

Abstract
In cancer, kinases are often activated and phosphatases suppressed, leading to aberrant activation of signaling pathways driving cellular proliferation, survival, and therapeutic resistance. Although pancreatic ductal adenocarcinoma (PDA) has historically been refractory to kinase inhibition, therapeutic activation of phosphatases is emerging as a promising strategy to restore balance to these hyperactive signaling cascades. In the current study, we hypothesized that phosphatase activation combined with kinase inhibition could deplete oncogenic survival signals to reduce tumor growth. We screened PDA cell lines for kinase inhibitors that could synergize with activation of PP2A, a tumor suppressor phosphatase, and determined that activation of PP2A and inhibition of mTOR synergistically increase apoptosis and reduce oncogenic phenotypes in vitro and in vivo. This combination treatment resulted in suppression of AKT/mTOR signaling coupled with reduced expression of c-MYC, an oncoprotein implicated in tumor progression and therapeutic resistance. Forced expression of c-MYC or loss of PP2A B56α, the specific PP2A subunit shown to negatively regulate c-MYC, increased resistance to mTOR inhibition. Conversely, decreased c-MYC expression increased the sensitivity of PDA cells to mTOR inhibition. Together these studies demonstrate that combined targeting of PP2A and mTOR suppresses proliferative signaling and induces cell death and implicate this combination as a promising therapeutic strategy for PDA patients.
Keywords: PP2A, SET, Pancreatic Cancer, mTOR, MYC
Precis
Findings present a combinatorial strategy targeting serine/threonine protein phosphatase PP2A and mTOR in PDA, a cancer for which there are currently no targeted therapeutic options.
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the United States and while chemotherapy can provide a survival benefit, the 5-year survival rate for pancreatic ductal adenocarcinoma (PDA) patients remains the lowest of all major cancers (1). Although KRAS mutations are an almost universal event in PDA, mutant KRAS continues to be a highly undruggable target and significantly contributes to therapeutic resistance (2, 3). Consistent with the high prevalence of mutant KRAS in PDA, single agent kinase inhibitors have had little clinical success in PDA patients, likely due to cellular plasticity and adaptation to alternative oncogenic signaling pathways (4, 5).
Protein Phosphatase 2A (PP2A) is a serine/threonine phosphatase that regulates multiple signaling cascades implicated in cancer progression, including downstream effectors of KRAS (6). Inhibition of PP2A contributes to oncogenesis in multiple tumor types, highlighting the importance of this protein in maintaining normal kinase activity (7). PDA cells have reduced PP2A activity and an upregulation of the PP2A inhibitors, CIP2A and SET (8, 9). Further, high CIP2A expression in PDA patients correlates with decreased overall survival (10), suggesting that suppression of PP2A may significantly contribute to PDA cell survival. As such, compounds that activate PP2A are emerging as promising cancer therapeutics (11). The majority of PP2A activating agents disrupt the interaction between PP2A and CIP2A or SET, indirectly increasing PP2A activation and reducing tumor growth (12–14). However, tricyclic neuroleptics have direct PP2A activating properties and our recent study by Sangodkar et. al. demonstrated that derivatives of these compounds, known as small-molecule activators of PP2A (SMAPs), specifically bind to the PP2A A subunit and facilitate PP2A activation resulting in reduced oncogenic phenotypes both in vitro and in vivo (15, 16). The specificity of these effects was demonstrated by loss of the therapeutic efficacy of SMAPs with the expression of the SV40 small T antigen, a known PP2A inhibitor, or expression of A subunit mutations. Thus, SMAPs directly bind the PP2A A subunit and predominately function through PP2A activation (16). Given the multiple oncogenic targets of PP2A, compounds that activate this phosphatase may prevent or suppress cancer cell signaling plasticity in response to kinase inhibitors.
Here we investigate the therapeutic efficacy of combining kinase inhibitors with phosphatase activators to synergistically attenuate oncogenic signaling and induce cell death in PDA cells. In order to identify kinases susceptible to PP2A activation, we initially assessed cell viability in a 120-kinase inhibitor screen in combination with an indirect PP2A activator, OP449. Results of this study led us to pursue mTOR inhibitor combinations with OP449 and DT1154, a direct SMAP. The PI3K/AKT/mTOR signaling node is activated downstream of KRAS and has been shown to be deregulated in a large percent of PDA patients (17–19). Clinically, mTOR inhibitors have shown little success as single agent compounds, primarily due to resistance mechanisms, making this node an ideal target for therapeutic combination strategies (20–22). INK128, an ATP-competitive mTORC1/2 inhibitor, was synergistic with PP2A activation and in combination with DT1154 resulted in a significant increase in apoptosis and reduced tumor growth in vivo over single agent treatment. mTOR inhibition alone suppressed AKT/mTOR signaling but was unable to drive a significant loss of the oncoprotein c-MYC (MYC) (MYCHigh/mTORLow). In contrast, the synergistic combination of INK128 and DT1154 reduced the activation of MYC and AKT/mTOR (MYCLow/mTORLow), identifying MYC signaling as a potential resistance mechanism by which pancreatic cancer cells survive mTOR inhibition. Together, these studies support the use of PP2A-activating compounds in combination with kinase inhibitors as a novel therapeutic strategy in PDA.
Materials and Methods
Cell culture, Knockdown, Adenoviral Transfection.
PANC1, HPAFII, MIAPACA2, ASPC1, PANC89 and HPNE pancreatic cells lines were obtained from Michel Ouellette (University of Nebraska Medical Center, Omaha NE). Cell lines were authenticated using short tandem repeat (STR) profiling. KPC and KPCMfl/+ cell lines were gifts from Drs. Jennifer P. Morton, Owen J. Sansom, and Michael A. Hollingsworth. All pancreatic cancer cell lines were tested for Mycoplasma using PCR-based strategies and grown in DMEM+10% FBS at 37°C in a 5% CO2 atmosphere. All experiments were performed within 6–8 cell passages after thaw. HPNE cells were grown as previously described (8). KPC or KP(R172H/+)C denotes cells generated from Pdx1-Cre;LSL-KrasG12D;LSL-TP53R172H mice and KP(fl/fl)C denotes cells generated from Pdx1-Cre;LSL-KrasG12D;LSL-TP53fl/fl mice. To generate primary human PDA cell lines, PDA tissue from patient derived xenografts was collected in collaboration with the Brenden-Colson Center for Pancreatic Care and the Oregon Pancreas Tissue Registry as previously described (23) and mechanically disassociated and cultured in DMEM +10% FBS. Each primary human cell line is denoted with a unique Oregon Pancreas Tissue Registry (OPTR) number. Samples generated from metastatic patient tissue are denoted with an “M”. Transient knockdowns were performed using siRNA to c-Myc (SI00300902, Qiagen) and PPP2R5A (PP2A B56α; L009352, Dharmacon) using Dharma-FECT1 (Dharmacon) or Lipofectamine 3000 (Invitrogen) transfection reagents, respectively. Nontargeting siRNA (siNT) (1027310, Qiagen) was used as a control. Cells were plated into viability assays or for western blot analysis 24 hours after being treated with siRNA. For overexpression studies, cells were treated for 24 hours with either adenovirus encoding GFP or an adenovirus encoding GFP and MycT58A in DMEM+2% FBS. Viral load was adjusted for equal expression of GFP. Transfected cells were then used in viability assays or western blot analysis.
Viability and Cytotoxicity Assays.
