartilage homeostasis, as the relative expression levels of the FGF2 isoforms in both adult human and murine healthy or OA cartilage is unknown. Though in fetal growth plate cartilage it was determined that LMW FGF2 was only expressed in resting cartilage, whereas HMW FGF2 was expressed by proliferating chondrocytes (40). This evidence further supports the idea that HMW FGF2 is the isoform contributing to OA, since during OA the normally quiescent chondrocytes begin to proliferate, recapitulating a process that resembles chondrocyte differentiation during endochondral ossification (41).
Finally, our observations provide insight into the varying roles of FGF2 and its isoforms in OA and why global deletion of FGF2 leads to the disease. Though the exact molecular basis of the involvement of FGF2 isoforms still requires further investigation, this data offers a more thorough understanding of the potential future treatment or management of OA.
Supplementary Material
Supplemental Figure 1. Fgf2ALLKO male develop spontaneous and mechanically-induced OA. A, H & E stained representative images of 6 months old Fgf2ALLKO samples show loss of articular cartilage and no inflammation. N=3/group. B, Histomorphometric parameters of Fgf2ALLKO femoral and tibial epiphyses reveal decreases in BV/TV, Tb.Th, and Tb.N and an increase in Tb.Sp compared to WT. Values are the means ± SD. *P <0.05, **P <0.01, ***P <0.001, N=5/group. C, Right tibial loaded knees of Fgf2ALLKO mice have pitting of the tibial surface (arrow) compared to the left contralateral control and WT. Sagittal images display decreased femoral subchondral bone (dashed arrow) and increased trabecular spacing and decreased trabecular number in femoral epiphyses compared to the contralateral control and WT. N=4–5/group.
Supplemental Figure 2. Histomorphometric analysis of subchondral bone of 2 months old male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates. A-C, Histomorphometric parameters of femoral and tibial epiphyses are similar between Fgf2ALLKO Fgf2HMWKO, Fgf2LMWKO, and their respective WT littermates. Values are the means ± SD, n=3/group.
ACKNOWLEDGMENTS
This project is supported in part by NIH grant R01 DK098566, NIH AR072985–05A1 and training grant NIH/NIDCR T90DE021989. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. MMH conceived and coordinated the study. MMH and PMB designed the study. PMB performed and analyzed the experiments shown and wrote the paper. LP provided technical assistance and contributed to the preparation of the figures. TD developed the FGF2 isoform mice. All authors reviewed the results and approved the final version of the manuscript. MMH is the guarantor of the manuscript.
REFERENCES
- 1.Heidari B 2011. Knee osteoarthritis prevalence, risk factors, pathogenesis and features: Part I. Caspian J. Intern. Med 2;205–212. [PMC free article] [PubMed] [Google Scholar]
- 2.Karvone RL, Negendank WG, Teitge RA, Reed AH, Miller PR, & Fernandez-Madrid F. 1994. Factors affecting articular cartilage thickness in osteoarthritis and aging. J. Rheumatol 21;1310–18. [PubMed] [Google Scholar]
- 3.Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. 1986. Development of criteria for the classification and reporting of osteoarthritis: classification of osteoarthritis of the knee. Arthritis Rheum 29;1039–49. [DOI] [PubMed] [Google Scholar]
- 4.Hayami T, Pickarski M, Zhuo Y, Wesolowski GA, Rodan GA, & Duong LT. 2006. Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone 38:234–43. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, et al. 1996. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J. Clin. Invest 97:761–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. 2005. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434:644–48. [DOI] [PubMed] [Google Scholar]
- 7.Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, et al. 2005. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 434:648–52. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Manner PA, Horner A, Shum L, Tuan RS, & Nuckolls GH. 2004. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage 12:963–73. [DOI] [PubMed] [Google Scholar]
