

Abstract
For mucinous ovarian cancer (MOC), standard platinum based therapy is largely ineffective. We sought to identify possible mechanisms of oxaliplatin resistance of MOC and develop strategies to overcome this resistance. A kinome-based small interfering RNA (siRNA) library screen was carried out using human MOC cells to identify novel targets to enhance the efficacy of chemotherapy. In vitro and in vivo validations of antitumor effects were performed using mouse MOC models. Specifically, the role of PRKRA/PACT in oxaliplatin resistance was interrogated. We focused on PRKRA, a known activator of PKR kinase, and its encoded protein PACT because it was one of the five most significantly downregulated genes in the siRNA screen. In orthotopic mouse models of MOC, we observed a significant anti-tumor effect of PRKRA siRNA plus oxaliplatin. In addition, expression of miR-515–3p was regulated by PACT-Dicer interaction, and miR-515–3p increased the sensitivity of MOC to oxaliplatin. Mechanistically, miR-515–3p regulated chemosensitivity, in part, by targeting AXL. The PRKRA/PACT axis represents an important therapeutic target in MOC to enhance sensitivity to oxaliplatin.
Keywords: PRKRA, PACT, Dicer, oxaliplatin, mucinous ovarian cancer, chemosensitivity, miRNA, siRNA
Introduction
Ovarian cancer represents multiple diseases with different clinicopathological and molecular features (1). Mucinous ovarian carcinoma (MOC) is a relatively rare subset (2–10% of epithelial ovarian cancer) (2, 3). While standard therapy for ovarian cancer includes taxane- and platinum-based chemotherapy (4, 5), such therapy is largely ineffective in patients with MOC. This is, in part, responsible for the poorer outcomes associated with MOC compared to other epithelial ovarian cancers (6–8). Therefore, new treatments are needed to improve MOC outcomes.
Recently reported evidence demonstrated that the pathogenesis of MOC might be similar to that of colorectal carcinomas (9–11). Most colorectal carcinomas are of the mucinous histological subtype, and patients with advanced colorectal cancer are treated with oxaliplatin (third-generation platinum compound) based chemotherapy (12). Oxaliplatin has proven efficacious against many types of cancers, but the development of resistance to this drug is a frequent obstacle (13). Therefore, to enhance the efficacy of oxaliplatin in the treatment of MOC, we performed a kinome-based small interfering RNA (siRNA) library screen in the presence of oxaliplatin to identify candidate targets that enhance oxaliplatin sensitivity. We found that knockdown of PRKRA, which is a known cellular protein activator of PKR kinase (14, 15), enhanced oxaliplatin sensitivity of MOC cells. Also, PACT, which is the protein encoded by PRKRA, is known to be a double stranded-RNA binding protein that impacts microRNA maturation in humans through interactions with Dicer. We further examined the role of PACT in MOC by identifying underlying mechanisms by which it promotes oxaliplatin resistance.
Materials and Methods
Reagents and antibodies
Oxaliplatin (Wyeth/Pfizer) was obtained from The University of Texas MD Anderson Cancer Center pharmacy. The primary antibodies used in Western blotting were anti-PACT (Santa Cruz Biotechnology), anti-Dicer (Bethyl Laboratories), anti-β-actin (Sigma-Aldrich), anti-Ki67 (Thermo Fisher Scientific), and anti-cleaved caspase 3 (Cell Signaling Technology) antibodies. The following secondary antibodies were used in Western blotting: horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G, horseradish peroxidase-conjugated rat anti-mouse immunoglobulin G2a (Serotec and Harlan Bioproducts for Science), and horseradish peroxidase-conjugated donkey anti-goat immunoglobulin G (Santa Cruz Biotechnology). The antibody concentrations are listed in Supplementary Table S1.
Cell lines and culture
RMUG-L-ip1 and RMUG-S-ip1 MOC cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 0.1% gentamicin at 37°C in 5% CO2 and 95% air. These cell lines were originally isolated from patients with MOC (16). Ovarian carcinoma cell lines (HeyA8, HeyA8-MDR, SKOV3, SKOV3-TR, A2780 and A2780-CP20), and the normal human surface epithelial cell line HIO180 were cultured in RPMI 1640 medium supplemented with 15% fetal bovine serum and 0.1% gentamicin at 37°C in 5% CO2 and 95% air. In vitro experiments were conducted at 80% cell confluence. All cell lines were purchased from the MDACC Characterized Cell Line Core, ATCC, or Japanese Collection of Research Bioresources (Ibaraki, Japan). All cell lines have been validated by STR fingerprint method through the MDACC Characterized Cell Line Core. Each cell line was never continuously passaged in culture for more than 3 months, and after that, a new vial of frozen cells was thawed. All cell lines were routinely tested to confirm the absence of Mycoplasma, and all in-vitro experiments were conducted with 60–80% confluent cultures.
SiRNA transfection
All siRNA transfections (Supplementary Table S2) were performed using RNAiMAX reagent (Invitrogen) with the forward transfection protocol from the manufacturer. Media was changed 6 hours after transfection to minimize toxicity.
Cell viability assay
Cells were plated in each well of a 96-well plate and maintained overnight. They were then exposed to oxaliplatin at a concentration of 0.1, 1, 3, 10, 30, or 100 μmol/L for 72 hours. The control groups were treated with an equal volume of vehicle. To assess cell survival, 50 μL of 0.15% MTT (Sigma-Aldrich) was added to each well, and the cells were incubated for 2 hours at 37°C. The medium containing MTT was removed from the wells, and 100 μL of dimethyl sulfoxide (Sigma-Aldrich) was added to the wells. The cells were then incubated at ambient temperature for 10 minutes. Absorbance in each well was read at 570 nmol/L using a 96-well Synergy HT microplate reader (Ceres UV 900C; BioTek). Cell survival was defined as the percentage of surviving cells relative to the percentage of control cells. The experiments were repeated at least three times.
For siRNA transfection, cells were transfected 6 hours and 48 hours after plating. Chemotherapy treatment was started 72 hours after plating the cells. Cells were incubated for 24, 48, 72 or 96 hours after chemotherapy treatment, and then the MTT assay was performed.
Colony formation assays
Colony formation assay was performed as described by Wang et al. (17); briefly, 500 untreated, or siRNA-treated RMUG-S-ip1 cells were placed into 12-well coated high-adhesion plates (Corning), and cultured for 8 days. Plates were then washed with 1 mL PBS, fixed and stained with methanol containing crystal violet solution for 5 minutes (0.025% w/v; Sigma). The number of colonies was counted in each well, excluding small (<50 cells) colonies.