Cells were plated in DMEM with 2% FBS in 96 well plates at 3,000 cells per well. 24 hours post-plating, cells were treated with a 9-point dose dilution of the indicated inhibitor, either as a single agent or in combination, with DMSO as a control. After 72 hours, viability was measured using [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) CellTiter 96 cell viability kit (Promega). Combination Indices (CI) were calculated using the Chou and Talalay method (24) with CalcuSyn software (CalcuSyn). Details of the cytotoxicity assays can be found in the Supplemental Methods. The therapeutic compounds used were as follows: OP449 (25) and DT1154 (16) were described previously. BEZ235, INK128, GDC-0068, PP242, CUDC101, VX680, XL880, and Dasatinib were purchased from Selleck Chem.
Quantitative PCR
RNA was isolated using the RNAeasy Isolation Kit (Qiagen) from PANC89 cells treated with either Vehicle, 10μM DT1154, 0.5 μM INK128, or the combination of DT1154 and INK128 for 6 hours. cDNA was made as described previously (8) and quantitative PCR (qPCR) was performed using SYBR Green reagent (Invitrogen). Primers: MYC (F: 5′-CAGTGGGCTGTGAGGAGGTT-3′, R: 5′-CAGGCTCCTGGCAAAAGGT-3′), NCL (Nucleolin) (5′-ACTGACCGGGAAACTGGGTC-3′, R: 5′-TGGCCCAGTCCAAGGTAACT-3′), E2F2 (F: 5′-ACAAGGCCAACAAGAGGCTG-3′, R: 5′-TCAGTCCTGTCGGGCACTTC-3′) and PPP2R5A (F: 5’-AGAGCCCTGATTTCCAGCCTA-3’, R: 5’-TTTCCCATAAATTCGGTGCAGA-3’). The relative fold change relative to Vehicle was analyzed using the ΔΔ(CT) method described previously(26).
Soft Agar Assay
For soft agar assays, 1.4% noble agar was mixed 1:1 with 2X DMEM with 4% FBS and plated in 12 well plates. Cells were harvested and 40,000 cells were resuspended in 2X DMEM with 4% FBS. The cell suspension was then mixed 1:1 with 0.7% noble agar and plated on top of the base agar layer. Plates were fed twice a week with DMEM medium containing 2% FBS and either Vehicle or the indicated doses of DT1154, INK128, or combination. Plates were stained with 0.5 ml 0.005% crystal violet over night at 4oC and the total number of colonies was quantified in triplicate wells using ImageJ.
Xenografts and Immunohistochemistry.
All animal studies were performed in compliance with OHSU animal use guidelines after approval by the OHSU IACUC. PANC89, KPC, and KPC Mycfl/+ cells were resuspended in 50/50 serum free DMEM/growth factor reduced Matrigel (BD Biosciences) and 0.5M cells were subcutaneously injected into the flank of NSG mice. Once tumors were palpable, mice were randomized into treatment arms (Vehicle, DT1154 (100mg/kg), INK128 (0.5 mg/kg) or combination). Each drug or Vehicle was administered once daily, six days a week by oral gavage (o.g.). For long-term studies, all mice were sacrificed when any tumor reached 2 cm in diameter. For short-term studies, all mice were sacrificed after 1 week of treatment and tumor tissue was either processed for formalin fixed paraffin embedding (FFPE) or snap frozen. For histological analysis, 6μM-thick FFPE sections were stained with hematoxylin and eosin (H/E). Full tumor H/E sections for each treatment condition were scanned using the Aperio (Leica Biosystems) and necrotic area was outlined and quantified using ImageJ. Apoptosis was detected using the ApopTag Plus Peroxidase In Situ Kit (Millipore, S7101). The number of TUNEL positive cells per 20X high power field (HPF) was quantified using ImageJ.
Western Blot, Phosphokinase Array, and Immunoprecipitation.
Pancreatic cancer cells were plated in DMEM with 2% FBS and 24 hours later treated with either Vehicle, 10μM DT1154, 0.5 μM INK128, or the combination of DT1154 and INK128 for 6 hours. Cells were lysed, run on an SDS page gel, and quantified using a LI-COR Odyssey as previously described (23). For tumor cell lysates, 50–150mg of tumor tissue was homogenized, lysed, and run as above. For the phosphokinase array (R&D Systems), PANC89 cells were treated and lysed as above and then 450 μg of protein per membrane was used per the kit protocol. Details of the MYC immunoprecipitation can be found in the Supplemental Methods. The following antibodies were used for western blot analysis: c-MYC (Y69 ab32072, Abcam; N-262 SC-764, Santa Cruz Biotechnology), pS62 MYC (ab78318, Abcam), pT58 MYC (Y011034, Applied Biological Materials), pAKT (CS4060, Cell Signaling Technology), AKT (CS2920, Cell Signaling Technology), pPRAS40 (CS2997, Cell Signaling Technology), PRAS40 (CS2691, Cell Signaling Technology), pS6 (CS4858, Cell Signaling Technology), 4EBP1 (CS9644, Cell Signaling Technology), GAPDH (AM4300, Applied Biosystems), IRDye800 (Rockland), and Alexa Fluor 680 (Molecular Probes). GAPDH and secondary antibodies were used at 1:10,000 and all other primary antibodies were used at 1:1000.
Statistical Analysis.
P values were determined by two-tailed Student t test or ANOVA, as denoted in the figure legends. A P value of <0.05 was used to determine statistical significance. Error bars represent the standard error of the mean or standard deviation, as denoted in the figure legend.
Results
PP2A activation increases the cytotoxic effects of select kinase inhibitors.
To identify oncogenic pathways susceptible to kinase inhibition and phosphatase activation, we analyzed the response of five human PDA cell lines in a kinase inhibitor screen with or without a low dose of OP449, a SET inhibitor that activates PP2A (27). Cell viability was assessed by MTS after three days and IC50s were calculated for both single agent and combination arms. PDA cell lines showed variable responses to single agent kinase inhibitors, consistent with intertumoral phenotypic heterogeneity found in PDA tumors (Fig. S1A, Supplemental Table 1)(5). We identified several kinase inhibitors that displayed increased sensitivity with PP2A activation (Fig. S1B). Select oncogenic pathways known to contribute to PDA progression were validated in 96-well format, where VX-680 (Aurora), Dasatinib (Src family kinase), and INK128 (mTOR) inhibition showed the highest synergy using the Chou-Talalay method (Confidence Interval (CI) values less than 0.7) (Fig. 1A)(17, 24, 28–30). We focused on the PI3K/AKT/mTOR pathway as INK128 showed the strongest synergy across cell lines, this pathway is regulated by PP2A at multiple points (31), and it is an important signaling node downstream of KRAS in PDA (19). We expanded our analysis to other kinase inhibitors within the PI3K/AKT/mTOR pathway to determine if the synergy between OP449 and INK128 is specific to mTOR inhibition. Combination of OP449 with GDC0068 (AKT) or BKM120 (PI3K) was synergistic in several lines, but compounds targeting mTOR (PP242 and INK128) resulted in the lowest CI values (Fig. 1B). These results suggest that mTOR inhibitors have increased efficacy in combination with PP2A activation compared to upstream inhibitors of the PI3K/AKT/mTOR pathway. Similar results were seen in a panel of breast cancer cell lines (Fig. S1C). Combination of INK128 and OP449 also resulted in increased cell death in PDA cells, as seen by flow cytometry for AnnexinV+ cells and a dose-dependent shift in viability curves (Fig. S1D, E).