- 9.Orito K, Koshino T, & Saito T. 2003. Fibroblast growth factor 2 in synovial fluid from an osteoarthritic knee with cartilage regeneration. J. Orthop. Sci 8:294–300. [DOI] [PubMed] [Google Scholar]
- 10.Im HJ, Li X, Muddasani P, Kim GH, Davis F, Rangan J, et al. 2009. Basic fibroblast growth factor accelerates matrix degradation via a neuro‐endocrine pathway in human adult articular chondrocytes. J. Cell Physiol 215:452–63. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Chia SL, Sawaji Y, Burleigh A, McLean C, Inglis J, Saklatvala J, et al. 2009. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheumatol 60:2019–27. [DOI] [PubMed] [Google Scholar]
- 12.Okada-Ban M, Thiery JP, & Jouanneau J. 2000. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol 32:263–7. [DOI] [PubMed] [Google Scholar]
- 13.Azhar M, Yin M, Zhou M, Li H, Mustafa M, Nusayr E, et al. 2009. Gene targeted ablation of high molecular weight fibroblast growth factor-2. Dev. Dynam 238:351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homer-Bouthiette C, Doetschman T, Xiao L, & Hurley MM. 2004. Knockout of nuclear high molecular weight FGF2 isoforms in mice modulates bone and phosphate homeostasis. J. Biol. Chem 289:36303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garmy-Susini B, Delmas E, Gourdy P, Zhou M, Bossard C, Bugler B, et al. 2004. Role of Fibroblast Growth Factor-2 Isoforms in the Effect of Estradiol on Endothelial Cell Migration and Proliferation. Circ. Res 94:1301–09. [DOI] [PubMed] [Google Scholar]
- 16.Xiao L, Liu P, Li X, Doetschman T, Coffin JD. Drissi H, et al. 2009. Exported 18-kDa isoform of fibroblast growth factor-2 is a critical determinant of bone mass in mice. J Biol Chem 284:3170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meo Burt P, Xiao L, Dealy C, Fisher MC, & Hurley MM. 2016. FGF2 High Molecular Weight Isoforms Contribute to Osteoarthropathy in Male Mice. Endocrinology 157:4602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, et al. 1998. Fibroblast growth factor 2 control of vascular tone. Nat. Med 4:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, et al. 2000. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Invest 105:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasson SS, Chambers MG, Van Den Berg WB, & Little CB. 2010. The OARSI histopathology initiative–recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18:S17–23. [DOI] [PubMed] [Google Scholar]
- 21.Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, & Haudenschild DR. 2012. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthritis Cartilage 20:773–82. [DOI] [PubMed] [Google Scholar]
- 22.Ko FC, Dragomir CL, Plumb DA, Hsia AW, Adebayo OO, Goldring SR, et al. 2016. Progressive cell‐mediated changes in articular cartilage and bone in mice are initiated by a single session of controlled cyclic compressive loading. J. Orthop. Res 34:1941–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gigout A, Guehring H, Froemel D, Meurer A, Ladel C, Reker D, Bay-Jensen AC, Karsdal MA, Lindemann S. 2017. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthritis Cartilage. 25(11):1858–1867. [DOI] [PubMed] [Google Scholar]
- 24.Guibert M, Gasser A, Kempf H, Bianchi A. 2017. Fibroblast Growth Factor 23 promotes terminal differentiation of ATDC5 Cells. PloS One April 13;12(4):e0174969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dreier R 2010. Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 12:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong L, Huang X, Karperien M, & Post JN. 2015. The regulatory role of signaling crosstalk in hypertrophy of MSCs and human articular chondrocytes. Int. J. Mol. Sci 16:19225–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karaliotas GI, Mavridis K, Scorilas A, & Babis GC. 2015. Quantitative analysis of the mRNA expression levels of BCL2 and BAX genes in human osteoarthritis and normal articular cartilage: An investigation into their differential expression. Mol. Med. Rep 12:4514–21. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Toury R, Hauchecorne M, & Balmain N. 1997. Expression of Bcl-2 protein in the epiphyseal plate cartilage and trabecular bone of growing rats. Histochem. Cell Biol 108:45–55. [DOI] [PubMed] [Google Scholar]