Apoptosis assay
Apoptosis of MOC cells was evaluated using an FITC Annexin V Apoptosis Detection Kit (BD Biosciences), as described previously (18). Briefly, cells were incubated in trypsin-ethylenediaminetetraacetic acid, and cell pellets were suspended in 200 μL of 1× annexin V binding buffer. Each cell suspension was incubated with 4 μL of FITC-Annexin V and 4 μL of propidium iodide at ambient temperature (25°C) in the dark for 30 minutes. Then, 400 μL of 1× binding buffer was added to each tube containing MOC cells, and these samples were analyzed using fluorescence-activated cell sorting. Each experiment was performed in triplicate.
Western blotting
Western blotting was performed, as previously described (18, 19). Briefly, cells were lysed with RIPA lysis buffer and centrifuged for 15 minutes at 4°C. Protein concentration was assessed using a Bio-Rad protein assay kit. The protein was loaded into gels (20 μg/well), and the bands were separated using electrophoresis. Bands were transferred to nitrocellulose membranes, blocked with 5% milk for 1 hour at ambient temperature, and incubated with primary antibodies against PACT or Dicer (1:1000 dilution) overnight at 4°C. Samples of proteins were incubated with horseradish peroxidase-conjugated anti-mouse and anti-rabbit antibodies (GE Healthcare) or an anti-goat antibody (Santa Cruz Biotechnology) for 1 hour at ambient temperature. Blots were developed and analyzed, as described previously (18, 19). Loading control (actin) was used; all experiments were performed in duplicate.
Quantitative real-time polymerase chain reaction
The QIAGEN RNeasy kit was used for quantifying mRNA, as described previously (19). For complementary DNA synthesis, a Verso cDNA kit (Thermo Fisher Scientific) was used per manufacturer’s instructions. mRNA levels were measured using a 7500 Fast Real-Time polymerase chain reaction (PCR) System (Applied Biosystems) with SYBR Green-based PCR for all genes as described previously (19). The specific primers used are listed in Supplementary Table S3. Semi-quantitative real-time PCR analysis of mRNA levels was performed with reverse-transcribed RNA and 1 μM sense and antisense primers in a total volume of 20 μL. For microRNA (miRNA) quantification, total RNA was isolated from cells using TRIzol extraction reagent (Invitrogen). For mature miRNA quantification, we used TaqMan miRNA assays (Life Technologies), and real-time reverse-transcription PCR was carried out per manufacturer’s instructions. Precursor miRNAs in cells were quantified using miScript precursor miRNA assays (QIAGEN). RNU6B (for mature miRNAs) and 18S (primary and precursor miRNAs) were used as housekeeping genes.
Target gene-binding sites and luciferase reporter assays for the AXL 3’-UTR
The putative miRNA binding sites on the AXL 3’-UTR were predicted via bioinformatics analysis using several algorithms for predicting miRNA targets using the following public sites: http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/index.html (miRWalk 2.0) (20). Candidate were tested for validation with real-time PCR. The pGL3 Luciferase Reporter Vectors for the predicted binding sites of the 3’-UTR region of AXL were obtained from Promega (Madison).
RMUG-S-ip1 cells were transfected with FuGene HD transfection reagent in a 96-well plate with scrambled control or miR-515–3p mimic (100 nm; Ambion) along with the 3’-UTR reporter gene and Renilla luciferase vector control construct (pTK-CLuc Vector). After 48 h of transfection, luciferase activity was determined with the Dual-Luciferase Reporter Assay System Kit using a microplate luminometer per the manufacturer’s guidelines (Biotek, Winooski). Luciferase activity was normalized against the pTK-CLuc Vector control construct, and an empty luciferase reporter vector was used as a negative control. The ratios obtained were further normalized according to the scrambled control.
Animal care
Nude mice (8–12 weeks old, athymic female, Ncr-nu) were obtained from Taconic Biosciences (Rensselaer, NY). The mice were housed and kept in a specific pathogen-free environment in the animal facility as described previously (18). The study protocols were approved and supervised by the Institutional Animal Care and Use Committee at MD Anderson.
Liposomal nanoparticle preparation
Therapeutic siRNA for in vivo experiments was incorporated into DOPC nanoliposomes as described previously (21). Briefly, the mixture was lyophilized and stored. Before in vivo administration, the lyophilized material was hydrated with phosphate-buffered saline at ambient temperature at a concentration of 200 μg of siRNA per kilogram per injection.
In vivo therapeutic experiments
To generate orthotopic models of MOC, RMUG-L-ip1 or RMUG-S-ip1 cells were injected into the peritoneal cavity of 40 nude mice (4 × 106 cells/mouse). The mice were randomly assigned to one of four groups of 10 mice each according to treatment (control siRNA DOPC, control siRNA DOPC plus oxaliplatin, PRKRA siRNA [siPRKRA] DOPC, and siPRKRA DOPC plus oxaliplatin), and treatment was initiated at 4 weeks after injection. Oxaliplatin (5 mg/kg/mouse) was given intraperitoneally twice weekly after being dissolved in phosphate-buffered saline. This dosage resulted in approximately 50% reduction in tumor growth in mucinous cancer models (22) and allowed for testing the effects of combinations of oxaliplatin with other biologically targeted drugs. A dose of 200 μg of siRNA per kilogram per mouse was packaged in DOPC nanoliposomes and delivered intraperitoneally twice weekly as described previously (21). Mice were monitored daily and weighed weekly. After 8 weeks of treatment, the mice were killed, and their total body weights, tumor locations and weights, and tumor nodule numbers were recorded. Tumor samples were fixed in 10% formalin and embedded in paraffin or with optimal cutting temperature compound in liquid nitrogen.
Statistical analysis
The distribution of continuous variables was assessed (Kolmogorov-Smirnov test) and expressed as appropriate (mean ± standard error or median with range). The Student t-test or the Mann-Whitney U test was conducted to determine statistical significance. Categorical variables were evaluated using the Fisher exact test (odds ratio and 95% confidence interval). P values less than 0.05 were considered statistically significant (all tests, two-tailed). The SPSS software program (version 18.0; IBM Corporation) was used for all statistical analyses.