Fig. 1:
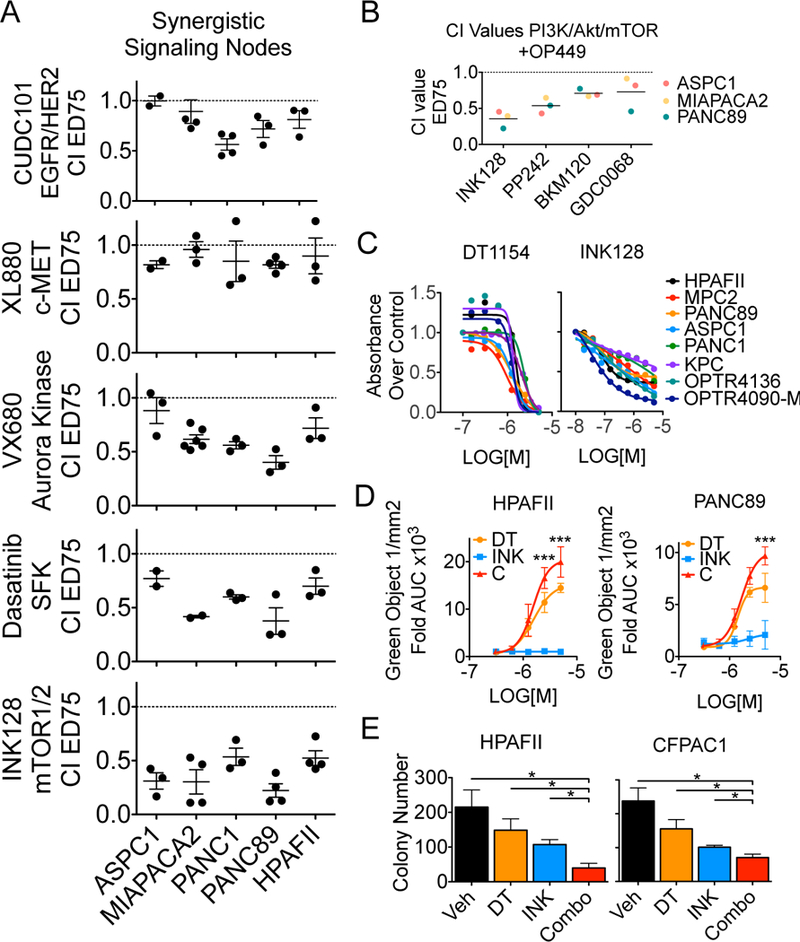
PP2A activation increases the cytotoxic effects of select kinase inhibitors. A. CI values at effective dose (ED) 75 for pancreatic cells lines treated with increasing concentrations of OP449 and select kinase inhibitors. Mean +/− SEM of biological replicates. B. CI values at ED75 of ASPC1, MIAPACA2, and PANC89 treated with INK128, PP242, BKM120, or GDC0068. Values for each cell line were generated from 2 (MIAPACA2- GDC0068) or 3 biological reps. Grand mean of all lines for each drug shown. C. PDA cell lines were treated with increasing doses of DT1154 (left) or INK128 (right) for 72 hours. Shown is the viability relative to Vehicle for each line. MIAPACA2 (MPC2) D. CellTox Green was added to HPAFII and PANC89 cells and then cells were treated with increasing concentrations of DT1154 and INK128. Fluorescent cells were imaged every 2 hours for 72 hours on an Incucyte Zoom. Shown is the area under the curve (AUC) generated from the green object count (1/mm2) over time relative to Vehicle control. Graph represents average of three biological replicates. ***P <0.001 by a 2-way ANOVA. E. HPAFII and CFPAC1 cells were plated in a soft agar colony assay and treated with Vehicle, 500nM DT1154, 10nM INK128, or combination. Colony number was quantified using ImageJ. Mean +/− SEM of three biological replicates. *p<0.05 by a two-tailed students t-test.
OP449 treatment reduces cell survival in several cancer types; however, SET inhibition is an indirect method of PP2A activation (8, 12, 26, 32). Consistent with our OP449 results, direct PP2A activation using the small-molecule activator, DT1154, reduced cell viability in all PDA cell lines tested, including two low-passage cell lines generated from patient derived xenograft tumors (OPTR4090-M and OPTR4136) (Fig. 1C, Fig. S2A)(16). INK128 reduced cell viability by 65% across PDA cell lines (Fig. 1C), but was unable to drive apoptosis, as determined by absence of CellTox Green (Fig. 1D). Combination of DT1154 and INK128 synergistically reduced viability in multiple PDA cell lines (Fig. S2B, C), but not in the immortalized, non-transformed pancreatic cell line, HPNE (Fig. S2B-open circles). The combination of DT1154 and INK128 also significantly increased apoptosis over single agent treatments (Fig. 1D) and resulted in a dose-dependent shift of the MTS viability curves (Fig. S2C). To determine if PP2A activation and mTOR inhibition attenuates the ability of PDA cells to grow in an anchorage-independent environment, HPAFII and CFPAC1 cells were grown in soft agar and treated with Vehicle, DT1154, INK128, or the combination. The combination of DT1154 and INK128 significantly reduced colony formation over single agent treatment (Fig. 1E). Together, these results suggest that phosphatase activation and mTOR inhibition can function synergistically to reduce PDA cell survival and transformed phenotypes in vitro.
Combination of DT1154 and INK128 synergistically attenuates AKT/mTOR oncogenic signaling.
To elucidate the signaling mechanisms underlying the synergy of PP2A activation and mTOR inhibition, PANC89 cells were treated with DT1154 and/or INK128 for 6 hours and the phosphorylation state of 38 kinases were screened using a phosphokinase array. The fold intensity of each phosphorylation site relative to Vehicle was calculated and visualized by KMeans clustering (Fig. 2A; Supplemental Table 2). β-Catenin, STAT5a, PRAS40, p70 S6 Kinase, STAT3, WNK1, HSP60, and p53 (Cluster 2) were the only proteins with phosphorylation sites significantly decreased in combination-treated cells compared to single agent treatments (Fig. 2A inset, Fig. S3A). Using Pathway Commons through the CBio Portal (33, 34) we determined that 7 of the 8 proteins in Cluster 2 are functionally linked to the PI3K/AKT/mTOR signaling pathway (Fig. 2B- Cluster 2 proteins noted with star), therefore we focused our analysis on this signaling node. Treatment with INK128 resulted in an almost complete loss of phosphorylation of the mTOR effectors S6 and 4EBP1, and reduced pAKT, but the combination of DT1154 and INK128 further decreased the levels of pAKT and pPRAS40, an mTOR inhibitor that is inactive in its phosphorylated state (Fig. 2C, S3B). Similar results were seen in a low-passage, primary human PDA cell line treated with DT1154 and INK128, as well as in PANC89 cells treated with OP449 and INK128 (Fig. S3C, D). Together, these results suggest that the combination of PP2A activation and mTOR inhibition more effectively suppresses the AKT/mTOR signaling node over single agent treatments.
