

Abstract
B7-H3 is a tumor promoting glycoprotein that is expressed at low levels in most normal tissues but is overexpressed in various human cancers which is associated with disease progression and poor patient outcome. Although numerous publications have reported the correlation between B7-H3 and cancer progression in many types of cancers, mechanistic studies on how B7-H3 regulates cancer malignancy are rare, and the mechanisms underlying the role of B7-H3 in drug resistance are almost unknown. Here we report a novel finding that upregulation of B7-H3 increases the breast cancer stem cell population and promotes cancer development. Depletion of B7-H3 in breast cancer significantly inhibits the cancer stem cells. By immunoprecipitation and mass spectrometry, we found that B7-H3 is associated with the major vault protein (MVP) and activates MEK through MVP enhancing B-RAF and MEK interaction. B7-H3 expression increases stem cell population by binding to MVP which regulates the activation of the MAPK kinase pathway. Depletion of MVP blocks the activation of MEK induced by B7-H3 and dramatically inhibits B7-H3 induced stem cells. This study reports novel functions of B7-H3 in regulating breast cancer stem cell enrichment. The novel mechanism for B7-H3-induced stem cell propagation by regulating MVP/MEK signaling axis independent of the classic Ras pathway may have important implications in the development of strategies for overcoming cancer cell resistance to chemotherapy.
Introduction
Cancer metastasis, recurrence, and drug resistance are the main causes of poor patient survival. Tumors are a composite of several heterogeneous cancer cell types. There is a small population of cancer cells called stem cell-like cancer cells (cancer stem cells, CSC), which have stemness properties similar to normal stem cells, are considered to be responsible for tumor development, drug and radiation resistance, metastasis, and recurrence [1]. Conventional chemotherapeutic drugs may only kill general cancer cells but spare the cancer stem cell population and lead to tumor recurrence[2]. Recently, researchers have found that there is a cellular transition between cancer cells and cancer stem cells to keep the cell population equilibrium, and breast cancer stem cells can even arise de novo from non-stem cells[3]. It is urgent to develop more effective agents to target cancer stem cells, and a combination therapy using conventional anticancer drugs with CSC-targeting agents may offer a promising strategy for curing cancer.
B7-H3, also known as CD276, is a member of the B7 family proteins. There are different two isoforms, one has 4 Ig-like domains (4Ig-B7-H3), and the other has only 2 Ig-like domains (2Ig-B7-H3). The predominant isoform in human tissue is 4Ig-B7-H3 while mice only have 2Ig-B7-H3 [4]. Deficiency of B7-H3 in mice leads to autoimmune disease [5]. The immunological function of B7-H3 is still contradictory and unclear in different models [6, 7]. It has been reported that miR-29 and miR-187 targeted the 3’UTR of B7-H3 and was correlated with better patient survival [8, 9]. Recently, B7-H3 is reported to be overexpressed in many types of tumor tissues and correlated with worse patient survival [10, 11].
The major vault protein (MVP) is a vault protein that is the largest intracellular ribonucleoprotein particle involved in RNA transportation. The function of MVP is still unclear. Recently, MVP is considered as a scaffold protein by binding to the C2 domain of PTEN in a Ca2+ dependent manner [12]. Several proteins have been reported to interact with MVP including the estrogen receptor, SHP2, COP1, Src, and inactive PERK, and MVP is dephosphorylated by the tyrosine phosphatase SHP-2as a substrate[13-16].It is also reported that MVP cooperates with Ras for EGF-induced Elk-1 activation, and the tyrosine phosphorylation of MVP is important for cell survival and proteins interaction [13]. MVP overexpression was related to insulin-like growth factor receptor-1 (IGF-1R) expression and patient survival [17].
In this study, we reveal that compared with general cancer cells, B7-H3 are overexpressed in the stem cell population. Overexpression of B7-H3 dramatically increased the cancer stem cell pool size through MEK activation. The correlation between B7-H3 and MEK activation was further confirmed in patient samples. Moreover, B7-H3 activated and increased the MEK/B-RAF complex by binding to MVP independently of the Ras mediated pathway. Deletion of the B7-H3 cytosolic domain dramatically decreased the interaction between MVP and B7-H3. Inhibition of MVP or MEK activation dramatically decreased the cancer stem cell population and cell invasiveness. Inhibition of MEK significantly re-sensitized B7-H3 overexpressing cancer cells to Taxol. Our findings elucidate a mechanism by which B7-H3 activates MEK to expand the stem cell population and drug resistance through B7H3-MVP interaction independent of the classical Ras mediated pathway revealing an important clinical implication for treatment of aggressive and drug resistance breast cancer by drug combination therapy.
Materials and Methods
Cells and cell culture
Human breast cancer cell lines MDA361, T47D, SKBR3, MDA-MB-468 (MDA-468), MCF-12A, and MCF-10A were purchased from American Type Culture Collection (ATCC). MDA361, T47D, MDA-468, and SKBR3 were cultured in DMEM/F-12 (Mediatech Inc.) supplemented with 10% FBS and penicillin/streptomycin. HMLE (kindly provided by Dr. R. A. Weinberg) cell lines were cultured in 1:1 Dulbecco’s Modified Eagle’s Medium (DMEM)/Ham’s F-12 medium (Mediatech Inc.) supplemented with 5% FBS (Clontech), 100 units/ml penicillin-streptomycin (Invitrogen), 2 mml-glutamine (Invitrogen), 10 ng/ml human epidermal growth factor (EGF) (Invitrogen), 0.5 μg/ml hydrocortisone (Sigma), and 10 μg/ml insulin (Sigma).MCF-12A and MCF-10Awere cultured in 1:1 Dulbecco’s Modified Eagle’s Medium (DMEM)/Ham’s F-12 medium (Mediatech Inc.) supplemented with 5% Horse serum (Clontech), 100 units/ml penicillin-streptomycin, 10 ng/ml human epidermal growth factor (EGF), 0.5 μg/ml hydrocortisone, and 10 μg/ml insulin .
Mammosphere formation assay
Cells were plated in ultra-low attachment 96-well plates (Corning) at different densities per well and grown in DMEM/F12 medium (serum-free) with 20 ng/ml EGF,0.4%BSA, and 20 ng/ml bFGF. 50 μl of fresh media was added every 3 days. Mammospheres were counted at 3–5 days after seeding and pictures were taken. Mammosphere (diameter >75 μm) number was counted.
3D growth assay and immunofluorescence staining
Seventy microliters of Cultrex 3-D Culture Matrix reduced growth factor basement membrane extract (R&D Systems) were added to triplicate wells of eight Chamber Polystyrene Vessels (BD Falcon). The vessels were placed in a 37°C 5% CO2 incubator for 30 min to allow the matrix to solidify. Cells (4000 per well) stably expressing vector control plasmid or B7-H3 overexpression plasmid were then seeded and allowed to grow for 8 days. The media was replaced with fresh growth media after 4 days. Acini ofMCF10A-Vector and MCF-10A-B7H3 were further fixed using a 1:1 mixture of methanol:acetone at −20°C for 10–12min. Acini were blocked with 2% FBS in PBS for 2 hours, then washed with PBS for 2 times. Acini were stain with anti-E-cadherin(Cell signaling) at 4°C overninght, and washed with PBS for 3 times. The Acini were stained with rhodamine-conjugated secondary antibody for 1–2 hr at room temperature in the dark, and the washed with PBS for 5 times. Slides were mounted with Prolong® Gold Antifade Reagent with DAPI.
Immunohistochemistry
Breast TMAs were purchased from US Biomax and Novus Biologicals and were immunohistochemically stained for p-MEK (Cell Signaling) and B7-H3(Santa Cruz) using the streptavidin–biotin complex method from an anti-rabbit (or mouse) HRP-DAB Cell and Tissue Culture Staining Kit (R&D Systems). A semi-quantitative scoring criterion was used in which both the staining intensity and number of positive areas were recorded. A final staining index (scores ranging from 0 to 9) was used to quantify the total positive staining. To achieve the final staining index, the intensity of the positive staining (scores: negative=0, weak=1, moderate=2, strong=3) was scored. Then, the number of positive-stained cells (scores: <10%=1, 10–50%=2, >50%=3) was calculated. Finally, the intensity of staining (1–3) was multiplied by the score for the number of positively stained cells (1–3) to achieve the final staining scores (1–9). The final scores were regarded as follows: 0=negative, 1=weak positive (score of 1–2), 2=moderate positive (score of 3–5), and 3=strong positive (score of 6–9).
Animal study
All animals were maintained and all procedures performed according to a protocol approved by the Institutional Animal Care and Use Committee at the University of South Alabama. Six-week-old female athymic nude mice were used in the experiments. Different concentration of cancer cells suspended in 200 ml of PBS and Matrigel mixture (1:1) were injected into the mammary fat pads of each mouse. One group of mice were treated with U0126 (50mg/kg) for three weeks. All mice were scarified at 100 days, and the tumors were taken out.