RNA-IP
RMUG-S-ip1 mucinous ovarian cells were plated at the amount of 1×10^6 cells per 15 cm plate. For the siRNA portion of the experiment, 4μg of non-targeting or PRKRA siRNA was transfected using the protocol mentioned above. For the overexpression portion, 4 μg of empty vector, PACT-overexpressing vector, and PACT-mDR3 mutant vector were transfected using FuGENE (Promega) at a ratio of 1 μg vector to 3 μL of FuGENE reagent. Cells were left in media containing the transfection mixture for 4 hours, then fresh complete media was added; 3 days later, the cells were harvested and subjected to the RIP-Assay kit (MBL). RNA was co-immunoprecipitated with AGO2 using the anti-AGO2 antibody (MBL). RNA Immunoprecipitates were then eluted and subject to Taqman qRT-PCR to detect the presence of the miRNAs of interest, using the protocol as described above. The fold enrichment was determined by comparing the fold enrichment of the AGO2-immunoprecipitated miRNAs relative to the control IgG-immunoprecipitated miRNAs.
High-throughput kinome siRNA screening and process for identifying target genes
For kinome siRNA screening, a total of 1278 siRNAs targeting 639 individual human kinase genes were preprinted on 384-well plates along with staggered negative and positive control siRNA, as described previously (23). The kinome screening experiments were performed in duplicate for RMUG-L-ip1 cells. For each experiment, plates preloaded with siRNA were thawed and 20 μL/well-diluted Dharmafect 3 (Dharmacon, Denver, CO) solution was added. After 30 minutes, 1000 RMUG-L-ip1 cells in 20 μL of medium were added to each well. Cell viability was determined at 96 hours by CellTiter-Glo luminescence assay read on a BMG Polarstar machine using excitation 544-nm/emission 590-nm filters. To identify target genes, we examined first that target gene-siRNA alone does not affect cell viability. Then, we checked whether the target gene-siRNA plus oxaliplatin showed greater inhibition of cell viability compared with target gene-siRNA alone. Through these criteria, we selected the 5 target genes.
Nanostring
For each sample, 100 ng of total RNA was analyzed with nCounter Human miRNA expression Assay Kit (NanoString Technologies). Negative control probes were also used. Raw data was analyzed with nSolve (NanoString Technologies). Data were normalized by calculating the geometric mean of the top 100 miRNAs in all samples, as recommended by NanoString. For calculating p-values, the LIMMA package (Linear Models for Microarray Data) from the Bioconductor R project was used. Raw data are available at NCBI GEO: GSE119845.
Results
PACT expression is increased in MOC cells and plays a role in chemoresistance
To identify candidate genes that modulate the response of MOC cells to oxaliplatin-based treatment, we performed a kinome-based siRNA library screen of 939 genes (Supplementary Table S4). We evaluated the efficacy of oxaliplatin in this study because prior studies demonstrated that it was more effective against MOC than were other platinum agents (22). For this screen, we selected RMUG-L-ip1 MOC cells, as they demonstrate oxaliplatin resistance (Supplementary Table S5). After we independently tested the efficacy of silencing the top five genes that sensitized MOC cells to oxaliplatin, we selected PRKRA as the target gene for combination therapy with oxaliplatin. That’s because siPRKRA plus oxaliplatin produced the greatest inhibition of MOC cell viability relative to control siRNA plus oxaliplatin (Fig. 1A; Supplementary Fig. S1A), while siPRKRA alone did not affect cell viability in MOC cells (Supplementary Fig. S1B). To demonstrate the silencing efficiency of siPRKRA, we transfected RMUG-L-ip1 and RMUG-S-ip1 cells with control siRNA or two different siPRKRA sequences. An expression analysis using quantitative real-time PCR and Western blotting demonstrated that PRKRA and its encoded protein, PACT, were silenced by both siPRKRA sequences (Supplementary Fig. S1C). To ascertain the involvement of PACT in chemoresistance, we performed colony formation assays and Western blot for analysis of cleaved caspase 3. We observed that treatment with siPRKRA plus oxaliplatin resulted in decreased colony formation (Fig 1B) and increased levels of cleaved caspase 3 (Supplementary Fig. S1D) more so than did treatment with control siRNA plus oxaliplatin in MOC cells.
Figure 1.
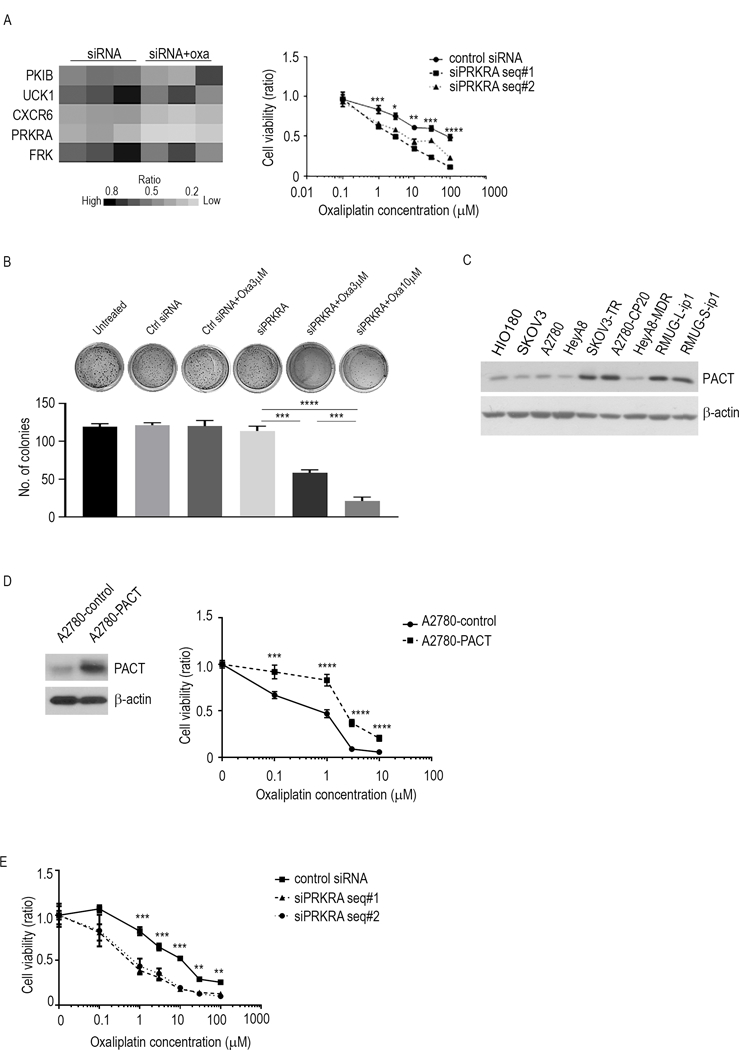
PRKRA expression and function in MOC. A, Results from the kinome-based siRNA library screen. Left, heat map with genes according to the ratio of MOC samples given siRNA to the samples given siRNA plus oxaliplatin. Right, cell viability assay for RMUG-L-ip1 cells given combination therapy with oxaliplatin and siPRKRA. The experiment was performed 72 hours after treatment. B, Colony formation assay for RMUG-S-ip1 cells given combination therapy with oxaliplatin and siPRKRA or control siRNA. C, Western blot analysis of PACT expression in non-transformed epithelial ovarian cells (HIO-180) and in ovarian cancer cell lines. D, Cell viability assay for A2780 cells treated with oxaliplatin. The cells were transfected with either a control vector or the pcDNA-PACT vector. The experiment was performed 72 hours after treatment. E, Cell viability assay for A2780-CP20 cells given combination therapy with oxaliplatin and siPRKRA. The experiment was performed 72 hours after treatment. The data are presented as means ± standard error of the mean for at least three experimental groups. **P < 0.01; ***P < 0.001; ****P < 0.0001 (Student t-test).
