

Abstract
Small cell lung cancer (SCLC) has a poor prognosis. Focal Adhesion Kinase (FAK) is a non-receptor tyrosine kinase regulating cell proliferation, survival, migration, and invasion, which is overexpressed and/or activated in several cancers, including SCLC. We wanted to determine whether FAK contributes to SCLC aggressive behavior. We first evaluated the effect of FAK small-molecule inhibitor PF-573,228 in NCI-H82, NCI-H146, NCI-H196, and NCI-H446 SCLC cell lines. PF-573,228 (0.1 to 5μM) inhibited FAK activity by decreasing phospho-FAK (Tyr397), without modifying total FAK expression. PF-573,228 decreased proliferation, DNA synthesis, induced cell cycle arrest in G2/M phases, and increased apoptosis in all cell lines. PF-573,228 also decreased motility in adherent cell lines. To make sure that these effects were not off-target, we then used a genetic method to inhibit FAK in NCI-H82 and NCI-H446, namely stable transduction with FAK shRNA and/or FAK-related non-kinase (FRNK), a splice variant lacking the N-terminal and kinase domains. While FAK shRNA transduction decreased total and phospho-FAK (Tyr397) expression, it did not affect proliferation, DNA synthesis, or progression through cell cycle. However, restoration of FAK-targeting (FAT) domain (attached to focal adhesion complex where it inhibits pro-proliferative proteins such as Rac-1) by FRNK transduction inhibited proliferation, DNA synthesis, and induced apoptosis. Moreover, while FAK shRNA transduction increased active Rac1 level, FRNK re‐expression in cells previously transduced with FAK shRNA decreased it. Therefore, FAK appears important in SCLC biology and targeting its kinase domain may have a therapeutic potential, while targeting its FAT domain should be avoided to prevent Rac1-mediated pro-tumoral activity.
Keywords: FAK inhibition; FRNK; PF-573,228; Rac1; FAK shRNA; small cell lung cancer
INTRODUCTION
Lung cancer is the most common cancer and the leading cause of cancer-related death worldwide, with a median five-year overall survival of 15%(1). Non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) account for 85% and 15% of all lung cancers respectively(2). SCLC is a neuroendocrine tumor and clinically the most aggressive type of lung cancer, characterized by a tendency for early dissemination and a five-year overall survival of 5%(3,4). Unlike NSCLC, there is currently no targeted therapy validated in SCLC, which is the consequence of a poor understanding of its biology.
Focal Adhesion Kinase (FAK) is a non-receptor cytoplasmic tyrosine kinase and scaffold protein localized in focal adhesions, mediating and regulating signals initiated by integrins and G-protein-coupled-receptors. FAK plays a role in various cellular functions, including proliferation, survival, adhesion, migration, and invasion. The protein is composed of an N-terminal four-point-one, ezrin, radixin, moesin (FERM) domain, a central kinase domain, and a C-terminal domain(5). Both the N-terminal and C-terminal domains mediate FAK interactions with other proteins critical for its kinase domain’s activation and different cellular functions’ regulation. FAK is maintained in an inactive state by the binding of the FERM domain to the kinase domain, which blocks access to the key autophosphorylation site tyrosine 397 (Tyr397)(6). Engagement of integrins with the extracellular matrix or stimulation of G-protein-linked receptors following the binding of growth factors leads to signals that displace the FERM domain, resulting in Tyr397 autophosphorylation, changes in conformation of FAK and/or binding partners, binding and/or regulation of downstream effectors such as Src, MAPK, PI3K, paxillin, and Rac(7,8). The C-terminal domain provides binding sites to proteins such as p130Cas and VEGFR3. It includes the focal adhesion targeting (FAT) sequence responsible for FAK’s localization to focal adhesions, promoting its co-localization with integrins through interactions with integrin-associated proteins such as paxillin. The FAT domain also associates with several Rho GTPases, such as p190RhoGEF.
FAK is overexpressed and activated in several cancers and contributes to cancer progression and metastasis through its important role in cell proliferation, survival, adhesion, spreading, migration, and invasion(5,9,10). A role of FAK in evasion of anti-tumor immunity, angiogenesis, epithelial-mesenchymal transition, regulation of cancer stem cells, DNA damage repair (DDR), and therapy resistance, including radioresistance, has also been described(11–15). This role of FAK in cancer progression stimulated the development of various approaches to inhibit FAK. The first approaches used antisense FAK oligonucleotides, FAK siRNA or shRNA, and overexpression of FAK-Related Non-Kinase (FRNK), a naturally occurring splice variant of FAK which lacks the N-terminal and kinase domains and inhibits FAK phosphorylation in a dominant negative fashion(11,16). Inhibition of FAK through these approaches induced apoptosis and inhibited migration and angiogenesis. Since these approaches have limitations for clinical applications, small-molecule inhibitors targeting FAK kinase domain have then been developed(11,16). They decreased Tyr397 phosphorylation and induced anti-tumoral effects in various cancer types in preclinical studies(17–20). Moreover, some of them (e.g. PF-562,271, VS-6063, and VS-4718) showed promising clinical activities in early-phase clinical trials in patients with selected solid cancers, including NSCLC but not SCLC(5,16,21–23). More recently, small-molecule inhibitors targeting different FAK scaffolding protein-protein interactions have been developed, such as inhibitors of FAK and VEGFR-3 interactions, and shown to induce anti-tumoral effects in preclinical studies(24).
However, FAK has been poorly studied in SCLC. We previously showed that it was amplified and overexpressed in SCLC tumors(25,26), and activated in SCLC cell lines(25). Based on these observations, we hypothesized that FAK activation in SCLC contributes to its aggressive behaviour and that FAK may represent a therapeutic target for SCLC. In the present study, we therefore evaluated FAK activity in four SCLC cell lines and evaluated the effects of FAK inhibition by pharmacological (PF-573,228, PF-562,271, FAK Inhibitor 14) and genetic approaches (FAK shRNA and/or FRNK stable transduction) on cellular functions relevant for cancer progression.
MATERIALS AND METHODS
Cell culture
Four SCLC cell lines, NCI-H82, NCI-H146, NCI-446, and NCI-H196 (ATCC, Manassas, VA) were cultured in RPMI (1:1) containing heat-inactivated fetal calf serum (FCS) (10%), L-glutamine (2mM), penicillin (100U/ml), and streptomycin (100μg/ml) (Lonza, Verviers, Belgium) at 37°C, 5% CO2. Tetracycline-free FCS (PAN-Biotech GmbH, Aidenbach, Germany) was used for FRNK transduction experiments. HEK 293FT (ATCC) cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with heat-inactivated FCS (10%), L-glutamine (2mM), penicillin (100U/ml), streptomycin (100μg/ml) (Lonza), and neomycin (500μg/ml) (Sigma, St. Louis, MO) at 37°C, 5% CO2. The experiments were carried out on cells whose passage was between 10 and 25. For the detection of Mycoplasma in cell culture, the MycoAlert Mycoplasma Detection Kit (Lonza) was used.
Drugs
PF-573,228 (PF-228) (Santa Cruz Biotechnology, Dallas, TX) and PF-562,271 (PF-271) (Sigma) were suspended in DMSO (Sigma), while FAK Inhibitor 14 (Inh14) (Sigma) was suspended in water to get 10mM, 3mM, and 35mM stocks, respectively. These stocks were diluted in culture medium just before experiments to get required concentrations (0.5 to 10μM for PF-228, 0.05 to 3 μM for PF-271, and 3 to 5μM for Inh14). In FRNK-transduced cell lines, FRNK expression was induced by doxycycline (1 to 100ng/ml) (Sigma).