- 29.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, & Firestein GS. 2001. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest 108:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Ji Q, Wang X, Kang L, Fu Y, Yin Y, et al. 2015. SOX9 is a regulator of ADAMTSs-induced cartilage degeneration at the early stage of human osteoarthritis. Osteoarthritis Cartilage 23:2259–6. [DOI] [PubMed] [Google Scholar]
- 31.Yan D, Chen D, Cool SM, Van Wijnen AJ, Mikecz K, Murphy G, et al. 2011. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res. Ther 13:R130. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Weng T, Yi L, Huang J, Luo F, Wen X, Du X, et al. 2012. Genetic inhibition of fibroblast growth factor receptor 1 in knee cartilage attenuates the degeneration of articular cartilage in adult mice. Arthritis Rheum 64:3982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davidson D, Blanc A, Filion D, Wang H, Plut P, Pfeffer G, et al. 2005. Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis. J. Biol. Chem 280:20509–15. [DOI] [PubMed] [Google Scholar]
- 34.Valverde-Franco G, Binette JS, Li W, Wang H, Chai S, Laflamme F, et al. 2006. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Hum. Mol. Genet 15:1783–92. [DOI] [PubMed] [Google Scholar]
- 35.Tang J, Su N, Zhou S, Xie Y, Huang J, Wen X, Wang Z, Wang Q, Xu W, Du X, Chen H, Chen L. 2016. Fibroblast Growth Factor Receptor 3 Inhibits Osteoarthritis Progression in the Knee Joints of Adult Mice. Arthritis Rheumatol. 68(10):2432–43. [DOI] [PubMed] [Google Scholar]
- 36.El-Seoudi A, El Kader TA, Nishida T, Eguchi T, Aoyama E, Takigawa M, et al. 2017. Catabolic effects of FGF-1 on chondrocytes and its possible role in osteoarthritis. J. Cell Commun. Signal 25:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li R, Wang B, He CQ, Yang YQ, Guo H, Chen Y, et al. 2015. Upregulation of fibroblast growth factor 1 in the synovial membranes of patients with late stage osteoarthritis. Genet. Mol. Res 14:11191–9. [DOI] [PubMed] [Google Scholar]
- 38.Bianchi A, Guibert M, Cailotto F, Gasser A, Presle N, Mainard D, et al. 2016. Fibroblast Growth Factor 23 drives MMP13 expression in human osteoarthritic chondrocytes in a Klotho-independent manner. Osteoarthritis Cartilage 24:1961–9. [DOI] [PubMed] [Google Scholar]
- 39.Liao S, Bodmer JR, Azhar M, Newman G, Coffin JD, Doetschman T, et al. 2010. The influence of FGF2 high molecular weight (HMW) isoforms in the development of cardiac ischemia–reperfusion injury. J.Mol. Cell Cardiol 48:1245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krejci P, Krakow D, Mekikian PB, & Wilcox WR. 2007. Fibroblast Growth Factors 1, 2, 17, and 19 Are the Predominant FGF Ligands Expressed in Human Fetal Growth Plate Cartilage. Pediatr. Res 61:267–72. [DOI] [PubMed] [Google Scholar]
- 41.Tchetina EV, Squires G, & Poole AR. 2005. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol 32:876–86. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Fgf2ALLKO male develop spontaneous and mechanically-induced OA. A, H & E stained representative images of 6 months old Fgf2ALLKO samples show loss of articular cartilage and no inflammation. N=3/group. B, Histomorphometric parameters of Fgf2ALLKO femoral and tibial epiphyses reveal decreases in BV/TV, Tb.Th, and Tb.N and an increase in Tb.Sp compared to WT. Values are the means ± SD. *P <0.05, **P <0.01, ***P <0.001, N=5/group. C, Right tibial loaded knees of Fgf2ALLKO mice have pitting of the tibial surface (arrow) compared to the left contralateral control and WT. Sagittal images display decreased femoral subchondral bone (dashed arrow) and increased trabecular spacing and decreased trabecular number in femoral epiphyses compared to the contralateral control and WT. N=4–5/group.
Supplemental Figure 2. Histomorphometric analysis of subchondral bone of 2 months old male Fgf2ALLKO, Fgf2HMWKO, Fgf2LMWKO, and wild-type littermates. A-C, Histomorphometric parameters of femoral and tibial epiphyses are similar between Fgf2ALLKO Fgf2HMWKO, Fgf2LMWKO, and their respective WT littermates. Values are the means ± SD, n=3/group.