We next evaluated PACT protein expression in eight ovarian cancer cell lines and one non-transformed human ovarian surface epithelial cell line (HIO180). The level of PACT expression in RMUG-L-ip1 and RMUG-S-ip1 cells was higher than that in chemosensitive ovarian cancer cell lines according to the ratio of PACT to β-actin expression (Fig. 1C). To ascertain the functional impact of PACT expression on chemoresistance of MOC, we performed a gain-of-function analysis of PACT in ovarian cancer cells. Because MOC is characterized by chemoresistance, we used the ovarian cancer cell line A2780, which is highly sensitive to oxaliplatin. We transfected these cells with a PACT expression vector or control empty vector and determined whether PACT overexpression promotes chemoresistance. We observed that the PACT-overexpressing (A2780-PACT) cells had 4.1-fold greater resistance to oxaliplatin than did the control (A2780-control) cells (Fig. 1D, Supplementary Fig. S1E). Moreover, we examined the viability of A2780-CP20 cells, which are resistant to oxaliplatin, to determine whether PRKRA knockdown causes oxaliplatin sensitivity in cells of other histological subtypes. The A2780-CP20 cells had greater sensitivity to oxaliplatin than the control upon knockdown of PRKRA expression (Fig. 1E, Supplementary Fig. S1F).
The combination of siPRKRA and oxaliplatin has enhanced anti-tumor efficacy against MOC
To identify potential tumor-promoting pathways regulated by PRKRA, we analyzed mRNA expression data from RNA sequencing of RMUG-S-ip1 cells transfected with siPRKRA or control siRNA. Heat maps of the expression of the 78 most downregulated genes (ratio, <0.8) and the 47 most upregulated genes (ratio, >1.3) in PRKRA-silenced RMUG-S-ip1 cells are shown in Supplementary Fig. S2A. Ingenuity Pathway Network analysis of mRNA sequencing data identified the “cell death and survival-related” pathway and the “cellular growth and proliferation-related” pathway as the two most impacted pathways of downregulated genes after siPRKRA treatment (Fig. 2A). Conversely, we identified the “cellular growth and proliferation-related” pathway as the most impacted pathway in the context of upregulated genes (Supplementary Fig. S2B).
Figure 2.
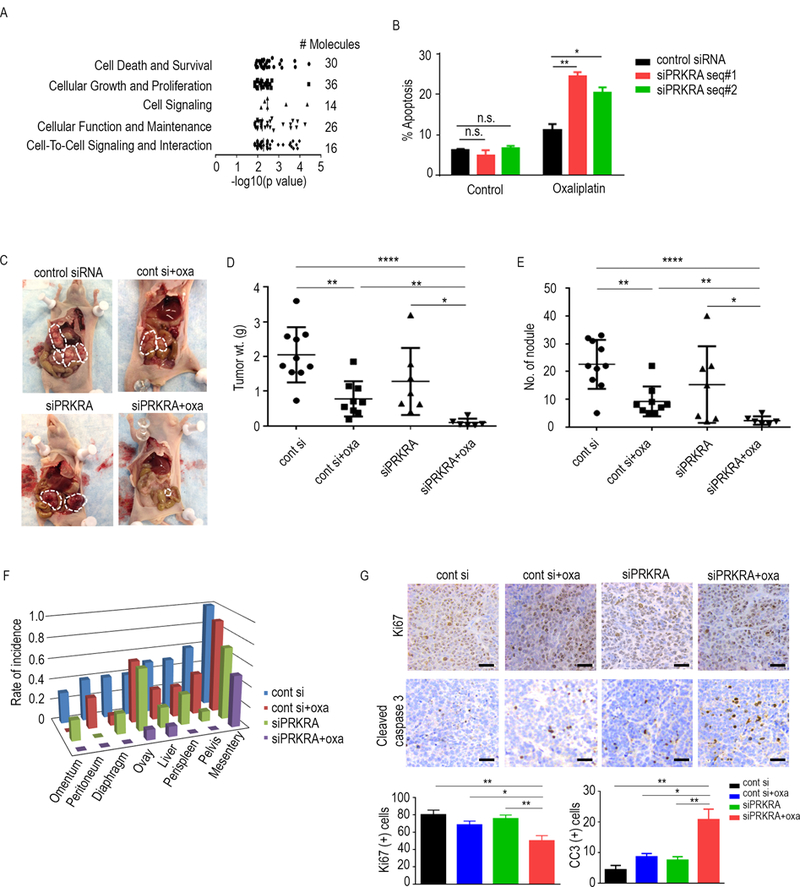
Anti-tumor effects of the combination of oxaliplatin and siPRKRA. A, The five molecular functions most significantly altered as a result of downregulated mRNAs under PRKRA-silenced conditions. Data are from RNA-sequencing of cells transfected with siPRKRA or control siRNA. The data were subjected to Ingenuity Pathway Network analysis. B, Apoptosis assay for oxaliplatin and siPRKRA in RMUG-L-ip1 cells at the end of 72-hour time point. Cells were transfected 6 hours and 48 hours after plating. Chemotherapy treatment was started 72 hours after plating the cells. The oxaliplatin dose used for the assay was 1 μM. C, Representative images of the extent of metastatic spread of MOC in mice given a control siRNA, control siRNA plus oxaliplatin, siPRKRA, or siPRKRA plus oxaliplatin (n = 10 per group). D and E, tumor weights (D) and nodule numbers (E) in each mouse group 43 days after RMUG-L-ip1 cell injection. F, Distribution of metastatic nodules in each mouse group. G, Immunohistochemical staining of MOC samples obtained from RMUG-L-ip1 orthotopic models for Ki67 and caspase 3. Scale bar=100μm. The in vitro data are presented as means ± standard error of the mean for at least three experimental groups. *P < 0.05; **P < 0.01; ****P < 0.0001 (Student t-test).