Flow Cytometry Analysis
Cells (1 × 106) were incubated with CD44-APC and CD24-PE (BD Biosciences) conjugated antibodies and placed on ice for 45 min then washed with blocking buffer (PBS with 0.1% Na2N3, 1%FBS). CD44/CD24 markers were analyzed using a FACSCanto Flow Cytometer (BD Biosciences).
Cell sorting
Parental HMLE cells (1 × 107) were in incubated with CD44-APC and CD24-PE (BD Biosciences)-conjugated antibodies and placed on ice for 45 min then washed with blocking buffer (PBS with 1% FBS) twice. HMLE CD24highCD44high and CD24lowCD44high cells were sorted from parental HMLE by FACSAria sorter (BD Biosciences) for further experiments. After sorting, the purity of CD24highCD44low and CD24lowCD44high population was analyzed by Flow Cytometry (99% and 95%, respectively).
Vector Construction and Establishment of Stable Cell Lines
Full length human B7-H3 was cloned into pMSCVpuro expression vector (Clontech). All cloned fragments were verified by sequencing. Infectious and replication-incompetent retroviral particles were produced according to the manufacturer’s protocol. MCF-10A, MCF-12A, SKBR3, MDA468, MDA361, T47D, and HMLE cells were infected with retroviral particles expressing B7-H3 or the puromycin resistance gene only (vector control). After retroviral infection and primary puromycin selection at 1μg/ml, cell pools were maintained for multiple culture passages under puromycin concentration (1ug/ml). Plasmids were transfected with Lipofactamine 2000 (Invitrogen) according to manufacturer’s protocol. LentiCrispr V2 vector was purchased from addgene. B7-H3 Crispr vector was constructed as per the protocol from Addgene. B7H3 SgRNA-1F: CACCGGTCCCTGAAGACCCAGTGG; B7H3 SgRNA-1R: AAACCCACTGGGTCTTCAGGGACC; B7H3 SgRNA-2F: CACCGACCCAGTGGTGGCACTGGT; B7H3 SgRNA-2R: AAACACCAGTGCC -ACCACTGGGTC. shRNA Vectors (pLKO.1) and MVP shRNA were kindly provided by the MCI Gene Expression, Engineering and Discovery (GEED) Facility. MVP shRNA targeted sequence is CCCATCAACCTCTTCAACACA.
siRNA Experiments
siRNA oligonucleotides for MVP, B7-H3, and MEK1/2 were purchased from Sigma with a scrambled siRNA (Sigma) serving as a control. Transfection was performed using Lipofectamine 2000 Transfection Reagent (Invitrogen) according to the manufacturer’s protocol. Forty-eight hrs after transfection, the cell lysates were prepared for further analysis by Western blotting.
Western blotting
Cells were harvested and lysed in NETN (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP40) for 10 min on ice. Lysates were cleared by centrifugation at 13,200 rpm at 4°C for 10 min. Supernatants were collected and protein concentrations were determined by the Bradford assay (Bio-Rad). The proteins were then separated with a SDS/polyacrylamide gel and transferred to a Nitrocellulose membrane (Bio-Rad). After blocking in TBS with 5% BSA (Sigma) for 1 hr, the membranes were incubated overnight at 4-8°C with the primary antibodies in TBS containing 1% BSA. The following antibodies were utilized: the β-actin antibody was purchased from Sigma; and the tubulin, B7H3, B-Raf, MEK and MVP antibody were purchased from Santa Cruz; MEK-P and Phospho-Tyrosine was purchased from cell signaling. Membranes were extensively washed with TBST and incubated with horseradish peroxidase conjugated secondary anti-mouse antibody or anti-rabbit antibody (dilution 1:2,500, Bio-Rad). After additional washes with TBST, antigen-antibody complexes were visualized with the enhanced chemiluminescence kit (Pierce).
MEK inhibition
Cells were plated in 6-well plates and treated with U0126 (70nM, Sigma) for 24hours. Cells (1 × 106) were incubated with CD44-APC and CD24-PE (BD Biosciences) conjugated antibodies and placed on ice for 45 min then washed with blocking buffer (PBS with 0.1% Na2N3, 1%FBS). CD44/CD24 markers were analyzed using a FACSCanto Flow Cytometer (BD Biosciences).
Ras activity assay
Ras activity assay (EMD Millipore) was performed according to the manufacture’s protocol with negative and positive controls.
Immunoprecipitation
Cells were harvested, washed with PBS, and lysed in TNN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.5% NP40) containing Halt protease and phosphatase inhibitor cocktail for 10 min on ice. Cell lysates (800-1000ug) were incubated with the respective antibodies and protein A/G plus agarose at 4°C for 3-5 hours. The immune complex were isolated via centrifugation and washed with TNN buffer four times. The proteins were then separated with a SDS/polyacrylamide gel.
Cell Viability Assay
A total of 8 × 103 cells/well were seeded in 96-well plates. Twenty-four hours later, the medium was replaced with fresh medium with or without Taxol (or Taxol plus U0126) and incubated for 48 h. Cell images were taken by microscope.
SmartFlare assay
SmartFlare assay was performed according to the manufacture’s protocol. Briefly, tumor tissues were minced into 1–2mm pieces and subjected to a combination of mechanical and enzymatic digestion. Digested tissue was filtered and resuspended in complete medium consisting of DMEM/F12 with 20% FBS. Single cell suspensions from fresh tumors were distributed into ultra-low adherent 6-well plates at a concentration of 1×106 cells per well.3μl of SmartFlare probe (EMDMillipore) prediluted into 50ul of PBS were added to the following wells:scramble-Cy5 background control probe, mRNA probe specific for Nanog-Cy5 and Sox2-Cy5.Cells were incubated overnight. After incubation, cells were analyzed by using a BDBiosciences FACSCantoII.
Statistical analyses
Statistical evaluation for data analysis was determined by Unpaired Student’s t-test unless otherwise indicated. All data are shown as the mean±standard error (S.E.M.). All experiments were performed with at least three independent replicates, and the data shown represent all available data. A P-value<0.05 is indicated by *, P<0.01 by **, P<0.001 by ***, and P<0.0001 by ****.
Results
Overexpression of B7-H3 enriches breast stem cells
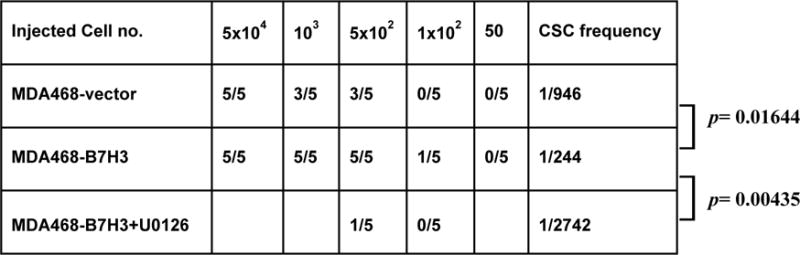
Overexpression of B7-H3 has been found in many types of cancer. However, the function of B7-H3 in tumor cells is still not completely understood. We previously reported that overexpression of B7-H3 confers cancer cell resistance to Taxol and reprograms glucose metabolism in cancer cells [18, 19]. It is well known that cancer stem cells are involved in drug resistance, metastasis, and relapse of cancers. Since cancer stem cells and drug resistance have a close correlation, we surmise that B7-H3 overexpressing cancer cells may induce drug resistance by enriching cancer stem cells. To examine the expression of B7-H3 in cancer stem cells, HMLE-CD24lowCD44high (cancer stem cell population) and HMLE-CD24highCD44low (non-stem cell population) cells were sorted from parental HMLE cells[20]. Compared with HMLE CD24highCD44lowcells, HMLE CD24lowCD44highcells showed a higher expression of B7-H3 (Figure. 1A). This result hints B7-H3 may play a role in maintaining the cancer stem cell population. To determine whether B7-H3 enhances the stem cell population, we exogenously expressed B7-H3 in HMLE, MCF-10A, MDA468, T47D, MA361, and SKBR3 cell lines. Compared with cells transfected with the vector control, B7-H3 overexpressing cells dramatically increased stem cell populations (CD24lowCD44high) (Figure 1B and Supplementary Figure 1A and B). To further confirm whether B7-H3 plays a role in regulating the stem cell population, we knocked down (or knocked out) the expression of B7-H3 in B7-H3 high-expressing MCF-10AT and HMLE CD24lowCD44high (stem cell population) cells by using two shRNA or two B7-H3 gRNAs via the CRISPR/cas9 system. The efficiency of knockdown or knockout was confirmed by western blot. Knockdown of B7-H3 in MCF-10AT dramatically decreased the stem cell population (CD24lowCD44high) (Figure 1C and Supplementary Figure 1C). Meanwhile, depletion of B7-H3 in HMLE CD24lowCD44highcells (stem cell population) significantly increased the non-stem cell population (Figure 1D and Supplementary Figure 1D). It is well known that cancer stem cells form mammospheres efficiently in comparison with non-stem cells [20]. To determine whether B7-H3 enhances mammosphere formation in breast cells, we seeded different numbers of HMLE-vector, HMLE-B7H3, SKBR3-vector, SKBR3-B7H3, MCF-10A vector, and MCF-10A-B7H3 cells in 96-well plates. Compared with the vector transfected cells, overexpression of B7-H3 dramatically increased mammosphere formation in SKBR3-B7H3, HMLE-B7H3, and MCF-10A-B7H3 cells (Figure 1E). These results support that B7-H3 is required for maintaining cancer stem cells. In vivo limited dilution assay is considered a gold standard for cancer stem cells [21]. In order to further test whether overexpressing B7-H3 in cells succeeded in altering the tumor-initiating frequency, we injected MDA468-vector and MDA468-B7H3 cells into immune-deficient nude mice. Similar to the in vitro mammosphere formation assay results, the mice injected with 102 MDA468-B7H3 cells formed a tumor while no tumor arose in mice with an equal number of cells expressing a control vector. While all the mice injected with 5×102MDA468-B7H3 cells formed tumors (5/5) but there are only 3 mice bearing tumors (3/5) with an equal number of cells expressing a control vector. The CSC frequency in MDA468-B7H3 (1/244) was dramatically increased in comparison with that of MDA468-vector (1/946) (Table 1). To further confirm that B7-H3 has an important function in maintaining the stem cell property in vivo, xenografted tumors were digested into single cells and stem cell markers (Smart Flare sox2 and nanog) were labeled and flow cytometry was performed to detect these markers. Compared to cells from MDA468-vector tumors, cells from MDA468-B7H3 tumors had significantly increased expression levels of sox2 and nanog (Supplementary Figure 2). All of the above data demonstrated that B7-H3 increased cancer stem cells in tumors.