Lentivirus construction
Total RNA was purified using TRlzol® Reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. RNA was then reverse-transcribed to complementary DNA (cDNA) using the Revertaid H minus first strand cDNA synthesis kit with random hexamer primer (Thermo-Fisher Scientific, St. Leon-Rot, Germany). The gateway cloning system (BP and LR) Clonase® II enzyme mix (Invitrogen) was used to generate doxycycline-inducible FRNK expression lentivector. To this end, AttB1 and AttB2 sequences were respectively added to FAK’s N-terminal and C-terminal by polymerase chain reaction (PCR) using the KAPA Hifi PCR kit (KapaBiosystems, Wilmington, MA) according to manufacturer’s instructions. The PCR product was then purified using the Macherey-Nagel PCR purification kit (Macherey-Nagel, Düren, Germany). Purified AttB-FRNK cDNA was cloned into the pJet1.2 vector using the CloneJET PCR cloning kit (Thermo-Fisher Scientific) and transformed into chemically competent E. Coli-plasmid DNA. The plasmid was isolated using Qiagen plasmid DNA mini-prep kit (Qiagen, Hilden, Germany) and sequenced in Beckman Coulter Genomics facility (Takeley, Essex, UK). BP recombination of pJet AttB-FRNK with donor vector (Addgene Plasmid #29634) was used to generate FRNK-entry vector. This last one was recombined with pCLX-pTF-R1-DEST-R2-EBR65 lentiviral vector (Addgene Plasmid #45952) by LR recombination to generate doxycycline-inducible FRNK expression lentivector (pCLX-pTF-B1-FRNK-B2-EBR65).
Lentivirus production and cell lines’ transduction with FAK shRNA and/or FRNK
HEK293T packaging cell lines were transfected with FAK shRNA, no-target (NT) shRNA (Sigma), FRNK plasmid, or empty vector (pCLX) using ProFection® Mammalian Transfection System (Promega, Madison, WI) in tetracycline-free medium. Virus-containing supernatants were collected 48h and 72h post-transfection and immediately used to transduce NCI-H82 and NCI-H446 at a density of 1×106 cells/ml for 48h in presence of polybrene (8μg/ml) (Sigma)(27). Transduced cells were selected with puromycin (2μg/ml) (Invivogen, Toulouse, France) and/or blasticidin (10μg/ml) (Invivogen) for at least ten days. The selective pressure with antibiotics was removed 24h before each experiment.
Western blot (WB)
SCLC cell lines cultured in 12-well flat-bottom plates in 3ml culture medium were collected (1×106 for NCI- H82, NCI-H146, and NCI-446; 1.5×105 for NCI-H196) and lysed during 0.5h on ice in 150μl lysis buffer (62.5mM Tris-HCl [pH 6.8], 2% lauryl sulfate sodium, 10% glycerol 50mM DTT). Equal amounts of lysates were separated by 12% SDS-PAGE, electrotransferred onto a nitrocellulose membrane, and immunoprobed with antibodies against phospho-FAK (Tyr397) (1/1000, rabbit monoclonal; Cell Signaling Technology, Danvers, MA), total FAK (1/200, rabbit polyclonal; Santa Cruz Biotechnology); PARPp85 (1/1000, rabbit polyclonal; Promega, Madison, WI), phospho-Paxillin (Tyr-118) (1/1000, rabbit polyclonal; Cell Signaling Technology), total Paxillin (1/1000, monoclonal mouse; BD Biosciences, San Diego, CA), and β-Actin (1/1000, mouse monoclonal; Sigma). Secondary antibodies consisted of HRP-conjugated goat anti-rabbit IgG (1:2000; Cell signaling Technology) or HRP-conjugated sheep anti-mouse IgG (1:10000; Sigma). Immunoreactive bands were developed using chemiluminescence (Amersham ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK),detected with a Chemidoc XRS apparatus (Bio-Rad, Hercules, CA), and densitometrically quantified using Quantity One software (Bio-Rad) (results shown in Supplementary Fig.S1).
Cell proliferation
NCI-H82, NCI-H146, NCI-H196, and NCI-H446 were seeded in 96-well plates in antibiotic-free medium at 6×104, 6×104, 4.5×104, and 0.5×103 cells per well respectively. For pharmacological experiments, PF-228 (0.5μM to 10μM), PF-271 (0.05 to 3μM) or Inh14 (3 to 15μM) was added 24h after seeding at various concentrations and cells were cultured for up to four days. Every day, WST-1 reagent (Roche, Mannheim, Germany) was added to each well and incubated during 3h. Wells’ absorbance was measured spectrophotometrically at 450 nm with iMark™ microplate absorbance reader (Bio-Rad). Experiments were performed in triplicates.
Cell cycle analysis
Cells were seeded into 12-well plates at 0.5×106 cells per well. After 24h, PF-228 (0.5μM to 5μM) or DMSO was added to culture medium. After 24h-treatment, cells were pulsed with bromodeoxyuridine (BrdU) (10μM) for 0.5h, centrifugated, pelleted, and fixed with ice-cold ethanol (70%). DNA denaturation was performed with 2N HCl/ 0.5%Triton X-100 solution for 30 min., followed by quenching with HCl (sodium tetraborate solution 0.1M pH8.5). Cells were incubated with FITC-conjugated anti-BrdU antibody (1/20; BD Biosciences), RNaseA (10μg/ml; Sigma), and propidium iodide (PI) (BD Biosciences) (20μg/ml). Stained nuclei from 10,000 cells were subjected to flow cytometry. Data were collected on a fluorescence-activated cell sorting (FACS) Canto II flow cytometer (BD Biosciences). Cell cycle analysis was performed with BD FACS Diva software and FlowJo (FlowJo LLC, Ashland, OR). Experiments were performed in triplicates.
Apoptosis assay
NCI-H82 and NCI-H446 were seeded in 12-well plates at 0.5×106 cells per well. After 24h-treatment with PF-228 (1, 3, and 5μM) or DMSO, cells were stained with antibodies against cleaved Caspase-3 (1:50; Cell Signaling Technology) or, after 48h-treatment, with BrdU via TUNEL assay (APO-BrdU Kit; BD Biosciences) according to manufacturer’s instructions. Staining was quantified by FACSCanto II. Data acquisition and analysis were performed with FlowJo. Experiments were performed in triplicates.
Wound healing assay associated with time-lapse video recording of cell motility
NCI-H196 and NCI-H446 were grown to confluence in 12-well plates. Cell monolayers were wounded using a micropipette tip and floating cells were washed off with PBS (Lonza). After overnight incubation, PF-228 or DMSO was added to culture medium. Cell movements within wounded area were monitored for 12h starting from the time drug was added using a Zeiss Axiovert 200M microscope (Zeiss, Thornwood, NY) at x200 magnification. Images were captured every 15-minutes from five different fields randomly selected in each well. About 100 individual cells were analyzed using the Tracking Analysis software. Individual cells were tracked manually using MTrackJ, an Image J (NIH, Bethesda, MD) plugin. Only non-dividing cells were analyzed to exclusively assess motility. Track’s full length (LEN) was determined from the first point to the last point of the track and represented the distance covered by the cell during the experiment. Migration velocity was obtained by dividing LEN with experiment duration (12h). Experiments were performed in triplicates.