To identify potential mechanisms by which the combination of siPRKRA and oxaliplatin exerts its anti-tumor activity, we tested its effects on MOC cell viability and apoptosis. Treatment with siPRKRA plus oxaliplatin consistently resulted in decreased viability of RMUG-L-ip1 and RMUG-S-ip1 cells more than treatment with control siRNA plus oxaliplatin (Fig. 1A; Supplementary Fig. S3A). The combination of siPRKRA and oxaliplatin induced similar late apoptosis in both cell lines more than treatment with control siRNA plus oxaliplatin (Fig. 2B; Supplementary Fig. S3B).
Because combination therapy with siPRKRA and oxaliplatin was effective against MOC cells in our in vitro study, we next evaluated the effects of this combination in RMUG-L-ip1 (Fig. 2C) and RMUG-S-ip1 (Supplementary Fig. S3C, D and E) mouse models. In these models, mice were given siPRKRA and oxaliplatin had 90% lower tumor weights (P < 0.0001) and 90% fewer tumor nodules (P < 0.0001) than did mice given control siRNA plus oxaliplatin (Fig. 2D and E; Supplementary Fig. S3C and D). The tumor metastasis rate, including at sites such as the mesentery and pelvis, was markedly lower in the combination treatment groups than in the other groups (Fig. 2F; Supplementary Fig. S3E). To determine the biological effects of the combination of siPRKRA and oxaliplatin, we used the RMUG-L-ip1 tumor model to examine proliferation (Ki67 staining) and apoptosis (caspase 3) rates. We observed fewer Ki67-positive cells in tumors treated with siPRKRA plus oxaliplatin than in tumors in the other groups. Also, the percentage of caspase 3-positive cells was higher in mice given siPRKRA plus oxaliplatin than in mice given control siRNA plus oxaliplatin (Fig. 2G), indicating both decreased proliferation, and greater apoptosis in the tumors treated with siPRKRA plus oxaliplatin.
PACT-mediated downregulation of miR-515–3p expression leads to increased chemoresistance
Because our preliminary data indicated that silencing of PRKRA upregulated the expression of some mature miRNAs, we sought to determine whether PACT’s binding affinity for Dicer affects the sensitivity of MOC cells to oxaliplatin. To determine which domain of PACT is required for Dicer binding in MOC cells, we created three point mutants of binding sites (mDR1, mDR2, and mDR3) as reported previously (24). Specifically, we performed point mutagenesis to alter the highly conserved amino acids in double-stranded RNA-binding domains (dsRBDs). DsRBD1 was mutated at the two conserved alanine residues (A91 and A92) to lysine residues in mDR1. Similarly, the conserved alanine residues at A185 and A186 of dsRBD2 and the alanine residues at A289 and A290 of dsRBD3 were converted to lysine residues in mDR2 and mDR3, respectively. We fused these mutants to a FLAG epitope at the N-termini and induced co-expression with myc-Dicer and performed co-immunoprecipitation experiments. The PACT mutants mDR1 and mDR2 efficiently bound to Dicer, whereas the C-terminal mutant mDR3 did not interact with Dicer (Supplementary Fig. S4A). Therefore, dsRBD3 of PACT may be involved in the interaction of PACT with Dicer in MOC cells.
We next performed a miRNA microarray analysis of MOC cells transfected with siPRKRA or control siRNA. We observed increased expression of certain miRNAs after silencing PRKRA (Fig. 3A, Supplementary TableS6). Using quantitative real-time PCR, we analyzed the expression of significantly upregulated miRNAs (Fig. 3A) in cells with silenced PRKRA. We found a marked increase in the levels of six of the seven miRNAs that we tested (Fig. 3B). We then aimed to determine whether PACT-Dicer interaction is responsible for biogenesis of these miRNAs. Upon our analysis of mature miRNA expression in MOC cells using quantitative real-time PCR, we observed that the wild-type PACT-containing plasmid inhibited mature miR-515–3p and miR-519–3p expression, whereas mDR3 expression did not result in any changes in the expression of mature miR-515–3p or miR-519–3p (Fig. 3C; Supplementary Fig. S4B). This result suggested that PACT negatively regulates miR-515–3p and miR-519–3p biogenesis by binding to Dicer in MOC cells. In addition, we performed RNA-IP to confirm the binding of miR-515–3p and miR-519–3p to AGO2 in the context of PRKRA knockdown as well as PACT wild-type and mDR3 overexpression. In all settings, we observed binding of these miRNAs and AGO2 (Fig. 3D, Supplementary Fig. S4C).
Figure 3.
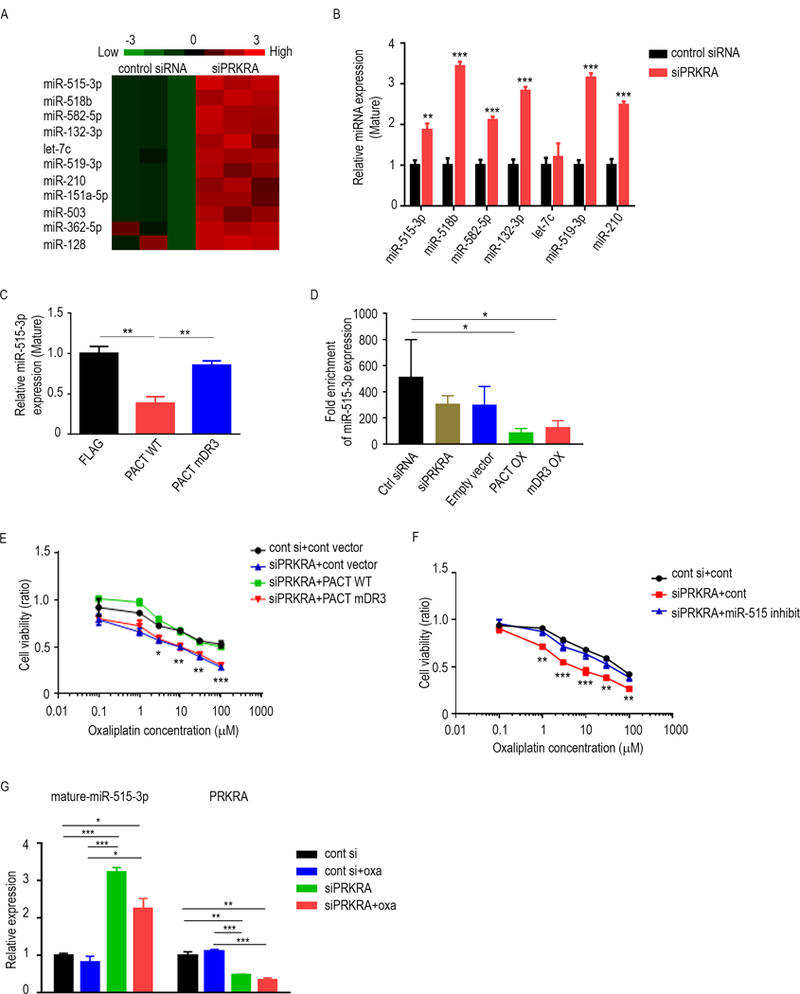
Downregulation of miR-515–3p expression by PACT leads to increased chemosensitivity of MOC. A, Heat map showing the 11 most upregulated mature miRNA levels under PRKRA-silenced conditions assessed using miRNA array data. B, Expression levels for significantly altered mature miRNAs in PRKRA-silenced in RMUG-L-ip1 cells. C, Mature miR-515–3p expression levels in RMUG-S-ip1 cells transfected with a plasmid containing FLAG, wild-type PACT, or PACT mDR3. D, RNA immunoprecipitation (RIP) analysis of miR-515–3p expression in RMUG-S-ip1 cells transfected with control siRNA, siPRKRA, empty vector, wild-type PACT plasmid or PACT mDR3 plasmid. E, Cell viability assay for PRKRA-silenced RMUG-L-ip1 cells transfected with a wild-type PACT-containing plasmid or mutant plasmid. The cells were treated with oxaliplatin. F, Cell viability assay of PRKRA-silenced RMUG-L-ip1 cells transfected with a miR-515–3p inhibitor or control antisense inhibitor. The cells were treated with oxaliplatin. G, Mature miR-515–3p and PRKRA expression levels in tumors obtained from RMUG-L-ip1 mouse models which treated with control siRNA, control siRNA plus oxaliplatin, siPRKRA, or siPRKRA plus oxaliplatin. Data are presented as means ± standard error of the mean for at least three experimental groups. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (Student t-test).
