

Abstract
Visualizing neural activity from deep brain regions in freely behaving animals through miniature fluorescent microscope (miniscope) systems is becoming more important for understanding neural encoding mechanisms underlying cognitive functions. Here we present our custom-designed miniscope GRadient INdex (GRIN) lens system that enables simultaneously recording from hundreds of neurons for months. This protocol includes miniscope design, the surgical procedure for GRIN lens implantation, miniscope mounting on the head of a mouse, and data acquisition and analysis. First, a target brain region is labeled with virus expressing GCaMP6; Second, a GRIN lens is implanted above the target brain region; Third, following mouse surgical recovery, a miniscope is mounted on the head of the mouse above the GRIN lens; Finally, neural activity is recorded from the freely behaving mouse. This system can be applied to recording the same population of neurons longitudinally, enabling the elucidation of neural mechanisms underlying behavioral control.
Keywords: miniscope, calcium imaging, GCaMP6, GRadient INdex lens, freely behaving mice
INTRODUCTION
Over the past half century, the development of calcium indicators, from synthetic dyes to genetically encoded fluorescent proteins, has enabled one to monitor in vivo neural activity using microscopy (Grienberger & Konnerth, 2012). Early generations of organic fluorescent calcium indicators provided important insights into the calcium-dependent neuronal processes (Hallett & Carbone, 1972; Llinas & Nicholson, 1975; Stinnakre & Tauc, 1973), though their utility for neural activity motoring in vivo was limited as these molecules needed to be introduced physically. An important breakthrough in neuroscience calcium imaging was achieved when genetically encoded calcium indicators (GECIs) were introduced (Miyawaki et al., 1997). Especially after the GCaMP-family GECIs were well developed by several groups (Akerboom et al., 2012; Chen et al., 2013; Nakai, Ohkura, & Imoto, 2001; Tian et al., 2009), neural activity monitoring using calcium imaging has become an important tool in neuroscience research.
In parallel to the development of the calcium indicators, instrumentation was developed to render calcium imaging possible. Since the realization of two-photon microscopy by Winfried Denk and colleagues in 1990 (Denk, Strickler, & Webb, 1990), there has been a dramatic increase in the research of calcium imaging using this technique.. Imaging cortical activity through cranial windows during many types of behavior in head-fixed animals sheds new light onto neural encoding of behavioral control (Komiyama et al., 2010; Yang, Pan, & Gan, 2009). Combined with the GRadient INdex (GRIN) lens which relays optical images from one end to the other, in vivo imaging has extended into deep brain regions more recently (Barretto, Messerschmidt, & Schnitzer, 2009). Moreover, with the development of miniature microscopes with epifluorescent (Ghosh et al., 2011) or two-photon light source (Helmchen, Fee, Tank, & Denk, 2001; Liang, Hall, Messerschmidt, Li, & Li, 2017; Sawinski et al., 2009; Zong et al., 2017), the accompanying behavior tests have expanded from head-fixed animals to freely-moving animals.
Calcium imaging enables recording of neural activity from specific population of neurons longitudinally at single-cell resolution. In 2011, Mark Schnitzer’s group first reported the miniature microscope with epifluorescent light source (Ghosh et al., 2011), which was light and small, and easy to mount on the head of a mouse for studying freely-behaving, complex behaviors. Combined with GRIN lens, the miniscope imaging system allows recording of calcium activity from hundreds of neurons in deep brain regions for months, facilitating the decoding of neural representations underlying specific behaviors.
In this unit, we present our custom miniscope GRIN lens system. Basic Protocol 1 briefly describes the miniature microscope design and assembly. Basic Protocol 2 introduces the surgical procedure for GRIN lens implantation to deep brain regions. Basic Protocol 3 presents the procedure for mounting the miniscope on the head of a mouse. Basic Protocol 4 shows data acquisition and analysis.
All the surgeries were performed in accordance with the guidelines of Institutional Animal Care and Use Committee, the Intramural Research Program, National Institute on Drug Abuse, National Institutes of Health.
BASIC PROTOCOL 1
BASIC PROTOCOL TITLE: Miniature microscope design and assembly
The miniature epifluorescent microscope (miniScope) system consists of miniScope main body, the data acquisition controller and the data transmission cable with commutator. The miniScope has a 2.4-gram weight and approximately 1.1 mm×1.1 mm maximum field of view. The dimensions of the miniScope are approximately 12 mm(L) × 12 mm(W) × 20 mm(H). The data acquisition frame rate is 10 Hz with 400 × 400 pixel resolution, or higher frame rate with smaller region of interest selection.
Materials
Tools for assembly
High Power UV Curing LED System (CS2010, Thorlabs), UV-curing optical adhesive (NOA81, Thorlabs), soldering iron, superglue (Professional, Loctite), plastic tweezers (7003A18, McMaster-Carr)
Optics
Excitation filter (470/40 nm, 3 mm × 3 mm × 1 mm, Chroma Technology), Emission filter (525/50 nm, 3 mm × 3 mm × 1 mm, Chroma Technology), Dichroic mirror (FF495, 5 mm × 5 mm × 1 mm, Semrock), blue LED (XPEBLU-L1, Cree), 4-mm diameter 6-mm focal length achromatic doublet lens (#63-690, Edmund optics), 4-mm diameter, 2.73-mm focal length aspherical lens (#83-605, Edmund optics)
MiniScope housing
Custom 3D printed miniScope housing, filter cube and base (Stereolithography with nickel plating, Protolabs, MN), #00-90 1/8” length screw (91781A410, McMaster-Carr) and #00-90 nut (92736A112, McMaster-Carr)
Cable
Omnetics polarized nano 10-pin connector (A79615-001, Omnetics), heat shrinking tube (6699T17, McMaster-Carr), slipring (1196, Adafruit), flexible wire (9564T2, McMaster-Carr), Harwin 10-pin connector (952-1199-ND, Digi-Key) and Harwin 2-pin connector (952-1189-ND, Digi-Key)
Electronics
Image sensor printed circuit board (PCB), data acquisition PCB, electronic components (the full BoM can be found at https://github.com/giovannibarbera/miniscope_v1.0), Opal Kelly XEM3010, data acquisition electronics housing (custom 3D printed), hook-up wires (Digikey, 2840/7BK005-ND), hot plate, soldering iron, solder paste, tweezers, fine gauge needle tip, epoxy, dissecting microscope.
Protocol steps
1. Construction of the miniScope
1.1). Filter cube assembly:
Use the plastic tweezers to place the dichroic mirror in the position illustrated in Fig. 1A. Apply a drop of optical adhesive NOA81 at each corner of the dichroic mirror, and use the high-power UV curing LED System to cure the optical adhesive for at least 30 seconds. Make sure that the coated edge of the dichroic mirror faces the excitation light direction. Similarly, use the plastic tweezers to place the emission filter in the position as illustrated in Fig. 1A. Apply a drop of optical adhesive NOA81 at each corner of the emission filter, and use the high-power UV curing LED System to cure the optical adhesive for at least 30 seconds. Make sure the coated edge of the emission filter faces the emission light direction.
Figure 1.