Fig. 1. B7-H3 increases breast stem cells.
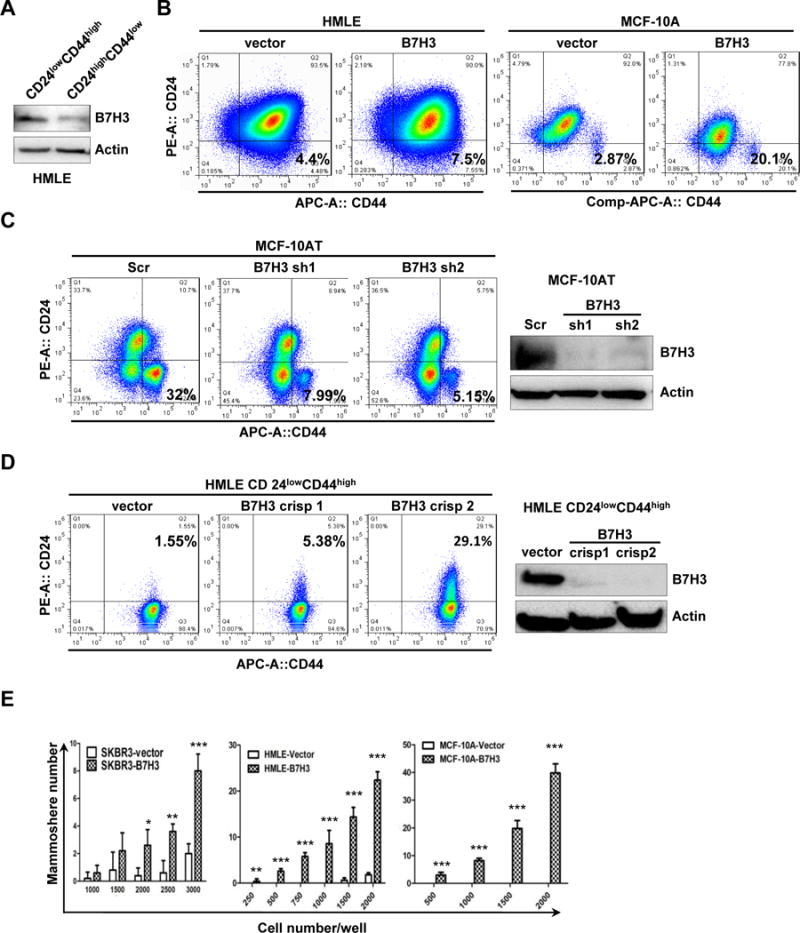
A. HMLE-CD24lowCD44high and HMLE-CD24highCD44low cells were sorted form parental HMLE cells. Cell lysates were prepared for Western blotting with an antibody against B7-H3, and β-actin was used as a loading control. B-D. 1 × 106 HMLE-vector, HMLE-B7H3, MCF-10A-vector, MCF-10A-B7H3, MCF-10AT-Scr, MCF-10AT-sh1, MCF-10AT-sh2, HMLE-CD24lowCD44high vector, HMLE-CD24lowCD44high crispr B7H3-1, and HMLE- CD24lowCD44high crispr B7H3-2 stable cells were incubated with CD24 and CD44 antibodies for 45 min, washed, and then analyzed by flow cytometry. E. SKBR3-vector, SKBR3-B7H3, HMLE-vector, HMLE-B7H3, MCF-10A-vector, and MCF-10A-B7H3 cells were seeded in ultralow attachment 96-well plates at different cell numbers with conditioned medium. After 5–10 days incubation, the mammosphere (diameter >75 μm) number was counted. X-axis represents the number of seeded cells per well. Columns represent the mean of three independent experiments; Bars represent S.E. **, p< 0.01; ***, p< 0.001.
Table 1.
B7-H3 overexpression increases breast cancer stem cells in vivo.
|
Cancer stem cell frequency was determined using extreme limiting dilution analysis (ELDA). n=5/group
B7-H3 overexpression disrupts cell polarity and acinar structure with increased invasion potential
Cell polarity is very important for cancer metastasis, tumor initiation, and invasiveness [22-26]. First, we used the 3D culture model system to study the acinar structure of breast cancer stem cells and non-stem cells. HMLE CD24lowCD44high (stem cell population) and HMLE CD24highCD44low (non-stem cell population) cells were seeded into Matrigel for 3D culture. The HMLE CD24highCD44low (non-stem cell population) cells formed typical acinar structures while HMLE CD24low CD44high (stem cell population) cells failed to form acinar structures and showed many invasive protrusions which indicate that the stem cells lost cell polarity (Figure 2A). To test the role of B7-H3 in cell polarity, HMLE-vector, HMLE-B7H3, MDA468-vector, MDA468-B7H3, MCF-12A-vector, and MCF-12A-B7H3 cells were seeded in Matrigel separately. Compared to cells with typical acinar structure, B7-H3 overexpressing cells formed irregular shaped structure with invasive protrusions (Figure 2B). To further confirm the function of B7-H3 in cellular invasive potential, we knocked out the expression of B7H3 by the CRISPR/cas9 technique in HMLE CD24lowCD44high cells. HMLE CD24lowCD44high with depletion of B7-H3 by CRISPR/cas9 rescued the acinar structure and inhibited the cell invasiveness (Figure 2C). Overexpression of B7-H3 also dramatically disrupted the expression of E-Cadherin in MCF-10A cells in 3D culture (Figure 2D). This result demonstrates that B7-H3 plays an important role in breast acinar structure formation, likely through controlling cell polarity.
Fig. 2. B7-H3 overexpression disrupts polarity and acinar structure with increased invasion potential.