Next, we asked whether the PACT-Dicer interaction and subsequent impact on miR-515–3p and/or miR-519–3p expression regulates chemosensitivity to oxaliplatin in MOC cells. First, we transfected mDR3 into RMUG-S-ip1 cells and performed a cell viability assay. While cells transfected with siPRKRA and a control vector had decreased viability after treatment with oxaliplatin, the viability of those transfected with siPRKRA and a wild-type PACT-containing vector was restored. In contrast, transfection of mDR3 did not restore cell viability after treatment with oxaliplatin alone (Fig. 3E, Supplementary Fig. S4D). These results suggest that PACT-Dicer binding affects chemoresistance of MOC.
Next, we tested whether miR-515–3p and miR-519–3p regulate chemosensitivity of MOC cells. Resistance to oxaliplatin was enhanced by exposure to a miR-515 inhibitor in MOC cells. Moreover, cells transfected with siPRKRA and the miR-515 inhibitor were more viable than cells transfected with siPRKRA alone (Fig. 3F, Supplementary Fig. S4E). In addition, we measured miR-515–3p expression in tumors obtained from RMUG-L-ip1 mouse models. Consistent with the in vitro results, miR-515–3p expression was higher in tumors from siPRKRA or siPRKRA plus oxaliplatin models than in tumors from other groups (Fig. 3G, Supplementary Fig. S4F) while siPRKRA or siPRKRA plus oxaliplatin did not affect Dicer expression in tumors (Supplementary Fig. S4G). These results suggest that miR-515–3p is a regulator of chemosensitivity in MOC cells.
MiR-515–3p regulates MOC chemosensitivity via AXL
To examine the mechanism by which miR-515–3p regulates chemosensitivity of MOC, we compared global differences in gene expression profiles between miR-515–3p–overexpressing RMUG-S-ip1 and control cells using a gene microarray. Heat maps of the expression of significantly downregulated and upregulated genes in miR-515–3p–overexpressing RMUG-S-ip1 cells are shown in Supplementary Fig. S5A. Using Ingenuity Pathway Analysis, we identified that for the downregulated genes, the most impacted cellular functions were related to “cell death and survival” (Fig. 4A) and for the upregulated genes, “DNA replication, recombination, and repair” (Supplementary Fig. S5B). We then specifically examined the genes related to these functions, as these pathways may hold relevance for chemoresistance. To determine whether the expression of these genes is regulated by PACT, we compared gene expression profiles between siPRKRA RMUG-S-ip1 cells and siControl cells using a gene array (Supplementary Fig. S2A). Upon integrating the list of downregulated genes in miR-515–3p–overexpressing cells and PRKRA-silenced cells, we identified AXL to be a potential mediator of chemosensitivity in MOC cells. To address this, we first determined whether miR-515–3p regulates AXL expression. We observed that cells transfected with a miR-515 mimic had lower AXL expression than control cells (Fig. 4B). We next examined whether PACT regulates AXL expression. We observed that cells transfected with siPRKRA also had lower AXL expression relative to the control (Fig. 4C). We then determined whether AXL regulates chemosensitivity of MOC cells. Cells transfected with AXL siRNA were less viable than cells transfected with control siRNA when combined with oxaliplatin (Fig. 4D). In addition, we examined whether miR-515–3p targets AXL in MOC cells. We performed a luciferase assay to examine the regulatory effects of miR-515–3p on the AXL 3’-UTR. When the reporter gene vector was co-expressed with miR-515 mimic, AXL 3’-UTR expression was reduced by 38% (Fig 4E). We also determined whether AXL regulates cell death in MOC cells. By using plasmid which does not have AXL-3’UTR, we performed apoptosis assay. When MOC cells were co-transected with the plasmid and siPRKRA, the effect of oxaliplatin was reduced compared with cells co-transfected siPRKRA and miR-515 inhibitor (Supplementary Fig. S5C).
Figure 4.