Fig. 2:
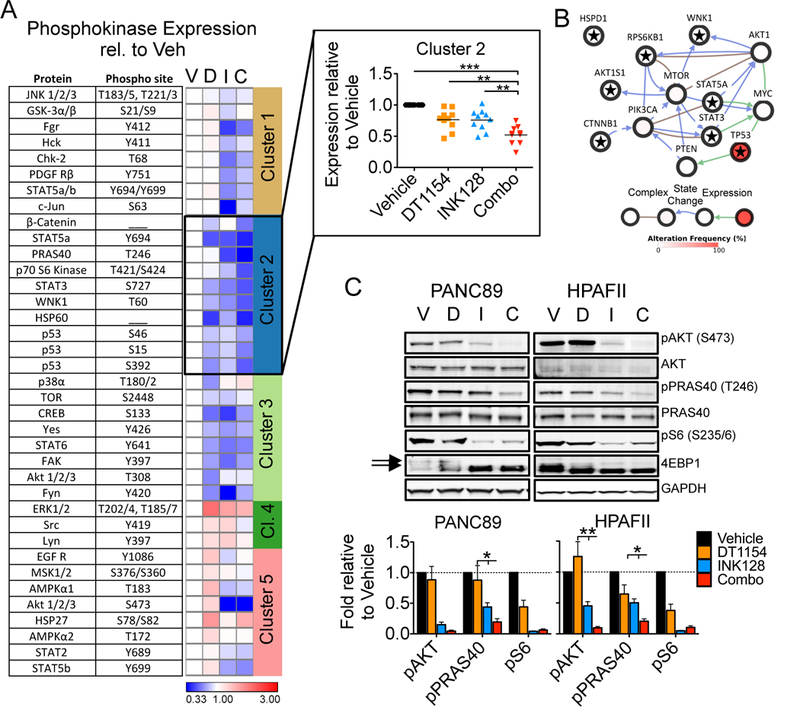
Combination of DT1154 and INK128 synergistically attenuates AKT/mTOR oncogenic signaling A. PANC89 cells were treated with Vehicle (V), DT1154 (D, 10μM), INK128 (I, 0.5μM), or the combination of DT1154 and INK (C) for 6 hours and cell lysates were probed using a phosphokinase array. Quantification of phospho-specific sites for each kinase relative to Vehicle control was used to generate a heat map using Morpheus and KMeans clustering was performed using one minus pearson correlation (Clusters 1–5). Phosphorylation sites, proteins, and expression values can be found in Supplemental Table 2. Inset shows the expression of the phosphorylated proteins in Cluster 2 relative to Vehicle control. Grand mean shown. ***p<0.001 **p<0.01 *p<0.05 by a 1-way ANOVA. B. A pathway Commons network map generated from targets in Cluster 2 using the QCMG, Nature 2016 dataset from CBio Portal. Complex, state change, expression, and alteration frequency between proteins are shown. Gene names of targets in Cluster 2 are denoted by a star. C. PANC89 and HPAFII cells were treated as in panel A and probed by western blot for pAKT (S473), pPRAS40 (T246), pS6 (S235/6) and totals. Representative blots and quantification of three biological replicates shown. Arrows indicate phosphorylated form of 4EBP1. pS6 quantified over GAPDH. Mean +/− SEM. *p<0.05 by two-tailed students t-test.
PP2A activation suppresses MYC mediated resistance to mTOR inhibition.
Several signaling mechanisms have been suggested to increase resistance to PI3K-mTOR inhibitors, including AKT feedback loops, mTOR mutation, and increased MYC signaling (35–37). We have previously shown that SET inhibition, results in decreased MYC expression in breast and pancreatic cancer cell lines (8, 26). Additionally, MYC, mTOR, CIP2A, SET, and PP2A are frequently deregulated in PDA and altered expression of these factors is associated with poor survival (Fig. S4A, B). To determine if the combination of DT1154 and INK128 alters MYC expression, PDA cells were treated with DT1154 and/or INK128 for 6 hours and total MYC levels were analyzed. As expected, DT1154 treatment decreased total MYC levels in PANC89 and HPAFII cells. INK128 had variable effects on MYC levels, but the combination of DT1154 and INK128 further decreased MYC expression relative to either single agent treatment alone, thus resulting in a MYCLow/mTORLow state in the PANC89 and HPAFII cells (Fig. 3A, B). In contrast, MYC levels remained relatively constant across treatment arms in MIAPACA2 (MPC2) and PANC1 cells; although these cell lines displayed decreased activation of AKT and mTOR signaling with combination treatment, they did not show combination synergy (CI= 0.89 and 0.88 respectively) (Fig. 3A, B, S2B and S3B). These results suggest that sustained MYC expression may provide a survival advantage to cells with reduced AKT/mTOR signaling and that the loss of both pathways is necessary for decreased cell survival.
Fig. 3:
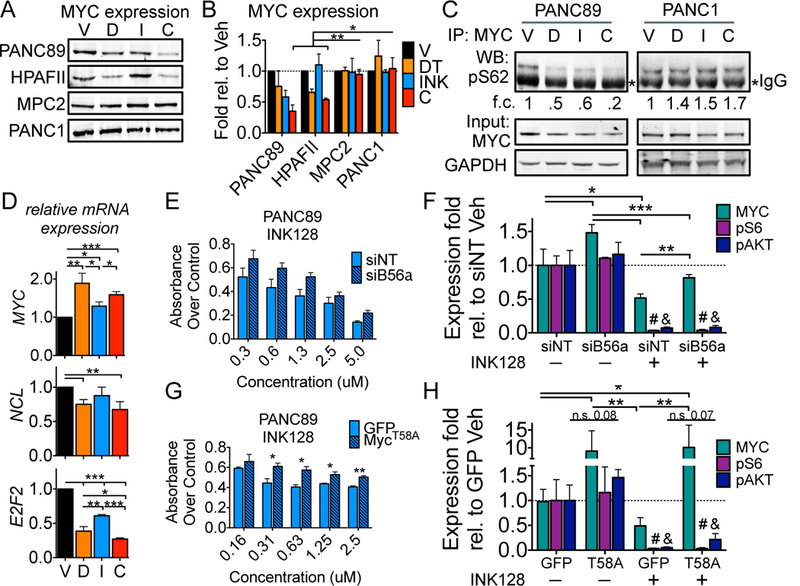
PP2A activation suppresses MYC mediated resistance to mTOR inhibition A and B. PANC89, HPAFII, MIAPACA2 (MPC2), and PANC1 cells were treated with Vehicle (V), DT1154 (D; 10μM), INK128 (I; 0.5μM), or the combination (C) for 6 hours and lysates were probed by western blot. Representative blot (A) and quantification over GAPDH (B) of three biological replicates shown. Mean +/− SEM C. PANC89 and PANC1 cells were treated as in Figure 3A. Total MYC was immunoprecipitated and lysates were probed for phosphoS62 MYC (pS62). Inputs were probed for total MYC and GAPDH *denotes IgG heavy chain. Quantification of pS62 levels over GAPDH relative to Vehicle shown D. qPCR of MYC, NCL (Nucleolin), and E2F2 from PANC89 cells treated as in Figure 3A. Mean +/− SEM of three biological replicates. E. PANC89 cells transfected with non-targeting siRNA (siNT) or a B56α targeting siRNA pool (siB56α) were treated with increasing concentrations of INK128. Viability was assessed by MTS 72 hours after drug treatment. Mean +/− SEM of three biological replicates. F. Cells from panel E were treated with either Vehicle or 0.5μM INK128 for 6 hours. Lysates were analyzed by western blot for MYC, pAKT (S473), pS6 (S235/6) and totals. Quantification from three biological replicates is shown. Mean +/− SD. G. PANC89 cells transfected with AdGFP (GFP) or AdMYCT58A (T58A) were treated with increasing concentrations of INK128. Viability was assessed by MTS 72 hours after drug treatment. Mean +/− SEM of three biological replicates. H. Cells from panel G were treated with either Vehicle or 0.5μM INK128 for 6 hours. Lysates were analyzed by western blot as above. Quantification from three biological replicates is shown. Mean +/−SD. For all panels ***p<0.001 **p<0.01 *p<0.05 by two-tailed students t-test; #, & denotes pS6 or pAKT levels, respectively, are significantly different in INK128 conditions compared to Vehicle.