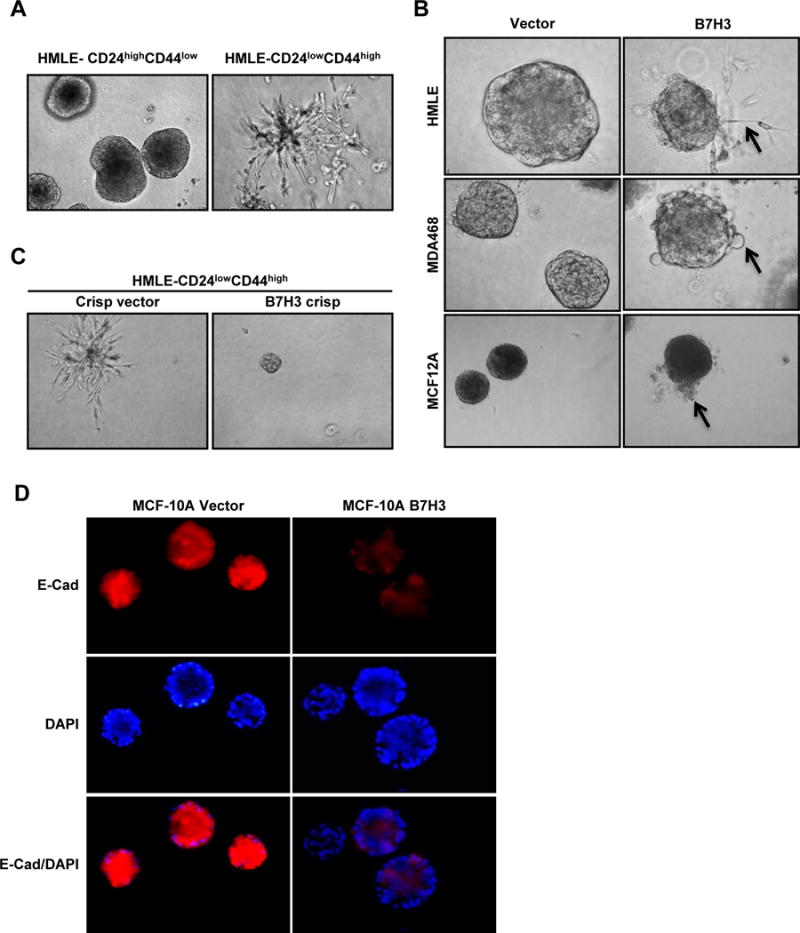
A-C. Cells were seeded in 3D Matrigel culture and grown for 8 days. Phase-contrast images show acinar morphology ofHMLECD24highCD44low, HMLECD24lowCD44high, HMLE-vector, HMLE-B7H3, MDA468-vector, MDA468-B7H3, MCF-12A-vector, MCF-12A-B7H3, HMLECD24lowCD44high crispr vector, and HMLECD24lowCD44high B7H3crispr cells. D. Cells were seeded in 3D Matrigel culture and grown for 8 days. Cells were stained with epithelial marker (E-cadherin, red) and DAPI (blue).
B7-H3 regulates breast cell stemness and acinar structure via MEK activation
It has been reported that MEK activation is required to maintain cancer stem cells [27]. To test whether overexpression of B7-H3 activates MEK, cell lysates of MDA468-vector, MDA468-B7H3, MCF-10A-vector, MCF-10A-B7H3, HMLE-vector, HMLE-B7H3, SKBR3-vector, and SKBR3-B7H3 cells were analyzed by immunoblotting. Compared to cells with the vector control, B7-H3 overexpressing cells dramatically increased the phosphorylation of MEK which is an indicator of MEK activation, and slightly increased total MEK (Figure 3A). To confirm this result, we knocked down the expression of B7-H3 in MCF-10AT cells with two different shRNAs. Depletion of B7-H3 decreased the expression of MEK phosphorylation in both shRNA-transfected cells (Figure 3B). To further confirm the relationship between B7-H3 and MEK activation in patients, immunohistochemistry was performed on tissue microarrays. The tissue microarrays were stained with B7-H3 and MEK phosphorylation specific antibodies. The expression level of B7-H3 had a positive correlation(R=0.4368) with MEK phosphorylation (Figure 3C and 3D, Table 2). These results indicate that B7-H3 activates MEK in breast cancer cells. To test whether MEK activation is required to maintain cancer stem cells in our model, the sorted HMLE CD24lowCD44high cells were treated with a MEK inhibitor (U0126) and control (DMSO). Compared to the control, the CD24high CD44low population (non-stem cells) dramatically increased in the sorted HMLE CD24lowCD44highcells treated with MEK inhibitor (Figure 4A and 4C). The results showed that MEK activation is required to block the stem cell population from shifting to non-stem cells. Next, we tested whether MEK activation is required for B7-H3 induced-stem cell populations. MCF-10A-B7H3 cells were treated with the MEK inhibitor (U0126) and control (DMSO). Compared with control cells, MCF-10A-B7H3 cells treated with MEK inhibitor dramatically increased the non-stem cell population (CD24high CD44low) (76.4% vs 94.9%)while significantly decreased the stem cell population (CD24low CD44high) (20.4% vs 0.58%) (Figure 4B-C). To further confirm B7-H3 induces the stemness property of breast cells via MEK activation, we knocked down MEK in HMLE-B7H3 and MCF-10A-B7H3 cells with siRNA. Compared to cells treated with scrambled oligo siRNA, depletion of MEK by siRNA significantly decreased the stem cell population in both HMLE-B7H3 and MCF-10A-B7H3 cells (Figure 4D-F). To further confirm the role of MEK in B7H3 induced breast cancer stem cells, in vivo different cell numbers of MDA468-B7H3 cells were injected into nude mice and then treated with U0126 (MEK inhibitor) for three weeks. Compared with mice that were injected with MDA468-B7H3 cells only, MEK inhibition dramatically decreased the tumor formation in vivo, and the CSC frequency of MDA486-B7H3 with U0126 also dramatically decreased (Table 1, P=0.00435). These in vitro and in vivo results showed that MEK activation plays a key role in B7-H3 induced cancer stem cell and tumor formation. Since stem cells disrupted acinar structure, we tested whether inhibition of MEK restored the typical acinar structure. The sorted HMLE CD24lowCD44high cells were treated with MEK inhibitor (U0126) or control (DMSO) and then seeded into Matrigel for 3D culture. Compared to cells treated with the control, HMLE CD24lowCD44high cells treated with U0126 lost invasive growth (Figure 4G).
Fig. 3. B7-H3 regulates MEK activation in vitro and in vivo.
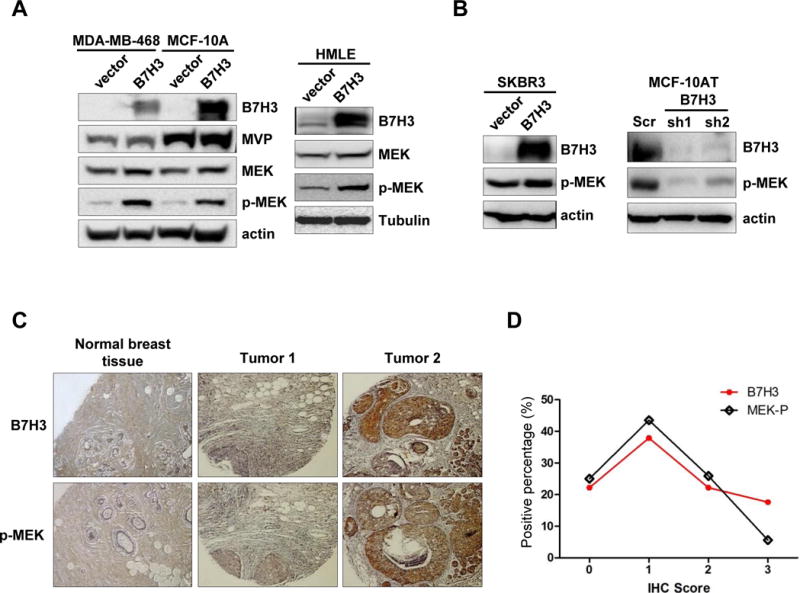
A-B. MDA468-vector, MDA468-B7H3, MCF-10A-vector, MCF-10-B7H3, HMLE-vector, HMLE-B7H3, SKB3-vector, SKBR3-B7H3, MCF-10AT-Scr, MCF-10AT-sh1(B7H3), and MCF-10AT-sh2 (B7H3) cells were collected. Cell lysates were prepared for Western blotting with an antibody against B7-H3, MVP, total MEK, MEK-p, and β-actin(or tubulin) was used as a loading control. C-D. Tissue microarrays were stained with B7-H3, MVP, and MEK-P antibodies. The intensity of the positive staining (scores: negative=0, weak=1, moderate=2, strong=3) was scored. Then, the number of positive-stained cells (scores: <10%=1, 10–50%=2,>50%=3) was calculated.
Table 2.
χ2 Analysis showing a positive correlation between expression of B7H3 and pMEK in patient samples.
| Score(IHC) | 0 | 1 | 2 | 3 | Statistic (Correlation) |
|---|---|---|---|---|---|
| B7H3 | 22.2%(24/108) | 37.9%(41/108) | 22.2%(24/108) | 17.6%(19/108) | R2= 0.4368 P<0.0001 |
| MEK-p | 25%(27/108) | 43.5%(47/108) | 25.9%(28/108) | 5.6%(6/108) |
Fig.4. MEK activation is required for B7-H3 induced breast stem cells and acinar disruption.