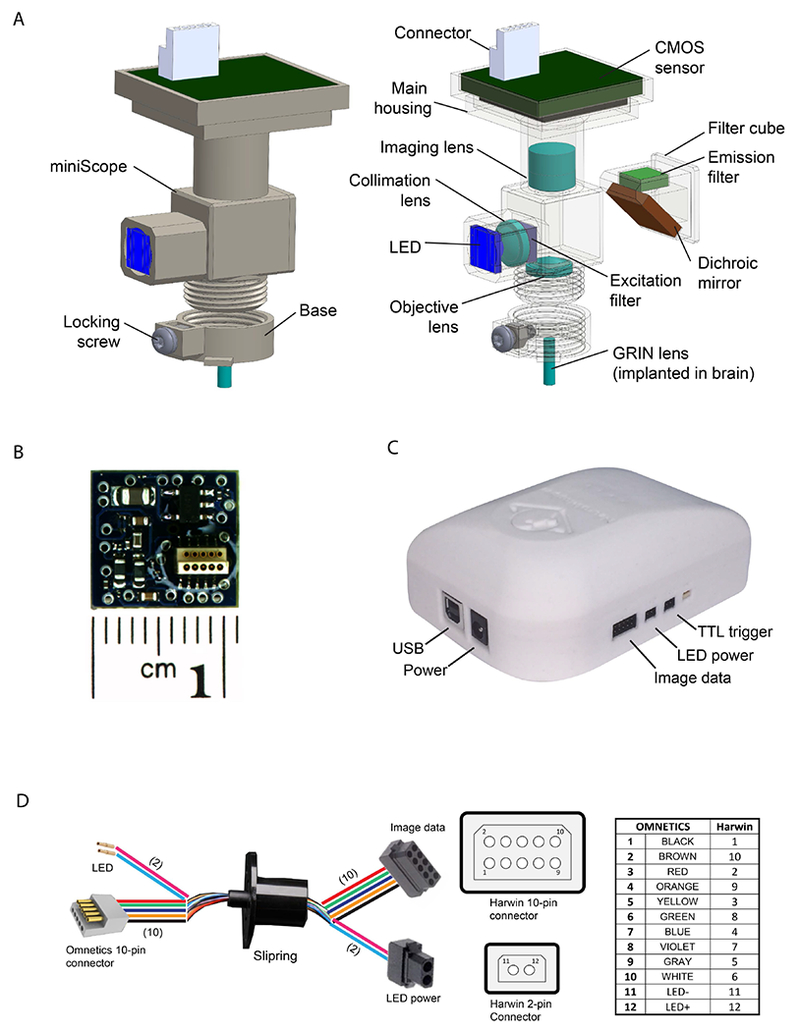
(A) Illustration of the miniScope main body and base. Left: 3D model of the integrated miniScope and base mounting mechanism. Right: Individual components consisting of the miniScope, including CMOS sensor (with connector), imaging lens, excitation/emission filters, dichroic mirror, collimation lens, LED, objective lens, main housing, filter cube, base, and locking screw (nut). (B) Illustration of the CMOS sensor PCB board. (C) Illustration of the data acquisition controller. (D) Illustration of the construction of the data transmission cable.
1.2). Housing body assembly:
(1) position the excitation filter in the position as illustrated in Fig. 1A. The coated edge faces the LED. Use optical adhesive to secure it. (2) Place the collimating lens (#83-605, Edmund optics) such that the flat side faces the excitation filter. Use optical adhesive to secure the lens. (3) Use plastic tweezers to insert the imaging lens (#63-690, Edmund optics) as illustrated in Fig. 1A. Use two drops of optical adhesive to secure the lens. (4) Use plastic tweezers to insert the objective lens (#83-605, Edmund optics) to the position illustrated in Fig. 1A. Use two drops of optical adhesive to secure the lens. (5) Insert the assembled filter cube into the main housing slot and apply superglue on the edges of the filter cube to secure them. (6) Insert the assembled image sensor PCB to the slot and use superglue to secure it.
1.3). LED soldering and assembly:
Use an electrical wire stripper(7294K15, McMaster-Carr) to cut two 1-inch 29-gauge wires and strip a 2-mm coating at the end of each side. Solder one side of one wire to the LED anode (+) and another side to a miniature connector pin. For the other wire, solder one side to the LED cathode (−) and another side to another miniature connector pin. Place the LED in the position as shown in Fig. 1A and use superglue to secure it.
1.4). Base assembly:
Insert the miniature nut (#00-90) to the slot on the base and use superglue to secure the nut on the base. The #00-90 1/8-inch screw is used to lock the miniScope in position.
All design files and associated details can be found in GitHub (https://github.com/giovannibarbera/miniscope_v1.0).
2. Assembly of the image sensor board
2.1) Place the image sensor board under the microscope, image sensor side facing up, and apply solder paste to the 52 ball grid array (BGA) and all exposed pads. For faster and more accurate results, use a stencil; if stencil is not available, use a fine gauge needle tip to apply solder paste to each individual BGA pad. Use only the minimum amount of solder paste required on each pad, to avoid bridging pads when mounting the image sensor.
2.2) Carefully place the image sensor on PCB. If the image sensor accidentally slides on the PCB, remove it, clean the PCB with IPA and re-apply solder paste.
2.3) Place the PCB, image sensor side up, on the hot plate, and turn it on to 300°C;. When the solder melts the image sensor will slightly tilt as the surface tension adjusts the BGA position. Remove from hot plate and allow to cool.
2.4) Flip PCB over and apply solder paste to all pads on the other side.
2.5) Place PCB under dissection microscope, and, with tweezers and soldering iron, manually place and solder all the electronic components on the PCB. Make sure the Omnetics connector has the right orientation.
2.6) Place epoxy around the Omnetics connector and let cure for 24 hours (Fig. 1B).
3. Assembly of the data acquisition system
The electronics for the data acquisition system are enclosed in a custom 3D printed electronics box which provides protection and easy access to connections between miniScope, host computer and other data acquisition systems. The mechanical design for top and bottom can be downloaded at https://github.com/giovannibarbera/miniscope_v1.0.
3.1) Apply solder paste to all pads, then, under dissection microscope, position components with tweezers and solder them. Repeat on the other side of the PCB for main PCB, connector PCB and LED PCB (optional). All design files, BoM and placement files can be found at https://github.com/giovannibarbera/miniscope_v1.0.
3.2) Apply epoxy around Omnetics connector for improved durability and let cure at room temperature for 24 hours in a clean, dust-free environment.
3.3) Tap 4 holes on the posts and 4 holes on the bottom side with a #2-56 bottoming tap.
3.4) Apply epoxy around connector PCB and place on the side of the top part of the electronics box. Let cure overnight.
3.5) Secure xem3010 board to posts with 1/4” #2-56 pan machine screws.
3.6) Connect LED PCB and connector PCB to main data acquisition board through WM9492-ND (Digikey) and WM6667-ND (Digikey), respectively.
3.7) Close secure top to bottom part of the box with 1/4” #2-56 flat machine screws and apply rubber bumpers (SJ5012-0-ND, Digikey) (Fig. 1C).
4. Assembly of the cable
4.1). MiniScope side:
(a) The Omnetics 10 pin connector is used to connect the 10-pin connector on the CMOS PCB board. Cut the wires of the pre-wired Omnetics 10-pin connector to ¼ inch. Strip approximately 1/8 inch of coating off the end of each wire.
(b) Cut the slipring shaft side lead to ¼ inch. Stripe approximately 1/8 inch of coating off the wire coating.
(c) Cut 12 18-inch long flexible wires (the length can be adjusted according to actual experimental requirement). Solder one side to the Omnetics connector and the other side to the slipring. Wires with same color code on each side are connecting. Use heat-shrink tubing to protect the solder exposures. Twist the white and orange wire pair before soldering.
(d) The slipring has a total of 12 pins. Two pins are soldered to two Harwin receptacle connector crimp (24-28 AWG). Use heat-shrink tubing to protect the solder exposures. These two connectors will be connected to the LED leads on the miniScope.
4.2). Controller side:
(a) On the flange side of the slipring, cut the lead to ¼ inch. Strip approximately 1/8 inch of coating off the wire.
(b) Cut 12 12-inch long flexible wires (the length can be adjusted according to actual experimental requirement). Solder one side to the Harwin 10-pin connector and the other side to the slipring. Use heat-shrink tubing to protect the solder exposures. Twist the white and orange wire pair before soldering.
(c) Solder the extra two wires to the Harwin 2-pin connector.