Matrigel invasion assay
Inserts separating the two chambers of 24-well invasion chambers (Corning, NY) were coated with Matrigel (0.3g/l) and incubated at 37°C for 2h. Lower chambers were filled with RPMI containing 10%-FBS. NCI-H196 and NCI-H446 were trypsinized, washed with PBS, suspended in 1%-FBS RPMI, plated in the upper chambers (25×103 and 10×104 cells per well for NCI-H196 NCI-H446, respectively), and incubated at 37°C for 3h. PF-228 (3 or 5μM) or DMSO was added in upper chambers 3h after seeding. After 12h incubation with the drug, cells remaining in the upper chamber were removed with cotton swabs and cells on the lower surface of the insert separating both chambers were fixed and stained with crystal violet. Image acquisition was performed with Axiovert 40 CFL Zeiss microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY). Images were obtained using ImageJ software (NIH, Bethesda, MD). Experiments were performed in duplicate.
Rac pull-down assay for activated GTPases
Active GTPases were pull-downed with Active Rac1 Pull-Down and Detection Kit (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s protocol. Cells were lysed with a lysis buffer containing a complete protease inhibitor cocktail. Equal amounts of proteins (800μg) were loaded into kit’s pull-down columns. Samples were incubated with Rac/Cdc42 PAK1 PAK-binding domain and rocked for 1h. Agarose beads were collected by centrifugation (30 sec. at 6,000 g and 4°C), washed, resuspended with 50μl 2xSDS sample buffer, and boiled for 5 min. Proteins were resolved by 12% SDS-PAGE and electrotransferred onto a membrane probed with mouse anti-Rac1 antibody (Thermo Fisher Scientific). GTP loading controls were incubated with GTP-γS (0.1mM) for 0.5h at 30°C.
Statistics
Statistical analyses were performed using the statistical analysis software JMP Pro version 12 (SAS Institute Inc., Cary, NC). Multiple linear regression analysis was used for WST-1 and Chi square test of independence for cell cycle and apoptosis data. Descriptive statistics were reported as mean ± standard deviation. Significance level was set at p<0.05 for each analysis.
RESULTS
Pharmacological inhibition of FAK has several anti-tumoral effects in SCLC
To investigate whether FAK is involved in the aggressive phenotype of SCLC, we tested the changes of cellular phenotype induced by the FAK small-molecule inhibitor PF-228 in four SCLC cell lines (two growing in suspension: NCI-H82 and NCI-H146, an adherent: NCI-H196, and a mixed-morphology: NCI-H446).
PF-228 inhibits FAK phosphorylation at Tyr397
Treatment with increasing concentrations of PF-228 (0.1 to 10μM) decreased FAK phosphorylation (Tyr397) in all tested cell lines dose-dependently, without modifying total FAK expression (Fig.1A). Phospho-FAK (Tyr397) decrease was less important in the adherent cell line NCI-H196, even at higher drug concentrations (0.5–10μM versus 0.1–3μM).
Figure 1: PF-573,228 (PF-228)’s effect on FAK expression/activity, cell proliferation, and cell cycle in SCLC cell lines.
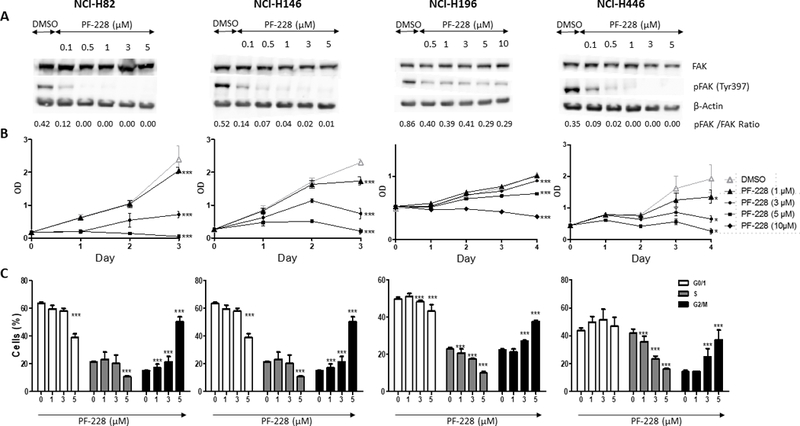
A. FAK expression and phosphorylation evaluation by Western blot (WB). Whole cell lysates from four SCLC cell lines treated with PF-228 or DMSO control for 90 min. were resolved by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blots were incubated with antibodies against total FAK (125 kD), phospho-FAK (Tyr397) (125 kD), and β-Actin (45 kD) for normalization. Dose-dependent inhibition of FAK phosphorylation (Tyr397) was observed by WB in cell lines treated with PF-228 as compared to those treated with DMSO, while total FAK expression was not modified. WB densitometric quantification is available in Supplementary Fig.S1. B. Cell proliferation evaluation by WST-1 assay. Four SCLC cell lines were cultured for three or four days in presence of PF-228 or DMSO. Dose-dependent inhibition of proliferation was observed by WST-1 assay in cells treated with PF-228 as compared to those treated with DMSO. Optical density (OD) in Y-axis reflects the proportion of metabolically active cells. Error bars represent mean +/− standard deviation (SD) (n=3). All the graphs represent one of three independent experiments with similar results. *** P ≤ 0.001. C. Cell cycle evaluation by flow cytometry. Four SCLC cell lines treated with PF-228 or DMSO for 24h were stained with anti-BrdU antibody and propidium iodide (PI), and the staining was quantified by fluorescence-activated cell sorting (FACS) analysis. Dose-dependent inhibition of DNA synthesis and induction of cell cycle arrest in G2/M phase was observed by flow cytometry in cell lines treated with PF-228 as compared to those treated with DMSO. Error bars represent mean +/− SD from three independent experiments. *** P ≤ 0.001.
PF-228 inhibits proliferation and progression through cell cycle in SCLC
Inhibition of FAK activity with 1 to 10μM PF-228 significantly decreased proliferation of the four SCLC lines dose-dependently (p<0.001 for all tested concentrations beside 1μM in NCI-H196) (Fig.1B). The effect was more pronounced in the suspension cell lines NCI-H82 and NCI-H146, which constitutively displayed higher proliferation rates. Cell cycle analysis showed that PF-228 inhibited progression through cell cycle by significantly reducing the S phase and inducing cell cycle arrest in G2/M phases in the four cell lines after 24h-treatment, dose-dependently (p<0.001 for all tested concentrations) (Fig.1C).
PF-228 induces apoptosis in SCLC
PF-228 at concentrations of 1 to 5 μM also significantly induced apoptosis in the four cell lines as demonstrated by a dose-dependent increase of PARP p85 expression by WB after 48h-treatment (Fig.2A). This was confirmed by flow cytometry in NCI-H82 and NCI-H446 cell lines, which showed a significant increase of BrdU-positive and activated Caspase 3-positive cells after 48h-treatment (p<0.001 for all tested concentrations except 1μM in NCI-H446 in the Caspase-3 assay) (Fig.2C).
Figure 2: PF-228’s effect on apoptosis in SCLC cell lines.

A. Apoptosis evaluation by PARP p85 WB. Whole cell lysates from four SCLC cell lines treated with PF-228 or DMSO for 48h were resolved by SDS-PAGE and blots were incubated with antibodies against PARP p85 (85 kD) and β-Actin (45 kD) for normalization. Dose-dependent increase of PARP p85 expression was observed by WB in cell lines treated with PF-228 as compared to those treated with DMSO. WB densitometric quantification is available in Supplementary Fig.S1. B and C. Apoptosis evaluation by flow cytometry. Two SCLC cell lines treated with PF-228 or DMSO for 24h (B) or 48h (C) were stained with anti-BrdU antibody and PI (B) or Pacific-Cleaved Caspase 3 and PI (C), and the staining was quantified by FACS. Cells were first gated in PI channel (PI-A and PI-H) to discard cells debris and doublets. Dose-dependent increase of apoptotic cells (BrdU-positive cells (B) or activated Caspase 3-positive cells (C)) was observed by flow cytometry in cell lines treated with PF-228 as compared to those treated with DMSO. Error bars represent mean +/− SD from three independent experiments. *** P ≤ 0.001.