MYC activity and stability is predominantly controlled by dynamic phosphorylation at S62 and T58, with dephosphorylation of S62 driving the proteosomal degradation of MYC (38). To determine if combination treatment decreased MYC phosphorylation, PANC89 and PANC1 cells were treated with DT1154 and/or INK128 for 6 hours and endogenous MYC was assayed for phosphorylation at S62 and T58. In PANC89 cells, both DT1154 and INK128 lead to the dephosphorylation of S62, but these levels were reduced further with the combination of DT1154 and INK128, suggesting that these compounds may regulate MYC function through non-redundant mechanisms. In contrast, pS62 levels remained unchanged in PANC1 cells (Fig. 3C). Similar results were seen with pT58 (Fig. S5), indicating that PANC89 cells have dynamic turnover of MYC, while PANC1 cells lack this regulation. The loss of MYC protein in PANC89 cells was not due to decreased MYC mRNA levels as MYC expression increased in response to PP2A activation, likely due to an alleviation of autoregulation feedback loops (39), supporting a posttranslational mechanism of regulation (Fig. 3D). Consistent with reduced MYC expression, MYC target genes, NCL (Nucleolin) and E2F2, were significantly decreased in combination-treated cells, suggesting that MYC function is suppressed (Fig. 3D).
Our lab has shown that the PP2A B’ subunit, PPP2R5A (B56α), targets PP2A to dephosphorylate MYC at S62 (40). To determine if attenuating PP2A results in increased MYC expression and resistance to mTOR inhibition, PANC89 cells were transfected with siRNA to B56α and then treated with increasing doses of INK128. Decreased expression of B56α (Fig. S6A) increased cell viability in INK128-treated cells compared to siNT (Fig. 3E). The increase in viability was not due a loss in INK128 function, as we saw no difference in AKT/mTOR signaling in siB56α cells (Fig. S6B, 3F). Consistent with the role of B56α in MYC stabilization, knockdown of B56α significantly increased MYC expression compared to siNT in Vehicle and INK128 treated cells (Fig. S6B, 3F). Thus, despite having an almost complete loss of AKT/mTOR signaling, siB56α cells treated with INK128 showed a MYCHigh/mTORLow cell state associated with increased viability (Fig. S6B, 3F). Consistent with these results, knockdown of B56α in PANC89 cells also decreased the efficacy of DT1154, suggesting that the effects of DT1154 and INK128 on MYC expression in this cell line are significantly mediated through PP2A’s posttranslational regulation of MYC (Fig. S6C). To determine if aberrant expression of MYC alone can increase resistance of PANC89 cells to INK128, we overexpressed MycT58A, a phosphorylation mutant resistant to PP2A (38, 41), and then treated cells with increasing concentrations of INK128. Similar to B56α knockdown, expression of MycT58A significantly increased the viability of INK128 treated PANC89 cells (Fig. 3G). mTOR activity was suppressed with INK128 irrespective of MYCT58A expression, resulting in a MYCHigh/mTORLow cell state, again associated with increased viability (Fig. S6D, 3H). Together, these results suggest that resistance to mTOR inhibition is mediated, in part, through stabilization of MYC.
Decreased MYC expression sensitizes PDA cells to mTOR inhibitors.
We next sought to determine whether direct suppression of MYC increases the efficacy of mTOR inhibition, particularly in lines where DT1154 was unable to decrease MYC expression. Suppression of MYC using siRNA significantly increased the sensitivity of PANC1 cells to INK128 (Fig. 4A). Sensitivity to INK128 was even more pronounced in cells with heterozygous expression of Myc (Pdx1-Cre;LSL-KrasG12D;LSL-TP53R172H;Mycfl/+ (KPC Mycfl/+)) compared to KPC cells (Fig. 4B) (42). INK128 maintained its ability to suppress mTOR signaling in both PANC1 siMYC or KPC Mycfl/+ cells, consistent with MYCLow/mTORLow signaling and reduced viability (Fig. S7A, S7B, 4C, 4D). Importantly, treatment of KPC Mycfl/+ cells with the combination of DT1154 and INK128 provided no additional benefit over INK128 alone (Fig. 4E right), as compared to KPC, where the combination was significantly more efficacious over either single agents (Fig 4E left). Treatment of KPC Mycfl/+ cells with a different mTOR inhibitor, BEZ235, resulted in a similar decrease in viability relative to KPC cells (Fig. S8A). In contrast, Mycfl/+ cells showed little to no increase in sensitivity in response to Dasatinib (SFK inhibitor) (Fig. S8B) and displayed increased resistance to Sunitinib (multi-RTK, VEGFR) (Fig. S8C), indicating that combined suppression of MYC and mTOR may result in a unique sensitivity. To determine whether the combined loss of Myc and mTOR results in decreased tumor growth in vivo, KPC and KPC Mycfl/+ cells were subcutaneously implanted and treated with either Vehicle or INK128. INK128 treatment had little to no effect on KPC tumors (Fig. 4F, G blue circles). In contrast, INK128 significantly reduced KPC Mycfl/+ tumor growth (Fig. 4F, G blue triangles).