(d) The overall wiring diagram is shown in Fig. 1D right panel.
BASIC PROTOCOL 2
BASIC PROTOCOL TITLE: GRIN Lens Implantation
For optical imaging in subcortical regions, the implanted GRIN lens implantation relays signals from deep brain targets to a height above the skull surface accessible for image acquisition. Two sizes of GRIN lens are typically used for deep brain imaging: 1mm diameter and 0.5 mm diameter. For brain regions such as dorsal striatum (Barbera et al., 2016), prelimbic cortex (Pinto & Dan, 2015) and hippocampus (Ziv et al., 2013), a 1mm diameter GRIN lens can be used to increase the field of view. For deeper regions, such as amygdala (Li et al., 2017), hypothalamus (Jennings et al., 2015), nucleus accumbens, etc., a 0.5 mm diameter GRIN lens will be a better choice to minimize brain tissue damage from lens implantation. To implant a 1mm diameter GRIN lens, tissue removal is necessary to make room for the lens, whereas a 0.5mm diameter GRIN lens can be directly implanted into the brain after a leading track is made with a blunt-end needle.
We present our procedure for GRIN lens implantation, which includes tissue removal for 1mm diameter GRIN lens implantation to the dorsal striatum and direct implantation of 0.5mm GRIN lens to the nucleus accumbens. Before implantation, virus injection to the target region is conducted. GRIN lens implantation is performed 7-14 days after viral injection.
Materials
Ibuprofen(Goodsense, 0113-0660-26), 100% oxygen, isoflurane (Henry Schein, 1182097), 4-0 wax coated braided silk suture (Roboz Surgical Store, SUT-1074-31), Buprenorphine (Buprenex, 12496-0757-5), Metabond kit (Parkell inc., S371, S396, S398), Dental acrylic cement (BASi, MD-1300), Carbon (Sigma-Aldrich, 484164-50g), Super glue, Gas with 5% CO2/95% O2, AAV virus (Upenn core facility), 20 ml vial of 0.9% Sodium Chloride Injection (Hospira, 0409-1966-05), 70% Alcohol, Antibiotic ointment (Neosporin), 2% Lidocaine (Fresenius Kabi, 63323-486-57), Lubricating ophthalmic ointment (Rugby, 0536-1086-91), Iodine(Betadine, 7.5%), Dexamethasone (Henry Schein, 002459).
Surgical instruments:
Autoclaved fine-tipped scissors (1, Fine Science Tools , 14081-09), fine-tipped thumb forceps (2, Fine Science Tools, 11370-42), Micro Spatula (1, Fine Science Tool, 10091-12), 0.5mm-drill bit (1, Fine Science Tools, 19007-05), 1.2mm-diameter drill bit (1, Widget Supply, D-EK09), bulldog serrefines (2, Fine Science Tool, 18051-28), UltraMicroPump (UMP3) with SYS-Micro4 Controller (1, World Precision Instruments, UMP3-1), cotton swabs (20, Fisher Scientific, 23-400-119), and 10ul syringe (1, Hamilton, 7653-01), Micro blade (1, Surgistar, 6900), Sterile packed 30 and 34 gauge small hub removable needles (Hamilton, 7762-03, 207434), 1 ml syringes (BD, 329654), and 26 or 30 gauge syringe needles (BD, 305109 or 305106) are also used.
Equipment:
Robotic surgical instrument (Fig. 2A): Detailed design file, assembly photos, and parts list for the robotic surgical instrument can be found at our Github website (https://github.com/liangbo/AutoStereota). Three arms of a stereotaxic instrument (Model 963, Kopf) are modified to connect three stepper motors(3326_0, Phidgets, Canada) individually; the motors are controlled by three USB-based motor controllers via MATLAB-based (Mathworks) GUI software. A syringe holding a 30-gauge blunt-end needle is parallelly mounted on the dorsal-ventral stereotaxic arm, and connected to a standard laboratory vacuum valve (pressure ~40 – 50 psi). . This robotic surgical instrument can automatically aspirate brain tissue layer by layer in a pre-defined diameter. A dissection scope (Stemi 2000, Zeiss), stereotaxic frame for mouse with nose cone for anesthesia, heat pad (TCAT-2DF, Physitemp), carbogen and oxygen tanks, and isoflurane vaporizer (VetEquip) are also needed.
Figure 2.
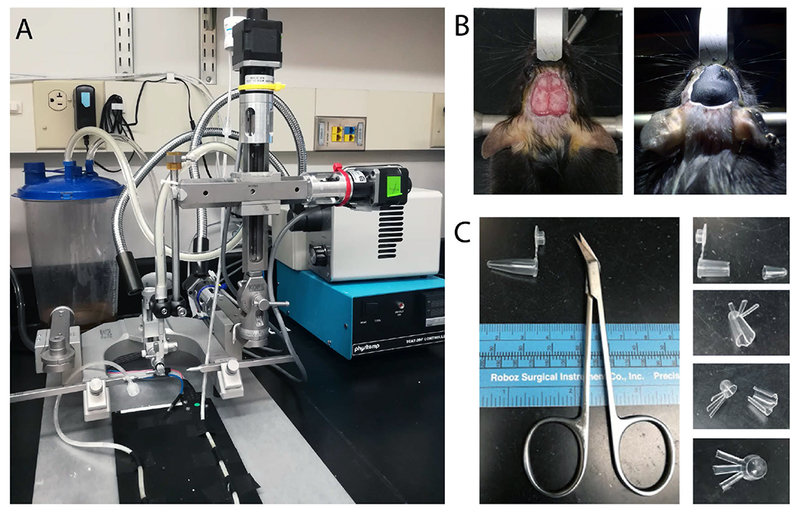
(A) Robotic surgical instrument, which includes three arms connecting three stepper motors individually; the motors are controlled by three USB-based motor controllers via MATLAB-based (Mathworks) GUI software. A syringe holding a 30-gauge blunt-end needle is parallelly mounted on the dorsal-ventral stereotaxic arm, and is connected to the vacuum pump. (B) Left, cleaned skull area; Right, GRIN lens is secured with two layers of dental cement. (C) Procedures for making GRIN lens cap from PCR tube bottom.
Adeno Associated Virus (AAV) virus injection
Work area(s) preparation:
The aseptic surgical field is the disinfected skin and exposed surgical wound on the dorsal skull. The aseptic surgical field is situated within a asceptic benchtop surgical area covered with fresh, absorbent paper and includes:
-
1)
The stereotaxic stage with a heating pad at its base to maintain 37°C mouse body temperature, and the UltraMicroPump (UMP3) with SYS-Micro4 Controller system (World Precision Instruments). A small paper towel, gauze pad, or clear plastic (e.g. Saran Wrap) sheet can be placed over the mouse’s neck and back to cover fur;
-
2)
A sterile instrument resting area (e.g. sterile petri dish, sterile glass beaker, small sterile drape or platform) to suspend sterile instrument tips above the clean surgical area;
-
3)
A virus handling/syringe loading area with aliquot of virus in a container of ice, virus aspirating pipette, parafilm sheet to load inoculum dose, 10% bleach solution container to deactivate non-injected virus, and a small MPW sharps box.
Protocol steps
Place mouse(2-3 months, ~25gram bodyweight) in an induction chamber (5% isoflurane, 1 liter/min oxygen flow rate) until mouse respiratory rate decreases to 1 breath per second, then transfer to an insulated paper towel separated from the surgical area, remove the fur with shaver from the top of the head and extend the furless area caudally to the first cervical vertebrae, making sure to leave lateral (side) margins large enough to prevent hair from entering the incision. Put the mouse back into the induction chamber if it wakes up during shaving, and repeate the anesthesia and shaving procedure.
- Switch the isoflurane toward the stereotaxic stage, using 2% isoflurane in oxygen at 0.4 liter/min. Mount the mouse on the stereotaxic stage, and maintain its body temperature at 37°C with a heating pad.