PF-228 inhibits migration and invasion in SCLC
Wound healing assay and modified Boyden chambers allowed the investigation of cellular migration and invasion in the two SCLC cell lines with an adherent component. PF-228 at a concentration of 3μM tended to decrease migration velocity from 5 to 2.5μm/min in NCI-H196 (p=0.0561) and from 9 to 4μm/min in NCI-H446 (p=0.0916) (Fig.3A; Supplementary VideoS1). PF-228 also inhibited invasion after 12h-treatment at 3μM, with the number of invasive cells able to migrate to the lower side of the insert separating the two Boyden chambers decreasing from 150 to 50 per field (20x magnification) for NCI-H196 and from 45 to 5 per field for NCI-H446 (Fig.3B).
Figure 3: PF-228’s effect on migration and invasion in SCLC cell lines.
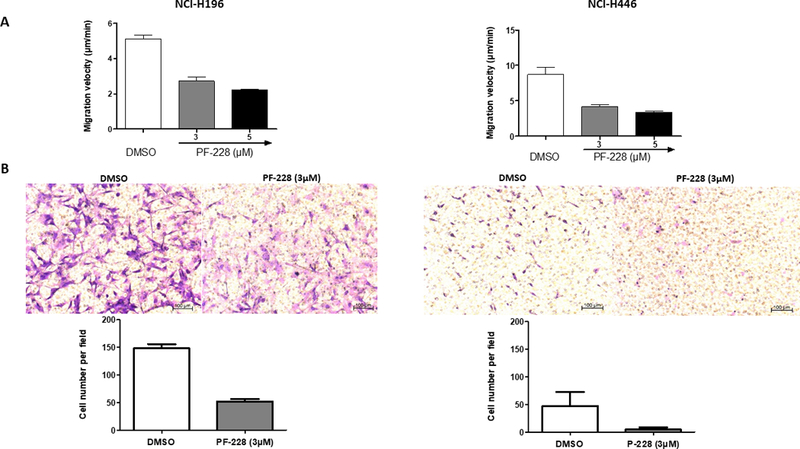
A. Migration evaluation by wound healing assay associated with time-lapse microscopy. Two adherent SCLC cell lines were grown to confluence, wounded, incubated overnight with culture medium, and then treated with PF-228 or DMSO for 12h. Cells were monitored during these 12h using a Zeiss Axiovert 200M microscope (Zeiss, Thornwood, NY). Images were captured every 15min. Velocity of cell migration was measured using ImageJ. Decreased motility was observed in cell lines treated with PF-228 as compared to those treated with DMSO. Error bars represent mean +/− SD from three independent experiments. B. Invasion evaluation by modified Boyden Chamber assay. Two adherent SCLC cell lines (one adherent and one with mixed-morphology) were seeded on the top of an insert pre-coated with matrigel and separating the two chambers of a transwell. Culture medium containing 1%-FBS was placed in the upper chamber and 10%-FBS in the lower chamber. After 12h-treatment with PF-228 or DMSO, cells that moved through the pores towards the bottom of the insert were stained with crystal violet, digitally pictured, and quantified by the free software ImageJ (NIH, Bethesda, MD, USA). Right panels are pictures of SCLC cell lines stained with crystal violet on the lower side of the insert which are representative of the numerous fields (x10 magnification) analyzed in two independent experiments performed in duplicate wells. Left panels represent quantification of the number of cells per field on the bottom of the insert. Decreased invasion was observed in cell lines treated with PF-228 for 12h as compared to those treated with DMSO. Error bars represent mean +/− SD from two independent experiments.
Inh14 and PF-271 also inhibit proliferation and induce apoptosis in SCLC
To verify that PF-228’s effects were related to FAK, we tested two other FAK inhibitors, Inh14 and PF-271, in NCI-H446. Similarly to PF-228, they both decreased FAK phosphorylation at Tyr397 and proliferation, and increased apoptosis as shown by increased PARP p85 expression (Supplementary Fig.S2).
Genetic inhibition of FAK leads to anti-tumoral effects only in presence of FRNK
Loss of total FAK following FAK shRNA transduction does not affect proliferation and progression through cell cycle in SCLC
Aiming to confirm the specificity of PF-228’s anti-tumoral effects in SCLC cell lines, experiments were carried out in NCI-H82 and NCI-H446 cells where FAK was inhibited by a genetic approach, namely the stable transduction of FAK shRNA (five clones). WB confirmed the almost complete loss of total FAK and phospho-FAK (Tyr397) expression following transduction with FAK shRNA as compared with NT shRNA (Fig.4A1). However, FAK shRNA transduction did not modify cell proliferation over three days and progression through cell cycle as evaluated by WST-1 and flow cytometry, respectively (Fig.4A2-4A3).
Figure 4: Effect of FAK shRNA and FAK-related non kinase (FRNK) transduction on FAK expression/ activity, cell proliferation, cell cycle, and apoptosis in SCLC cell lines.

A. Two SCLC cell lines were stably transduced with FAK shRNA or no-target (NT) shRNA as control, and submitted to puromycin selection for two weeks. 1. FAK expression and activity evaluation by WB. Whole cell lysates from these two cell lines were resolved with SDS-PAGE and blots were incubated with antibodies against total FAK (125 kD), phospho-FAK (Tyr397) (125 kD), and β-Actin (45 kD) for normalization. Significant decrease of FAK expression and phosphorylation (Tyr397) was observed by WB in SCLC cell lines transduced with FAK shRNA as compared to those transduced with NT shRNA. WB densitometric quantification is available in Supplementary Fig.S1. 2. Cell proliferation evaluation by WST-1 assay. SCLC cell lines were cultured for three days. No significant difference in cell proliferation was observed by WST-1 between cells transduced with FAK shRNA and those transduced with NT shRNA. Optical density (OD) in Y-axis reflects the proportion of metabolically active cells. Error bars represent mean +/− SD (n=5). All the graphs represent one of five independent experiments with similar results. *** P ≤ 0.001. 3. Cell cycle evaluation by flow cytometry. SCLC cell lines were stained with anti-BrdU antibody and PI, and the staining was quantified by FACS. No significant difference in cell cycle was observed between cells transduced with FAK shRNA and those transduced with NT shRNA transfection. Error bars represent mean +/− SD from three independent experiments. ***P ≤ 0.001. B. NCI-H446 cell lines were stably transduced with doxycycline-inducible FRNK-expression plasmid or empty vector (pCLX) as control, and submitted to blasticidin-selection for two weeks. Cells were treated with doxycycline for 48h before the experiments. 1. FAK and PARP p85 expression/activity evaluation by WB. Whole cell lysates from SCLC cell lines were resolved with SDS-PAGE and blots were incubated with antibodies against total FAK (125 kD), FRNK (41 kD), phospho-FAK (Tyr397) (125 kD), PARP p85 (85 kD), and β-Actin (45 kD) for normalization. Significant increase of FRNK expression was confirmed by WB in SCLC cell lines transduced with FRNK and treated with doxycycline as compared to those not treated with doxycycline or transduced with pCLX empty vector. Significant increase of PARP p85 expression, a marker of apoptosis, was also observed by WB in cells expressing FRNK, while total FAK and phospho-FAK (Tyr397) expression remained unchanged. 2. Cell proliferation evaluation by WST-1 assay. SCLC cell lines were cultured for five days. Inhibition of proliferation was observed by WST-1 in cell lines expressing FRNK as compared to those not expressing it. Optical density (OD) in Y-axis reflects the proportion of metabolically active cells. Error bars represent mean +/− SD (n=5). All the graphs represent one of five independent experiments with similar results. *** P ≤ 0.001 3. Cell cycle evaluation by flow cytometry. SCLC cell lines were stained with anti-BrdU antibody and PI, and the staining was quantified by FACS. DNA synthesis was decreased in cell lines expressing FRNK as compared to those not expressing it. Error bars represent +/− SD from five independent experiments. *** P ≤ 0.001.