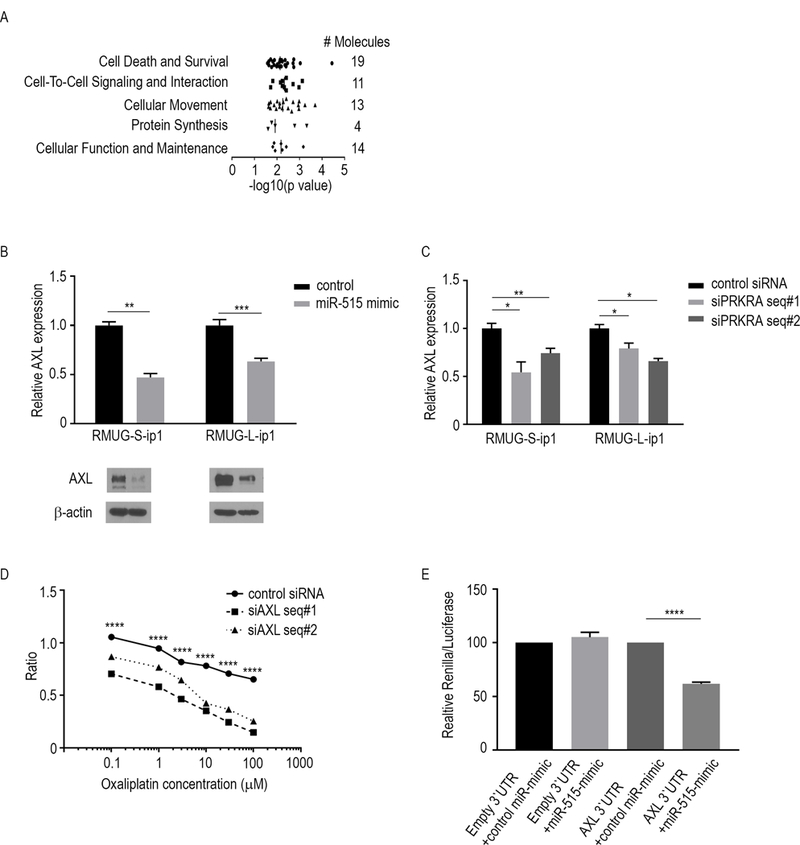
MiR-515–3p regulates the chemosensitivity of MOC via AXL. A, The five molecular functions most significantly altered as a result of downregulated mRNA expression following miR-515-transfection. The data are from RNA-sequencing of MOC cells transfected with a miR-515 mimic or control mimic. The data were subjected to Ingenuity Pathway Network analysis. B, AXL mRNA (top) and protein (bottom) expression levels in RMUG-L-ip1 and RMUG-S-ip1 cells transfected with a miR-515 mimic or control mimic. C, AXL mRNA expression levels in RMUG-L-ip1 and RMUG-S-ip1 cells transfected with siPRKRA or a control siRNA. D, Cell viability assay for oxaliplatin sensitivity in RMUG-S-ip1 cells transfected with AXL siRNA (siAXL). The experiment was performed for 72 hours. E, Untreated RMUG-S-ip1 cells were co-transfected with wild-type or AXL 3’UTR and the miR-515 mimic or negative control. After 48 h, luciferase/Renilla activity was measured. The data are presented as means ± standard error of the mean for at least three experimental groups. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (Student t-test).
Discussion
The key finding in this work is that treatment with siPRKRA plus oxaliplatin had a significant anti-tumor effect on MOC cells. This is important because MOC is known to have poorer outcomes and is more chemoresistant than other subtypes of ovarian carcinoma (2, 3, 22, 25).
Previously published work has reported that in complex with Dicer, PACT suppresses the processing of precursor miRNA substrates (26). In addition, PACT can affect various sizes of miRNAs, thereby influencing target-binding properties. Experiments with PACT variants showed that the two N-terminal RNA-binding domains of this protein have differences in double-stranded RNA substrate identification and processing of the Dicer–double-stranded RNA-binding protein complex. The full role of PACT in the miRNA pathway remains to be determined (27).
In the present study, we demonstrated that PACT regulates the chemosensitivity of MOC cells. Using a binding site mutant that cannot bind to Dicer, we also found that PACT-Dicer interaction affects PACT-mediated chemoresistance. Additionally, using this binding site mutant, we demonstrated that PACT negatively regulates miRNA processing in MOC cells, and that knockdown of PACT expression results in increased levels of mature miR-515–3p. Decreased expression of Dicer in cancer cells is associated with poor clinical outcomes (28–31). Recent reports described an association between Dicer expression and increased metastasis in the context of a Dicer knockout model (32). Furthermore, changes in mature miRNA expression observed in hypoxic environments have been linked to decreased expression of Dicer. Taken together, changes in Dicer expression and processing are involved in promoting tumor growth and progression (19). Here, we have added to this understanding by demonstrating that changes to Dicer-regulated processing of a subset of miRNAs by PACT is another mechanism by which Dicer is involved in cancer pathogenesis. These results could have clinical implications. Specifically, the combination therapy of siPRKRA and oxaliplatin may benefit patients with MOC. Moreover, because some chemoresistant ovarian cancer cell lines have higher expression of PACT than do chemosensitive cell lines, siPRKRA-based therapy may benefit patients with other histological subtypes of ovarian cancer as well.
Another important finding of this study is that miR-515–3p can affect chemosensitivity in MOC cells. Recent studies have suggested that some miRNAs, such as miR-199b-5p, miR-506, and miR-484, are associated with chemoresistance of serous ovarian cancer (33–35). However, whether certain miRNAs are related to chemoresistance of MOC is not well understood. In addition, although miR-515–3p expression is reported to be increased in plasma during pregnancy, the role of this miRNA in cancer is unclear (36, 37). In the present study, we found that miR-515–3p also plays an important role in chemosensitivity of MOC.
In summary, this study demonstrated that PACT is frequently overexpressed in MOC cells and may be a therapeutic target for enhancing chemotherapeutic efficacy in this disease. In light of the results presented here, the combination of siPRKRA and oxaliplatin may be a candidate for further development in mucinous ovarian cancer.
Supplementary Material
Acknowledgments
T. Hisamatsu was supported by Uehara Memorial Foundation Research Fellowships for Research Abroad (201440169). S. Y. Wu was supported by Cancer Prevention & Research Institute of Texas (CPRIT) training grants (RP101502 and RP101489). This work was also supported, in part, by National Institutes of Health grants (P50CA217685, UH3TR000943, R35 CA209904, and CA016672), the Frank McGraw Memorial Chair in Cancer Research, the Meyer and Ida Gordon Foundation, and the American Cancer Society Research Professor Award. This research was also supported, in part, by the Blanton-Davis Ovarian Cancer Research Program. J. M. Hansen was supported by a NIH T32 Training Grant CA101642. This manuscript was reviewed and edited by the Department of Scientific Publications at MD Anderson.
Footnotes
Conflict of Interest Statement: The authors declare that they have no conflicts of interest.