-
A)Full anesthesia is confirmed by absence of response to a toe pinch, and is monitored frequently during the surgery.
-
B)Apply a lubricating ophthalmic ointment with a clean swab to mouse eyes to keep the eyes moist during surgery.
-
C)Disinfect the exposed scalp and ears with surgical scrub (iodine or chlorhexidine based detergent)) and rinse with 70% ethanol or sterile water using sterile swabs.
-
A)
Using a 1-ml syringe equipped with a 26-G needle, inject a small volume of 2% lidocaine under the surface of the dorsal scalp to provide local analgesia. Using a scalpel, make an incision through the scalp down the midline of the head to expose the skull, pull the skin edges laterally to expose the lambda and bregma landmarks, apply bulldog serrefines to either side of the skin incision to keep the skull surface exposed, and carefully peel/remove the fascia from the skull using fine-tipped forceps or sterile cotton swabs.
Confirm that the head of the mouse is level in the stereotaxic frame by comparing bregma and lamda with micro drill mounted on the dorsal-ventral stereotaxic arm, locate the drill bit tip to the bregma, write down the coordinates, then move the tip above lamda, adjust the nose holder position to keep the bregma and lambda at the same horizontal level. [Acceptable tolerance in D/V measurement differences at bregma and lamda can be empirically determined through separate dye infusion experiments].
- Locate the drill bit tip above the target area according to stereotaxic coordinates.
-
A)Keep the skull moist and burring surfaces “cool” with applications of sterile saline. Burr a round, 0.5mm-diameter hole in the skull at the coordinates (for dorsal striatum, AP: −0.6mm, M/L: +2mm, with top of syringe holder angled 30° caudally; for NAc, AP: +1.2mm, M/L: +1mm with vertical syringe holder).
-
B)After carefully removing bone fragments, puncture the dura with a 30-gauge needle under dissection microscope.
-
A)
Using a 34 gauge needle, load virus into a 10μl syringe. First pre-fill with 5μl saline, then withdraw a 500nl air bubble, and finally withdraw 1000nl virus. The air/saline interface serves as the proxy for virus/air interface when visually tracking infusion. Infusions are made by tracking this meniscus and ejecting 50nl per min.
Locate the needle tip at bregma, write down the coordinates, then move the needle tip to the injection coordinates above the skull hole. Visually confirm that the needle is not clogged by pushing out 100nl virus, then very slowly (at an approximate rate of 1mm/1minute) position the needle through the skull hole to targeted coordinates within the brain (for dorsal striatum, AP: −0.6mm, M/L: +2mm, D/V: −3mm, syringe holder angled 30° caudally; for NAc, AP: +1.2mm, M/L: +1mm, D/V: −4.2mm). Lower the needle slightly beyond the targeted Z-coordinate (0.1mm) then withdraw to the targeted position, creating a small pocket into which the viral solution can be infused and minimizing unintended diffusion of the vector up the track created by the injector.
Set up the micropump and initiate the injection of 500nl volume at 50nL/min. Wait another 5 min after injection is completed to let the virus diffuse and prevent efflux of virus during removal, then very slowly remove the needle from the brain (same rate as above). [The slow delivery rate of small volumes enables more homogenous viral delivery and reduces tissue damage by minimizing the injection pressure associated with smaller lumens. The post-infusion rest period helps to minimize diffusion of the virus up the needle track.] Clean the needle with a bleach wetted cotton swab; push out all remaining virus from the syringe into bleach solution.
Suture the skin edges. Apply Neosporin ointment to the closed skin incision line, and administer 0.5 to 1 ml pre-warmed sterile saline subcutaneously.
Remove the mouse from the stereotaxic frame. Return the mouse to its home cage to recover from anesthesia in a 37 °C isothermal chamber and monitor the mouse until it is ambulatory.
Administer analgesic and monitor the health of the mouse daily for at least three days post-surgery. For post-operative analgesia, administer buprenorphine (0.05-0.10 mg/kg) after the mouse is ambulatory, or use an NSAID analgesic such as meloxicam or carprofen.
Wait for 7-14 days before GRIN lens implantation.
GRIN (Gradient Index) lens implantation
Direct implantation of 0.5mm-diameter GRIN lens to NAc
-
1.Follow steps 1 through 5 as described above for the AAV injection procedure, with the following changes:
-
A)Administer dexamethasone (2mg/kg, intramuscular or subcutaneously) to minimize surgery related tissue swelling and inflammation.
-
B)Excise (remove) a round (along the skull edge and beside the eyes ~ 5 mm diameter) section of dorsal scalp sufficient to expose bregma and its sutures. Use a small surgical blade and fine forceps to peel and scrape periosteum tissue from the skull.
-
C)Apply a thin layer of super glue along the edges of the skin excision to bond skin to cleaned skull. Wait for 5 min to let the tissue adhesive dry. A hand-held pipette tip may be used as a precise glue applicator.
-
D)Clean the periosteum-free skull area using a micro blade and cotton swabs. This will enhance adherence of dental cement to secure the implanted lens. (Fig. 2B)
-
A)
-
2.
Load 10μl syringe (with 30 gauge needle) with 5μl saline, the needle will be used to make a leading track for subsequent GRIN lens implantation.
-
3.
Locate the needle tip to the bregma, write down the coordinate, then move the needle tip to above the hole for implantation, then very slowly (at an approximate rate of 1mm/1minute) insert the needle through the hole to target brain region coordinates (for NAc, AP: +1.2mm, M/L: +1mm, D/V: −4.2mm). Wait for 5 min then very slowly remove the needle from the brain (same rate as above).
-
4.
Secure the top part (about 1.5mm long) of 0.5mm-diameter GRIN lens into the glass tube (0.6mm ID, 0.84mm OD, cut to 2 cm long, VITROCOM) with super glue, make sure the GRIN lens parallels the glass tube. Wait for 10min to let the glue get dry.
-
5.
Load the glass tube holding the GRIN lens in the holder (Model 1770, Kopf), mount on the dorsal-ventral arm of robotic surgical instrument, locate the lens tip to bregma, write down the coordinates, clean the hole with aCSF, make sure there is no blood around the hole, then move the lens tip above the hole for implantation. Very slowly (at an approximate rate of 1mm/1minute) insert the lens through the hole towards the target stereotaxic coordinates in the brain, using the same coordinates used for the leading track but pushing 0.1mm further downward.
-
6.
Blot excess aCSF and blood from around GRIN lens with a sterile paper towel. Wait for 5 min to let the skull dry.
-
7.
Apply a layer of Metabond dental cement over the exposed skull and all sides of the GRIN lens. Wait for 5 min to allow Metabond to cure. Then put on another layer of dental acrylic cement (mixed with 1/5 Carbon, to make the dental acrylic cement black in order to reduce LED light reflection during imaging). Wait 10 min to allow the dental cement to cure.
-
9.
Fill the glass tube with acetone to resolve the super glue, wait for 5 min. Loosen the holder and remove the glass tube. Dip the cotton tip into the acetone, then slightly remove extra super glue on the GRIN lens with the cotton tip.
-
10.
Cover the GRIN lens top with a cap (we use a hand-trimmed PCR tube bottom) affixed to the dental cement using super glue to protect the GRIN lens from damage. (Fig. 2B). Do not let glue touch the GRIN lens. If the glue gets onto GRIN lens, drip acetone onto the glue to get it soft, then remove the glue with a cotton tip.
-
11.
Administer 1 ml of pre-warmed sterile saline subcutaneously.
-
12.
Remove the mouse from the stereotaxic frame. Return the mouse to its home cage to recover from anesthesia in a 37 °C isothermal chamber and monitor until it is ambulatory.
-
13.
Administer analgesic and monitor mouse as described above.