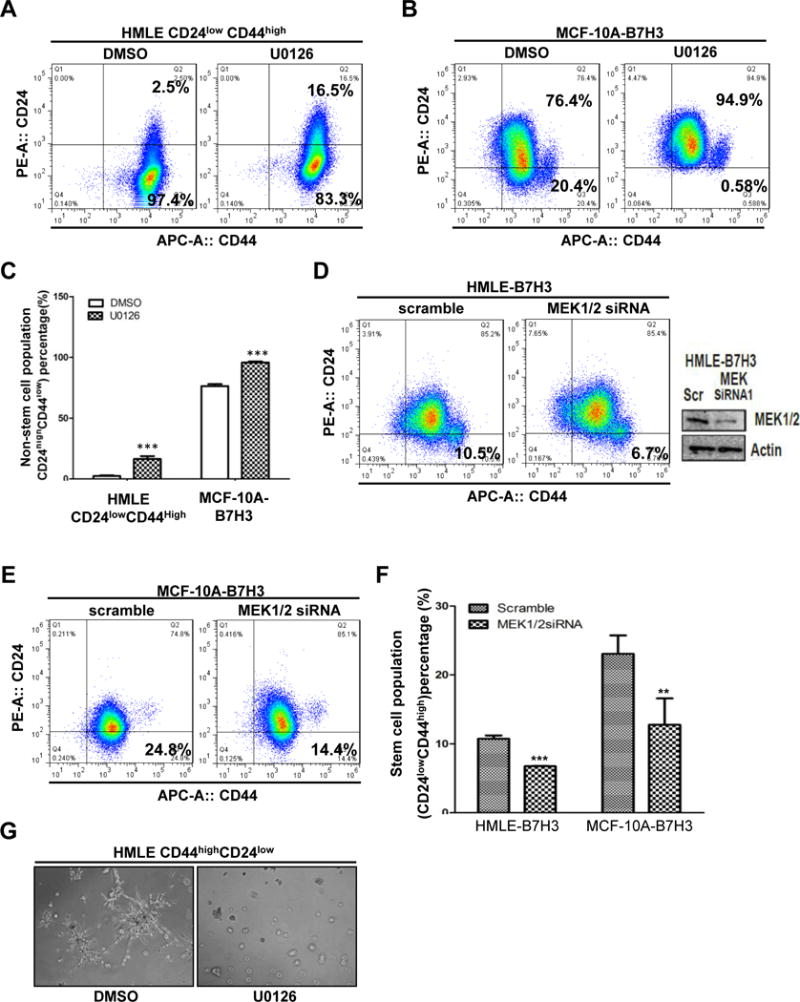
A-C. HMLECD24lowCD44high and MCF-10A-B7H3 were treated U0126 and DMSO(control) for 24 hours. Cells were incubated with CD24 and CD44 antibodies for 45 min, washed, and then analyzed by flow cytometry. D-E. HMLE-B7H3 and MCF-10A-B7H3 cells were transfected with scramble and MEK1/2 siRNA respectively. 48 hours later after transfection, cells were incubated with CD24 and CD44 antibodies for 45 min, washed, and then analyzed by flow cytometry. G. Cells were seeded in 3D Matrigel culture and treated with U0126 or DMSO as control. F. Phase-contrast images showing acinar morphology ofHMLECD24highCD44low with U0126 and DMSO.
B7-H3 regulates MEK activation and breast cell stemness through MVP
Since B7-H3 regulates the cancer stem cells and acinar structure via MEK activation, we further investigated how B7-H3 regulates MEK activity. It is well known that MEK was activated by the Raf-Ras signal pathway [28]. First, we hypothesized that overexpression of B7-H3 increased MEK phosphorylation by increasing the activity of Ras. To test this hypothesis, we examined Ras activation in B7-H3 high and low expressing cells. MDA468-vector, MDA468-B7H3, MCF-10AT-scr, MCF-10AT B7H3 shRNA1 andMCF-10AT B7H3 shRNA2 cells were collected, and whole cell lysates were subjected to pull down by GST-Raf1-RBD protein. We found B7-H3 did not affect Ras activity (Supplementary Figure 3A). Furthermore, by immunoprecipitation we also found there is no protein interaction between Ras and B7-H3 (Supplementary Figure 3B). Based on these results, we concluded that B7-H3 activates MEK independent of Ras activity. To further explore other alternative mechanisms involving MEK activation by B7-H3, immunoprecipitation with antibodies against B7-H3 was conducted using whole cell lysates extracted from MDA468-B7H3 cells to identify potential proteins that interact with B7-H3. An SDS–polyacrylamide gel electrophoresis (SDS–PAGE) was done, and the immunoprecipitated proteins were stained with sypro ruby gel stain. Visible protein bands were cut and extracted from the gel then subjected to mass spectrometry (Supplementary Figure 4).We analyzed and screened the massspectrometry results with a high score and more matched peptides. In our immunoprecipitated protein sample, we found one protein with a molecular weight of around 100 kDa named MVP (major vault protein) which was first described in non–small cell lung cancer cells involved in drug resistance and EGFR signaling[13, 29]. By IHC on tissue microarray with 108 patient samples, we found that the expression level of B7-H3 had no correlation with the expression of MVP (data not shown) in patient samples. This is consistent with our in vitro data that overexpression of B7-H3 did not change the expression level of MVP (Figure 3A). Since both B7-H3 and MVP are involved in drug resistance and MVP is involved in EGF activated ERK phosphorylation, we hypothesized that B7-H3 activates MEK via MVP. To test this, we examined whether B7-H3 associates with MVP. Immunoprecipitation using anti-B7H3 antibody followed by western blotting for MVP, and reverse immunoprecipitation using anti-MVP antibody followed by western blotting for B7-H3 were performed in MDA468-B7H3 cells. The results showed that both proteins can be detected by western blotting when whole cell lysates were immunoprecipitated with antibodies against the other protein (Figure 5A, B). It has been reported that EGF can enhance MVP-Src interaction[15]. The breast contains fat tissue, mainly composed of adipose cells which secret leptin. To explore whether the EGF and other potential factors such as leptin affect the interaction between B7-H3 and MVP, we treated MDA468-B7H3 with EGF or leptin. Surprisingly, we found that leptin, but not EGF enhanced the interaction between MVP and B7-H3 (Supplementary Figure 5). How leptin regulates the MVP and B7-H3 interaction needs to be further explored.
Fig. 5. B7-H3 regulates MEK activation through MVP.
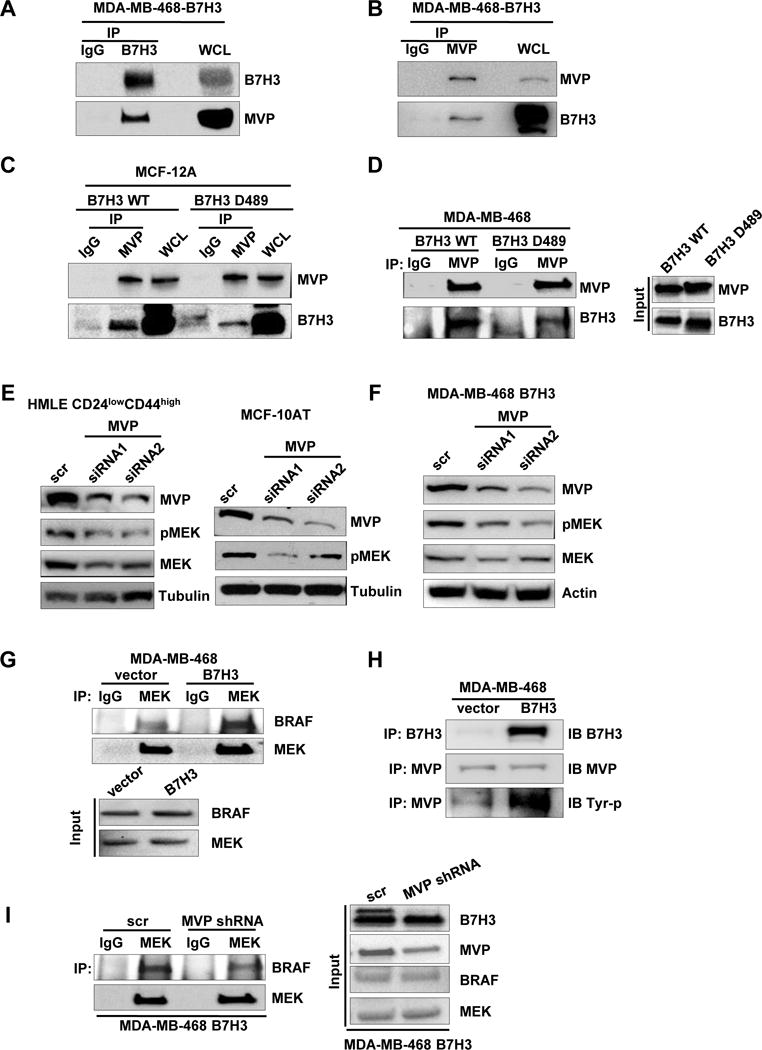
A-B. IP of B7-H3 (or MVP) and control mouse IgG followed by immunoblot analysis of MDA468-B7H3 with whole cell lysates as positive control. C-D. Immunoblot analysis of WCL or IP of MVP and control mouse IgG from lysates from MCF-12A-wt-B7H3, MCF-12A-B7H3-D489(cytosolic domain deletion), MDA468-wt-B7H3, MDA468-B7H3-D489(cytosolic domain deletion). E-F. Cells were transfected with scramble and MVP siRNAs respectively. 48 hours later after transfection, cell lysates were prepared for Western blotting with an antibody against B7-H3, MVP, total MEK, MEK-p, and β-actin(or tubulin) was used as a loading control. G. Immunoblot analysis of IP of MEK and control mouse IgG of lysates from MDA468-vector and MDA468-B7H3. H. Immunoblot analysis of IP of MVP (or B7-H3) and control mouse IgG of lysates from MDA468-vector and MDA468-B7H3. I. Immunoblot analysis of IP of MEK and control mouse IgG of lysates from MDA468-B7H3(scramble) and MDA468-B7H3(MVP shRNA).