Fig. 4:
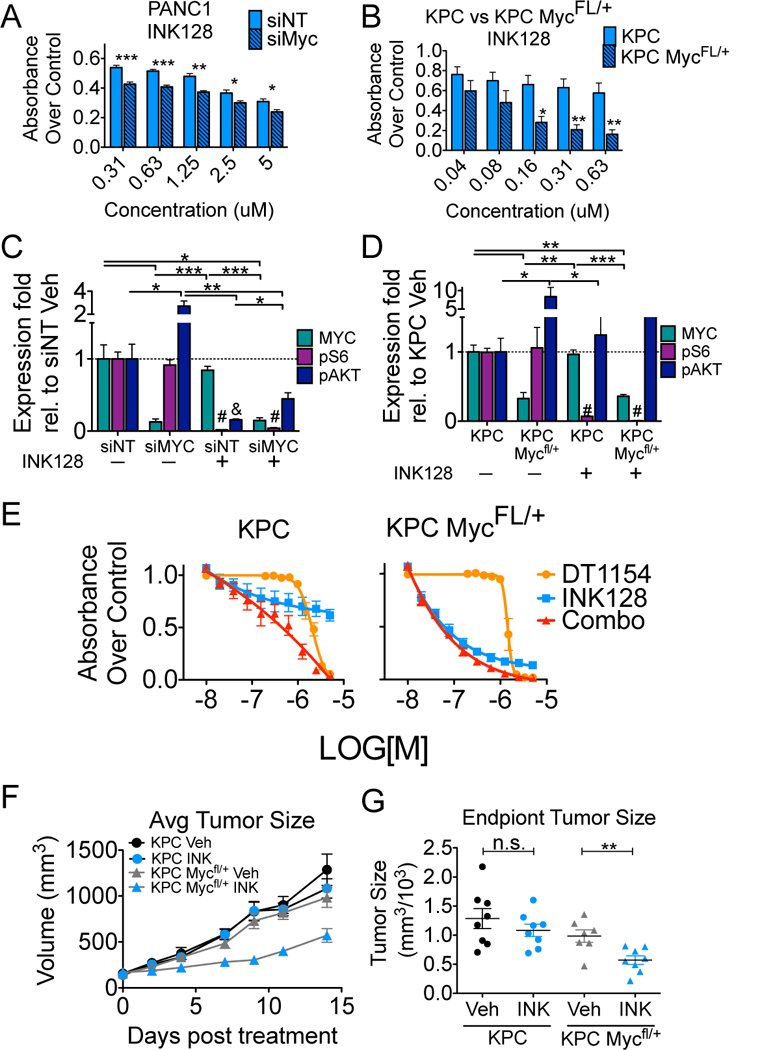
Decreased Myc expression sensitizes PDA cells to mTOR inhibition in vitro and in vivo A. PANC1 cells transfected with non-targeting siRNA (siNT) or MYC targeting siRNA (siMYC) were treated with increasing concentrations of INK128. Viability was assessed by MTS 72 hours after drug treatment. Mean +/−SEM of three biological replicates. B. Cells derived from KPC or KPC Mfl/+ mice were treated with increasing concentrations of INK128. Viability was assessed by MTS 72 hours after drug treatment. Mean +/−SEM of three biological replicates. C. Cells from panel A (siNT or siMYC) and D. cells from panel B (KPC or KPC Mfl/+) were treated with either Vehicle or 0.5μM INK128 for 6 hours. Lysates were analyzed by western blot for MYC, pAKT (S473), pS6 (S235/6) and totals. Quantification from three biological replicates is shown. Mean +/−SEM. E. KPC (left) and KPC Mfl/+ (right) cells were treated with either Vehicle or increasing concentrations of DT1154, INK128, or the combination for 72 hours. Viability relative to Vehicle was assessed by MTS. Average of 4 replicates across 2 biological experiments shown. Mean +/−SEM. For all panels ***p<0.001 **p<0.01 *p<0.05 by two-tailed students t-test. #, & denotes pS6 or pAKT levels, respectively, are significantly different in INK128 conditions compared to Vehicle. F. KPC and KPC Mfl/+ cells were injected subcutaneous and then treated with either Vehicle or INK128 (0.5 mg/kg oral gavage, once a day/6 days a week). Tumor volume was measured across time. Mean +/− SEM. n= 8 KPC, 8 KPC+INK128, 7 KPC Mfl/+, 8 KPC Mfl/++INK128. G. Quantification of end point tumor size from panel F. **p<0.01 by two-tailed students t-test
PP2A activation combined with mTOR inhibition decreases tumorigenic properties in vivo.
We next determined whether the combination of DT1154 and INK128 would have anti-tumorigenic properties in vivo. PANC89 cells were injected subcutaneously and mice were treated with Vehicle, DT1154, INK128, or the combination. Consistent with our results in vitro, combination treatment significantly reduced tumor growth compared to either drug alone (Fig. 5A, B). In addition to decreased tumor size, histological analysis revealed a significant increase in tumor cell necrosis in combination-treated tumors relative to single agent or Vehicle-treated tumors (Fig. 5C, S9A). To assess apoptosis during therapeutic treatment, PANC89 cells were injected subcutaneously and tumors were harvested after 7 days of treatment with Vehicle, DT1154, INK128, or the combination. Similar to long-term treatment, short-term treatment with the combination of DT1154 and INK128 significantly decreased tumor growth and tumor weight, while simultaneously increasing necrosis (Fig. S9B-D). Combination treated tumors also displayed a significant increase in apoptotic (TUNEL+) cells within non-necrotic areas (Fig. 5D, S9E). To determine the effects of combination treatment on oncogenic pathways, tumors were lysed and analyzed by western blot. Consistent with our results in vitro, single agent treatment with DT1154 reduced MYC expression, but was unable to significantly alter AKT/mTOR signaling. Conversely, INK128 was able to reduce mTOR signaling, but had minimal effects on MYC expression. In contrast, the combination of DT1154 and INK128 resulted in a significant loss of both MYC and AKT/mTOR signaling pathway (Fig 5E, F).
Fig. 5:
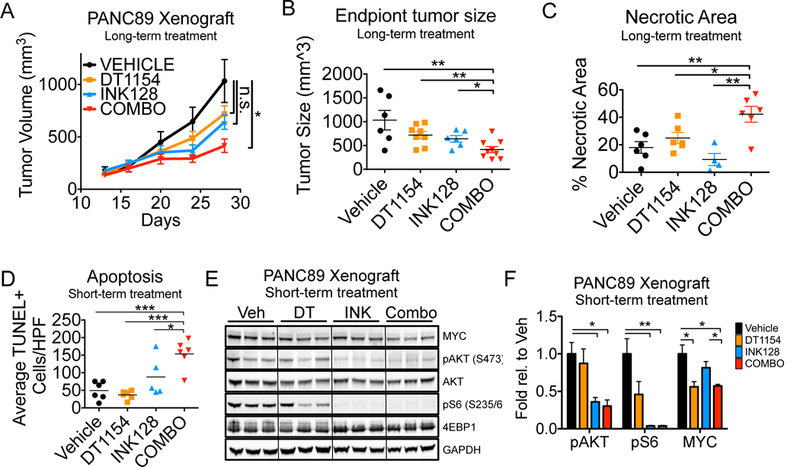
PP2A activation combined with mTOR inhibition decreases tumorigenic properties in vitro and in vivo A. PANC89 were grown subcutaneously and mice were treated with Vehicle, DT1154 (15mg/kg), INK128 (0.5 mg/kg), or combination. Tumors were calipered and tumor volume was plotted across time. Mean +/− SEM shown. n= 6 Vehicle, 8 DT1154, 6 INK128, and 9 combination treated tumors. Area under the curve was analyzed for each treatment arm, *p<0.05 by a 1-way ANOVA B. Quantification of end point tumor size from panel B. Mean +/− SEM shown. C. Quantification of necrotic tumor area from H/E images using ImageJ. Mean +/− SEM shown. D. PANC89 xenograft tumors were grown as in panel A and harvested after 7 days of treatment with Vehicle (Veh), DT1154 (DT, 15mg/kg), INK128 (INK, 0.5 mg/kg), or combination (Combo). Tissues sections were stained with the ApopTag Plus Peroxidase In Situ Kit and the number of TUNEL positive cells was quantified per high-powered field (HPF, 20x). ***p<0.001 *p<0.05 by a 1-way ANOVA. E. Lysates from tumors grown in panel D were analyzed by western blot for MYC, pAKT (S473), pS6 (S235/6), 4EBP1, and totals. F. Quantification of 3 tumors in each treatment group from panel E shown. Mean +/−SEM. **p<0.01 *p<0.05 by two-tailed students t-test
Together, these data suggest that inhibition of mTOR signaling can reduce PDA cell survival through a loss of the AKT/mTOR pathway; however, cells can circumvent this loss by maintaining MYC signaling (Fig. 6A- MYCHigh/mTORLow). Combined loss of MYC and mTOR, through the use of PP2A activators and mTOR inhibitors, results in a more complete loss of oncogenic signaling and a synergistic decrease in cell viability (Fig. 6B- MYCLow/mTORLow).