Once again aiming to evaluate PF-228’s specificity, we treated SCLC cell lines transduced with FAK or NT shRNA with PF-228. As expected, we observed a significantly less important inhibition of proliferation in cell lines transduced with FAK shRNA. We also showed that PF-228 induced apoptosis, as demonstrated by increased PARP p85 expression, only in cells transduced with NT shRNA (Supplementary Fig.S3).
FRNK overexpression following transduction inhibits proliferation and survival in SCLC
In order to address the apparent discrepancy between PF-228’s effects and those of FAK shRNA transduction, we used a second genetic approach to inhibit FAK in NCI-H446, namely the stable transduction of a doxycycline-inducible FRNK vector. FRNK, which lacks FAK’s N-terminal and kinase domains, is a known physical repressor of FAK signaling(28). WB confirmed a significant and dose-dependent (doxycycline) increase of FRNK expression in NCI-H446 transduced with doxycycline-inducible FRNK vector and treated with doxycycline, as compared with those not treated with doxycycline or transduced with pCLX empty vector, while total FAK and phospho-FAK (Tyr397) expression remained unchanged (Fig.4B1).
Interestingly, FRNK overexpression significantly decreased cell proliferation over five days (p<0.001) and DNA synthesis after 48h-treatment with doxycycline (p<0.001) as evaluated by WST-1 and flow cytometry, respectively (Fig.4B2-4B3). FRNK overexpression also significantly induced apoptosis as shown by increased PARP p85 expression by WB after 48h-treatment with doxycycline (Fig.4B1). The effects on proliferation, DNA synthesis, and apoptosis were proportional to doxycycline concentrations and FRNK expression levels. As opposed to FAK inhibition by FAK shRNA transduction, FAK inhibition by FRNK overexpression induced anti-tumoral effects similar to FAK pharmacological inhibition.
FRNK overexpression following transduction in SCLC cell lines previously transduced with FAK shRNA inhibits proliferation and survival
Facing different results with the two genetic approaches used to inhibit FAK, we wondered whether the loss of FRNK was responsible for the absence of effect of FAK shRNA transduction on survival. To test this, we overexpressed FRNK in NCI-H446 cells stably transduced with FAK shRNA by transducing them with doxycycline-inducible FRNK vector. FRNK overexpression did not modify total FAK and phospho-FAK (Tyr397) expression, which were both downregulated by FAK shRNA transduction (Fig.5A1). However, in these double-transduced cells, with FAK shRNA and then FRNK, we observed an inhibition of cell growth over four days as evaluated by WST-1 (p<0.001) (Fig. 5A2), and an induction of apoptosis as shown by increased PARP p85 expression by WB (Fig.5A1). The effects on proliferation and apoptosis were both proportional to doxycycline concentrations and FRNK expression levels.
Figure 5: Effect of FRNK transduction on FAK expression/activity, proliferation, apoptosis, and Rac1 expression in SCLC cell lines transduced with FAK shRNA.

NCI-H446 cell lines were first stably transduced with FAK shRNA or no-target (NT) shRNA and then stably transduced with doxycycline-inducible FRNK-expression plasmid or pCLX empty vector as control. After transduction, they were submitted to puromycin and blasticidin-selection for two weeks. Finally, they were treated with doxycycline for 48h before the experiments.A. FAK and PARP p85 expression/activity evaluation by WB. Whole cell lysates from SCLC cell lines were resolved with SDS-PAGE and blots were incubated with antibodies against total FAK (125 kD), FRNK (41 kD), phospho-FAK (Tyr397) (125 kD), PARP p85 (85 kD), and β-Actin (45 kD) for normalization. Decreased FAK and phospho-FAK (Tyr397) expression was observed by WB in cell lines double-transduced with FAK shRNA and FRNK as compared to those transduced with NT shRNA and PCLX. Increased FRNK expression was observed in cell lines double-transduced with FAK shRNA and FRNK when treated with doxycycline. Increased PARP p85 expression, a marker of apoptosis, was observed in cells expressing FRNK. WB densitometric quantification is available in Supplementary Fig.S1. B. Cell proliferation evaluation by WST-1 assay. SCLC cell lines were cultured for four days. Inhibition of proliferation was observed in cell lines double-transduced with FAK shRNA and FRNK and expressing FRNK after treatment with doxycycline. Optical density (OD) in Y-axis reflects the proportion of metabolically active cells. Error bars represent mean +/− standard deviation (SD) (n=5). All the graphs represent one of five independent experiments with similar results. *** P ≤ 0.001. C. Effects of FRNK on Rac1 activity. NCI-H446 SCLC cell lines were double-transduced with FAK shRNA and a doxycycline-inducible FRNK vector or with no-target (NT) shRNA and pCLX empty vector as control. 1. FAK and FRNK expression evaluation by WB. Whole cell lysates from SCLC cell lines were resolved with SDS-PAGE and blots were incubated with antibodies against total FAK (125 kD), FRNK (41 kD), and β-Actin (45 kD) for normalization. Decreased FAK expression was observed by WB in cell lines double-transduced with FAK shRNA and FRNK as compared to those transduced with NT shRNA and PCLX. Increased FRNK expression was observed in cell lines double-transduced with FAK shRNA and FRNK when treated with doxycycline. WB densitometric quantification is available in Supplementary Fig.S1. 2. Rac1 activation evaluation by Rac pull down assay for activated GTPases. Whole cell lysates from SCLC cell lines were enriched in activated GTPases using a GST-PAK affinity assay (GTPases pull down assay). Enriched eluates were resolved with SDS-PAGE and blots were incubated with antibodies against Rac1 (21 kD) and β-Actin (45 kD) for normalization. In control cells double-transduced with NT shRNA and PCLX, WB revealed low activated Rac1 expression at baseline, while treating them with GTP significantly increased its expression. In cells double-transduced with FAK shRNA and FRNK, activated Rac1 expression was present in the absence of doxycycline, while doxycycline-induced FRNK expression significantly decreased its expression. WB densitometric quantification is available in Supplementary Fig.S1.
FRNK keeps Rac1 GTPase inactivated in SCLC
Based on a previous report in endothelial cells which also showed that different methods of FAK inhibition result in different functional outcomes and that this occurs through the regulation of Rac activation(29), we evaluated activated Rac1 level in NCI-H446 cell lines double-transduced with FAK shRNA and doxycycline-inducible FRNK vector using Rac pull-down assay for activated GTPases (Fig.5B1). In NT shRNA and pCLX double-transduced cells used as control, with no FRNK expression, activated Rac1 level was low at baseline, while treating them with GTP significantly increased it as expected (Fig.5B2). In cells transduced with FAK shRNA and doxycycline-inducible FRNK vector, activated Rac1 was present at baseline in cells without FRNK expression, while FRNK overexpression significantly decreased activated Rac1 level (Fig.5B2). These results indicate that the loss of FRNK following the physical loss of total FAK increases activated Rac1 level. As Rac1 is a pro-proliferative protein(30,31), we have an explanation to why SCLC cell lines transduced with FAK shRNA remain proliferative (Fig.4A2): the pro-proliferative effect of Rac1 activation counterbalances the anti-proliferative effect induced by the absence of FAK phosphorylation at Tyr397.