To test how B7-H3 binds to MVP, we depleted the cytosolic domain in wild type B7-H3 (mutant named as B7H3-D489) and transfected it into MDA468 cells. Comparing with the wild type B7H3, B7H3-D489 markedly decreased the interaction between B7-H3 and MVP by immunoprecipitation assay (Figure 5C, D). The results showed that the cytosolic domain of B7-H3 plays a key role in B7-H3 interaction with MVP. To test whether MVP regulates the activation of MEK, we knocked down MVP in HMLE CD24lowCD44high and MCF-10AT cells with two different siRNAs. Knockdown of MVP significantly decreased MEK phosphorylation in both cell lines (Figure 5E). Since MVP regulates MEK activation, we tested whether B7-H3 activated MEK via MVP. We knocked down MVP with two different siRNAs in MDA468-B7H3 cells. Depletion of MVP significantly decreased MEK activation which was induced by B7-H3 in MDA468-B7H3 cells (Figure 5F). Based on these results, we concluded that B7-H3 activates MEK through MVP. It is still unknown how MVP regulates the activation of MEK. It is well known that B-RAF is a key protein kinase to phosphorylate MEK [30]. First, we determined whether B7-H3 regulates the interaction between B-RAF and MEK. The whole cell lysate of MDA468-vector and MDA468-B7H3 were immunoprecipitated by MEK antibody and then blotted by B-RAF antibody. Overexpression of B7-H3 dramatically increased the interaction between B-RAF and MEK (Figure 5G). Since B7-H3 regulates MEK activation via MVP, we then determined whether B7-H3 regulates the activation of MVP by affecting the tyrosine phosphorylation of MVP. The whole cell lysate of MDA468-vector and MDA468-B7H3 were immunoprecipitated using MVP antibody and then blotted by a tyrosine phosphorylation antibody. Overexpression of B7-H3 dramatically increased tyrosine phosphorylation of the MVP protein (Figure 5H). Next, we determined whether B7-H3 increased the interaction between B-RAF and MEK through MVP activation. We depleted MVP in MDA468-B7H3 cells by transfecting with MVP shRNA. Depletion of MVP in MDA468-B7H3 significantly decreased the interaction between B-RAF and MEK (Figure 5I). Taken together, we concluded that B7-H3 induced MEK activation by enhancing B-RAF and MEK interaction via MVP activation.
Since MEK activation is required for B7-H3 induced cell stemness, we next determined whether MVP is required for maintaining stem cells and cell polarity. First, we knocked down the expression of MVP in HMLE sorted stem cell population (CD24lowCD44high) with three specific siRNAs. Compared to cells with scramble siRNA, depletion of MVP significantly increased the non-stem cell population in HMLE sorted stem cells (3.1% vs. 14.6 %, 8.5 %, and 12.5%) (Figure 6A). The results showed that MVP is required for breast cells to maintain their stem cell property. To determine whether MVP is required for B7-H3 induced breast stem cells, we knocked down the expression of MVP in MCF-10A-B7H3 and HMLE-B7H3 cells using two different specific MVP siRNAs (Figure 6B). Compared to cells treated with scramble siRNA, depletion of MVP significantly decreased the CD24lowCD44high cell population in both MCF-10A-B7H3 and HMLE-B7H3. Since breast stem cells are required for cancer invasiveness and recurrence, we further tested the role of MVP in cell invasiveness with a 3D culture system. We knocked down MVP in HMLE sorted stem cell population (CD24lowCD44high) with two different specific siRNAs and scramble siRNA as control. Depletion of MVP completely inhibited the cellular invasion potential and restored the acinar formation in HMLE sorted stem cell population (CD24lowCD44high) while cells transfected with scramble siRNA had disrupted acinar structure (Figure 6C). This result showed MVP plays a major role in acinar formation and cell invasion. To determine whether MVP is required for B7-H3 induced acinar disruption, we knocked down the expression of MVP in HMLE-B7H3 with two different siRNAs. Compared to cells with scramble siRNA, HMLE-B7H3 cells with depletion of MVP restored acinar structure with a smooth surface (Figure 6D). Based on these data, we conclude that B7-H3 induced breast cancer stem cell and invasion occurs via MVP by inducing MEK activation.
Fig.6. MVP is required for B7-H3 induced breast stem cells and acinar disruption.
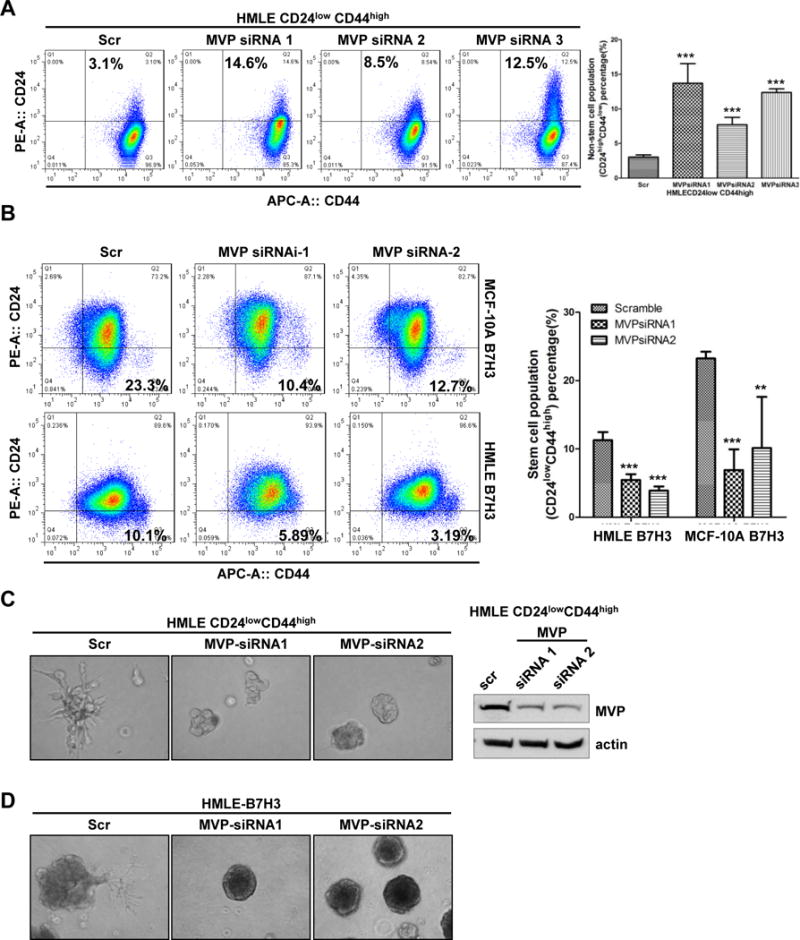
A-C. Cells were transfected with scramble and MVP siRNAs respectively. 48 hours later after transfection, cells were incubated with CD24 and CD44 antibodies for 45 min, washed, and then analyzed by flow cytometry.D. Cells were transfected with scramble and MVP siRNAs respectively. 24 hours later after transfection, cell were seeded in 3D Matrigel culture and grown for 8 days. Phase-contrast images show acinar morphology.