Implantation of 1mm-diameter GRIN lens
-
1.Follow step 1 as described above for the direct implantation of 0.5mm-diameter GRIN lens procedure with these changes:
-
A)Do not proceed with surgery if body weight is less than 22 g, as mice below this weight do not recover well from the surgery.
-
B)Prepare fresh aCSF, and bubble with carbogen gas (95% oxygen 5% carbondioxide).
-
A)
-
2.Locate the brain area to be imaged using stereotaxic coordinates and mark the skull location for making an opening (craniotomy).
-
A)Keep the skull moist and burring surfaces “cool” with applications of sterile saline. Use a dental drill to burr a round, 1.1 mm-diameter hole in the skull at the coordinates (A/P: + 0.9 mm, M/L: +2mm for dorsal striatum in vivo imaging; D/V: −2 mm).
-
B)After carefully removing bone fragments, puncture the dura with a 30-gauge needle tip and use a fine thumb forceps to remove dura shreds and bone fragments, potential obstacles for the aspiration needle.
-
A)
-
3.
Locate the needle tip to bregma, write down the coordinate, then move the needle tip to be centered above the desired craniotomy region for aspiration of brain tissue. The aspiration needle is a 30-gauge, blunt tip needle with a sharpened tip wall.
-
4.
Open vacuum pulling through the sharpened needle, and start aCSF irrigation of the craniotomy site. Pre-warmed aCSF (37 °C) filtered through a 0.22μm filter is administered via a 30-gauge needle using 1-meter gravity difference.
-
5.Set program parameters to aspirate the brain tissue above the brain imaging region of interest. For software introduction, please visit our Github website (https://github.com/liangbo/AutoStereota).
-
A)Aspiration is performed slowly, layer by layer, by a robotic surgical instrument until the desired brain region is reached. For dorsal striatum, reaching striatum is visually confirmed by the change from the white color of corpus callosum to the dark color of striatum.
-
B)Since the aspiration needle tip starts at the level of Bregma, about 300μm above the brain surface, the first z-step may be as much as 500μm. Subsequently, increase the aspiration depth by just 200μm per layer until 200-300μm from the desired brain region, then adjust the z-step to 100μm per layer and double the aspiration resolution.
-
C)Pay attention to debris from bone, dura, hair, scar tissue or large blood vessels or clots during aspiration to prevent cacuum needle blockage. Even partial occlusion of the needle can interfere with vacuum strength and prevent successful tissue removal. Monitor the craniotomy sit for these obstructions through a dissection scope, and carefully remove them with fine forceps. Frequency of needle blockage depends on the clearance of debris pieces. Once the needle gets clogged, stop the aspiration and aCSF irrigation, remove the needle and clear it by pushing through saline, then reconnect the needle, and repeat step 5.
-
D)Bleeding from vessel rupture during aspiration is expected - continue aCSF irrigation and aspiration. Once aspiration is done (needle tip reached target z-coordinate, for dorsal striatum, D/V: −1.75mm), wait for 5 minutes to let small, ruptured vessels seal. Subsequently, lower the needle to 200μm above the craniotomy floor to clean the site, then return the needle to a height near the skull surface. Repeat this cleaning step until blood is no longer aspirated.
-
A)
-
6.
Remove the needle and stop aCSF irrigation. Gently lower and seat the disinfected GRIN lens (pre-cleaned by 15 min immersion in 70% alcohol followed by saline immersion until placed) into the hole. Be sure the lens is surrounded by aCSF with no air bubbles trapped beneath it as it is lowered. Use gentle pressure from a soft, sterilize paper towel to press the GRIN lens to assure that its tip is in contact with the brain tissue at the bottom of the tissue well. Wick excess aCSF from around GRIN lens with a sterile paper towel.
-
7.
Apply melted agarose (1%, melted in microwave, maintained in 40-degree water bath) onto the skull around the GRIN lens to seal the gap between skull and GRIN lens. Remove excess agarose with micro blade after it hardens.
-
8.
Apply a layer of Metabond dental cement over the exposed skull and all sides of the GRIN lens. Wait for 5 min to let the Metabond cure. Then add a layer of dental acrylic cement (mixed with 20% Carbon powder, to make the dental acrylic cement black to reduce LED light reflection during imaging), wait 10 min to let the dental cement cure. (Fig. 2B)
-
7.
Cover the GRIN lens top with a cap(customized PCR tube bottom, Fig, 2 C), and glue the cap on the dental cement to protect from damage (Fig. 2 B). Do not let glue touch the GRIN lens.
-
8.
Follow steps 11-13 from the direct implantation of 0.5mm-diameter GRIN lens procedure for post-operative care.
BASIC PROTOCOL 3
BASIC PROTOCOL TITLE: Miniscope mounting on the head of a mouse
After the mouse recovers from GRIN lens implantation for 3-4 weeks, a miniscope can be mounted to the head of the mouse following careful optical alignment. Our miniscopes rest on a threaded cylinder, and are reversibly screw-mounted to the head of the mouse via a compatible, threaded base that must be fixed irreversibly onto the head with dental cement. The optical path of the screw-mounted miniscope is aligned with the GRIN lens prior to cementing this base in place. After mounting, the miniscope can be disconnected by unscrewing from the base, and reconnected for imaging prior to subsequent tests.
Materials
Mounting bottom component of Miniscope
Three weeks after surgical GRIN lens implantation, the miniscope base is mounted above the previously implanted GRIN lens for further in vivo imaging. This procedure is not a surgical procedure, but cleanliness remains important for good imaging.
Two potential problems should be kept in mind: 1) risk of accidental death from anesthesia, and 2) trauma during removal of protective cap from cranial platform. The former is addressed by the use of isoflurane, a readily titrated-to-effect anesthetic. The latter is addressed by avoiding rigid, stereotaxic apparatus restraint and control of force during cap removal.
Equipment:
Miniscope (Github), miniscope base (Github), set screws, #00-90 hex nut, PIFE tape (Taegatech, 131N MH7532), dissection microscope, stereotax, goose-neck lamp (AmScope, LED-50W), computer with custom-designed NeuView software (Github), control box and associated cables (Github), screw driver (Moody tools, 2089), thumb forceps (2), air duster (THEMCCONNELLGROUP,INC), 1.2mm-diameter drill bit, Micro Spatula, cotton tip applicators. A dissection scope, stereotaxic device for mouse with nose cone for anesthesia, heat pad, oxygen tank, and isoflurane vaporizer are also needed.
Miniscope mounting station (Fig. 3A): The custom built miniscope mounting station has three motorized translation stages (Thorlabs, MTS50-Z8) for 3-dimension adjustment, combined with goniometer (Thorlabs, GN05) and continuous rotation stage (Thorlabs, CR1) to allow fine angle adjustment in all directions. The miniscope holder is mounted on the goniometer.
Figure 3.

(A) Miniscope mounting station has three motorized translation stages for 3-dimension adjustment, combined with continuous rotation to allow fine angle adjustment in all directions, the miniscope holder is mounted on the goniometer. (B) Left, parts for miniscope base assembly; Right, to prepare the bottom part, place the #00-90 hex nut in the slot of the base, then use a small dab of super glue to secure the nut in the slot
The procedures are performed within a clean benchtop area which is covered with fresh, absorbent paper and contains:
-
A)
The stereotaxic stage with a heating pad at its base to maintain 37°C mouse body temperature and with an anesthesia nose cone;
-
B)
Clean anesthesia induction chamber.
Protocol steps
-
1)
Place the mouse within an induction chamber and introduce isoflurane (3% in oxygen at 1 liter/min air flow) until the animal is non-ambulatory for > 1 minute and respiration rate drops to approximately 60 breaths per minute.