Since FAK is known to phosphorylate Paxillin and phospho-Paxillin to activate Rac1 via the adaptor protein CrkII(32), we evaluated phospho-Paxillin (Tyr118) expression in NCI-H446 by WB and immunofluorescence. The two methods showed that FAK inhibition, either with PF-228 or double-transduction with FAK shRNA and FRNK, did not modify phospho-Paxillin (Tyr118) expression, which was however low even at baseline (Suppl. Fig.S4).
DISCUSSION
In this study, we evaluated whether FAK, known to be overexpressed in SCLC tumors(25,26) and activated in SCLC cell lines(25), contributes to the aggressive behavior of SCLC and is a potential therapeutic target in SCLC. In a previous study, we showed that FAK was constitutively activated in SCLC cell lines, with high levels of FAK phosphorylation at Tyr397, and that the pharmacological inhibition of FAK with PF-228 decreased cell adhesion and modified cell phenotype(25). Here, we explored the role of FAK in cellular functions relevant for cancer progression and showed for the first time that inhibition of FAK activity with PF-228 decreased proliferation, induced cell cycle arrest, increased apoptosis, and decreased motility and invasion. All these important anti-tumoral effects of PF-228 suggest that FAK is important in SCLC biology and may have a therapeutic potential. The inhibitory effect of PF-228 on SCLC motility and invasion was similar to the results reported in other cancer types or in normal cells(20). Of note, we tested migration and invasion only in the two adherent cell lines as these functions are difficult to evaluate in suspension cell lines. Interestingly, we observed an effect of PF-228 on proliferation and apoptosis already at low drug concentrations. The first study which tested PF-228 showed an effect on migration and focal adhesion turnover but failed to demonstrate an effect on proliferation and survival in prostate cancer cell line PC3, fibroblastic cell line REF52, and canine kidney cell line MDCK(20). Another study showed that PF-228 inhibited proliferation and induced apoptosis in endometrial cancer cell lines but, as opposed to our study, much higher concentrations of PF-228 were used (50μM). The fact that low concentrations of PF-228 inhibited proliferation and survival in SCLC cell lines suggest the specificity of the drug and the importance of FAK in pro-proliferative and pro-survival signaling pathways. Other FAK inhibitors induced inhibition of proliferation or survival in vitro in various cell types but were not specific of FAK (e.g.: TAE-226 inhibits FAK, PYK2, and IGF-1R)(33–35). In this study, we additionally tested two other FAK inhibitors, Inh14 and PF-271(18,36), and observed that they also inhibited proliferation and induced apoptosis in SCLC. This strengthened us in the idea that PF-228’s effects were related to FAK, even though Inh14 and PF-271 are both less FAK-specific than PF-228, known to have the highest FAK-specificity among FAK inhibitors.
In order to better address the specificity of PF-228’s effects on proliferation and survival in SCLC cell lines, we evaluated the consequence of FAK inhibition by a genetic method, namely FAK shRNA stable transduction. Surprisingly, the physical loss of FAK did not impact on proliferation and cell cycle. But interestingly, treatment of FAK shRNA-transduced cells with PF-228 did not induce apoptosis and had only a limited effect on proliferation. The Absent/limited effect of PF-228 in cells with no/low FAK expression also suggests the drug’s specificity.
To address the apparent discrepancy between PF-228’s effects and those of FAK shRNA transduction, we used a second genetic approach to inhibit FAK, namely the stable transduction of doxycycline-inducible FRNK vector leading to the overexpression of FRNK, a truncated protein including only FAK’s carboxy-terminal non-catalytic domain and a well-known physical repressor of FAK signaling(28,37). We observed that, as with PF-228, FRNK overexpression inhibited cell proliferation and DNA synthesis and increased apoptosis in SCLC cell lines. At this step, we hypothesized that the opposite results obtained with the two genetic approaches we used to inhibit FAK were related to FRNK, absent in cells transduced with FAK shRNA while present in those transduced with doxycycline-inducible FRNK vector and expressing FRNK. Our hypothesis was confirmed in double-transduced SCLC cell lines, first with FAK shRNA and then with FRNK, which revealed anti-tumoral effects in cells overexpressing FRNK. In a similar way, it has previously been reported that different methods of FAK inhibition result in different functional outcomes in endothelial cells: approaches inhibiting FAK phosphorylation at Tyr397 (such as FAK small-molecule inhibitors or FRNK transduction) inhibited proliferation and migration, while those abolishing FAK expression (such as FAK shRNA or siRNA) did not impact on these cellular processes(38). Also supporting the importance of FRNK in the regulation of proliferation, a previous report showed that expressing FRNK with a C1034S mutation disrupted focal adhesion binding but had no effect on proliferation(39).
In endothelial cells, FAK has been proposed as a phospho-regulated repressor of the activation of Rac(38), a pro-proliferative GTPase present in focal adhesions(31,40). This was based on the observation that FRNK expression, FAK Tyr397F mutation (simple substitution of Tyr397 with a non-phosphorylated residue), or treatment with a FAK kinase inhibitor decreased Rac activation induced by complete growth medium, while the physical loss of FAK following FAK shRNA transduction did not affect it(38). Similarly, in cells double-transduced with FAK shRNA and doxycycline-inducible FRNK vector, we found high level of activated Rac1 in cells overexpressing FRNK, while it was low in the absence of FRNK expression. Based on these results, we propose the following model in SCLC, schematized in Fig.6: 1/ In normal conditions (absence of FAK inhibition) in SCLC, FAK constitutive activation results in FAK phosphorylation at Tyr397, leading to the activation of downstream phosphorylation-dependent signaling and to changes in the conformation of FAK and/or its binding partners, which allow Rac1 activation in the focal adhesion complex. 2/ PF-228 and FRNK overexpression both inhibit FAK phosphorylation at Tyr397, leading to the inhibition of downstream signaling and the absence of change in conformation of FAK and/or its binding partners, which prevents Rac1 activation. This results in anti-tumoral effects, as observed in our experiments. 3/ In contrast, the physical loss of FAK after FAK shRNA transduction induces an inhibition of FAK phosphorylation-dependent signals but allows the activation of Rac1 because of the absence of repression by FAK. This last event results in pro-tumoral effects counterbalancing the anti-tumoral effects of FAK phosphorylation inhibition, explaining why FAK shRNA transduction did not affect proliferation and survival in the SCLC cell lines we tested.
Figure 6: A model of FAK as a regulator of Rac1 activation in SCLC.

A. “Normal” conditions in SCLC with FAK constitutively activated. FAK phosphorylation at Tyr397 leads to the activation of downstream phosphorylation-dependent signaling and to changes in the conformation of FAK and/or its binding partners, which allow Rac1 activation in the focal adhesion complex. These two events result in pro-tumoral effects. B. FAK inhibition by PF-228 and FRNK overexpression. These two methods inhibit FAK phosphorylation at Tyr397, leading to the inhibition of downstream signaling and the absence of change in conformation of FAK and/or its binding partners, which prevents Rac1 activation. These two events result in anti-tumoral effects. C. FAK inhibition by FAK shRNA transduction. The physical loss of FAK induces an inhibition of FAK phosphorylation-dependent signals but allows the activation of Rac1 because of the absence of repression by FAK. The anti-tumoral effects related to the absence of FAK are counterbalanced by the pro-tumoral effects related to Rac1 activation.