Fig. 6:
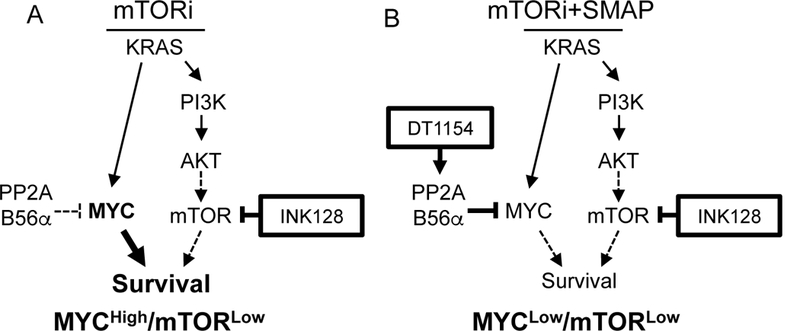
PP2A activation combined with mTOR inhibition as a therapeutic strategy to reduce PDA survival. A. Oncogenic KRAS drives the activation of both the MYC and PI3K/AKT/mTOR pathways. Upon mTOR inhibition, PDA cells capitalize on low PP2A levels, signaling through MYC for survival, creating a MYCHigh/mTORLow cell state. B. Activation of PP2A combined with mTOR inhibition results in low MYC and mTOR signaling in a MYCLow/mTORLow cell state, decreasing PDA cell survival.
Discussion
Pancreatic tumors have a diverse mutational profile that contributes to the redundant activation of survival pathways, rendering single agent targeted therapies clinically ineffective and supporting combination therapeutic strategies (5). Suppression of phosphatases significantly contributes to oncogenic progression in multiple cells types and we have demonstrated that PDA cells have reduced PP2A activity compared to normal cells (8, 43, 44). Attenuated PP2A activity may facilitate the rewiring of oncogenic pathways in response to kinase inhibition, leading to therapeutic resistance. Our current results suggest that restoring PP2A function can impact multiple oncogenic pathways, potentially restricting cell plasticity and increasing dependency on specific kinase pathways. We show that PP2A activation can synergistically increase the efficacy of several kinase inhibitors in PDA cells. We focus on mTOR inhibition and demonstrate that combination with PP2A activation results in synergistic cell death of pancreatic cancer cells in vitro and in vivo. This synergy is associated with the suppression of key pathways attributed to cancer cell survival and therapeutic resistance, indicating that PP2A activation may prevent signaling plasticity in response to targeted kinase inhibition.
In our studies, PP2A activation displayed synergy with inhibitors of the PI3K/AKT/mTOR signaling node, a pathway that is regulated at several points by PP2A. Specifically, PP2A has been shown to dephosphorylate AKT, mTOR, and the mTOR effectors 4EBP1 and S6K and conversely, mTOR is able to suppress PP2A activity (6). Further, mTOR and PP2A have been shown to function in a reciprocal regulatory network with CIP2A (a PP2A inhibitor) and c-MYC (a PP2A target), where CIP2A attenuates PP2A activity, increasing signaling through mTOR and MYC and subsequently activating feedback loops that increase expression of mTOR, MYC, and CIP2A (45). Together, these studies suggest that cellular alterations that shift the balance of these factors can drive molecular changes that contribute to oncogenesis. Indeed, several studies have highlighted the importance of mTOR signaling in PDA progression and aggressiveness, with components of the PI3K/AKT/mTOR pathway being altered in the majority of PDA patients (46).
While mTOR has been identified as an important target in PDA, several resistance mechanisms have been identified that decrease the clinical efficacy of mTOR inhibitors (21). Treatment with mTORC1 inhibitors, such as Rapamycin, can cause a significant increase in pAKT signaling, leading to therapeutic resistance. As such, compounds that target mTORC1 and 2 have displayed increased clinical efficacy, underscoring the importance of AKT inhibition to tumor cell survival. Here, we demonstrate that treating PDA cells with INK128, an mTORC1/2 inhibitor, results in almost a complete loss of mTORC1 signaling (pS6 and 4EBP1) and a suppression of mTORC2 signaling (pAKT). Consistent with the role of PP2A in regulating AKT phosphorylation, DT1154 in combination with INK128 further reduced pAKT signaling over INK128 treatment alone. Additionally, AKT has been shown to phosphorylate the mTOR inhibitor, PRAS40, at T246, inhibiting PRAS40 function. We found a significant decrease in phospho-PRAS40 in combination treated cells, indicating that AKT function is more completely suppressed. Together these studies suggest that increased PP2A activity in response to DT1154 can repress signaling plasticity and resistance that occurs in response to mTOR inhibition. These results are consistent with reports demonstrating that KPC mice are refractory to single agent mTOR inhibition and that combination therapy may be necessary to prolong survival (30).
The MYC oncoprotein plays an important role in tumor progression and therapeutic resistance in multiple cancer types (23, 47). In breast and prostate cancer, deregulated MYC expression increases resistance to PI3K/mTOR inhibitors (35, 36, 48). However, despite the known roles of MYC in resistance, therapeutic targeting of MYC remains a significant challenge. Here, treatment with DT1154, effectively reduced MYC levels in HPAFII and PANC89 cells. Furthermore, DT1154 combined with INK128 had a greater suppression of MYC in these cell lines. Interestingly, the combination of INK128 and DT1154 was not synergistic in MIAPACA2 and PANC1 cells, where MYC expression was refractory to treatment, despite efficient suppression of pAKT/mTOR signaling, suggesting that a MYCHigh/mTORLow cell state represents a resistant subpopulation. Consistent with this hypothesis, suppression of PP2A B56α or expression of MycT58A, resulted in an increased resistance to INK128 and a MYCHigh/mTORLow cell state in INK128 treated PANC89 cells. Conversely, transient knockdown or genetic loss of one copy of MYC was able to restore a MYCLow/mTORLow cell state in PANC1 and KPC cells and sensitize these cells to INK128 both in vitro and in vivo. Our previous studies demonstrated that OP449 was unable to decrease MYC pS62 in PANC1 cells (8). This line also happened to have the highest expression of CIP2A and SET out of the panel of PDA cell lines we analyzed, relative to the normal pancreatic epithelial line HPNE, indicating that this line has acquired mechanisms to suppress PP2A function. Future experiments will explore resistance mechanisms to DT1154 and investigate strategies to re-activate PP2A in these settings. Together, these results suggest that PP2A activating compounds can function synergistically with mTOR inhibitors and that negative regulation of MYC is important in this context to suppress MYC-driven resistance pathways and create a MYCLow/mTORLow cell state that is less viable.
In conclusion, we demonstrate that therapeutic activation of PP2A can increase the efficacy of kinase inhibitors, in part by limiting signaling crosstalk and plasticity. Further, we identify MYC as a potential resistance mechanism by which PDA cells escape mTOR inhibition. While mTOR inhibitors are not currently used as standard of care in pancreatic cancer patients, future studies will determine if MYC expression may be a biomarker of response to mTOR inhibitors in solid tumors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Michel Ouellette (University of Nebraska Medical Center) for providing cell lines, Drs. Marc Loriaux and Jeff Tyner (Oregon Health and Science University) for technical advice and the use of equipment to complete the kinase inhibitor screen, and all members of the RCS laboratory for editing the manuscript and other helpful suggestions. B. L. Allen-Petersen was supported by Tartar Research Fellowship Award and F32 CA192769. R.C. Sears was supported by R01 CA100855, CA186241, CA196228; U54 CA209988; DOD BC061306; Komen BCTR0706821; and the Brenden-Colson Foundation. Histopathology and Flow Cytometry Core services were supported by the OHSU Knight Cancer Institute Cancer Center Support Grant P30 CA69533. Finally, we thank the Brenden-Colson Center for Pancreatic Care for primary patient material and clinical support.