-
2)
Adjust the isoflurane to 2-3% and 0.4 liter/min and immediately transfer the mouse to the stereotaxic apparatus to reinstate anesthesia through the nose cone. Transfer between chamber and fixation in stereotaxic stage should take no more than 15 seconds to maintain anesthesia; leave animal’s head “lightly” secured on the stereotaxic apparatus until after removing the protective cap covering GRIN lens. Apply ophthalmic ointment to each eye. Maintain mouse body temperature at 37 °C using a temperature control system. Regularly perform toe pinch and observe the regularity of anesthesia to monitor depth of anesthesia.
-
3)
Remove the plastic protective cap from the dental cement. Use one pair of forceps to push the head’s dental cement downward and the other pair of forceps to pry and lift off the cap.
-
4)
Tighten the nose piece and ear bars to secure the mouse in the apparatus. Polish the super glue remnant off the dental cement on the head of the mouse, and clear debris with the air duster. Clean the GRIN lens with a wet cotton tip applicator immersed in lens detergent.
-
5)Prepare the miniscope and base.
- To prepare the base, place the #00-90 hex nut in the slot of the base, then use a small dab of super glue to secure the nut in the slot (Fig. 3B).
- Cover the thread on the miniscope with PIFE tape, then attach the base onto miniscope, and use a lockin screw to secure the bottom part on the miniscope body.
- Connect the miniscope to control box with cable, open NeuView software, and start image streaming from the miniscope sensor.
-
6)Gently but firmly clamp the miniscope into a holder and manipulate its position above the GRIN lens until the correct focus is found. With the brain surface in focus (visible through the data acquisition software display), fix the miniscope base on the head of the mouse with dental cement.
- First, align the bottom of the miniscope (screwed into the detachable base) with the GRIN lens by eyes, visually checking that the miniscope and GRIN lens are parallel when viewed from any side. Further adjustment is done according to the streaming live image.
- Adjust the motor to slowly bring the miniscope bottom/base closer to the GRIN lens surface. At the same time, check the signal on the computer screen to find the best focus. Because dental cement will shrink slightly when cured, it’s best to lift the miniscope slightly (~50μm) after finding the best focal position, and before fixing the miniscope base to the head of the mouse with dental cement (mixed with carbon). Care should be taken not to cement the miniscope body to its detachable base. When finished, briefly power on the miniscope LED to check by eye for leakage of light indicating gaps between miniscope base and the mouse head; seal any gaps with dental cement.
- Wait about 5min for the dental cement to cure and securely fix the base to the head of the mouse.
-
7)
Remove the mouse from the stereotaxic stage, turn off the isoflurane, and let the mouse recover in the home cage while still wearing the miniscope.
-
8)
Verify live-streaming images after the mouse awakens. Adjust the scope height subtly to obtain the best focus by raising or lowering the miniscope inside its threaded base, and note the ideal angle for each scope and mouse. The set screw is used to tighten this configuration for imaging.
-
9)
Once you are finished, breifly anesthetize the mouse again in the induction chamber, quickly detach the miniscope body from the base, put on a protection cap, and tighten the set screw on the base so it will not come loose. Place the mouse into the home cage to monitor anesthesia recovery.
Miniscope mounting for daily in vivo imaging
Once the base for the miniScope is mounted on the head of the mouse, in vivo imaging can be performed daily after attachment of the miniscope body to the fixed base component.
The primary anticipated problems are risk of anesthesia and drying of eyes. These are addressed respectively by the use of isoflurane, a readily titrated-to-effect anesthetic, and by the application of Puralube ophthalmic ointment to mouse eyes quickly after anesthesia induction.
Materials
Miniscope, screw driver, PIFE tape, thumb forceps (2), air duster, cotton swabs, lens detergent (Fisherbrand, 22-143974), Puralube ophthalmic ointment , isoflurane inhalation chamber and oxygen tank.
Protocol steps
-
1)Anesthetize the mouse with isoflurane in the induction chamber (3% at 1 liter/min) until the mouse is non-ambulatory for 1 minute and breathing slows to approximately 60 breaths per minute. Quickly remove mouse from the chamber for manipulation, apply Puralube ophthalmic ointment to eyes, and return to chamber if chemical restraint is not adequate to complete these steps:
-
A)Loosen the set screw on the base with screwdriver, remove the protective cap and clear GRIN lens surface with air duster,
-
B)Gently clean the GRIN lens surface with cotton swab immersed with lens detergent.
-
C)Wrap screw threads on the bottom of the miniscope with PIFE tape, then screw into the base.
-
D)Connect the miniscope with cable, and open the software to adjust the focus.
-
E)Tighten the set screw with screwdriver to fix the miniscope on the head of the mouse and detach the cable.
-
A)
-
2)
Place the mouse into home cage to monitor anesthesia recovery. After 30 min recovery from isoflurane, the cable can be reconnected, and the mouse is ready for behavioral testing. See movie S1 and S2 for dorsal striatum and nucleus accumbens in vivo calcium imaging examples.
BASIC PROTOCOL 4
BASIC PROTOCOL TITLE: Data acquisition and analysis
In this protocol we discuss the data acquisition procedure and how to interpret and analyze the collected data. For data acquisition any computer running Windows (Vista or higher) or Mac (OS × 10.5 or higher) can be used, but for better performance at least 8GB of RAM is recommended.
All the image pre-processing, cell identification, calcium trace extraction and statistical analyses are performed in Matlab.
Materials
MiniScope, 5 V power source, USB type B cable, data acquisition hardware, computer (Windows or Mac) with NeuView software installed (github).
Data Acquisition System
The data acquisition system is based on a field-programmable gate array (FPGA; XEM3001v2, Opal Kelly), which controls the CMOS image sensor, the excitation LED and the communication and data transfer to the host computer. A custom control program (NeuView) is developed in C++ to communicate with the FPGA board and set parameters such as pixel integration time and LED power through a USB port. The software also allows the user to record time stamps for each frame and provides I/O 3.3 V TTL signals for synchronization with external behavioral devices such as cameras or other data acquisition systems.
Calcium Images Analysis
The recorded images are analyzed using custom MATLAB scripts.
Image Registration: A Fourier based phase correlation image registration algorithm (Kuglin, 1975) is applied to all background subtracted image sequences to correct for image translations due to mouse movement.
Neuron Identification: a gradient-based automatic cell detection algorithm (Lindeberg, 1998, Barbera, 2016), the Spatio-Temporal Gradient Matching (STGM) method, is used to iteratively generate a cell map of all active neurons in each session (Figure S1). First, average background for each session is subtracted from all frames. Then images are smoothed through convolution with Gaussian kernel of 3×3 pixels standard deviation. Next, the x and y gradients (Gx and Gy) of the smoothed image Ixy are independently calculated, and a threshold is applied to Gx and Gy as follows:
where the thresholds Tx and Ty are set to 4 times the root mean square (RMS) of Gx and Gy, respectively. Subsequently, a simple template matching algorithm is applied to detect all sequences of positive and negative gradient peaks (indicating the edges of in-focus neurons) within a maximum distance D = 30 μm (12 pixels). If a spatial (along x and y) and temporal (for 3 consecutive frames) match is detected, a 2×2 pixel mask is added as a new cell to the overall cell map of the current recording session. Finally, for each session, the cumulative cell map is segmented, and each cell’s location is calculated as the centroid of each segmented region.
Cell map registration across different days: neuron maps are manually registered across days using standard methods (Pinto and Dan, 2015; Ziv et al., 2013) to compensate for translations and rotation caused by repositioning of the microscope. An overall neural map is created by merging together all cell maps.