Altogether our results suggest that, in order to induce proliferation and survival in SCLC cell lines, the physical presence of FAK is not required because the physical loss of FAK release the repressive signal on Rac and allows its activation, which induces proliferation and survival. In contrast, when the FRNK region of the FAT domain is present, FAK phosphorylation at Tyr397 seems necessary to induce proliferation and survival. Importantly, in a natural setting, there is no FAK shRNA; normal or cancer cells express total FAK (including FRNK) and FAK phosphorylation at Tyr397 is required for their proliferation and survival. Therefore, we can conclude that FAK plays an important role in various pro-tumoral properties of SCLC through its kinase domain and that inhibiting FAK phosphorylation at Tyr397 may have a therapeutic potential. Even though the discoveries made with FAK shRNA correspond to an artificial setting, they suggest that FAK small-molecule inhibitors should target the kinase domain but not FAK’s regions which play a repressive role on pro-proliferative proteins, such as FRNK on Rac. Recently, small-molecule inhibitors targeting different FAK scaffolding protein-protein interactions have been developed and shown to induce anti-tumoral effects in preclinical studies(16), but further development of such inhibitors should take into account the complexity of FAK in order to be successful.
Of note, we did not find any phospho-Paxillin (Tyr118) expression modification in SCLC cells where FAK was inhibited with PF-228 or FAK shRNA +/− FRNK transduction. Since FAK is known to phosphorylate Paxillin and phospho-Paxillin to activate Rac1 via the adaptor protein CrkII(32), we expected to find an inhibition of Paxillin phosphorylation following FAK inhibition. However, similarly to our observation, previous studies also reported that FAK did not affect Paxillin tyrosine phosphorylation level(41,42). Further investigations are required to better understand these observations.
To be mentioned, while PF-228 induced cell cycle arrest in G2 and S phases, FRNK transduction induced cell cycle arrest in S phase only, which was however sufficient to impact on proliferation and apoptosis. We assume that this discrepancy is related to an off-target effect of PF-228, which often happens with small-molecule inhibitors, even the specific ones. Nevertheless, this does not change the conclusion that FAK plays a role in SCLC proliferation and survival since we showed that PF-228 and FRNK transduction both inhibited cell proliferation and induced apoptosis in SCLC cell lines.
More in depth investigation of FAK’s role in cell cycle, apoptosis, and specifically DDR in SCLC may be relevant. Indeed, a recent study showed that FAK regulates DDR and that FAK inhibition by PF-271, RNA interference, or CRISPR/CAS9 gene editing induces persistent DNA damage and radiosensitizes KRAS-mutated NSCLC cell lines and xenografts(15). In parallel, another study showed that nuclear FAK stimulates gene transcription favoring DDR and that FAK ablation by CRISP/Cas9 editing induces DNA damage and increased radiosensitivity in NSCLC cells(13). Similarly, it has recently been demonstrated that FAK overexpression is a radioresistance biomarker in locally-advanced HPV-negative head and neck squamous cell carcinoma (HNSCC), also a smoking-related malignancy, and that its inhibition with PF-271 radiosensitized HNSCC cell lines, with increased G2/M arrest and DNA damage(14). In this context, combining FAK small-molecule inhibitor with radiotherapy in SCLC certainly deserves further investigations.
In summary, experiments using PF-228, a FAK small-molecule inhibitor, showed that inhibition of FAK phosphorylation at Tyr397 decreased proliferation, induced cell cycle arrest, increased apoptosis, and decreased motility and invasion in SCLC cell lines. FAK inhibition by FRNK overexpression after transduction of a doxycycline-inducible FRNK vector also induced inhibition of proliferation and survival, suggesting the specificity of PF-228. In contrast, FAK inhibition by FAK shRNA transduction did not affect proliferation and survival, probably because the physical absence of FRNK released a repressive signal on Rac, a pro-proliferative protein. Taken collectively, these data demonstrate that FAK is important in SCLC biology and that targeting its kinase domain may have a therapeutic potential in SCLC, while targeting its FAT domain should be avoided or done carefully to prevent pro-proliferative proteins from counter-balancing the anti-tumoral effects of FAK inhibition. Further studies in SCLC xenograft models are required to better understand the complexity of FAK in SCLC. Ultimately, this may lead to the evaluation of FAK inhibitors in clinical trials of patients suffering from SCLC, a deadly disease which still lacks efficient targeted therapies.
Supplementary Material
ACKNOWLEDGEMENTS
Dr. F. Aboubakar Nana was supported by Fonds Spécial de Recherche (FSR) (Communauté Française de Belgique), Télévie (Fonds National de la Recherche Scientifique (FNRS)) (7.4624.15), and Fondation Willy and Marcy De Vooght, Belgium. Dr. P. P. Massion’s effort was supported by the Veterans Administration I01CX001425, USA. Dr. C. Pilette was supported by FNRS (1.R016.18) and WELBIO (CR-2012S-05). Dr. Ocak was supported by grants from Fondation Mont-Godinne (FMG-2011-BR-02, FMG-2013-BR-02, FMG-2014-BR-01, FMG-2015-BR-02, FMG-2016-BR-02, and FMG-2017-BR-04), Télévie (FNRS) (7.4588.10F and 7.4624.15), FSR, and Secteurs des Sciences de la Santé, Université catholique de Louvain (UCL), Belgium.
We thank the Pole of Pediatry of Institut de Recherche Expérimentale et Clinique (IREC) of UCL for sharing their flow cytometry facility, particularly Dr. Catherine Lombard for her assistance. We thank the Pole of Microbiology of IREC for sharing their molecular biology facility. Finally, we thank L. Desmet (Plateforme Technologique de Support en Méthodologie et Calcul Statistique, UCL) for the statistical analyses.
Financial support
Dr. F. Aboubakar Nana was supported by Télévie (Fonds National de la Recherche Scientifique (FNRS)) (7.4624.15), Fonds Spécial de Recherche (FSR) (Communauté Française de Belgique), and Fondation Willy and Marcy De Vooght, Belgium. Dr. P. P. Massion’s effort was supported by the Veterans Administration I01CX001425, USA. Dr. C. Pilette was supported by FNRS (1.R016.18) and by WELBIO (CR-2012S-05). Dr. Ocak was supported by grants from Fondation Mont-Godinne (FMG-2011-BR-02, FMG-2013-BR-02, FMG-2014-BR-01, FMG-2015-BR-02, FMG-2016-BR-02, and FMG-2017-BR-04), Télévie (FNRS) (7.4588.10F and 7.4624.15), FSR, and Secteurs des Sciences de la Santé, Université catholique de Louvain (UCL), Belgium.
Footnotes
Conflicts of interest
The authors declare no potential conflict of interest.
REFERENCES
- 1.GLOBOCAN 2012 (IARC) SoCS. GLOBOCAN 2012 (IARC), Section of Cancer Surveillance (31/7/2014).