References
- 1.Cho KR, Shih Ie M. Ovarian cancer. Annu Rev Pathol 2009; 4: 287–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hess V, A’hern R, Nasiri N, King DM, Blake PR, Barton DP, et al. Mucinous epithelial ovarian cancer: a separate entity requiring specific treatment. J Clin Oncol 2004; 22: 1040–4. [DOI] [PubMed] [Google Scholar]
- 3.Frumovitz M, Schmeler KM, Malpica A, Sood AK, Gershenson DM. Unmasking the complexities of mucinous ovarian carcinoma. Gynecol Oncol 2010; 117: 491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pectasides D, Fountzilas G, Aravantinos G, Kalofonos HP, Efstathiou E, Salamalekis E, et al. Advanced stage mucinous epithelial ovarian cancer: the Hellenic Cooperative Oncology Group experience. Gynecol Oncol 2005; 97: 436–41. [DOI] [PubMed] [Google Scholar]
- 5.Pisano C, Greggi S, Tambaro R, Losito S, Iodice F, Di Maio M, et al. Activity of chemotherapy in mucinous epithelial ovarian cancer: a retrospective study. Anticancer Res 2005; 25: 3501–5. [PubMed] [Google Scholar]
- 6.Bamias A, Psaltopoulou T, Sotiropoulou M, Haidopoulos D, Lianos E, Bournakis E, et al. Mucinous but not clear cell histology is associated with inferior survival in patients with advanced stage ovarian carcinoma treated with platinum-paclitaxel chemotherapy. Cancer 2010; 116: 1462–8. [DOI] [PubMed] [Google Scholar]
- 7.Alexandre J, Ray-Coquard I, Selle F, Floquet A, Cottu P, Weber B, et al. Mucinous advanced epithelial ovarian carcinoma: clinical presentation and sensitivity to platinum-paclitaxel-based chemotherapy, the GINECO experience. Ann Oncol 2010; 21: 2377–81. [DOI] [PubMed] [Google Scholar]
- 8.Naik JD, Seligmann J, Perren TJ. Mucinous tumours of the ovary. J Clin Pathol 2012; 65: 580–4. [DOI] [PubMed] [Google Scholar]
- 9.Heinzelmann-Schwarz VA, Gardiner-Garden M, Henshall SM, Scurry JP, Scolyer RA, Smith AN, et al. A distinct molecular profile associated with mucinous epithelial ovarian cancer. Br J Cancer 2006; 94: 904–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquez RT, Baggerly KA, Patterson AP, Liu J, Broaddus R, Frumovitz M, et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin Cancer Res 2005; 11: 6116–26. [DOI] [PubMed] [Google Scholar]
- 11.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319: 525–32. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, et al. Colorectal cancer. Lancet 2010; 375: 1030–47. [DOI] [PubMed] [Google Scholar]
- 13.Almendro V, Ametller E, Garcia-Recio S, Collazo O, Casas I, Auge JM, et al. The role of MMP7 and its cross-talk with the FAS/FASL system during the acquisition of chemoresistance to oxaliplatin. PLoS One 2009; 4: e4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 1998; 17: 4379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem 1999; 274: 15427–32. [DOI] [PubMed] [Google Scholar]
- 16.Sakayori M, Nozawa S, Udagawa Y, Chin K, Lee SG, Sakuma T, et al. Biological properties of two newly established cell lines (RMUG-S, RMUG-L) from a human ovarian mucinous cystadenocarcinoma. Hum Cell 1990; 3: 52–6. [PubMed] [Google Scholar]
- 17.Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res 2014; 74: 4922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JW, Han HD, Shahzad MM, Kim SW, Mangala LS, Nick AM, et al. EphA2 immunoconjugate as molecularly targeted chemotherapy for ovarian carcinoma. J Natl Cancer Inst 2009; 101: 1193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rupaimoole R, Wu SY, Pradeep S, Ivan C, Pecot CV, Gharpure KM, et al. Hypoxia-mediated downregulation of miRNA biogenesis promotes tumour progression. Nat Commun 2014; 5: 5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dweep H, Sticht C, Pandey P, Gretz N. miRWalk--database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform 2011; 44: 839–47. [DOI] [PubMed] [Google Scholar]
- 21.Landen CN Jr., Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res 2005; 65: 6910–8. [DOI] [PubMed] [Google Scholar]
- 22.Matsuo K, Nishimura M, Bottsford-Miller JN, Huang J, Komurov K, Armaiz-Pena GN, et al. Targeting SRC in mucinous ovarian carcinoma. Clin Cancer Res 2011; 17: 5367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiedemann RE, Zhu YX, Schmidt J, Yin H, Shi CX, Que Q, et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 2010; 115: 1594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO J 2006; 25: 522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimada M, Kigawa J, Ohishi Y, Yasuda M, Suzuki M, Hiura M, et al. Clinicopathological characteristics of mucinous adenocarcinoma of the ovary. Gynecol Oncol 2009; 113: 331–4. [DOI] [PubMed] [Google Scholar]
- 26.Lee HY, Zhou K, Smith AM, Noland CL, Doudna JA. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res 2013; 41: 6568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15: 509–24. [DOI] [PubMed] [Google Scholar]
- 28.Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med 2008; 359: 2641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dedes KJ, Natrajan R, Lambros MB, Geyer FC, Lopez-Garcia MA, Savage K, et al. Down-regulation of the miRNA master regulators Drosha and Dicer is associated with specific subgroups of breast cancer. Eur J Cancer 2011; 47: 138–50. [DOI] [PubMed] [Google Scholar]
- 30.Lin RJ, Lin YC, Chen J, Kuo HH, Chen YY, Diccianni MB, et al. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res 2010; 70: 7841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 2005; 96: 111–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 2009; 23: 2700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu MX, Siu MK, Liu SS, Yam JW, Ngan HY, Chan DW. Epigenetic silencing of microRNA-199b-5p is associated with acquired chemoresistance via activation of JAG1-Notch1 signaling in ovarian cancer. Oncotarget 2014; 5: 944–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu G, Yang D, Rupaimoole R, Pecot CV, Sun Y, Mangala LS, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst 2015; 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vecchione A, Belletti B, Lovat F, Volinia S, Chiappetta G, Giglio S, et al. A microRNA signature defines chemoresistance in ovarian cancer through modulation of angiogenesis. Proc Natl Acad Sci U S A 2013; 110: 9845–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miura K, Higashijima A, Mishima H, Miura S, Kitajima M, Kaneuchi M, et al. Pregnancy-associated microRNAs in plasma as potential molecular markers of ectopic pregnancy. Fertil Steril 2015; 103: 1202–8e1. [DOI] [PubMed] [Google Scholar]
- 37.Morisaki S, Miura K, Higashijima A, Abe S, Miura S, Hasegawa Y, et al. Effect of labor on plasma concentrations and postpartum clearance of cell-free, pregnancy-associated, placenta-specific microRNAs. Prenat Diagn 2015; 35: 44–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.