Discussion
Recently, more and more data showed that B7-H3 plays an important role in tumor progress, tumor metastasis, and patient survival[10, 11, 31-37]. B7-H3 was considered as a good marker for tumor prognosis [34, 38, 39]. Compared to normal tissue, breast tumor tissue has high B7-H3 positive rates (78.1% vs 36.8%). Moreover; most of the metastatic carcinoma have high positive rates of B7-H3 (82.8%). Our previous data showed B7-H3 regulated cancer drug resistance via Jak2/Stat3 signaling pathway[19]. It has been reported that Jak2 and Stat3 play an important role in cancer stem cells [40, 41]. Here we found that overexpressing B7-H3 increased breast stem cells. We first surmised that B7-H3 increased cancer stem cell population through Stat3/Jak2 activation. Unfortunately, we did not observe that the stem cell population was decreased when we knocked down Stat3 or Jak2 in B7-H3 overexpressing breast cancer cells (data not shown). These results hint that B7-H3 may increase the cancer stem cell population through another molecular pathway besides Stat3 or Jak2 signal transduction. So far, it is still unclear how B7-H3 regulates cancer stem cells, tumor development, and drug resistance. Understanding the mechanism of B7-H3 induced tumor progression and drug resistance will provide insight into developing a molecular biomarker and appropriate therapeutic strategies for B7-H3 overexpressing tumors.
It is well known that the Raf-MEK-ERK pathway is activated by Ras which is very important for tumor development and stem cell proliferation[42, 43]. We found that overexpressing B7-H3 increased BRAF-MEK interaction and subsequently MEK activation. We surmised that B7H3 activated BRAF-MEK through interaction with Ras signaling. Unfortunately, we found that B7-H3 has no interaction and does cause activation of Ras. This hints that B7-H3 activated MEK independent of Ras activation[44-46]. We found that B7-H3 interacts with MVP by immunoprecipitation and mass spectrometry. Deletion of the cytosolic domain in B7-H3 markedly decreased the interaction between B7-H3 and MVP. This result hinted that the cytosolic domain of B7-H3 is important but not the only site for MVP and B7-H3 interaction. Furthermore, depletion of MVP blocked B7H3 induced MEK activation and decreased B-Raf-MEK interaction. We have shown for the first time that MVP is important for B-Raf-MEK interaction and MEK activation. How MVP regulates B-Raf-MEK interaction needs to be further explored. Taken together, we have shown that B7-H3 regulated MEK-BRAF interaction and MEK activation via MVP activation independent of Ras activation.
Recent studies showed that MVP also promotes cancer progression and migration [47]. The function of MVP in cancer progression is still not completely understood. To understand the function of MVP in cancer stem cells, we knocked down MVP in HMLECD24lowCD44highcells. Depletion of MVP increased the non-stem population in sorted stem cell population. This hints MVP is required for maintaining stem cells. Furthermore, depletion of MVP also dramatically decreased B7-H3-induced breast cancer stem cell population and restored B7-H3-disrupted acinar structure. Furthermore, inhibition of MEK activity significantly also decreased B7-H3-induced breast cancer stem cells in vivo and in vitro, and re-sensitized B7-H3 overexpressing breast cancer cells to Taxol (Figure 7). We also found that MVP regulates Stat3, Jak2, Nanog and c-MYC (data not shown). This hints MVP maybe a key upstream regulator of Stat3, Jak2, Nanog, and c-MYC. Our new findings depict a clear molecular model of how B7-H3 can induce breast cancer stem cell and drug resistance.
Fig. 7. MEK inhibitor resensitized B7-H3 overexpressing breast cancer cells to Taxol.
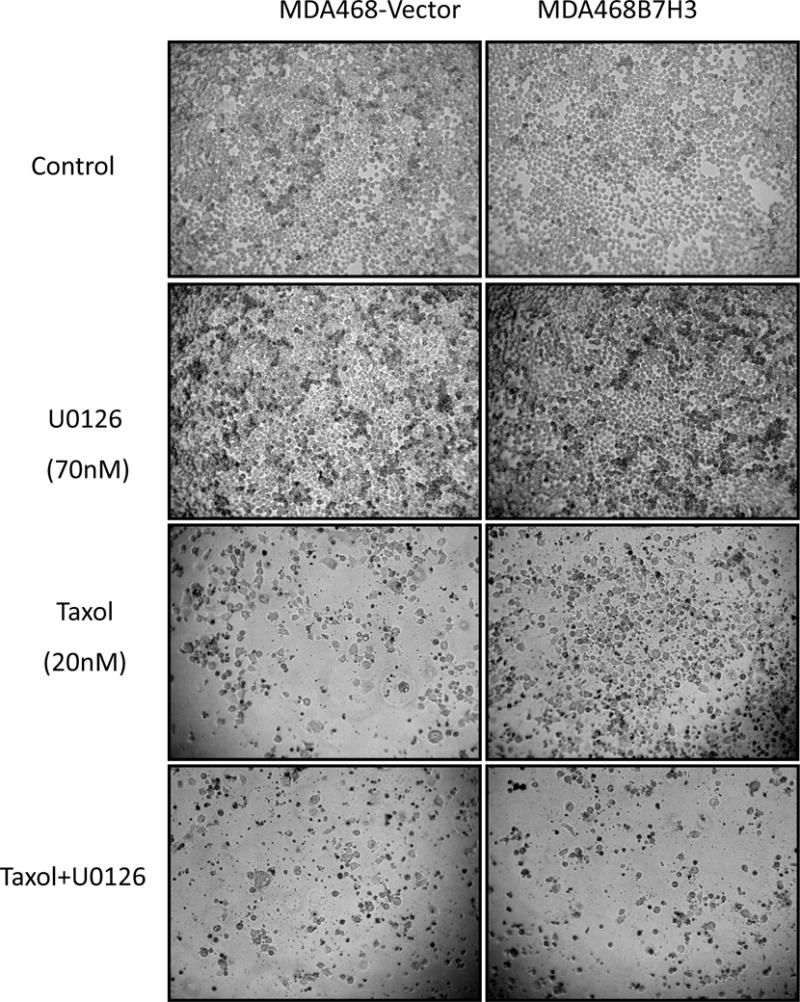
MDA468-vector and MDA468-B7H3 cells were seeded in 96 well plate at 8×103 cells/well. Cells were treated with medium, U0126, Taxol, and Taxol plus U0126 respectively for 48 hours. Cell images were taken by microscope.
In summary, we report thatB7-H3 enriches the cancer stem cell population and drug resistance via activating MEK through MVP, independent of classical the Ras-mediated pathway. We also demonstrate that the combination of Taxol and MEK inhibition overcomes B7-H3induced cancer drug resistance. This study provided important information for clinical study design of combined therapeutics targeting B7-H3 overexpressing breast cancers.
Supplementary Material
Acknowledgments
This study was supported by NIH grants R01CA149646 (M. Tan), the Vincent F. Kilborn, Jr. Cancer Research Foundation (M. Tan). NIH CA148629 to RWS. RWS is an Abraham A. Mitchell Distinguished Investigator.
Footnotes
The authors declare no potential conflicts of interest.
Author contributions
ZXL and MT conceived and designed this study. ZXL, WLZ, JBP, RA and SM performed experiments. JFL, SM and RWS provided reagents and advice. ZXL, MT and JHK analyzed the data and wrote the manuscript.