- 2.Houston KA, Henley SJ, Li J, White MC, Richards TB. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004–2009. Lung cancer 2014;86(1):22–8 doi 10.1016/j.lungcan.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, et al. Changing Epidemiology of Small-Cell Lung Cancer in the United States Over the Last 30 Years: Analysis of the Surveillance, Epidemiologic, and End Results Database. Journal of Clinical Oncology 2006;24(28):4539–44 doi 10.1200/jco.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- 4.Behera M, Ragin C, Kim S, Pillai RN, Chen Z, Steuer CE, et al. Trends, predictors, and impact of systemic chemotherapy in small cell lung cancer patients between 1985 and 2005. Cancer 2015. doi 10.1002/cncr.29674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer 2014;14(9):598–610 doi 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene 0000;23(48):7906–9. [DOI] [PubMed] [Google Scholar]
- 7.Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mechanisms of CAS substrate domain tyrosine phosphorylation by FAK and Src. Molecular and cellular biology 2001;21(22):7641–52 doi 10.1128/mcb.21.22.7641–7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siesser PM, Hanks SK. The signaling and biological implications of FAK overexpression in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2006;12(11 Pt 1):3233–7 doi 10.1158/1078–0432.CCR-06–0456. [DOI] [PubMed] [Google Scholar]
- 9.Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Frontiers in bioscience : a journal and virtual library 2003;8:d982–96. [DOI] [PubMed] [Google Scholar]
- 10.Parsons JT. Focal adhesion kinase: the first ten years. Journal of cell science 2003;116(Pt 8):1409–16. [DOI] [PubMed] [Google Scholar]
- 11.Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacology & therapeutics 2015;146:132–49 doi 10.1016/j.pharmthera.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Eke I, Cordes N. Focal adhesion signaling and therapy resistance in cancer. Seminars in cancer biology 2015;31:65–75 doi 10.1016/j.semcancer.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Constanzo JD, Tang K-j, Rindhe S, Melegari M, Liu H, Tang X, et al. PIAS1-FAK Interaction Promotes the Survival and Progression of Non-Small Cell Lung Cancer. Neoplasia 2016;18(5):282–93 doi 10.1016/j.neo.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skinner HD, Giri U, Yang L, Woo SH, Story MD, Pickering CR, et al. Proteomic Profiling Identifies PTK2/FAK as a Driver of Radioresistance in HPV-negative Head and Neck Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(18):4643–50 doi 10.1158/1078–0432.ccr-15–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang K-J, Constanzo JD, Venkateswaran N, Melegari M, Ilcheva M, Morales JC, et al. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clinical Cancer Research 2016;22(23):5851–63 doi 10.1158/1078–0432.ccr-15–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Frontiers in bioscience (Landmark edition) 2014;19:687–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell cycle (Georgetown, Tex) 2009;8(15):2435–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, et al. Antitumor Activity and Pharmacology of a Selective Focal Adhesion Kinase Inhibitor, PF-562,271. Cancer research 2008;68(6):1935–44 doi 10.1158/0008–5472.can-07–5155. [DOI] [PubMed] [Google Scholar]
- 19.Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Molecular carcinogenesis 2007;46(6):488–96 doi 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 20.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, et al. Cellular Characterization of a Novel Focal Adhesion Kinase Inhibitor. Journal of Biological Chemistry 2007;282(20):14845–52 doi 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 21.Shanthi E, Krishna MH, Arunesh GM, Venkateswara Reddy K, Sooriya Kumar J, Viswanadhan VN. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: a patent review. Expert opinion on therapeutic patents 2014;24(10):1077–100 doi 10.1517/13543776.2014.948845. [DOI] [PubMed] [Google Scholar]
- 22.Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30(13):1527–33 doi 10.1200/jco.2011.38.9346. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu T, Fukuoka K, Takeda M, Iwasa T, Yoshida T, Horobin J, et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer chemotherapy and pharmacology 2016;77:997–1003 doi 10.1007/s00280–016-3010–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA, Cance WG. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. Journal of medicinal chemistry 2009;52(15):4716–24 doi 10.1021/jm900159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ocak S, Yamashita H, Udyavar AR, Miller AN, Gonzalez AL, Zou Y, et al. DNA copy number aberrations in small-cell lung cancer reveal activation of the focal adhesion pathway. Oncogene 2010;29(48):6331–42 doi 10.1038/onc.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ocak S, Chen H, Callison C, Gonzalez AL, Massion PP. Expression of focal adhesion kinase in small-cell lung carcinoma. Cancer 2012;118(5):1293–301 doi 10.1002/cncr.26382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protocols 2006;1(1):241–5. [DOI] [PubMed] [Google Scholar]
- 28.Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature 1996;380(6574):538–40 doi 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- 29.Bryant PW, Zheng Q, Pumiglia KM. Focal adhesion kinase is a phospho-regulated repressor of Rac and proliferation in human endothelial cells. Biology Open 2012. doi 10.1242/bio.20121008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gastonguay A,J Berg T, Hauser A, Schuld N,L Lorimer E, L Williams C The role of Rac1 in the regulation of NF-kB activity, cell proliferation, and cell migration in non-small cell lung carcinoma. 2012. 647–56 p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014;5:e29019 doi 10.4161/sgtp.29019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valles AM, Beuvin M, Boyer B. Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. The Journal of biological chemistry 2004;279(43):44490–6 doi 10.1074/jbc.M405144200. [DOI] [PubMed] [Google Scholar]
- 33.Kurio N, Shimo T, Fukazawa T, Takaoka M, Okui T, Hassan NM, et al. Anti-tumor effect in human breast cancer by TAE226, a dual inhibitor for FAK and IGF-IR in vitro and in vivo. Experimental cell research 2011;317(8):1134–46 doi 10.1016/j.yexcr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 34.Otani H, Yamamoto H, Takaoka M, Sakaguchi M, Soh J, Jida M, et al. TAE226, a Bis-Anilino Pyrimidine Compound, Inhibits the EGFR-Mutant Kinase Including T790M Mutant to Show Anti-Tumor Effect on EGFR-Mutant Non-Small Cell Lung Cancer Cells. PLoS One 2015;10(6):e0129838 doi 10.1371/journal.pone.0129838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang ZG, Fukazawa T, Nishikawa T, Watanabe N, Sakurama K, Motoki T, et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncology reports 2008;20(6):1473–7. [PubMed] [Google Scholar]
- 36.Golubovskaya V, Curtin L, Groman A, Sexton S, Cance WG. In vivo toxicity, metabolism and pharmacokinetic properties of FAK inhibitor 14 or Y15 (1, 2, 4, 5-benzenetetramine tetrahydrochloride). Archives of toxicology 2015;89(7):1095–101 doi 10.1007/s00204–014-1290-y. [DOI] [PubMed] [Google Scholar]
- 37.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol 1999;71(3–4):435–78. [DOI] [PubMed] [Google Scholar]
- 38.Bryant PW, Zheng Q, Pumiglia KM. Focal adhesion kinase is a phospho-regulated repressor of Rac and proliferation in human endothelial cells. Biology Open 2012;1(8):723–30 doi 10.1242/bio.20121008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryant P, Zheng Q, Pumiglia K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Molecular and cellular biology 2006;26(11):4201–13 doi 10.1128/mcb.01612–05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell communication and signaling : CCS 2010;8:23 doi 10.1186/1478–811x-8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moissoglu K, Sachdev S, Gelman IH. Enhanced v-Src-induced oncogenic transformation in the absence of focal adhesion kinase is mediated by phosphatidylinositol 3-kinase. Biochemical and biophysical research communications 2005;330(3):673–84 doi 10.1016/j.bbrc.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 42.Roy S, Ruest PJ, Hanks SK. FAK regulates tyrosine phosphorylation of CAS, paxillin, and PYK2 in cells expressing v-Src, but is not a critical determinant of v-Src transformation. Journal of cellular biochemistry 2002;84(2):377–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.