Calcium traces calculation: calcium traces are extracted for each identified neuron using the Annular Region Subtraction (ARS) method, according to the following procedure: first a region of interest (ROI) is assigned to each neuron as the circular region corresponding to the cell soma (diameter = 15 μm). Next the baseline fluorescence F0 is calculated as the average of the minimum pixel value for all pixels in the ROI during each recording session. The ROI fluorescence FROI is then calculated as the average of the background subtracted images over the ROI: FROI = mean(FRAW − F0)ROI, where FRAW is the raw pixel value. Next we adopted a commonly used method (Chen et al., 2013b; Kerlin et al., 2010; Pinto and Dan, 2015) to correct for the potential contamination from out-of-focus neurons or/and neuropil fluorescence, i.e. FSIG = FROI − γ FCON where FSIG is the true fluorescence signal, FCON is contamination fluorescence and is calculated as the minimum value of the background subtracted images over an annular region (d1 = 20 μm, d2 = 30 μm) surrounding the soma ROI (FCON = min(FRAW − F0)ANNULAR), and γ is the contamination factor, empirically estimated by the ratio of fluorescence in blood vessel region and fluorescence in the annular region. A binary representation of the neural activity is generated by applying a threshold of three times the root mean square (RMS) of each neuron’s baseline fluorescence. A calcium transient onset is defined as FSIG crossing the threshold of three times the RMS of the calcium trace baseline. For each calcium transient, the local maxima within a 3 s window from the onset is identified, and the amplitude and time decay constant can be estimated by fitting the transient decay data (3 s window from the local maxima) with an exponential function A(−t/τ0) , where A is the amplitude and τ0 is the time decay constant.
REAGENTS AND SOLUTIONS
Artificial cerebrospinal fluid (aCSF)
119 mM NaCl
26.2 mM NaHCO3
2.5 mM KCl
1 mM NaH2PO4
1.3 mM MgCl2
10 mM glucose
2.5 mM CaCl2.
Ketamine (diluted to 10mg/ml) and xylazine (diluted to 1.5 mg/ml) within one bottle
1% Agarose (type III, Sigma-Aldrich) in ddH2O (melted in microwave, maintain in 40 degree water bath for use)
COMMENTARY
Background Information
The proper choice of method for a given study will be determined by specific behavioral and biological questions, and a variety of methods will ultimately complement each other in unraveling neural mechanisms controlling animal behaviors.
The miniature microscope in vivo calcium imaging method has advantages and disadvantages compared with electrophysiological single-unit recording. Miniature microscopes enable large-scale neural activity to be recorded from specific, genetically-defined population(s) of neurons longitudinally at single-cell resolution. By visually establishing the location of studied neurons and registering activity at their spatial location across days, miniscopes provide more certainty in the longitudinal study of individual neurons than electrophysiological recordings. Moreover, the spatial relationships among neurons recorded by miniscope can be studied (Barbera et al. 2016), whereas spatial information is poor when electrically monitoring single units. However, limited by calcium indicators’ kinetics, calcium imaging methods cannot record high frequency signals. Single-unit recording has the advantage of high temporal resolution, and can record high frequency electrical signals, such as sharp-wave ripple events, which could not be faithfully resolved by existing calcium indicators. However, single-unit recording monitors a population of neurons with neuronal type presumed based on action potential characteristics and is therefore unable to distinguish neuronal types with similar firing characteristics, such as D1 neurons and D2 neurons in the striatum.
In addition to miniature epifluorescent microscopes, two other calcium imaging methods are available for in vivo neural activity recording: two-photon microscopy and fiber photometry. Two-photon microscopes can image several hundreds of micrometers into brain tissues, allowing 3D optical imaging of deep brain tissues via a GRIN lens. Two-photon microscopy is usually combined with head-fixed behavior. Recently, use of a miniature two-photon microscope is reported for mouse in vivo calcium imaging, though it has the limitation of an optical fiber that limits the choices of behavior test. Fiber photometry is a bulk-recording method that can be combined with freely-moving behavior, but it only records the integrated signals from a population of neurons around the fiber tip and has no cellular resolution. Therefore fiber photometry conveys much less detailed behaviorally relevant information.
Future miniature microscope development will generate even more powerful tools for neuroscience research. They may better reveal intrinsic neural activity by using faster neural activity indicators, such as voltage sensors. The action potential cycle usually lasts 1~2ms and can occur hundreds of times a second. GCaMP6f has a kinetic constant of 100ms which limits the recording of high frequency signals, especially for fast-spiking neurons. Voltage sensors have much faster response times; for instance, accelerated sensor of action potential 1 (St-Pierre et al., 2014) has a response constant of ~2 ms, which offers the potential for in vivo imaging of single AP. To captalize on high requency signals, a high frequency CMOS sensor (500~1000Hz) is also needed, which would produce a huge amount of data: 1min imaging at 500 Hz will produce 14.6 GB data of 16-bit, 512 × 512 pixels tiff image. To be compatible with freely-moving behavior, light weight miniature microscopes can be paired with ultra-lightweight cables or made wireless. Future miniscopes will also build on the ability to detect genetically identified neurons by distinguishing multiple fluorophores. The combined potential for high frequency recording from multiple identified populations longitudinally in freely-behaving animals makes the miniscope an excellent tool for neuroscience research.
Here we share our developments of the Spatio-Temporal Gradient Matching (STGM) cell identification algorithm and Annular Region Subtraction(ARS) method to detect individual active neurons and calcium transients for further analysis(Barbera et al., 2016). Our results suggest that ARS algorithm performance is comparable to the CNMF method and is better than PCA/ICA method. More importantly, unlike PCA/ICA and CNMFE methods, our STGM/ARS algorithm is suitable for real time implementation, comprising only Discrete Fourier Transforms and simple pixel operations and template matching algorithms. This may enable online neuronal pattern identification and closed-loop control through optogenetics or brain-machine interfaces.
Critical Parameters
The choice between direct implantation of a 0.5mm-diameter GRIN lens and implantation of a 1mm-diameter GRIN lens with tissue aspiration is based on the studied brain region and biological questions. For hippocampus CA1 imaging, implantation of GRIN lens with aspiration is advisable, since it would be difficult for a GRIN lens to be directly inserted through the overlying corpus callosum without damaging the CA1 region.
Due to toxicity of commercially available GRIN lenses, we developed a new method to coat toxic lenses commonly used in industry (1mm diameter, ILW-100-P0460-055-NC; 0.5mm diameter, ILW-050-P1460-055-NC; Go!Foton). We coated the GRIN lens with a 10-μm layer of optically clear parylene-C (VSI Parylene, Broomfield, Colorado). The estimated cost for coating is approximately $1 per GRIN lens. When compared with a non-toxic GRIN lens from Grintech, currently the leading GRIN lens choice for deep brain in vivo imaging, the coated lens has similar in vivo imaging performance.
In the protocol of direct implantation of 0.5mm-diameter GRIN lens to NAc, the 0.5mm-diameter Go!Foton GRIN lens (ILW-050-P1460-055-NC) is coated with parylene-C. The lens should be automatically coated with a layer of proteins when inserting through the brain tissue towards the target brain region.
We collected calcium data at the frame rate of 10 Hz, based on the fact that the GCaMP6 family has the temporal resolution for fluorescent response to a minimum inter spike interval of 50~150ms (Chen et al., 2013). The images are saved in tiff format with a 400×400 pixel image covering a 1.1 mm×1.1 mm field of view.
Troubleshooting
Injection of virus into dorsal striatum will cause scar tissue in the corpus callosum. If the virus injection tract is vertical (perpendicular to the striatum), scar tissue will be created directly above the target region. This will block the vacuum needle during subsequent brain tissue aspiration. Therefore, we choose to inject the virus starting at a point on the skull caudal to the target, and with the needle tip angled rostrally by 30 degrees to prevent scar tissue along the track of subsequent tissue aspiration. But in other brain regions, such as the prefrontal cortex, there is no significant risk of scar tissue formation.
For 1mm diameter GRIN lens implantation, removing brain tissue overlying the field of view using a robotic surgical instrument produces a cylindrical hole beneath the craniotomy window with a flat bottom, such that the tissue bottom matches lens bottom very well. This can significantly increase neuron number in the field of view compared with using a hand-held vacuum needle to aspirate brain tissue. Before placing the lens into the hole, care should be taken to ensure bleeding has stopped, as blood at the bottom of GRIN lens will increase scar tissue formation that interferes with imaging.