References
- 1.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine. 2012;7(4):597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 4.Vigdorovich V, Ramagopal UA, Lazar-Molnar E, Sylvestre E, Lee JS, Hofmeyer KA, et al. Structure and T cell inhibition properties of B7 family member, B7-H3. Structure. 2013;21(5):707–717. doi: 10.1016/j.str.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo L, Zhu G, Xu H, Yao S, Zhou G, Zhu Y, et al. Promotes Pathogenesis of Autoimmune Disease and Inflammation by Regulating the Activity of Different T Cell Subsets. PloS one. 2015;10(6):B7–H3. e0130126. doi: 10.1371/journal.pone.0130126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo L, Chapoval AI, Flies DB, Zhu G, Hirano F, Wang S, et al. B7-H3 enhances tumor immunity in vivo by costimulating rapid clonal expansion of antigen-specific CD8+ cytolytic T cells. Journal of immunology. 2004;173(9):5445–5450. doi: 10.4049/jimmunol.173.9.5445. [DOI] [PubMed] [Google Scholar]
- 7.Leitner J, Klauser C, Pickl WF, Stockl J, Majdic O, Bardet AF, et al. B7-H3 is a potent inhibitor of human T-cell activation: No evidence for B7-H3 and TREML2 interaction. European journal of immunology. 2009;39(7):1754–1764. doi: 10.1002/eji.200839028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang ZS, Zhong M, Bian YH, Mu YF, Qin SL, Yu MH, et al. MicroRNA-187 inhibits tumor growth and invasion by directly targeting CD276 in colorectal cancer. Oncotarget. 2016;7(28):44266–44276. doi: 10.18632/oncotarget.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Cheung IY, Guo HF, Cheung NK. MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: potential implications for immune based therapy of human solid tumors. Cancer research. 2009;69(15):6275–6281. doi: 10.1158/0008-5472.CAN-08-4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingebrigtsen VA, Boye K, Tekle C, Nesland JM, Flatmark K, Fodstad O. B7-H3 expression in colorectal cancer: nuclear localization strongly predicts poor outcome in colon cancer. International journal of cancer. 2012;131(11):2528–2536. doi: 10.1002/ijc.27566. [DOI] [PubMed] [Google Scholar]
- 11.Bin Z, Guangbo Z, Yan G, Huan Z, Desheng L, Xueguang Z. Overexpression of B7-H3 in CD133+ colorectal cancer cells is associated with cancer progression and survival in human patients. The Journal of surgical research. 2014;188(2):396–403. doi: 10.1016/j.jss.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 12.Minaguchi T, Waite KA, Eng C. Nuclear localization of PTEN is regulated by Ca(2+) through a tyrosil phosphorylation-independent conformational modification in major vault protein. Cancer research. 2006;66(24):11677–11682. doi: 10.1158/0008-5472.CAN-06-2240. [DOI] [PubMed] [Google Scholar]
- 13.Kolli S, Zito CI, Mossink MH, Wiemer EA, Bennett AM. The major vault protein is a novel substrate for the tyrosine phosphatase SHP-2 and scaffold protein in epidermal growth factor signaling. The Journal of biological chemistry. 2004;279(28):29374–29385. doi: 10.1074/jbc.M313955200. [DOI] [PubMed] [Google Scholar]
- 14.Yi C, Li S, Chen X, Wiemer EA, Wang J, Wei N, et al. Major vault protein, in concert with constitutively photomorphogenic 1, negatively regulates c-Jun-mediated activator protein 1 transcription in mammalian cells. Cancer research. 2005;65(13):5835–5840. doi: 10.1158/0008-5472.CAN-05-0423. [DOI] [PubMed] [Google Scholar]
- 15.Kim E, Lee S, Mian MF, Yun SU, Song M, Yi KS, et al. Crosstalk between Src and major vault protein in epidermal growth factor-dependent cell signalling. The FEBS journal. 2006;273(4):793–804. doi: 10.1111/j.1742-4658.2006.05112.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Neo SP, Gunaratne J, Poulsen A, Boping L, Ong EH, et al. Feedback regulation on PTEN/AKT pathway by the ER stress kinase PERK mediated by interaction with the Vault complex. Cellular signalling. 2015;27(3):436–442. doi: 10.1016/j.cellsig.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Henriquez-Hernandez LA, Moreno M, Rey A, Lloret M, Lara PC. MVP expression in the prediction of clinical outcome of locally advanced oral squamous cell carcinoma patients treated with radiotherapy. Radiation oncology. 2012;7:147. doi: 10.1186/1748-717X-7-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. Immunoregulatory Protein B7-H3 Reprograms Glucose Metabolism in Cancer Cells by ROS-Mediated Stabilization of HIF1alpha. Cancer research. 2016;76(8):2231–2242. doi: 10.1158/0008-5472.CAN-15-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H, Tekle C, Chen YW, Kristian A, Zhao Y, Zhou M, et al. B7-H3 silencing increases paclitaxel sensitivity by abrogating Jak2/Stat3 phosphorylation. Molecular cancer therapeutics. 2011;10(6):960–971. doi: 10.1158/1535-7163.MCT-11-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rycaj K, Tang DG. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer research. 2015;75(19):4003–4011. doi: 10.1158/0008-5472.CAN-15-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandalovicova A, Vomastek T, Rosel D, Brabek J. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget. 2016;7(18):25022–25049. doi: 10.18632/oncotarget.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halaoui R, McCaffrey L. Rewiring cell polarity signaling in cancer. Oncogene. 2015;34(8):939–950. doi: 10.1038/onc.2014.59. [DOI] [PubMed] [Google Scholar]
- 24.Macara IG, McCaffrey L. Cell polarity in morphogenesis and metastasis. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2013;368(1629):20130012. doi: 10.1098/rstb.2013.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godde NJ, Galea RC, Elsum IA, Humbert PO. Cell polarity in motion: redefining mammary tissue organization through EMT and cell polarity transitions. Journal of mammary gland biology and neoplasia. 2010;15(2):149–168. doi: 10.1007/s10911-010-9180-2. [DOI] [PubMed] [Google Scholar]
- 26.Wodarz A, Nathke I. Cell polarity in development and cancer. Nature cell biology. 2007;9(9):1016–1024. doi: 10.1038/ncb433. [DOI] [PubMed] [Google Scholar]
- 27.Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer cell. 2011;19(1):86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer cell. 2014;25(5):697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheffer GL, Schroeijers AB, Izquierdo MA, Wiemer EA, Scheper RJ. Lung resistance-related protein/major vault protein and vaults in multidrug-resistant cancer. Current opinion in oncology. 2000;12(6):550–556. doi: 10.1097/00001622-200011000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer cell. 2014;26(3):402–413. doi: 10.1016/j.ccr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Zhang Q, Chen W, Shan B, Ding Y, Zhang G, et al. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PloS one. 2013;8(8):e70689. doi: 10.1371/journal.pone.0070689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemke D, Pfenning PN, Sahm F, Klein AC, Kempf T, Warnken U, et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(1):105–117. doi: 10.1158/1078-0432.CCR-11-0880. [DOI] [PubMed] [Google Scholar]
- 33.Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23(8):1104–1112. doi: 10.1038/modpathol.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arigami T, Narita N, Mizuno R, Nguyen L, Ye X, Chung A, et al. B7-h3 ligand expression by primary breast cancer and associated with regional nodal metastasis. Annals of surgery. 2010;252(6):1044–1051. doi: 10.1097/SLA.0b013e3181f1939d. [DOI] [PubMed] [Google Scholar]
- 35.Chavin G, Sheinin Y, Crispen PL, Boorjian SA, Roth TJ, Rangel L, et al. Expression of immunosuppresive B7-H3 ligand by hormone-treated prostate cancer tumors and metastases. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15(6):2174–2180. doi: 10.1158/1078-0432.CCR-08-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zang X, Thompson RH, Al-Ahmadie HA, Serio AM, Reuter VE, Eastham JA, et al. B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19458–19463. doi: 10.1073/pnas.0709802104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X, Inman BA, et al. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer research. 2007;67(16):7893–7900. doi: 10.1158/0008-5472.CAN-07-1068. [DOI] [PubMed] [Google Scholar]
- 38.Parker AS, Heckman MG, Sheinin Y, Wu KJ, Hilton TW, Diehl NN, et al. Evaluation of B7-H3 expression as a biomarker of biochemical recurrence after salvage radiation therapy for recurrent prostate cancer. International journal of radiation oncology, biology, physics. 2011;79(5):1343–1349. doi: 10.1016/j.ijrobp.2010.01.061. [DOI] [PubMed] [Google Scholar]
- 39.Zhang G, Xu Y, Lu X, Huang H, Zhou Y, Lu B, et al. Diagnosis value of serum B7-H3 expression in non-small cell lung cancer. Lung cancer. 2009;66(2):245–249. doi: 10.1016/j.lungcan.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 40.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. The Journal of clinical investigation. 2011;121(7):2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abubaker K, Luwor RB, Zhu H, McNally O, Quinn MA, Burns CJ, et al. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC cancer. 2014;14:317. doi: 10.1186/1471-2407-14-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bender RH, Haigis KM, Gutmann DH. Activated k-ras, but not h-ras or N-ras, regulates brain neural stem cell proliferation in a raf/rb-dependent manner. Stem cells. 2015;33(6):1998–2010. doi: 10.1002/stem.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galuppo R, Maynard E, Shah M, Daily MF, Chen C, Spear BT, et al. Synergistic inhibition of HCC and liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and WNT/beta-catenin pathways. Anticancer research. 2014;34(4):1709–1713. [PMC free article] [PubMed] [Google Scholar]
- 44.Drosten M, Sum EY, Lechuga CG, Simon-Carrasco L, Jacob HK, Garcia-Medina R, et al. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(42):15155–15160. doi: 10.1073/pnas.1417549111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt M, Goebeler M, Posern G, Feller SM, Seitz CS, Brocker EB, et al. Ras-independent activation of the Raf/MEK/ERK pathway upon calcium-induced differentiation of keratinocytes. The Journal of biological chemistry. 2000;275(52):41011–41017. doi: 10.1074/jbc.M003716200. [DOI] [PubMed] [Google Scholar]
- 46.Rizzo MA, Shome K, Watkins SC, Romero G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. The Journal of biological chemistry. 2000;275(31):23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 47.Teng Y, Ren Y, Hu X, Mu J, Samykutty A, Zhuang X, et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nature communications. 2017;8:14448. doi: 10.1038/ncomms14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.