Footnotes
Conflict of Interest Statement:Dale J. Christensen is a shareholder in Oncotide Pharmaceuticals. Goutham Narla and Michael Ohlmeyer were on the Board of Dual Therapeutics, which had licensed DT1154 at the time of this study. All other authors have no conflicts of interest or competing financial interests to disclose. The Icahn School of Medicine at Mount Sinai, on behalf of Goutham Narla and Michael Ohlmeyer, has filed patents covering composition of matter on DT1154 and like small molecules for the treatment of human cancer and other diseases (International Application Numbers: PCT/US15/19770, PCT/US15/19764; and US Patent: US 9,540,358 B2). Rappta Therapeutics, Inc. (established September 7th, 2017) has licensed this intellectual property on 12/8/2017 for the clinical and commercial development of this series of small molecule PP2A activators. Dale J. Christensen, Goutham Narla, and Rosalie C. Sears are founders with ownership interest in Rappta Therapeutics, Inc. with no or minor compensation (under $10,000). Brett Sheppard has an advisory relationship with Pfizer with minor compensation (under $10,000).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015;2(1):4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chand S, O’Hayer K, Blanco FF, Winter JM, Brody JR. The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms. Int J Biol Sci. 2016;12(3):273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruvolo PP. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016;6:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14(4):152–60. [DOI] [PubMed] [Google Scholar]
- 8.Farrell AS, Allen-Petersen B, Daniel CJ, Wang X, Wang Z, Rodriguez S, et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol Cancer Res. 2014;12(6):924–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130(1):51–62. [DOI] [PubMed] [Google Scholar]
- 10.Xu P, Yao J, He J, Zhao L, Wang X, Li Z, et al. CIP2A down regulation enhances the sensitivity of pancreatic cancer cells to gemcitabine. Oncotarget. 2016;7(12):14831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connor CM, Perl A, Leonard D, Sangodkar J, Narla G. Therapeutic Targeting of PP2A. Int J Biochem Cell Biol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res. 2014;20(8):2092–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enjoji S, Yabe R, Fujiwara N, Tsuji S, Vitek MP, Mizuno T, et al. The therapeutic effects of SET/I2PP2A inhibitors on canine melanoma. J Vet Med Sci. 2015;77(11):1451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CY, Huang TT, Huang CT, Hu MH, Wang DS, Wang WL, et al. EGFR-independent Elk1/CIP2A signalling mediates apoptotic effect of an erlotinib derivative TD52 in triple-negative breast cancer cells. Eur J Cancer. 2017;72:112–23. [DOI] [PubMed] [Google Scholar]
- 15.Gutierrez A, Pan L, Groen RW, Baleydier F, Kentsis A, Marineau J, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest. 2014;124(2):644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest. 2017;127(6):2081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellizzi AM, Bloomston M, Zhou XP, Iwenofu OH, Frankel WL. The mTOR pathway is frequently activated in pancreatic ductal adenocarcinoma and chronic pancreatitis. Appl Immunohistochem Mol Morphol. 2010;18(5):442–7. [DOI] [PubMed] [Google Scholar]
- 18.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–20. [DOI] [PubMed] [Google Scholar]
- 19.Iriana S, Ahmed S, Gong J, Annamalai AA, Tuli R, Hendifar AE. Targeting mTOR in Pancreatic Ductal Adenocarcinoma. Front Oncol. 2016;6:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conciatori F, Ciuffreda L, Bazzichetto C, Falcone I, Pilotto S, Bria E, et al. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers (Basel). 2018;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guri Y, Hall MN. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer. 2016;2(11):688–97. [DOI] [PubMed] [Google Scholar]
- 22.Hassan Z, Schneeweis C, Wirth M, Veltkamp C, Dantes Z, Feuerecker B, et al. MTOR inhibitor-based combination therapies for pancreatic cancer. Br J Cancer. 2018;118(3):366–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farrell AS, Joly MM, Allen-Petersen BL, Worth PJ, Lanciault C, Sauer D, et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat Commun. 2017;8(1):1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. [DOI] [PubMed] [Google Scholar]
- 25.Christensen DJ, Chen Y, Oddo J, Matta KM, Neil J, Davis ED, et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: a predictor of aggressive disease and a new treatment target. Blood. 2011;118(15):4150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc Natl Acad Sci U S A. 2014;111(25):9157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen DJ, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, et al. Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J Immunol. 2011;186(4):2535–42. [DOI] [PubMed] [Google Scholar]
- 28.Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9(3):991–7. [PubMed] [Google Scholar]
- 29.Shields DJ, Murphy EA, Desgrosellier JS, Mielgo A, Lau SK, Barnes LA, et al. Oncogenic Ras/Src cooperativity in pancreatic neoplasia. Oncogene. 2011;30(18):2123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Driscoll DR, Karim SA, Sano M, Gay DM, Jacob W, Yu J, et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016;76(23):6911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. Febs J. 2016;283(6):1004–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Garcia C, Fazli L, Gleave M, Vitek MP, Jansen M, et al. Inhibition of Pten deficient Castration Resistant Prostate Cancer by Targeting of the SET - PP2A Signaling axis. Sci Rep. 2015;5:15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108(37):E699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7(11):787–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan J, Yu Q. Molecular mechanisms of tumor resistance to PI3K-mTOR-targeted therapy. Chin J Cancer. 2013;32(7):376–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14(19):2501–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Facchini LM, Chen S, Marhin WW, Lear JN, Penn LZ. The Myc negative autoregulation mechanism requires Myc-Max association and involves the c-myc P2 minimal promoter. Mol Cell Biol. 1997;17(1):100–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol. 2006;26(7):2832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6(4):308–18. [DOI] [PubMed] [Google Scholar]
- 42.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. [DOI] [PubMed] [Google Scholar]
- 43.Lambrecht C, Libbrecht L, Sagaert X, Pauwels P, Hoorne Y, Crowther J, et al. Loss of protein phosphatase 2A regulatory subunit B56delta promotes spontaneous tumorigenesis in vivo. Oncogene. 2017. [DOI] [PubMed] [Google Scholar]
- 44.Janghorban M, Langer EM, Wang X, Zachman D, Daniel CJ, Hooper J, et al. The tumor suppressor phosphatase PP2A-B56alpha regulates stemness and promotes the initiation of malignancies in a novel murine model. PLoS One. 2017;12(11):e0188910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De P, Carlson J, Leyland-Jones B, Dey N. Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A (CIP2A): an oncoprotein with many hands. Oncotarget. 2014;5(13):4581–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou L, Yuan D, Zhang ZG, Liang ZY, Zhou WX, Yang JY, et al. Expression of key mTOR pathway components in pancreatic ductal adenocarcinoma: A multicenter study for clinicopathologic and prognostic significance. Cancer Lett. 2017. [DOI] [PubMed] [Google Scholar]
- 47.Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan J, Yu Q. PDK1-driven Myc signaling regulates cellular response to mTOR inhibitors. Cell Cycle. 2011;10(7):1019–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.