The most important thing during daily imaging is to ensure the focus between different days remains the same. To accomplish this, the focus of the miniscope is adjusted to obtain the best image on the first day of imaging, and the position of the miniscope main body is marked on the permanently affixed base. On all subsequent imaging days, the miniscope is repositioned on the same mark, and subtle adjustment of focus is further performed during image streaming and compared to the image acquired on day one. This ensures the focus and image angle remain the same. When the interval between different days is more than two weeks, there are notable changes, such as blood vessels shifting, in the field of view for some mice. In this situation, several landmarks in the image should be chosen and matched between any two subsequent imaging sessions.
In order to facilitate later analysis with behavior, it is important to synchronously trigger collection of calcium imaging and behavioral data, and to match the frame rates.
Statistical Analyses
Understanding Results
Raw data depicting calcium events are initially saved as a series of tiff images. Calcium events appear as small areas that suddenly exhibit a strong rise in pixel brightness (i.e. a burst of fluorescence), and then rapidly decay (movie S1). Analysis procedures are described in basic protocol 4. Quantitative calcium dynamics coupled with behavior recording and analysis should provide powerful insights into neural mechanisms underlying animal behaviors.
Time Considerations
It typically takes 1 hour to perform virus injection for one mouse. After virus injection, one should wait for 7-14 days to allow GCaMP6 to express. For direct implantation of a 0.5mm diameter GRIN lens, 1 hour is sufficient for the entire procedure. It will take longer for the 1mm diameter GRIN lens implantation, usually 2-3 hours for a surgery from start to finish depending on target depth.
GRIN lens implantation surgery will cause damage to the brain tissue, especially for the 1mm diameter GRIN lens implantation procedure during which tissue removal is necessary to make room for the GRIN lens. After surgery, we usually wait for 3-4 weeks to let the brain tissue recover from surgery before imaging. If behavior training is necessary before imaging, behavior training for mice could start one week after surgical recovery. With proper application of methods detailed within this protocol, the same population of neurons can be monitored for at least a month.
Supplementary Material
Movie S1 GCaMP6s fluorescence signal from dorsal striatum D2 neurons through the miniScope when mouse was in free locomotion.
Movie S2 GCaMP6f fluorescence signal from nucleus accumbens neurons through the miniScope when mouse was in free locomotion.
Significance Statement:
Neurons in the brain display electrical activities, through which they communicate with each other to control different kinds of behaviors. Activity of neurons can be monitored using a recently developed imaging technology, the miniature microscope (miniscope), that records calcium activity from hundreds of neurons simultaneously. Combined with an implanted GRIN lens that relays signals from deep brain regions to cranial surface, the miniscope can record neuronal activity from deep brain regions in freely behaving animals.
ACKNOWLEDGEMENT
(mandatory for NIH, optional for all others)
The work is supported by NIH NIDA IRP.
LITERATURE CITED
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, … Looger LL (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci, 32(40), 13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera G, Liang B, Zhang L, Gerfen CR, Culurciello E, Chen R, … Lin DT (2016). Spatially Compact Neural Clusters in the Dorsal Striatum Encode Locomotion Relevant Information. Neuron, 92(1), 202–213. doi: 10.1016/j.neuron.2016.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto RP, Messerschmidt B, & Schnitzer MJ (2009). In vivo fluorescence imaging with high-resolution microlenses. Nat Methods, 6(7), 511–512. doi: 10.1038/nmeth.1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, … Kim DS (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature, 499(7458), 295–300. doi: 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, & Webb WW (1990). Two-photon laser scanning fluorescence microscopy. Science, 248(4951), 73–76. [DOI] [PubMed] [Google Scholar]
- Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, & Schnitzer MJ (2011). Miniaturized integration of a fluorescence microscope. Nat Methods, 8(10), 871–878. doi: 10.1038/nmeth.1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C, & Konnerth A (2012). Imaging Calcium in Neurons. Neuron, 73(5), 862–885. doi: 10.1016/j.neuron.2012.02.011 [DOI] [PubMed] [Google Scholar]
- Hallett M, & Carbone E (1972). Studies of calcium influx into squid giant axons with aequorin. J Cell Physiol, 80(2), 219–226. doi: 10.1002/jcp.1040800208 [DOI] [PubMed] [Google Scholar]
- Helmchen F, Fee MS, Tank DW, & Denk W (2001). A miniature head-mounted two-photon microscope. high-resolution brain imaging in freely moving animals. Neuron, 31(6), 903–912. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, … Stuber GD (2015). Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell, 160(3), 516–527. doi: 10.1016/j.cell.2014.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama T, Sato TR, O’Connor DH, Zhang YX, Huber D, Hooks BM, … Svoboda K (2010). Learning-related fine-scale specificity imaged in motor cortex circuits of behaving mice. Nature, 464(7292), 1182–1186. doi: 10.1038/nature08897 [DOI] [PubMed] [Google Scholar]
- Li Y, Mathis A, Grewe BF, Osterhout JA, Ahanonu B, Schnitzer MJ, … Dulac C (2017). Neuronal Representation of Social Information in the Medial Amygdala of Awake Behaving Mice. Cell, 171(5), 1176–1190 e1117. doi: 10.1016/j.cell.2017.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WX, Hall G, Messerschmidt B, Li MJ, & Li XD (2017). Nonlinear optical endomicroscopy for label-free functional histology in vivo. Light-Science & Applications, 6. doi:ARTN e17082 10.1038/lsa.2017.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, & Nicholson C (1975). Calcium role in depolarization-secretion coupling: an aequorin study in squid giant synapse. Proc Natl Acad Sci U S A, 72(1), 187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, & Tsien RY (1997). Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature, 388(6645), 882–887. doi: 10.1038/42264 [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, & Imoto K (2001). A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol, 19(2), 137–141. doi: 10.1038/84397 [DOI] [PubMed] [Google Scholar]
- Pinto L, & Dan Y (2015). Cell-Type-Specific Activity in Prefrontal Cortex during Goal-Directed Behavior. Neuron, 87(2), 437–450. doi: 10.1016/j.neuron.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawinski J, Wallace DJ, Greenberg DS, Grossmann S, Denk W, & Kerr JN (2009). Visually evoked activity in cortical cells imaged in freely moving animals. Proc Natl Acad Sci U S A, 106(46), 19557–19562. doi: 10.1073/pnas.0903680106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre F, Marshall JD, Yang Y, Gong Y, Schnitzer MJ, & Lin MZ (2014). High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat Neurosci, 17(6), 884–889. doi: 10.1038/nn.3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinnakre J, & Tauc L (1973). Calcium influx in active Aplysia neurones detected by injected aequorin. Nat New Biol, 242(117), 113–115. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, … Looger LL (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods, 6(12), 875–881. doi: 10.1038/nmeth.1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, & Gan WB (2009). Stably maintained dendritic spines are associated with lifelong memories. Nature, 462(7275), 920–924. doi: 10.1038/nature08577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Burns LD, Cocker ED, Hamel EO, Ghosh KK, Kitch LJ, … Schnitzer MJ (2013). Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci, 16(3), 264–266. doi: 10.1038/nn.3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W, Wu R, Li M, Hu Y, Li Y, Li J, … Cheng H (2017). Fast high-resolution miniature two-photon microscopy for brain imaging in freely behaving mice. Nat Methods, 14(7), 713–719. doi: 10.1038/nmeth.4305 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1 GCaMP6s fluorescence signal from dorsal striatum D2 neurons through the miniScope when mouse was in free locomotion.
Movie S2 GCaMP6f fluorescence signal from nucleus accumbens neurons through the miniScope when mouse was in free locomotion.